
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAqueous up-conversion organic phosphorescence and tunable dual emission in a single-molecular emitter†
Yang
Li
 ab,
Zizhao
Huang
a,
Aixing
Shao
a,
Zhiqin
Wu
a,
Zhenyi
He
a,
He
Tian
a and
Xiang
Ma
*a
ab,
Zizhao
Huang
a,
Aixing
Shao
a,
Zhiqin
Wu
a,
Zhenyi
He
a,
He
Tian
a and
Xiang
Ma
*a
aKey Laboratory for Advanced Materials and Joint International Research Laboratory of Precision Chemistry and Molecular Engineering, Frontiers Science Center for Materiobiology and Dynamic Chemistry, School of Chemistry and Molecular Engineering, East China University of Science and Technology, Shanghai 200237, China. E-mail: maxiang@ecust.edu.cn
bSchool of Chemical Engineering & Pharmacy, Pharmaceutical Research Institute, Hubei Key Laboratory of Novel Reactor and Green Chemical Technology, Wuhan Institute of Technology, Wuhan 430205, China
First published on 3rd March 2025
Abstract
Materials exhibiting up-conversion room-temperature phosphorescence (RTP) with multi-emissive properties in aqueous solutions hold significant potential for optical imaging and sensing applications. However, achieving such photophysical materials within a molecular emitter remains a formidable challenge. Herein, we report a series of single-molecule chromophores demonstrating aqueous tunable up-conversion RTP and fluorescence dual emission. The RTP and fluorescence emission could be finely adjusted by manipulating the excitation wavelength within the visible and near-infrared range, enabling dynamic color modulation across the entire visible spectrum from blue to orange-red. Furthermore, we utilized the up-conversion RTP capability of a single-molecular emitter to achieve two-photon and time-resolved imaging. More importantly, through ratiometric regulation of phosphorescence by temperature combined with stable fluorescence as an internal reference, the RTP molecule enabled reliable temperature sensing in living cells. This study unveils a highly efficient strategy for fabricating intelligent organic RTP materials and sensors featuring dynamically controlled multi-emission.
Introduction
Organic room temperature phosphorescent (RTP) materials have emerged as a prominent area of research in materials science chemistry over the past decade owing to their immense potential for applications in biological imaging, sensing, electrochemical devices, and anti-counterfeiting.1–5 In particular, aqueous-phase organic RTP materials have garnered significant interest owing to their low toxicity, extended lifetime, and capability to distinguish from background fluorescence in bio-species.6–10 These characteristics hold great promise for highly sensitive time-resolved imaging and chemo-/bio-sensing.11–14 However, the dissolved oxygen and unrestricted rotation and vibration of organic luminescent molecules in aqueous solutions significantly facilitate non-radiative transitions from the triplet excited state to the ground state, leading to the quenching of RTP emission.15–17 To date, numerous strategies have been employed for the advancement of aqueous organic RTP materials, including fabricating phosphorescent nanoparticles and harnessing supramolecular assembly.18–22 Despite achieving aqueous RTP emission in organic luminescent materials,23–27 developing a single-molecule emitter capable of exhibiting phosphorescence-based ratiometric multi-emission behavior in response to variations in biological microenvironmental factors, such as temperature, light, oxygen, and pH, remains an arduous challenge.Currently, the excitation wavelength of most aqueous organic RTP materials is relatively short (<400 nm, ultraviolet light), which not only raises potential safety concerns in biomedical applications but also restricts their tissue penetrability and results in diminished imaging contrast.28–30 Recently, considerable attention has been focused on pure organic compounds that exhibit long-wavelength phosphorescence emission upon excitation using a visible or even near-infrared (NIR) light.31,32 Up-conversion luminescent (UCL) materials are a distinctive class of luminophores, which are capable of converting low-energy excitation into high-energy emission.33 These UCL materials possess several advantageous features, including a large anti-Stokes shift, excellent penetration through biological tissues, and a high signal-to-background (SBR) imaging ratio.34,35 However, there are only a few reported examples of UCL organic dyes, which are used for constructing aqueous RTP materials.36,37 Moreover, the presently accessible organic up-conversion RTP materials exclusively emit a single color within the visible or NIR spectrum.38 Their applications are somewhat restricted owing to poor photostabilities, non-tunable emissions, or complex preparation processes. The development of a single-molecular chromophore possessing up-conversion RTP capabilities and tunable multi-emission is thus imperative.
In this study, we synthesized a single-molecular emitter Yn with multiple heteroatoms and carbonyl groups to enhance the rate of intersystem crossing (ISC), thereby promoting triplet excitons and achieving molecular RTP in water (Fig. 1). Additionally, flexible alkyl chains were incorporated into the design to facilitate the formation of a folding configuration, thus improving the generation of exciplex complexes through charge transfer (CT) processes that favor phosphorescence emission. Significantly, a broad range of excitation wavelengths, encompassing both visible and NIR light, can be employed to precisely control the up-conversion dual-emission phenomenon of blue fluorescence (455 nm) and orange-red RTP (580 nm) exhibited by the Y3 molecule in aqueous solutions. The single-molecular chromophore exhibited a distinct temperature-insensitive fluorescent emission and a temperature-dependent RTP signal, rendering it highly suitable for the ratiometric quantification of organismal temperatures. Moreover, the Y3 molecule possessed several remarkable features, including exceptional pH and photostability, as well as superb biocompatibility. Finally, the Y3 molecule was employed in multifunctional optical imaging and sensing applications encompassing two-photon phosphorescence imaging, time-resolved bioimaging, and ratiometric sensing of cellular temperature. Therefore, this single-molecular platform not only facilitates the rational design of smart luminescent materials with adjustable RTP/fluorescence emission but also enables high-contrast bioimaging and ratiometric sensing.
 | ||
| Fig. 1 Rational design of a single-molecular emitter with RTP/fluorescence dual emission. (A) Previous strategies for constructing aqueous organic RTP materials. (B) Working principle of engineering a single-molecule chromophore for aqueous organic RTP. (C) Simplified Jablonski diagram of the up-conversion organic phosphorescence and regulatable dual emission in Y3 molecule. (D) Controllable aqueous organic RTP/fluorescence and multiple advantages in the Y3 molecular platform. | ||
Results and discussion
We synthesized a series of Yn (n = 1–3) molecules through facile synthetic routes involving nucleophilic substitution and Suzuki coupling reactions (Fig. S1†). These Yn compounds were further characterized by 1H and 13C NMR spectroscopy, high-resolution mass spectrometry (Fig. S2–S14†), and optical property analysis. The UV-vis absorption spectra of the Yn molecules (n = 1–3) displayed maximum absorption wavelengths at 323 nm, 318 nm, and 385 nm, respectively (Fig. 2A). The absorption peaks within the 318–323 nm range can be attributed to the π–π* electronic transitions in these three compounds, showing characteristic shifts. This observation suggests that the presence of an amine group induces a redshift in the absorption wavelength of these Yn molecules. Moreover, we conducted a comparative analysis of the emission wavelengths and intensities of Y1–Y3, revealing a significantly diminished emission intensity for Y1 in contrast to both Y2 and Y3. This outcome implies that the presence of the tert-butyl carbonyl group may impede the rotation of the amino group, thereby diminishing non-radiative decay processes and resulting in an amplified emission intensity39 (Fig. 2B). Interestingly, both Y2 and Y3 molecules exhibited distinct dual emissions. The dual emission peaks of Y3 were observed at 455 nm and 580 nm, respectively. Similarly, for Y2, the dual emission peaks were found at 450 nm and 550 nm. Notably, the maximum emission wavelength of Y2 is redshifted by 30 nm compared with that of Y3 due to the introduction of an oxygen moiety, resulting in stronger charge transfer. | ||
| Fig. 2 Photophysical properties of Yn molecules. (A) UV-vis absorption spectra of Yn (n = 1–3) at concentrations of 1 × 10−4 M in water. (B) Photoluminescence (PL) spectra of Yn (n = 1–3) (excited at 400 nm and 410 nm) aqueous solutions at concentrations of 1 × 10−4 M. (C) Phosphorescence intensity of Y3 in aqueous solution (delay time = 0.1 ms) at different temperatures (273 K to 77 K). (D) Lifetime of Y3 at 580 nm in aqueous solution at different temperatures (273 K to 77 K). (E) Lifetime of Y3 (1 × 10−4 M) at room temperature in water. (F) PL spectra of Y3 (1 × 10−4 M) under deaeration conditions. | ||
In order to determine the nature of the 580 nm emission peak of Y3 in water, we obtained the temperature-dependent time-resolved photoluminescence spectra. At a temperature of 77 K, Y3 exhibited pronounced delayed emission with a luminescence lifetime of 85 ms (Fig. 2C). With increasing temperature (77 K to 273 K), the luminescence intensity of Y3 gradually diminishes while its lifetime progressively shortens (Fig. 2D), indicating that the long-wavelength emission from Y3 is attributed to phosphorescence rather than the thermal activation-delayed fluorescence. Furthermore, the lifetime of the 580 nm emission peak of Y3 in water at ambient temperature conditions was determined to be 20 μs (Fig. 2E), while the lifetime of Y3 at the 455 nm emission peak in water under the same conditions was less than 5 ns (Fig. S15†). This demonstrated that Y3 exhibits dual emissions consisting of fluorescence at 355 nm and phosphorescence at 580 nm. The emission intensity was significantly enhanced upon deoxygenation through nitrogen bubbling, in accordance with the phenomenon of oxygen quenching triplet-state electrons (Fig. 2F). The phosphorescence quantum yields of Y2 and Y3 in aqueous solutions were further determined to be 1.1% and 1.5%, respectively (Fig. S16†).
Previously, molecular luminophores exhibiting pronounced dual emissions of both fluorescence and phosphorescence, with exceptional tunable luminescent properties, were reported.40–43 The PL spectra of Y2 and Y3 were further measured in water under different excitation wavelengths. As depicted in Fig. 3A, Y3 exhibited a switchable phosphorescence and fluorescence emission that were dependent on the excitation wavelength within the range of 360–440 nm. The emission peak wavelength predominantly occurred at 455 nm fluorescence when excited with irradiation below 390 nm, whereas it shifted to 580 nm phosphorescence for excitation wavelengths above this threshold. Additionally, the Commission International del'Eclairage (CIE) coordinates were determined to illustrate the color change of the PL emission of Y3 upon changing the excitation wavelength. Fig. 3B clearly demonstrates a transition from blue to orange-red, passing through a white-light region. Interestingly, up-conversion emission is observed when the excitation wavelength falls within the NIR range (Fig. 3C). Specifically, by utilizing the NIR excitation in the 700–850 nm range, precise manipulation of the emission color of Y3 can also be achieved (Fig. 3D).
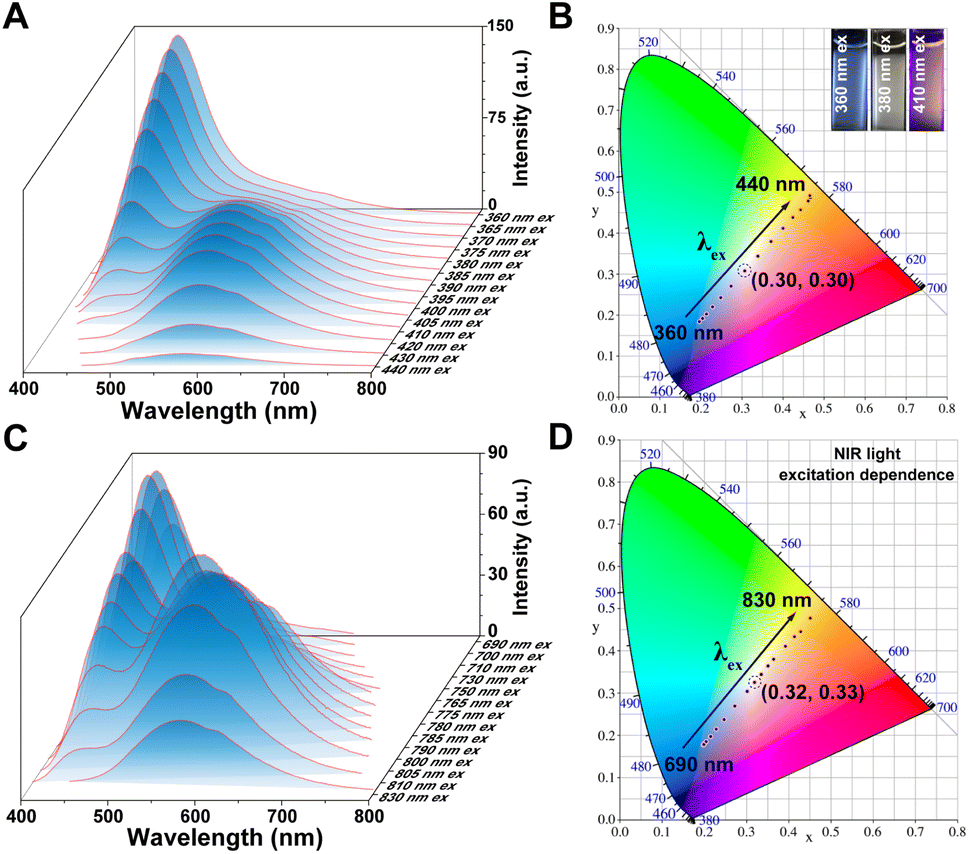 | ||
| Fig. 3 Excitation-wavelength spectra of Y3. (A) PL spectra of Y3 (1 × 10−4 M, excited at 360 nm to 440 nm) in aqueous solutions. (B) Chromaticity coordinates (CIE) of Y3 with varying excitation wavelengths in aqueous solution in accordance with (A). Inset images: the luminescent color of the Y3 molecule upon excitation at wavelengths of 360 nm, 380 nm, and 410 nm, respectively. (C) PL spectra of Y3 (1 × 10−4 M, excited at 690 nm to 830 nm) in aqueous solutions. (D) CIE of Y3 with varying NIR excitation wavelengths in aqueous solution in accordance with (C). | ||
The wide-range excitation wavelength-dependent RTP/fluorescence emission was also observed in the Y2 molecule (Fig. S17†). Within the excitation wavelength range of 350 nm to 430 nm and 700 nm to 820 nm, the emission of Y2 underwent a conversion from 450 nm fluorescence to 550 nm phosphorescence, resulting in a change in luminescent color from blue to yellow. The above results demonstrated that the fluorescence and phosphorescence emission in the aqueous solution of Y2 and Y3 molecules can be modulated by adjusting the excitation wavelength across the UV, visible light, and NIR regions. Moreover, a dual emission of phosphorescence/fluorescence within a specific excitation wavelength range can be achieved, enabling simultaneous adjustment of multiple colors. These findings highlight the potential applications of aqueous up-conversion organic phosphorescence and adjustable phosphorescence/fluorescence dual emission from the Y2 and Y3 molecules in optical imaging and ratiometric sensing.
After identifying the luminescent center, a plausible mechanism was proposed (Scheme S1†).44 The donor, acceptor, and CT exciplex moieties can be independently excited. When the excitation wavelength is below 360 nm, the bromo-substituted phenylpyridinium and trimethyl ester pyridine salt moieties are preferentially activated, resulting in a predominantly blue emission from the Y3 solution. At wavelengths above 360 nm, the CT complexes become preferentially excited, leading to an increase in the intensity of the orange-red emission. In order to ascertain the presence of CT features within the molecule itself or in neighboring molecules, we conducted measurements for concentration-dependent UV-vis absorption (Fig. S18†) and PL spectra (Fig. S19†) of the Y3 molecule. The absorption spectra exhibited no conspicuous bathochromic shift upon increasing the concentration of Y3. Moreover, the absorption intensity at 328 nm displayed a linear increase with rising concentration, conforming precisely to Beer–Lambert's law and indicating negligible interactions among solute molecules. The position and shape of the phosphorescence/fluorescence dual emission in the PL spectra remained nearly unchanged across a concentration range from 0.001 mM to 0.1 mM. The RTP and fluorescence emissions were observed even in a highly dilute solution, indicating that the luminescent properties come solely from the Y3 molecule rather than intermolecular interactions (Fig. S19†). Moreover, based on the distinct maximum excitation wavelengths at 455 nm and 580 nm of Y3 and 450 nm and 550 nm of Y2, as shown in Fig. S20,† it can be deduced that the pyridinium salt monomer lacks the ability to induce orange-red or yellow phosphorescence emission from its excimer state. Therefore, we can infer that this unique phosphorescent property originates from the Y3 molecule rather than any molecular aggregates, thus confirming its classification as a single-molecule luminescence system.
The subsequent investigation focused on the analysis of structural parameters in this single-molecule system. The Yn structure comprises a propane chain that connects two phenylpyridine moieties, potentially resulting in a flexible conformation. Previous studies conducted by our research group and other relevant reports have substantiated the existence of folded or stretched conformations for such molecules,45–47 contingent upon their surrounding environment. The interproton nuclear Overhauser effect (NOE) in this molecular system was investigated using the 2D ROESY NMR method. As depicted in Fig. 4A, distinct NOE signals were observed between protons H3 of the pyridine group and protons H8 of the other pyridine moieties, as well as between protons H1,2 of the bromophenyl group and protons H9 of the pyridine moieties, demonstrating the close spatial proximity of the two phenylpyridine units, which is a characteristic feature commonly associated with folded conformations. The enhanced stability of the folded conformation can be further explained by employing the independent gradient model, which is based on Hirshfeld partition analysis (IGMH), for evaluating Y2 and Y3 (Fig. 4B and S21†). This analysis reveals evident π–π stacking effects and van der Waals interactions between the benzene units (green lamellar regions), which are absent in the stretched conformation.
 | ||
| Fig. 4 Characterization of single-molecular luminescent properties. (A) Sectional 2D-ROESY-NMR spectrum of Y3 (1 mM in D2O and at 298 K). (B) IGMH isosurfaces and folded conformations of Y2 and Y3. (C) PL spectra of adding equivalent amounts of CB[7] and CB[8] into the Y3 aqueous solution. (D) Schematic of the supramolecular regulation method for the Y3 molecular conformation variation. (E) Normalized PL spectra of Y3 solutions at various volume fractions of MeOH and water (from 0 to 100%, fw), the fluorescence intensity at 455 nm was unified. (F) Phosphorescence/fluorescence (phos.580/fluo.455) intensity ratio as a function of water fraction fw. | ||
In view of the high affinity for alkyl dipyridinium and the presence of a rigid cavity, cucurbit[7]uril (CB[7]) and cucurbit[8]uril (CB[8]) macrocycles were employed as host molecules for binding the Y3 molecule. The structure of the host–guest system was investigated using 2D ROESY NMR spectroscopy. Notable NOE signals were observed between protons H3,4,6,8,9 of Y3 and the inward proton Hx of CB[7], confirming the threading of CB[7] onto Y3 at the alkyl dipyridinium site (Fig. S22†). The rigid cavity provided by CB[7] and precise self-assembly prevented any folding of Y3, resulting in an elongated conformation without any charge transfer. The subsequent PL spectra also revealed that upon combination with CB[7], the 580 nm phosphorescence peak vanished, while the fluorescence peak intensity at 455 nm exhibited a significant increase (Fig. 4C). This observation is consistent with the optical changes induced by the alteration in its conformational state. In contrast, when Y3 was complexed with CB[8], the fluorescence peak at 455 nm disappeared, whereas there was a remarkable enhancement in the intensity of the 580 nm phosphorescence peak. Consequently, it can be inferred that CB[7], possessing a small rigid cavity, hinders the folding configuration of the Y3 molecule; conversely, CB[8], featuring a larger rigid cavity, sustains the folded configuration of the Y3 molecule (Fig. 4D).
Tunable dual emission of phosphorescence and fluorescence, as well as conformational transition, can be conveniently achieved by controlling the charge transfer efficiency of the Y3 molecule. One approach to achieve this is by using mixed solvents of methanol (MeOH) and water (H2O), where MeOH is a suitable solvent for dissolving extended π systems. The normalized photoluminescence spectra of Y3 in different ratios of MeOH to H2O solution are shown in Fig. 4E. However, when the water content decreases, the phosphorescence emission of Y3 also decreases. At a water ratio (fw) below 40% (Fig. 4F), only mono emission is observed due to the absence of the self-folded conformation. This can be attributed to an increase in the distance between the donors and acceptors in high MeOH fractions, which hinders and restricts the CT process as molecules preferentially adopt a stretched state.
To further reveal the possible reason for the aqueous molecular phosphorescence of Y2 and Y3, we conducted density functional theory (DFT) and time-dependent density functional theory (TD-DFT) calculations using the Gaussian G09 program with the B3LYP/6-311G* set. Fig. 5A demonstrates the S0 → S1 transition in all Yn (n = 1–3) compounds, which can be attributed to the HOMO–LUMO transition. The LUMO distribution and energy level were not significantly affected by variations in the substituent type due to their similar molecular skeleton and electron-donating functional group (amino). The LUMOs of these compounds exhibited a consistent distribution primarily located on the amino and phenylpyridine segments, with no noticeable changes in energy levels. However, there was a notable alteration in both the distribution and energy level of the HOMO when considering different amide groups (medium electron-donating functional group). In comparison to the electron distribution of HOMO, Y2 and Y3 exhibit a higher concentration of n electrons on the carbonyl group, indicating an increased likelihood for n–π* transition.
 | ||
| Fig. 5 Quantum chemical calculation of Yn. (A) Transition configurations (%), energy level diagrams, and frontier molecular orbital of the S0–S1 transition of Yn (n = 1–3). (B) SOC coefficients and energy-level diagrams in Y1 and Y3. | ||
The transition energy from the S0 state to the S1 state is 1.791 eV in the Y3 molecule (Fig. 5B), which is slightly larger than that in the Y1 molecule (1.480 eV). However, the energy gap corresponding to the single–triplet excited states (ΔEST) for the Y3 molecule (S1–T1, 0.296 eV) is smaller than that for the Y1 molecule (S1−T1, 0.355 eV). In addition, the Y3 molecule has a significantly larger spin–orbit coupling (SOC) coefficient (〈S1|HSO|T1〉, 1.85 cm−1) than that of the Y1 molecule (〈S1|HSO|T1〉, 0.87 cm−1). It is widely acknowledged that the rate of intersystem crossing (kisc) primarily depends on both ΔEST and SOC values. Consequently, by reducing ΔEST significantly and increasing SOC substantially, the kisc of the Y3 molecule could be significantly higher than that of the Y1 molecule, which was a requisite element for efficient RTP. The Y2 and Y3 single-molecule system enables more efficient ISC and phosphorescence emission by collectively benefiting from the carbonyl group, various heteroatoms, significant CT, and a favorable radiative pathway.
In light of the exceptional up-conversion phosphorescence and fluorescence dual emission demonstrated by the Y3 molecule in an aqueous solution, we subsequently investigated its potential application in biological imaging and sensing. The cytotoxicity of the Y3 molecule was initially evaluated using the MTT assay. The results revealed that even at high concentrations, the Y3 molecule exhibited no toxicity towards HeLa cells (Fig. S23†). To assess the photostability of Y3-incubated cells, they were exposed to a 405 nm laser for 6 min under a confocal high-resolution microscope. Importantly, no noticeable fluctuations in the red phosphorescence intensity of Y3 were observed after exposure to the 405 nm laser for 6 min (Fig. S24†). Furthermore, across various pH values (pH 5.0–9.0) in a PBS buffer, the luminescent intensity of Y3 remained consistently stable, indicating its excellent pH stability (Fig. S25†).
The NIR two-photon absorption performance of the Y3 molecule was further evaluated, considering their potential for enhanced tissue penetration and reduced photodamage in biological applications. Fig. 6A demonstrates that Y3 exhibited the NIR two-photon absorption in water. Additionally, the Y3 molecule exhibited maximum emission peaks at 455 nm and 580 nm when excited by femtosecond pulsed lasers with 720 nm and 800 nm wavelengths, respectively (Fig. S26†). These results are consistent with previous findings on NIR excitation-dependent spectra, further confirming the dual emission of up-conversion phosphorescence and fluorescence in the Y3 molecule. The confocal images of HeLa cells after incubation with Y3 (50 μM) for only 6 hours are shown in Fig. 6B. Compared with one-photon imaging, two-photon luminescent imaging excited by a NIR femtosecond pulsed laser displayed better performance with an improved SBR ratio. Additionally, the strong NIR emission (>650 nm) observed in the emission spectrum of the Y3 molecule prompted us to investigate its potential for NIR imaging in cells. As depicted in Fig. 6C, our results demonstrated that the Y3 molecule exhibits remarkable suitability for NIR phosphorescence cellular imaging.
 | ||
| Fig. 6 Cellular two-photon and time-resolved imaging. (A) Two-photon excitation spectra of Y3 (0.1 mM) in water. (B) HeLa cells incubated with Y3 (50 μM). One-photon imaging: λex = 405 nm, λem = 580–700 nm. Two-photon imaging: λex = 800 nm, λem = 580–700 nm, Scale bar: 25 μm. (C) NIR phosphorescence image of HeLa cells after incubation with Y3 (50 μM), λex = 405 nm, λem = 650–800 nm. (D) Schematic of organic RTP avoiding autofluorescence. (E) Time-gated luminescent images with various delay times of HeLa cells treated with Y3 for 4 h. Scale bar: 20 μm. (F) Phosphorescence intensity analysis of images in (E) with ImageJ software. | ||
Phosphorescence effectively mitigates the issue of spontaneous fluorescence in biological systems due to its prolonged emission lifetime, rendering it suitable for time-resolved imaging techniques with extended temporal scales (Fig. 6D). Subsequently, the phosphorescence lifetime imaging microscopy (PLIM) technique was employed to achieve precise microsecond time-gated imaging at the cellular level using the Y3 molecule. Fig. 6E demonstrates the detection of phosphorescent signals from the Y3 molecule in HeLa cells. Moreover, a long-lasting phosphorescent signal with a lifetime exceeding 100 μs was clearly detectable, indicating that Y3 functions as an effective imaging tool unaffected by background fluorescent signals. By extending the delay time beyond 1 μs (20 μs, 40 μs, 60 μs, 80 μs and 100 μs), we achieved stable detection of the long-life phosphorescent group signal with higher SBR values (SBR = 10.0, 8.1, 7.3 and 6.3) (Fig. 6F). The chromophore Y3, therefore, emerges as an exceptionally versatile organic RTP molecule with immense potential to serve as a robust tool for time-resolved bioimaging in the detection of intricate physiological environments.
The temperature, being a crucial and fundamental determinant for the survival of living organisms, plays a pivotal role in the physiological states of organisms.48,49 Subsequently, we evaluated the quantitative ratiometric temperature sensing capability of the Y3 molecule based on its dual RTP/fluorescent emissions. Under irradiation at 405 nm, Y3 exhibited simultaneous blue fluorescence and orange-red phosphorescence in its emission spectra. Significantly, the emission of orange-red phosphorescence at 580 nm exhibited a gradual and sensitive decline as the temperature decreased. In contrast, the red fluorescence at 455 nm remained relatively stable (Fig. 7A), making it an excellent reference signal for internal ratiometric analysis. The luminescent color of Y3 transitioned from orange-red to white light with increasing temperature (Fig. S26†). Furthermore, leveraging the temperature-dependent characteristic, we quantitatively evaluated the ability of the Y3 molecule for ratiometric temperature measurements. The results presented in Fig. 7B demonstrate a strong linear correlation between phos.580/fluo.455 and the temperature level (R2 = 0.988), indicating the precise quantitative ratiometric sensing of the phosphorescence capability of the Y3 molecule. The RTP/fluorescence ratios exhibit linear relationships with temperature within the physiological and therapeutic range, achieving a thermal relative sensitivity (Sa)50 of 1.9% °C−1 at 35 °C. These findings highlight the accuracy of the temperature monitoring ability of the phosphorescence/fluorescence ratiometric thermometry. These results underscore the precision of phosphorescence/fluorescence ratiometric thermometry in monitoring temperature.
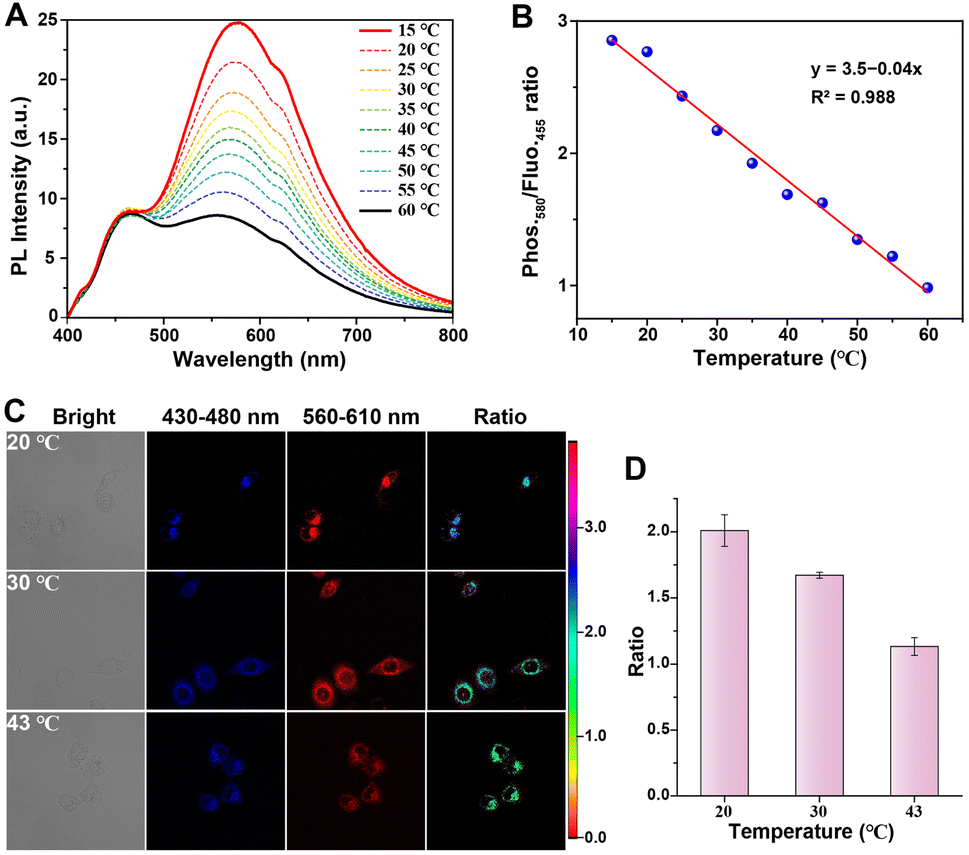 | ||
| Fig. 7 Ratiometric RTP/fluorescence dual emission for temperature sensing. (A) PL spectra of the Y3 molecule in aqueous solution (405 nm excitation) at different temperatures (15–60 °C). (B) Linear relationship between the temperature and the ratio of 580 nm phosphorescence intensity/455 nm fluorescence intensity from (A). (C) Ratiometric and confocal images of HeLa cells after treatment with Y3 molecule (50 μM) at different temperatures (approximately 20, 30, and 43 °C) for a duration of 1 hour. The blue channel fluorescence was collected within the range of 430 to 480 nm, and the red channel phosphorescence was collected within the range of 560 to 610 nm. Ratiometric images constructed using both the blue and red channels with an excitation wavelength of λex = 405 nm. (D) Ratios of mean luminescent intensity at various temperature levels. | ||
Considering the ability of the Y3 molecule to quantitatively sense temperature levels using a distinctive internal reference from fluorescent and RTP emissions, we further investigated its suitability for ratiometric imaging and temperature monitoring in live cells through confocal microscopy. Following incubation with the Y3 molecule for 1 hour under relatively high-temperature conditions, HeLa cells exhibited a remarkable fluorescence signal within the wavelength range of 430–480 nm, along with a moderate phosphorescence signal between 560–610 nm due to the non-radiative effects caused by elevated temperatures (Fig. 7C). As the temperature decreased, the fluorescence signal remained relatively stable, while there was a significant enhancement in the phosphorescence signal observed in HeLa cells, thus confirming that the Y3 molecule could effectively trace and monitor temperature levels within live cells. Notably, the ratiometric imaging of intracellular temperature was obtained by calculating the ratio between the phosphorescence and fluorescence intensities (Iphos./Ifluor.). The average ratios Ifluor./Iphos. in cells treated at approximately 20 °C and 43 °C were determined to be 2.1 and 1.1, respectively. A significant two-fold decrease in the ratio was observed (Fig. 7D). Thus, the Y3 molecule demonstrated its capability for the quantitative monitoring of temperature levels in live cells.
Conclusions
In summary, we have successfully synthesized a novel luminescent molecule, Yn, with a dual up-conversion phosphorescence and fluorescence emission based on an alkyl chain-bridged diphenylpyridinium through precise molecular engineering. The incorporation of amino groups within the Yn molecular scaffold serves as an effective luminescence regulatory site, while the introduction of carbonyl groups promotes enhanced phosphorescence and fluorescence emission. Importantly, by precisely controlling the excitation wavelength, we can regulate the emission color of the Y3 molecule in the aqueous phase between red and blue, even achieving white emission. Furthermore, this water-soluble Y3 molecule exhibits excellent biocompatibility and stability, making it highly suitable for applications such as autofluorescence-free time-resolved imaging and high-contrast two-photon imaging in cells. Additionally, the Y3 molecule exhibits temperature-sensitive phosphorescence and another temperature-insensitive fluorescence, enabling a distinctive ratiometric evaluation of temperature levels within living tumor cells. Importantly, our study not only presents a rational design principle for achieving a dual emission of aqueous up-conversion phosphorescence and fluorescence in a single-molecular emitter but also provides an opportunity to develop a sophisticated molecular platform for multiplexed biological imaging and sensing, ratiometric sensing, two-photon imaging, time-resolved imaging, anti-counterfeiting, and numerous other applications.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Y. Li, H. Tian and X. Ma designed the materials and conceived the project. Y. Li and Z. Wu conducted the synthesis and characterizations of the molecules. Y. Li performed imaging experiments. Y. Li, H. Tian and X. Ma wrote the manuscript. All authors engaged in discussions and conducted data analysis regarding the results.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the financial support from the National Natural Science Foundation of China (22125803, T2488302, 22020102006 and 22307036), Science and Technology Commission of Shanghai Municipality (grant no. 24DX1400200), China Postdoctoral Science Foundation (2023M731079), and the Fundamental Research Funds for the Central Universities.Notes and references
- E. Hamzehpoor, C. Ruchlin, Y. Tao, C. H. Liu, H. M. Titi and D. F. Perepichka, Nat. Chem., 2022, 15, 83–90 Search PubMed.
- C. Chen, Z. Chi, K. C. Chong, A. S. Batsanov, Z. Yang, Z. Mao, Z. Yang and B. Liu, Nat. Mater., 2021, 20, 175–180 Search PubMed.
- W. Ye, H. Ma, H. Shi, H. Wang, A. Lv, L. Bian, M. Zhang, C. Ma, K. Ling, M. Gu, Y. Mao, X. Yao, C. Gao, K. Shen, W. Jia, J. Zhi, S. Cai, Z. Song, J. Li, Y. Zhang, S. Lu, K. Liu, C. Dong, Q. Wang, Y. Zhou, W. Yao, Y. Zhang, H. Zhang, Z. Zhang, X. Hang, Z. An, X. Liu and W. Huang, Nat. Mater., 2021, 20, 1539–1544 Search PubMed.
- K. Chang, L. Xiao, Y. Fan, J. Gu, Y. Wang, J. Yang, M. Chen, Y. Zhang, Q. Li and Z. Li, Sci. Adv., 2023, 9, eadf6757 Search PubMed.
- J. Song, Y. Zhou, Z. Pan, Y. Hu, Z. He, H. Tian and X. Ma, Matter, 2023, 6, 2005–2018 CrossRef CAS.
- B. Chang, J. Chen, J. Bao, T. Sun and Z. Cheng, Chem. Rev., 2023, 123, 13966–14037 CrossRef CAS PubMed.
- B. Chang, D. Li, Y. Ren, C. Qu, X. Shi, R. Liu, H. Liu, J. Tian, Z. Hu, T. Sun and Z. Cheng, Nat. Biomed. Eng., 2022, 6, 629–639 Search PubMed.
- L. Liang, J. Chen, K. Shao, X. Qin, Z. Pan and X. Liu, Nat. Mater., 2023, 22, 289–304 Search PubMed.
- W. Zhao, Z. He and B. Z. Tang, Nat. Rev. Mater., 2020, 5, 869–885 CrossRef CAS.
- H. J. Yu, Q. Zhou, X. Dai, F. F. Shen, Y. M. Zhang, X. Xu and Y. Liu, J. Am. Chem. Soc., 2021, 143, 13887–13894 Search PubMed.
- J. Zhou, J. Li, K. Y. Zhang, S. Liu and Q. Zhao, Coord. Chem. Rev., 2022, 453, 214334 Search PubMed.
- X.-Y. Dai, M. Huo and Y. Liu, Nat. Rev. Chem, 2023, 7, 854–874 CrossRef CAS PubMed.
- L. Ma and X. Ma, Sci. China:Chem., 2022, 66, 304–314 CrossRef.
- D. Li, F. Lu, J. Wang, W. Hu, X. M. Cao, X. Ma and H. Tian, J. Am. Chem. Soc., 2018, 140, 1916–1923 CrossRef CAS PubMed.
- B. Ding, X. Ma and H. Tian, Acc. Mater. Res., 2023, 4, 827–838 CrossRef CAS.
- T. Zhang, X. Ma, H. Wu, L. Zhu, Y. Zhao and H. Tian, Angew. Chem., Int. Ed., 2020, 59, 11206–11216 CrossRef CAS PubMed.
- X. Jia, C. Shao, X. Bai, Q. Zhou, B. Wu, L. Wang, B. Yue, H. Zhu and L. Zhu, Proc. Natl. Acad. Sci. U.S.A., 2019, 116, 4816–4821 CrossRef CAS PubMed.
- X. Ma, J. Wang and H. Tian, Acc. Chem. Res., 2019, 52, 738–748 Search PubMed.
- H. Sun and L. Zhu, Aggregate, 2022, e253 Search PubMed.
- Y. Jiang and K. Pu, Chem. Rev., 2021, 121, 13086–13131 CrossRef CAS PubMed.
- L. Gu, H. Shi, L. Bian, M. Gu, K. Ling, X. Wang, H. Ma, S. Cai, W. Ning, L. Fu, H. Wang, S. Wang, Y. Gao, W. Yao, F. Huo, Y. Tao, Z. An, X. Liu and W. Huang, Nat. Photonics, 2019, 13, 406–411 CrossRef CAS.
- X. Wang, H. Shi, H. Ma, W. Ye, L. Song, J. Zan, X. Yao, X. Ou, G. Yang, Z. Zhao, M. Singh, C. Lin, H. Wang, W. Jia, Q. Wang, J. Zhi, C. Dong, X. Jiang, Y. Tang, X. Xie, Y. Yang, J. Wang, Q. Chen, Y. Wang, H. Yang, G. Zhang, Z. An, X. Liu and W. Huang, Nat. Photonics, 2021, 15, 187–192 CrossRef CAS.
- H. Chen, Y. Sun, M. Liu, F. Li, Q. Peng and H. Huang, Angew. Chem., Int. Ed., 2023, 62, e202302629 CrossRef CAS PubMed.
- W. Li, Q. Huang, Z. Mao, J. Zhao, H. Wu, J. Chen, Z. Yang, Y. Li, Z. Yang, Y. Zhang, M. P. Aldred and Z. Chi, Angew. Chem., Int. Ed., 2020, 59, 3739–3745 CrossRef CAS PubMed.
- H. Sun, M. He, G. V. Baryshnikov, B. Wu, R. R. Valiev, S. Shen, M. Zhang, X. Xu, Z. Li, G. Liu, H. Ågren and L. Zhu, Angew. Chem., Int. Ed., 2024, 63, e202318159 CrossRef CAS PubMed.
- J. Li, K. Chen, J. Wei, Y. Ma, R. Zhou, S. Liu, Q. Zhao and W.-Y. Wong, J. Am. Chem. Soc., 2021, 143, 18317–18324 CrossRef CAS PubMed.
- Z. Man, Z. Lv, Z. Xu, M. Liu, J. He, Q. Liao, J. Yao, Q. Peng and H. Fu, J. Am. Chem. Soc., 2022, 144, 12652–12660 CrossRef CAS PubMed.
- J. Han, J. Zhang, T. Zhao, M. Liu and P. Duan, CCS Chem., 2021, 3, 665–674 CrossRef CAS.
- L. Wu, J. Liu, P. Li, B. Tang and T. D. James, Chem. Soc. Rev., 2021, 50, 702–734 RSC.
- H. Zhou, X. Zeng, A. Li, W. Zhou, L. Tang, W. Hu, Q. Fan, X. Meng, H. Deng, L. Duan, Y. Li, Z. Deng, X. Hong and Y. Xiao, Nat. Commun., 2020, 11, 6183 CrossRef CAS PubMed.
- Y. Fan, S. Liu, M. Wu, L. Xiao, Y. Fan, M. Han, K. Chang, Y. Zhang, X. Zhen, Q. Li and Z. Li, Adv. Mater., 2022, 34, e2201280 CrossRef PubMed.
- S. Sun, L. Ma, J. Wang, X. Ma and H. Tian, Natl. Sci. Rev., 2022, 9, nwab085 CrossRef CAS PubMed.
- B. Ding, L. Ma, Z. Huang, X. Ma and H. Tian, Sci. Adv., 2021, 7, eabf9668 CrossRef CAS PubMed.
- J. Wang, Z. Huang, X. Ma and H. Tian, Angew. Chem., Int. Ed., 2020, 59, 9928–9933 CrossRef CAS PubMed.
- W. L. Zhou, W. Lin, Y. Chen and Y. Liu, Chem. Sci., 2022, 13, 7976–7989 RSC.
- X. F. Wang, H. Xiao, P. Z. Chen, Q. Z. Yang, B. Chen, C. H. Tung, Y. Z. Chen and L. Z. Wu, J. Am. Chem. Soc., 2019, 141, 5045–5050 CrossRef CAS PubMed.
- I. Bhattacharjee, N. Acharya, S. Karmakar and D. Ray, J. Phys. Chem. C, 2018, 122, 21589–21597 CrossRef CAS.
- X. K. Ma, X. Zhou, J. Wu, F. F. Shen and Y. Liu, Adv. Sci., 2022, 9, e2201182 CrossRef PubMed.
- C. Yan, Z. Guo, W. Chi, W. Fu, S. A. A. Abedi, X. Liu, H. Tian and W. H. Zhu, Nat. Commun., 2021, 12, 3869 CrossRef CAS PubMed.
- H. Bhatia, S. Dey and D. Ray, ACS Omega, 2021, 6, 3858–3865 CrossRef CAS PubMed.
- S. Karmakar, R. Deka, S. Dey and D. Ray, J. Lumin., 2023, 263, 120029 CrossRef CAS.
- R. Deka, S. Dey, M. Upadhyay, S. Chawla and D. Ray, J. Phys. Chem. A, 2024, 128, 581–589 CrossRef CAS PubMed.
- H. Bhatia and D. Ray, J. Phys. Chem. C, 2019, 123, 22104–22113 CrossRef CAS.
- H. Bhatia, I. Bhattacharjee and D. Ray, J. Phys. Chem. C, 2018, 9, 3808–3813 CAS.
- D. Li, W. Hu, J. Wang, Q. Zhang, X. M. Cao, X. Ma and H. Tian, Chem. Sci., 2018, 9, 5709–5715 RSC.
- L. Mao, Y. Han, Q. W. Zhang and Y. Tian, Nat. Commun., 2023, 14, 1419 CrossRef CAS PubMed.
- X. K. Ma, W. Zhang, Z. Liu, H. Zhang, B. Zhang and Y. Liu, Adv. Mater., 2021, 33, e2007476 CrossRef PubMed.
- K. Xue, C. Wang, J. Wang, S. Lv, B. Hao, C. Zhu and B. Z. Tang, J. Am. Chem. Soc., 2021, 143, 14147–14157 CrossRef CAS PubMed.
- J. Wang, X. Yao, Y. Liu, H. Zhou, W. Chen, G. Sun, J. Su, X. Ma and H. Tian, Adv. Opt. Mater., 2018, 6, 1800074 CrossRef.
- N. Acharya, M. Upadhyay, R. Deka and D. Ray, J. Phys. Chem. C, 2024, 128, 8750–8758 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc08330j |
| This journal is © The Royal Society of Chemistry 2025 |