
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCatalytic insertion of nitrenes into B–H bonds†
Nikita M.
Ankudinov
 *a,
Nikita V.
Alexeev
ab,
Evgeniya S.
Podyacheva
ac,
Denis A.
Chusov
ac,
Konstantin A.
Lyssenko
bc and
Dmitry S.
Perekalin
*ac
*a,
Nikita V.
Alexeev
ab,
Evgeniya S.
Podyacheva
ac,
Denis A.
Chusov
ac,
Konstantin A.
Lyssenko
bc and
Dmitry S.
Perekalin
*ac
aA.N. Nesmeyanov Institute of Organoelement Compounds, Russian Academy of Sciences, 28 Vavilova Str., Moscow 119334, Russia. E-mail: nickankudinov@gmail.com; dsp@ineos.ac.ru
bLomonosov Moscow State University, Leninskie Gory 1 Bld. 3, Moscow 119991, Russia
cNational Research University Higher School of Economics, 7 Vavilova Str., Moscow 117312, Russia
First published on 4th March 2025
Abstract
Organic compounds with boron–nitrogen bonds are widely used as fluorescent sensors and semiconducting materials. This paper presents a new approach for the formation of B–N bonds via catalytic insertion of nitrenes into B–H bonds. The reaction proceeds most selectively for cyclic boranes with a 2-phenylpyridine framework and nitrenes generated in situ by the oxidation of sulfonamides and sulfamates. The most effective catalysts for this process are the readily available rhodium and ruthenium carboxylates of [M2(OOCR)4]X type. Complexes with carboxylate ligands NTTL derived from S-tert-leucine provide unique chiral products with stereogenic boron atoms. The developed method can be used for the introduction of boron heterocycles into biologically active molecules.
Introduction
Boron–nitrogen bonds are isoelectronic to ubiquitous carbon–carbon bonds, but due to charge polarization and higher reactivity, they can be even more useful for a variety of applications.1,2 For example, while alkanes remain relatively inert, their aminoborane analogues RH2B–NH2R can undergo facile catalytic dehydrogenation, giving polymers that serve as precursors for boron nitride ceramics (Scheme 1a).3,4 Introduction of B–N fragments in polycyclic molecules remarkably enhances charge transport and other properties of organic optoelectronic materials.5–9 | ||
| Scheme 1 Research background and design of this study. | ||
Despite the potential utility of compounds with covalent B–N bonds, the methods for their synthesis are essentially confined to two approaches. The first one relies on the direct reaction between boranes R2BH and amines R2NH to give R2B–NR2via hydrogen evolution, which is mostly used for strongly acidic heterocyclic amines. A more general strategy uses the reaction of boron halides R2BHal with nucleophilic amines. However, it has notably restricted functional group tolerance because of the high reactivity and hydrolytic instability of boron halides. Therefore, the development of new, selective methods for the formation of B–N bonds is highly desirable.
One of the powerful methods for the construction of C–N bonds in complex organic molecules is based on catalytic reactions of nitrenes (Scheme 1b).10,11 In recent years, there has been a significant progress in the asymmetric insertion of nitrenes into C–H bonds, sparking the development of new catalysts and synthetic pathways for the synthesis of valuable chiral amines.12 Surprisingly, the analogous insertion of nitrenes into much more reactive B–H bonds has remained essentially unexplored.13–15
Herein, we report the first detailed investigation of this reaction, which provides a new method for the construction of B–N bonds (Scheme 1c). Due to the high nucleophilicity of boranes, they can react under mild conditions with electrophilic nitrenes generated in situ from the corresponding sulfonamides and sulfamates. Insertion of nitrenes into prochiral boranes with the 2-aryl-pyridine framework in the presence of chiral rhodium and ruthenium carboxylates gives access to unique compounds with stereogenic boron atoms. Notably, this nitrene insertion is the only method for the asymmetric formation of covalent B–N bonds.16,17
Results and discussion
Reaction optimization
We started our investigation by testing the reactions of the common nitrene precursor PhI![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NTs with various borane adducts L-BH3 (1) (Table 1). Rh2esp2 catalyst (esp = tetramethyl-1,3-benzenedipropionate) was employed as a natural choice, known for its exceptional robustness in nitrene chemistry, in contrast to other dirhodium(II,II) carboxylates like Rh2(OAc)4 (ref. 18 and 19) The reaction of PhINTs with the electron-rich carbene–borane 1a in the presence of Rh2(esp)2 (2 mol%) immediately gave a mixture of two insertion products – mono- (2a) and bis-amide (3a) (entry 1). However, further attempts to enhance the selectivity for the formation of 2a by reducing the loading of the nitrene precursor and lowering the temperature were unsuccessful. When the sterically hindered borane adduct 1b bearing two 2,6-diisopropyl-phenyl substituents was used instead of 1a, no conversion of the borane was observed, despite the prompt full consumption of the starting nitrene precursor. Similarly, the reactions of PhINTs with triethylamine and tributyl-phosphine boranes 1c and 1d did not provide the desired products, possibly because of the lower nucleophilicity attributed to the higher strength of B–H bonds.20,21 To our delight, the pyridine–borane complex 1e afforded the desired mono-amide 2e with exceptionally high selectivity (entry 5), although we were unable to isolate it in pure form because of its low stability. Interestingly, nitrenes reacted faster with NHC–borane 1a than pyridine–borane 1e, while the opposite was observed for the carbene insertion reactions.20
NTs into various B–H bonds
NTs with various borane adducts L-BH3 (1) (Table 1). Rh2esp2 catalyst (esp = tetramethyl-1,3-benzenedipropionate) was employed as a natural choice, known for its exceptional robustness in nitrene chemistry, in contrast to other dirhodium(II,II) carboxylates like Rh2(OAc)4 (ref. 18 and 19) The reaction of PhINTs with the electron-rich carbene–borane 1a in the presence of Rh2(esp)2 (2 mol%) immediately gave a mixture of two insertion products – mono- (2a) and bis-amide (3a) (entry 1). However, further attempts to enhance the selectivity for the formation of 2a by reducing the loading of the nitrene precursor and lowering the temperature were unsuccessful. When the sterically hindered borane adduct 1b bearing two 2,6-diisopropyl-phenyl substituents was used instead of 1a, no conversion of the borane was observed, despite the prompt full consumption of the starting nitrene precursor. Similarly, the reactions of PhINTs with triethylamine and tributyl-phosphine boranes 1c and 1d did not provide the desired products, possibly because of the lower nucleophilicity attributed to the higher strength of B–H bonds.20,21 To our delight, the pyridine–borane complex 1e afforded the desired mono-amide 2e with exceptionally high selectivity (entry 5), although we were unable to isolate it in pure form because of its low stability. Interestingly, nitrenes reacted faster with NHC–borane 1a than pyridine–borane 1e, while the opposite was observed for the carbene insertion reactions.20
NTs into various B–H bonds
|
|
||||
|---|---|---|---|---|
| Entry | Borane | Conv.a,b (%) | Yield 2a (%) | Yield 3a (%) |
| a Conversion of the starting boranes and yields of the products were estimated by 1H NMR spectra of the crude mixtures using 1,3,5-tribromobenzene as the internal standard. b Unaccounted amounts of the reacted boranes probably correspond to the formation of polymeric insoluble materials. | ||||
| 1 | 1a | 88 | 24 | 30 |
| 2 | 1b | <5 | <5 | <5 |
| 3 | 1c | 14 | <5 | <5 |
| 4 | 1d | 9 | <5 | <5 |
| 5 | 1e | 52 | 50 | <5 |
| 6 | 1f | 77 | 69 | <5 |
Since only the pyridine–borane selectively yielded the mono-insertion product, we decided to enhance its stability by employing the chelated 2-phenylpyridine borane 1f.22 Indeed, the reaction of 1f with PhINTs also gave only the mono-amide 2f, which, unlike pyridine derivative 2e, can be isolated and purified by standard column chromatography without decomposition. Later inspection revealed that 2f only slowly decomposes upon standing in air in CDCl3 solution (ca. 5% decomposition over 3 days at 20 °C) and can be stored indefinitely as a solid in a freezer. The obtained NHC-amidoboranes 2a and 3a are even more stable and show no decomposition after one week in solution at ambient temperature in air. We were able to confirm the structures of 2a, 3a, and 2f by X-ray diffraction analysis; the structure of 2a is shown in Fig. 1.
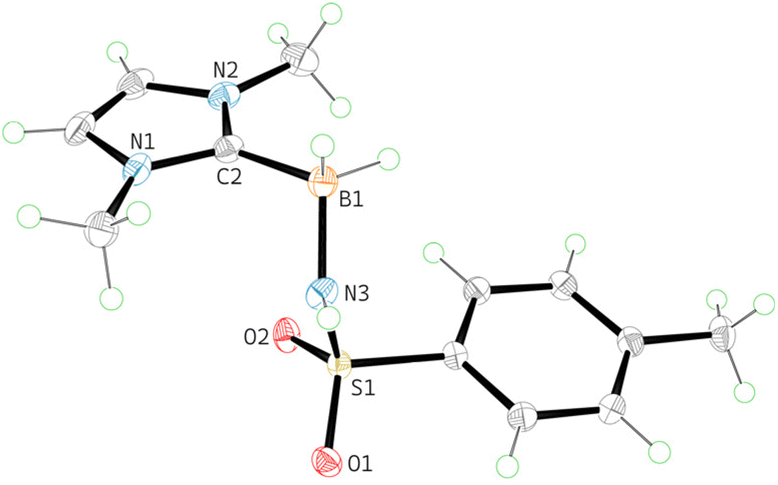 | ||
| Fig. 1 X-ray structure of 2a. Atoms are shown as 50% thermal ellipsoids. BH2 and NH hydrogens were located from the Fourier density synthesis. Selected distances [Å]: B1–C2 1.612(2), B1–N3 1.553(2). | ||
Before proceeding with the investigation of the reaction scope, we explored the activity of other catalysts, which have previously been used in the nitrene insertion reactions, such as Co(TPP) (TPP = tetraphenylporphyrin), [Cu(MeCN)4]+, and [Au(PPh3)]+.12 These complexes also provided the desired product 2f, albeit in significantly lower yields than Rh2(esp)2 (see the ESI†). At the same time, we were pleased to find out that in the case of the Rh2(esp)2 catalyst, the nitrene precursor PhINTs can be generated in situ from tosylamide and PhIO oxidant, despite the presence of the potentially oxidizable borane 1f. The resulting mixture provides 2f in 54% yield, which is only slightly lower than that obtained by the reaction with the preformed iodinane. The control experiments showed that borane 1f does not react with TsNH2 or PhIO alone, while its reaction with TsNIPh in the absence of a catalyst proceeded slowly and unselectively (only ca. 30% of 2f was formed, see the ESI†).
Scope and limitations
With the optimized conditions in hand, we explored the reactivity of various substituted nitrenes in the B–H insertion reaction (Scheme 2). Aryl sulfonamides reacted similarly to TsNH2, giving the desired insertion products 2g–l in ca. 55–65% yields, regardless of the presence of electron-rich (2l, Ar = naphthyl) or electron-poor (2j, Ar = p-nitrophenyl) groups. Even more electron-rich alkyl sulfonamides also reacted smoothly and gave products 2m–o, although the reaction time for these substrates had to be extended up to 24 hours (aryl sulfonamides usually reacted within 1 hour).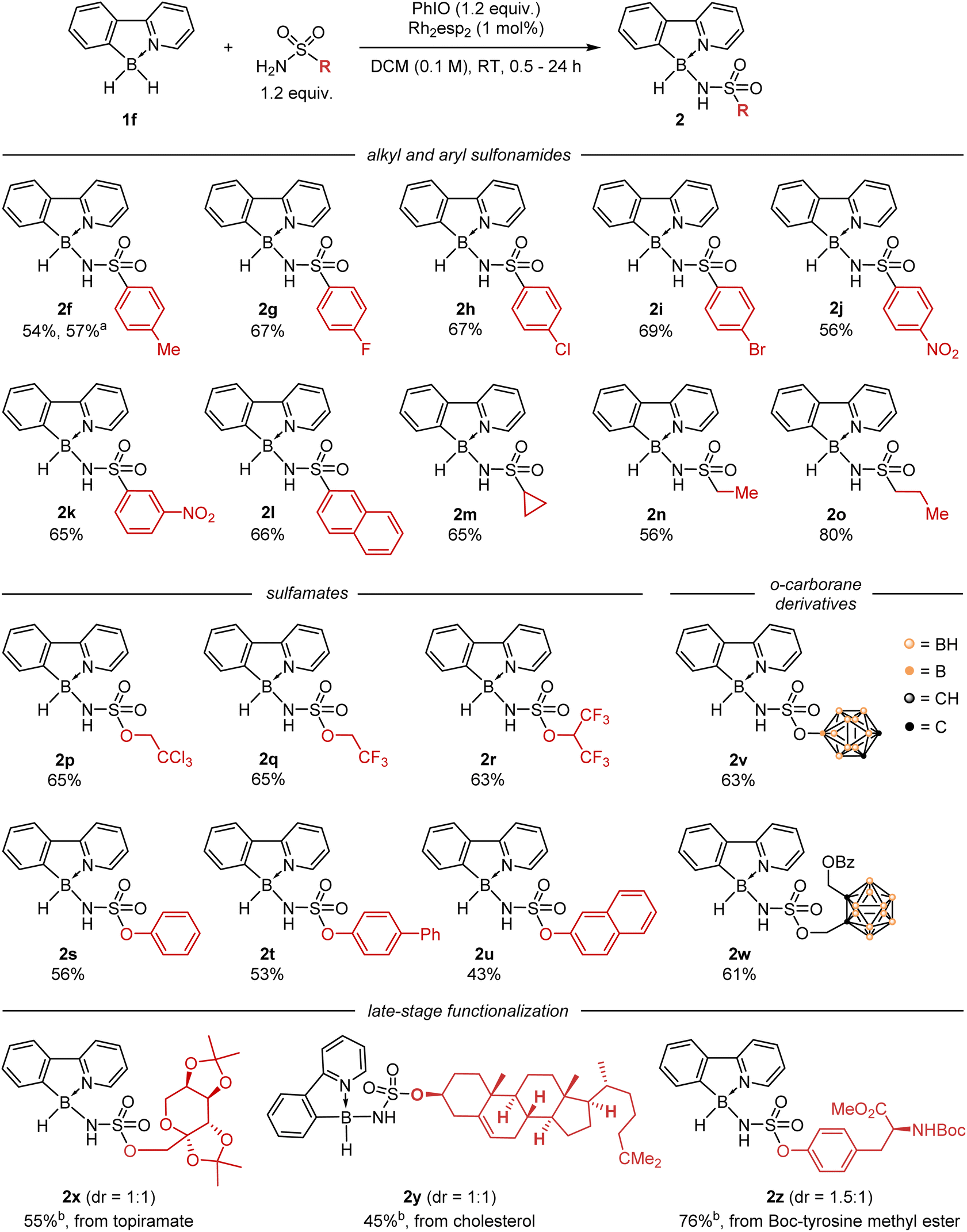 | ||
| Scheme 2 Catalytic insertion of nitrenes derived from sulfonamides and sulfonates into 2-phenylpyridine–borane 1f. Reaction conditions: 1f (0.2 mmol), sulfonamide or sulfonate (0.24 mmol), PhIO (0.24 mmol), Rh2esp2 (1 mol%), DCM (2 ml), RT, 0.5 to 24 h. Isolated yields are given. a Performed on a 6 mmol scale. b Performed on a 0.1 mmol scale. | ||
Then we explored the reactivity of the more electron-deficient sulfamates ROSO2NH2. The classic trichloroethyl sulfamate (TcesNH2)23 reacted with 1f much faster than TsNH2, reaching the full conversion within a few minutes and giving the corresponding product 2p in 65% yield. Other polyhalogenated alkyl sulfamates, as well as aryl sulfamates, provided the expected products 2q–u in comparable yields. It should be noted that the resulting compounds are somewhat less stable than sulfonamide insertion products 2f–o, and some decomposition was observed upon standing in solution in air for 24 hours. Given the interest in carboranes as unique structural pharmacophores for drug design,24,25 we also carried out the reaction with two carborane sulfamate derivatives, which gave the target products 2v and 2w in ca. 60% yields. It is noteworthy that intramolecular insertion of nitrene into less electron-rich B–H bonds of the carborane core was not observed, although similar reactions are known in the literature.14 Finally, we examined the possibility of using the developed strategy for the late-stage functionalization of sulfamates derived from natural compounds. It was found that primary (2x, topiramate derivative), secondary (2y, cholesterol derivative), and aryl sulfamates (2z, tyrosine derivative) reacted smoothly with 1f to give the desired products. This approach may be useful for boron labelling of biologically active molecules.
Overall, this scope demonstrates that borane 1f can react with a variety of nitrene precursors. This situation is markedly different from the catalytic nitrene insertion into C–H bonds, which usually require some specific nitrene precursors such as TcesNH2. These results are consistent with the fact that B–H bonds are, in general, much more reactive than C–H bonds.20 However, nitrenes with low electrophilicity seem to be not suitable for selective B–H insertion. Thus, no reaction occurs upon heating 1f with benzyl azide at 60 °C, while heating with tosyl azide converts only 50% of the borane into 2f after 120 h. The use of the rhodium catalyst Rh2esp2 did not improve the rate and apparently decreased the selectivity of this reaction. No conversion of 1f was observed in the reaction with 3-phenyl-1,4,2-dioxazolone-5 in the presence of the [Cp*RhCl2]/NaBArF4 catalyst26 as well as in the reaction with BocNH2 and PhI(OAc)2 in the presence of Rh2esp2.27 The activated trichloroethyl carbamate TrocNH2 quickly reacted with 1f under similar conditions, however we could not isolate the corresponding insertion product in the pure form due to its low stability.
We further expanded the scope of accessible products by varying the structure of starting boranes (Scheme 3). The introduction of substituents into the phenylpyridine framework did not hamper the insertion of nitrene generated from TcesNH2 and the corresponding products 2aa–ae were obtained in good yields (55–70%). The reaction also proceeded well for the quinoline and dihydroisoquinoline derivatives giving the amidoboranes 2af and 2ag. As noted above, NHC–borane 1a reacts with PhINTs giving a mixture of mono- and bis-insertion products. However, when 1a was first modified by the insertion of methyl diazoacetate into the B–H bond,28,29 further insertion of nitrenes proceeded smoothly and gave the amides 2ah and 2ai in 43% and 58% yields.
 | ||
| Scheme 3 Catalytic insertion of nitrenes into various boranes. Reaction conditions: borane (0.2 mmol), TsNH2 or TcesNH2 (0.24 mmol), PhIO (0.24 mmol), Rh2esp2 (1 mol%), DCM (2 ml). Isolated yields are given. | ||
Mechanistic studies
To get a deeper understanding of the reaction, we performed several mechanistic studies (Scheme 4). In particular, the competitive reaction 1f with PhINTs in neat styrene (91 equiv.) gave mostly the corresponding aziridine (81%) along with only minor amounts of 2f (10%) due to low conversion of 1f (14%). At the same time, a similar reaction in tetralin, which is a common substrate for nitrene insertion, led to the products of amination of B–H and C–H bonds in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio. Considering the ratio of the starting borane to tetralin (1:73) and the number of potentially reactive hydrogen atoms, the B–H bond in 1f appears to be ca. 150 times more reactive than the benzylic C–H bond. Similarly, the competitive reaction of 1f with PhINTs in the presence of triethylsilane revealed that the B–H bond is at least 50 times more reactive than the Si–H bond.
:1 ratio. Considering the ratio of the starting borane to tetralin (1:73) and the number of potentially reactive hydrogen atoms, the B–H bond in 1f appears to be ca. 150 times more reactive than the benzylic C–H bond. Similarly, the competitive reaction of 1f with PhINTs in the presence of triethylsilane revealed that the B–H bond is at least 50 times more reactive than the Si–H bond.
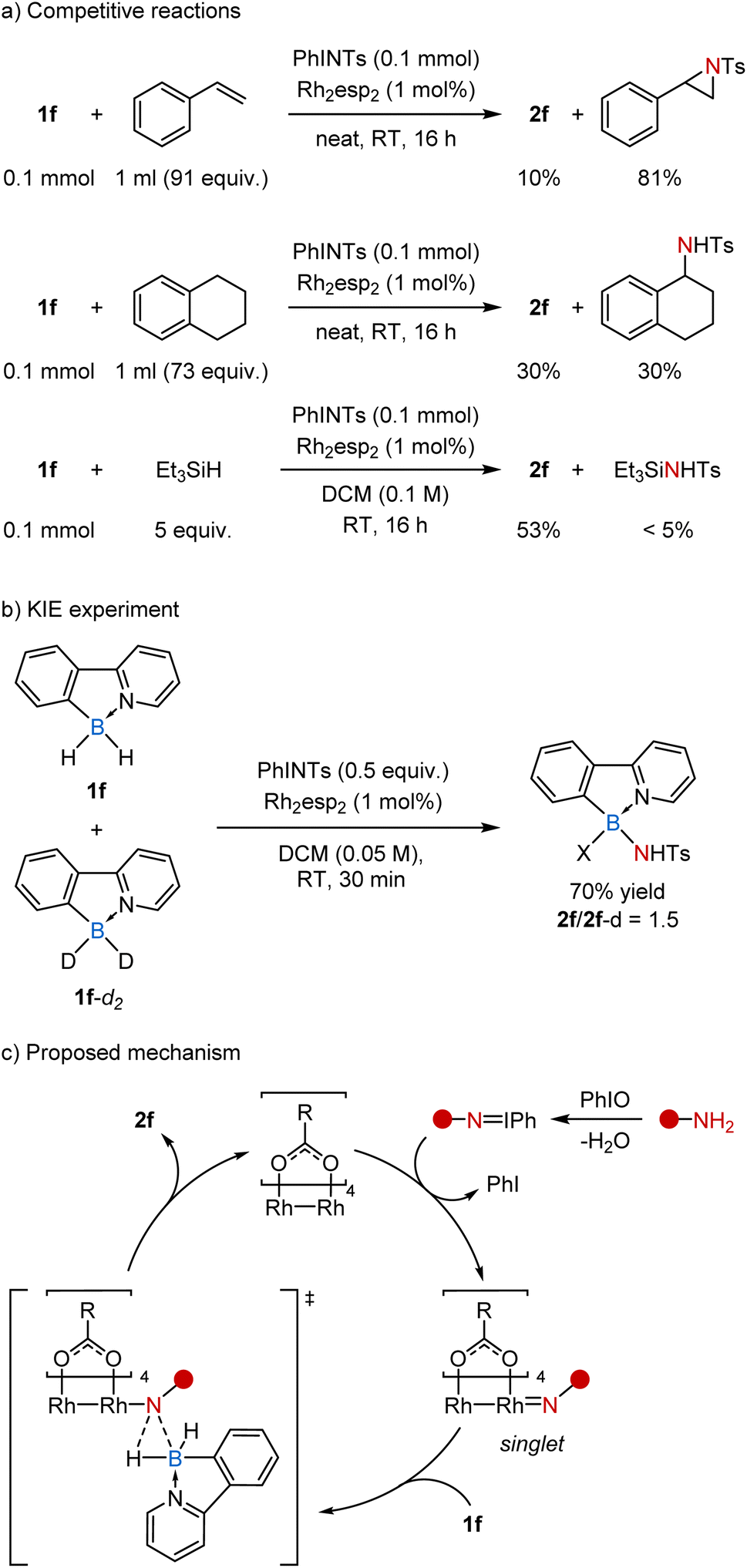 | ||
| Scheme 4 Mechanistic studies. | ||
The competitive reaction of 1f and its deuterated analog 1f-d2 with PhINTs provided a mixture of products 2f and 2f-d in a 1.5:1 ratio, regardless of the conversion of starting materials (Scheme 4b). This kinetic isotopic effect (KIE) is similar to that previously observed for Rh-catalyzed carbene insertion into B–H bonds (KIE = 1.5)30 and somewhat lower than the one obtained for the Rh-catalyzed nitrene insertion into C–H bonds (KIE = 2.6).31 It is generally assumed that KIE values lower than 3 indicate a concerted insertion mechanism involving metal complexes with singlet nitrenes.32 Thus, the reaction appears to follow the classical mechanism, similar to the one proposed for the well-studied C–H insertion reactions (Scheme 4c).33 Initially, amide reacts with PhIO to form the imino–iodane. Next it coordinates with the rhodium catalyst, releases PhI, and forms a singlet nitrene complex. Finally, this electrophilic complex reacts with the nucleophilic borane in a concerted fashion to form the desired B–N bond.
Asymmetric insertion
The cyclic amidoboranes 2 obtained in this work contain a stereogenic boron atom with four different substituents. While chiral stereogenic boron compounds sporadically appeared in the literature, the protocols for their synthesis have not been developed until very recently.16,34 Within the last decade, M.-H. Xu et al.,35–37 S.-F. Zhu et al.,38–40 and several other groups41,42 demonstrated that rhodium and copper-catalyzed insertion of carbenes into B–H bonds can produce boranes with stereogenic carbon atoms. More recently, Yu and Song et al. used a similar reaction of 2-arylpyridine boranes to synthesize compounds with stereogenic boron atoms43,44 (see also a related work on NHC–boranes).45 Therefore, we assumed that similar nitrene insertion can afford the chiral amidoboranes.For our study, we strategically chose the dimethyl-substituted prochiral borane 1g (Table 2) as the starting material over the unsubstituted 1f for two reasons. First, the presence of two methyl groups makes the benzene ring sterically more different from the pyridine ring, which was expected to provide better asymmetric induction. Second, ortho-substituents can prevent potential racemization of the stereogenic boron center via the dissociation of the B–Py bond and the rotation of the BHNHTs group.46
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Catalyst | Yielda, % | erb | Entry | Catalyst | Yielda, % | erb |
| a Isolated yields are given. b Determined by chiral HPLC. c PhCl as a solvent, −30 °C for 24 hours. d o-DCB as a solvent, +4 °C for 6 hours. | |||||||
| 1 | Rh2[(S)-NTTL]4 | 85 | 62:38 |
8 | Ru2[(S)-NTTL]4BArF | 71 | 70:30 |
| 2 | Rh2[(S)-4-Br-NTTL]4 | 78 | 59:41 |
9 | Ru2[(S)-4-Br-NTTL]4BArF | 60 | 68:32 |
| 3 | Rh2[(S)-4-Ph-NTTL]4 | 77 | 67:33 |
10 | Ru2[(S)-4-Ph-NTTL]4BArF | 44 | 60:40 |
| 4 | Rh2[(S)-4-PhPh-NTTL]4 | 69 | 66:34 |
11 | Ru2[(S)-NTTL]4N3 | 69 | 71:29 |
| 5 | Rh2[(S)-4-Np-NTTL]4 | 64 | 65:35 |
12 | Ru2[(S)-NTTL]4NO3 | 63 | 72:28 |
| 6 | Rh2[(S)-4-ArF-NTTL]4 | 71 | 55:45 |
13 | Ru2[(S)-NTTL]4Cl | 63 | 72:28 |
| 7 | Rh2[(S)-4-Ph-NTTL]4c | 91 | 82:18 |
14 | Ru2[(S)-NTTL]4Cl d | 81 | 83:17 |

Since rhodium carboxylates were shown to be the most effective catalysts for the nitrene B–H insertion, we tested the selectivity of different chiral paddlewheel Rh(II,II) complexes in the reaction of 1g with TcesNH2 and PhIO. However, among 12 tested catalysts only one, namely Rh2[(S)-NTTL]4, gave the insertion product 2ga with notable enantioselectivity 62:38 er. We continued catalyst screening by varying the substituents in the naphthalimide core of the NTTL ligand (Table 2). Previously, the Davies group has demonstrated that rhodium complexes with a bromo-substituted TPCP ligand can be easily modified by the classic Suzuki–Miyaura reaction.29 We expanded this approach to NTTL complexes. Thus, fourfold cross-coupling reactions between Rh2[(S)-4-Br-NTTL]4 and various boronic acids gave a series of aryl-substituted Rh2[(S)-4-Ar-NTTL]4 complexes in good yields. Among those, the simple phenyl-substituted catalyst Rh2[(S)-4-Ph-NTTL]4 was found to be the most effective and gave the desired insertion product 2ga with 67:33 er (Table 2, entry 3). Subsequent optimization of the reaction conditions, in particular, lowering the temperature to −30 °C and changing the solvent to chlorobenzene, allowed us to synthesize 2ga in 91% yield and with a decent 82:18 er (entry 7).
In an attempt to enhance the enantioselectivity we decided to replace rhodium carboxylates with their ruthenium analogs [Ru2(OOCR)4]X. Recently, the Matsunaga group has shown that these complexes can outperform classic rhodium carboxylates in terms of robustness and stereoselectivity.47,48 We explored the catalytic activity of several ruthenium carboxylates bearing NTTL-ligands and found that they reacted more slowly than rhodium analogs, but somewhat more selectively (Table 2, entries 8–10). Interestingly, there was no direct succession of the substituent effects in the NTTL core upon the transition from rhodium to ruthenium complexes. For instance, the 4-phenyl-substituted rhodium complex showed the highest enantioselectivity, which was not the case for ruthenium analogs. We briefly investigated the role of the counter-ion X in [Ru2(OOCR)4]X complexes, since Davies et al. recently have shown that it may significantly influence selectivity.49 However, in our case changing X from BArF to Cl, N3, or NO3 had only little effect (entries 11–13). On the other hand, lowering the reaction temperature to +4 °C and changing the solvent to o-C6H4Cl2 allowed us to obtain the target amidoborane 2ga in 81% yield and with 83:17 er using the simple [Ru2((S)-NTTL)4]Cl catalyst (entry 14). Noteworthily, the control experiment revealed that borane 1f can react with highly electrophilic TcesNH2 and PhIO even in the absence of the rhodium or ruthenium catalysts and this side-process can diminish the selectivity at 20 °C.
We then explored the scope of this asymmetric reaction and the effects of substituent in the phenyl ring of phenylpyridine boranes 1g–j on the enantioselectivity (Scheme 5). In contrast to our expectation, the introduction of the bulky tBu group in the ortho-position to the boron atom (2ha) had no positive effect on selectivity and only decreased the overall yield, apparently, because of the excessive steric repulsion. We were also surprised to find that the unsubstituted borane 1f undergoes nitrene insertion with decent enantioselectivity, giving the product R-2m with 83:17 er. Both phenyl and bromo-substituted boranes were converted into the target products with good enantioselectivities 91:9 er.
 | ||
| Scheme 5 Scope of 2-phenylpyridines. | ||
Finally, we explored the asymmetric insertion of different nitrenes into the bromo-substituted borane 1j using the [Ru2((S)-NTTL)4]Cl catalyst (Scheme 6). Two factors were found to be necessary to achieve high efficiency under the optimized conditions: (1) high electrophilicity of the nitrene species (for high yields), and (2) the presence of –OCH2– moiety next to the sulfur (for high stereoselectivity). Thus, electron-deficient sulfamates, such as CF3CH2OSO2NH2, C3F7CH2OSO2NH2, and C6F5CH2OSO2NH2, which were previously found to be effective nitrene precursors,50,51 gave the desired products 2jb–jd in 60–70% yields with about 90:10 er. Other sulfamates and sulfonamides gave amidoborane products 2je–jg with much lower enantioselectivity. In particular, electron-deficient hexafluoroisopropyl sulfamate gave almost a racemic product 2jf, indicating the importance of the –OCH2– group. We also investigated the catalytic activity of the rhodium complex Rh2[(S)-4-Ph-NTTL]4, but it gave products 2ja–je with lower selectivity.
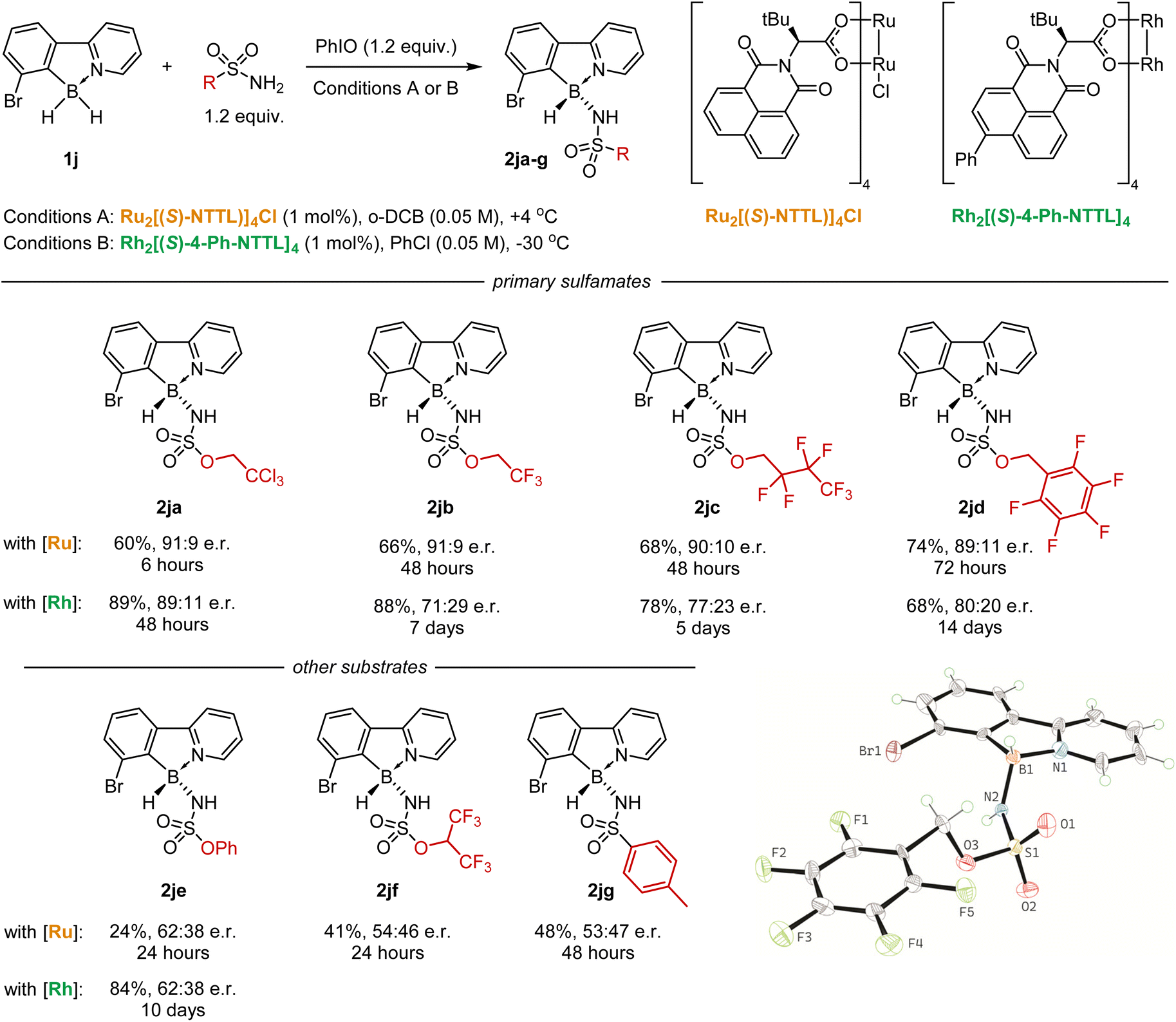 | ||
| Scheme 6 Scope of suitable nitrenes and crystal structure of the chiral product 2jd. | ||
The absolute configuration of the boron stereocenter (R) was established by X-ray diffraction analysis of the product 2jd. Surprisingly, the crystal unit cell contains two independent molecules, the one being the R-enantiomer and the other being the superposition of R- and S-enantiomers with 77% and 23% contributions, respectively. Thus, the overall enantiomeric purity of the crystalline sample closely corresponds to the enantiomeric excess measured in solution by chiral HPLC.
Conclusions
The initial goal of this project was to explore the reactivity of classical nitrene species with non-classical substrates. Interestingly, the least active C–H bonds are in fact the most studied in nitrene insertion reactions. Undoubtedly, catalytic amination of hydrocarbons is a valuable tool in organic synthesis, but why more reactive boranes have not been tested? This question haunted our mind for quite some time, because, from a fundamental viewpoint, nothing should interfere with the existence of such a reaction. As we began our project on B–H nitrene insertion reactions, we quickly realized the main problem was not the reactivity, but the stability of the products. After adjusting the surroundings of the boron center, we found the cyclic arylpyridine boranes to be the most suitable substrates. As a result of these studies, the first method for the catalytic insertion of nitrenes into B–H bonds was developed.Since the resulting cyclic amidoboranes contained a stereogenic boron center, we decided to explore an enantioselective version of this reaction. The only thing to be done was to find a suitable chiral catalyst. This looked like a rather simple task, since many different rhodium carboxylates were introduced since the pioneering studies of Davies and Doyle.52,53 However, only M2[NTTL]4X (M = Rh or Ru) phthalimide carboxylates were found to be effective in terms of stereoselectivity, allowing for the synthesis of chiral-at-boron products with er up to 91:9. Owing to the exceptionally high stability of dirhodium and diruthenium cores, the structures of the catalysts can be tuned by the cross-coupling reactions of the preformed bromo-substituted complexes M[(S)-4-Br-NTTL]4. This approach can certainly be useful for post-modification of other carboxylate complexes. The reasons why only NTTL-bearing catalysts provide reasonable enantioselectivity for nitrene insertion into B–H bonds remain unclear. It is well known that dirhodium(II,II) carboxylates have exceptionally high conformational flexibility. While X-ray analysis of crystals has been previously considered to be helpful for rationalization of the observed selectivity, today it has become clear that one must have information about the three-dimensional shape of the catalyst in solution.54,55 Significant progress has been made to address this issue in the recent studies of Davies and Sigman groups.56–59 However, the proposed descriptor models are still unintuitive and computationally expensive.
Following Matsunaga et al.,47 we confirmed that ruthenium carboxylates can be used as more selective alternatives for the classic rhodium catalysts despite their significantly lower activity. The exceedingly high reactivity of dirhodium paddlewheel complexes allows one to replace them with less active analogs and still carry out reactions under mild conditions.
We believe that the proposed method for nitrene insertion in B–H bonds will be useful for the synthesis of new organoboron compounds, which can find application in materials chemistry and biochemistry. Further efforts may be focused on the improvement of the thermal and chemical stability of the resulting amidoboranes. However, the products obtained in this study are already stable enough to be studied in the context of various applications.60
Data availability
All the data supporting this article have been included in the ESI.†Author contributions
N. M. A. and D. S. P. – conceptualization; N. M. A. and N. V. A. – investigation (lead); E. S. P., D. A. C, K. A. L., and D. S. P. – investigation (supporting); N. M. A. and D. S. P. – writing original draft.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Russian Science Foundation (grant no. 23-13-00345). Analytical data were collected using the equipment of the Center for Collective Use of INEOS RAS with financial support from the Ministry of Science and Higher Education of the Russian Federation (contract no. 075-00276-25-00). X-ray data were collected using the equipment (Bruker D8 QUEST) provided by the Lomonosov Moscow State University and supported by the State budget for scientific research (project no. 121031300090-2).Notes and references
- J. P. M. Antonio, G. D. V. Farias, F. M. F. Santos, R. Oliveira, P. M. S. D. Cal and P. M. P. Gois, in Non-covalent Interactions in the Synthesis and Design of New Compounds, Wiley, 2016, pp. 23–48 Search PubMed.
- Z. Liu and T. B. Marder, Angew. Chem., Int. Ed., 2008, 47, 242–244 CrossRef PubMed.
- A. L. Colebatch and A. S. Weller, Chem.–Eur. J., 2019, 25, 1379–1390 CrossRef PubMed.
- A. Staubitz, M. E. Sloan, A. P. M. Robertson, A. Friedrich, S. Schneider, P. J. Gates, J. S. A. Der Günne and I. Manners, J. Am. Chem. Soc., 2010, 132, 13332–13345 CrossRef PubMed.
- P. G. Campbell, A. J. V Marwitz and S. Liu, Angew. Chem., Int. Ed., 2012, 51, 6074–6092 CrossRef PubMed.
- R. Zhao, N. Wang, Y. Yu and J. Liu, Chem. Mater., 2020, 32, 1308–1314 Search PubMed.
- X. Shao, M. Liu, J. Liu and L. Wang, Angew. Chem., Int. Ed., 2022, 61, e202205893 CrossRef PubMed.
- T. Sakamaki, T. Nakamuro, K. Yamashita, K. Hirata, R. Shang and E. Nakamura, Chem. Mater., 2021, 33, 5337–5344 CrossRef.
- C.-L. Deng, A. D. Obi, B. Y. E. Tra, S. K. Sarkar, D. A. Dickie and R. J. Gilliard, Nat. Chem., 2024, 16, 437–445 CrossRef.
- H. M. L. Davies and J. R. Manning, Nature, 2008, 451, 417–424 CrossRef PubMed.
- S. Y. Hong, Y. Hwang, M. Lee and S. Chang, Acc. Chem. Res., 2021, 54, 2683–2700 CrossRef.
- M. Ju and J. M. Schomaker, Nat. Rev. Chem, 2021, 5, 580–594 CrossRef.
- A. M. Rodríguez, J. Pérez-Ruíz, F. Molina, A. Poveda, R. Pérez-Soto, F. Maseras, M. M. Díaz-Requejo and P. J. Pérez, J. Am. Chem. Soc., 2022, 144, 10608–10614 CrossRef.
- H. Lyu, Y. Quan and Z. Xie, J. Am. Chem. Soc., 2016, 138, 12727–12730 CrossRef.
- A. Grünwald, B. Goswami, K. Breitwieser, B. Morgenstern, M. Gimferrer, F. W. Heinemann, D. M. Momper, C. W. M. Kay and D. Munz, J. Am. Chem. Soc., 2022, 144, 8897–8901 CrossRef.
- X. Li, G. Zhang and Q. Song, Chem. Commun., 2023, 59, 3812–3820 RSC.
- B. Zu, Y. Guo, L. Ren, Y. Li and C. He, Nat. Synth., 2023, 2, 564–571 CrossRef.
- C. G. Espino, K. W. Fiori, M. Kim and J. Du Bois, J. Am. Chem. Soc., 2004, 126, 15378–15379 CrossRef PubMed.
- D. N. Zalatan and J. Du Bois, J. Am. Chem. Soc., 2009, 131, 7558–7559 CrossRef PubMed.
- T. H. Allen and D. P. Curran, J. Org. Chem., 2016, 81, 2094–2098 CrossRef.
- M. Horn, L. H. Schappele, G. Lang-Wittkowski, H. Mayr and A. R. Ofial, Chem.–Eur. J., 2013, 19, 249–263 CrossRef.
- N. Ishida, T. Moriya, T. Goya and M. Murakami, J. Org. Chem., 2010, 75, 8709–8712 CrossRef.
- K. W. Fiori and J. Du Bois, J. Am. Chem. Soc., 2007, 129, 562–568 CrossRef PubMed.
- A. Marfavi, P. Kavianpour and L. M. Rendina, Nat. Rev. Chem, 2022, 6, 486–504 CrossRef.
- E. G. Tse, S. D. Houston, C. M. Williams, G. P. Savage, L. M. Rendina, I. Hallyburton, M. Anderson, R. Sharma, G. S. Walker, R. S. Obach and M. H. Todd, J. Med. Chem., 2020, 63, 11585–11601 CrossRef.
- K. M. van Vliet and B. de Bruin, ACS Catal., 2020, 10, 4751–4769 CrossRef.
- M. Zenzola, R. Doran, R. Luisi and J. A. Bull, J. Org. Chem., 2015, 80, 6391–6399 CrossRef CAS.
- X. Li and D. P. Curran, J. Am. Chem. Soc., 2013, 135, 12076–12081 CrossRef CAS.
- T. H. Allen, T. Kawamoto, S. Gardner, S. J. Geib and D. P. Curran, Org. Lett., 2017, 19, 3680–3683 CrossRef CAS.
- Y. Pang, Q. He, Z.-Q. Li, J.-M. Yang, J.-H. Yu, S.-F. Zhu and Q.-L. Zhou, J. Am. Chem. Soc., 2018, 140, 10663–10668 CrossRef CAS PubMed.
- M. E. Harvey, D. G. Musaev and J. Du Bois, J. Am. Chem. Soc., 2011, 133, 17207–17216 CrossRef CAS PubMed.
- K. W. Fiori, C. G. Espino, B. H. Brodsky and J. Du Bois, Tetrahedron, 2009, 65, 3042–3051 CrossRef CAS.
- E. Azek, M. Khalifa, J. Bartholoméüs, M. Ernzerhof and H. Lebel, Chem. Sci., 2019, 10, 718–729 RSC.
- A. Abdou-Mohamed, C. Aupic, C. Fournet, J.-L. Parrain, G. Chouraqui and O. Chuzel, Chem. Soc. Rev., 2023, 52, 4381–4391 RSC.
- D. Chen, X. Zhang, W.-Y. Y. Qi, B. Xu and M.-H. Xu, J. Am. Chem. Soc., 2015, 137, 5268–5271 CrossRef CAS PubMed.
- W. Xu, T. Yamakawa, M. Huang, P. Tian, Z. Jiang and M.-H. Xu, Angew. Chem., Int. Ed., 2024, 63, e202412193 CrossRef CAS PubMed.
- J.-G. Liu, B. Liu, Z. Li and M.-H. Xu, CCS Chem., 2024, 1–12 Search PubMed.
- Q. Cheng, S. Zhu, Y. Zhang, X. Xie and Q. Zhou, J. Am. Chem. Soc., 2013, 135, 14094–14097 CrossRef CAS.
- Y. Zhao, Y. Su, X. Li, L. Yang, M. Huang and S. Zhu, Angew. Chem., Int. Ed., 2021, 60, 24214–24219 CrossRef PubMed.
- M.-Y. Huang and S.-F. Zhu, Chem. Sci., 2021, 12, 15790–15801 RSC.
- N. M. Ankudinov, D. A. Chusov, Y. V Nelyubina and D. S. Perekalin, Angew. Chem., Int. Ed., 2021, 60, 18712–18720 CrossRef CAS PubMed.
- L. Li, K. Yu, H. An, X. Cai and Q. Song, Chem. Sci., 2024, 15, 7130–7135 RSC.
- G. Zhang, Z. Zhang, M. Hou, X. Cai, K. Yang, P. Yu and Q. Song, Nat. Commun., 2022, 13, 2624 CrossRef CAS PubMed.
- G. Zhang, X. Cai, J. Jia, B. Feng, K. Yang and Q. Song, ACS Catal., 2023, 13, 9502–9508 CrossRef CAS.
- N. M. Ankudinov, A. A. Komarova, E. S. Podyacheva, D. A. Chusov, A. A. Danshina and D. S. Perekalin, Chem. Commun., 2024, 60, 8601–8604 RSC.
- B. Zu, Y. Guo and C. He, J. Am. Chem. Soc., 2021, 143, 16302–16310 CrossRef CAS PubMed.
- T. Miyazawa, T. Suzuki, Y. Kumagai, K. Takizawa, T. Kikuchi, S. Kato, A. Onoda, T. Hayashi, Y. Kamei, F. Kamiyama, M. Anada, M. Kojima, T. Yoshino and S. Matsunaga, Nat. Catal., 2020, 3, 851–858 CrossRef CAS.
- K. Makino, Y. Kumagai, T. Yoshino, M. Kojima and S. Matsunaga, Org. Lett., 2023, 25, 3234–3238 CrossRef CAS PubMed.
- J. K. Sailer, J. C. Sharland, J. Bacsa, C. F. Harris, J. F. Berry, D. G. Musaev and H. M. L. Davies, Organometallics, 2023, 42, 2122–2133 CrossRef CAS PubMed.
- A. Fanourakis, B. D. Williams, K. J. Paterson and R. J. Phipps, J. Am. Chem. Soc., 2021, 143, 10070–10076 CrossRef CAS.
- A. Nasrallah, V. Boquet, A. Hecker, P. Retailleau, B. Darses and P. Dauban, Angew. Chem., Int. Ed., 2019, 58, 8192–8196 CrossRef CAS PubMed.
- M. P. Doyle, W. R. Winchester, J. A. A. Hoorn, V. Lynch, S. H. Simonsen and R. Ghosh, J. Am. Chem. Soc., 1993, 115, 9968–9978 CrossRef CAS.
- H. M. L. Davies, P. R. Bruzinski, D. H. Lake, N. Kong and M. J. Fall, J. Am. Chem. Soc., 1996, 118, 6897–6907 CrossRef CAS.
- H. M. L. Davies and D. Morton, Chem. Soc. Rev., 2011, 40, 1857 RSC.
- F. Adly, Catalysts, 2017, 7, 347 CrossRef.
- Z. Ren, D. G. Musaev and H. M. L. Davies, ChemCatChem, 2021, 13, 174–179 CrossRef CAS.
- R. C. Cammarota, W. Liu, J. Bacsa, H. M. L. Davies and M. S. Sigman, J. Am. Chem. Soc., 2022, 144, 1881–1898 CrossRef CAS.
- Y. T. Boni, R. C. Cammarota, K. Liao, M. S. Sigman and H. M. L. Davies, J. Am. Chem. Soc., 2022, 144, 15549–15561 CrossRef CAS PubMed.
- L. W. Souza, B. R. Miller, R. C. Cammarota, A. Lo, I. Lopez, Y.-S. Shiue, B. D. Bergstrom, S. N. Dishman, J. C. Fettinger, M. S. Sigman and J. T. Shaw, ACS Catal., 2024, 14, 104–115 CrossRef CAS PubMed.
- For example, strong fluorescence properties have been recently discovered for very similar aryl-pyridine borane derivatives. K. Škoch, M. Buziková, D. Hnyk, M. Litecká, M. Kloda, K. Kirakci and K. Lang, Chem.–Eur. J., 2024, e202403263 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: experimental procedures, NMR spectra, HPLC traces, and crystallographic data. CCDC 2347444–2347448. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5sc00723b |
| This journal is © The Royal Society of Chemistry 2025 |