
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDual-atomic Cu–Ag pairs boosting selective electroreduction of CO2 to acetate†
Zemin
Feng
,
Chenghong
Hu
,
Huangcong
Tang
,
Kui
Shen
 ,
Liyu
Chen
* and
Yingwei
Li
*
,
Liyu
Chen
* and
Yingwei
Li
*
Guangdong Provincial Key Laboratory of Fuel Cell Technology, School of Chemistry and Chemical Engineering, South China University of Technology, Guangzhou 510640, China. E-mail: liyuchen@scut.edu.cn; liyw@scut.edu.cn
First published on 21st April 2025
Abstract
Single-atomic Cu catalysts show promise for the electrochemical CO2 reduction (CO2RR) to acetate, but their efficiency is limited by the difficulty in generating the CO intermediate needed for C–C coupling. While co-catalysts can enhance CO generation, weak interaction between co-catalytic and single-atom Cu sites hinders CO spillover, resulting in low acetate yield. Herein, we design atomic Cu–Ag pairs to enhance CO generation and facilitate CO spillover from Ag to Cu in the CO2RR to enhance acetate production. The Cu–Ag/NC catalyst shows a high faradaic efficiency of 50% for acetate and 72% for C2 products at −0.5 V versus a reversible hydrogen electrode, significantly outperforming single-atomic Cu catalysts. Theoretical calculations and in situ characterization demonstrate that the Cu–Ag bonding can facilitate the *CO spillover from Ag to Cu sites, while the electronic modification of Cu by Ag accelerates the subsequent formation of acetate on Cu sites.
1 Introduction
The electrocatalytic CO2 reduction reaction (CO2RR) offers a promising way to utilize renewable electricity to produce value-added products while reducing CO2 emissions.1–8 The CO2RR to single carbon (C1) products (e.g., CO, CH4, CH3OH) has achieved significant progress,9–14 while the production of multi-carbon compounds (ethylene, ethanol, and acetate) with higher value and energy density is less developed.15–18 Among multi-carbon products, acetate is an important bulk chemical, which is widely used in the production of polymers, organic solvents, and medicines.19–23To date, Cu-based catalysts have shown promising results in the CO2RR to ethanol and ethylene, but less success has been achieved in acetate production.24–26 Experimental and theoretical studies suggest that the conversion of CO2 to ethylene and ethanol requires C–C coupling between *CO and *CO or *CHO, requiring the cooperation of two adjacent catalytic sites.27–31 In contrast, acetate formation follows a C–C coupling pathway involving *CH3 and CO2, where isolated active sites may be more beneficial.32,33 However, monoatomic Cu sites have limited intrinsic activity in converting CO2 into *CO, leading to low yields of acetate.34
Two-pot tandem catalytic systems, which consist of two independent electrolyzers to separate CO2-to-CO and CO-to-acetate processes, could afford a high yield of acetate. However, these systems require removal of CO2 to yield high-purity CO for electroreduction of CO to acetate. Integration of CO2-to-CO and CO-to-acetate catalysts in a single system can alleviate this issue, but balancing the CO formation rate with C–C coupling remains a challenge.35 Excess CO reduces the faradaic efficiency (FE) of acetate, while insufficient CO generation lowers acetate yield. This imbalance arises from poor contact between the CO generation sites and C–C coupling sites, limiting CO spillover and acetate production.
In this study, we design dual-atomic metal pairs to facilitate CO generation and spillover between two active sites, improving both activity and selectivity for the CO2RR to acetate. As a proof-of-concept, atomic Ag sites with high CO activity are bonded with atomic Cu sites to promote CO spillover. The catalyst of dual-atomic Cu–Ag pairs anchored on nitrogen-doped carbon (Cu–Ag/NC) achieves a FE of 50% for acetate and 72% for C2 products, significantly outperforming single-atomic Cu catalysts. Theoretical studies and in situ characterization reveal that Cu–Ag bonding shortens the distance of *CO spillover and modifies the electronic structure of Cu, enhancing *CO conversion into acetate.
2 Results and discussion
The synthesis of the Cu–Ag atomic pair catalyst is shown in Fig. 1a. ZIF-8(Cu/Ag) was prepared by doping Cu2+ and Ag+ ions into the ZIF-8 framework. Scanning electron microscopy (SEM) images reveal that ZIF-8(Cu/Ag) has a dodecahedral morphology with an average size of 150 nm (Fig. 1b). Powder X-ray diffraction (PXRD) patterns of ZIF-8(Cu/Ag) confirm that the introduction of Cu2+ and Ag+ metal ions does not disrupt the structure of ZIF-8 (Fig. S1†).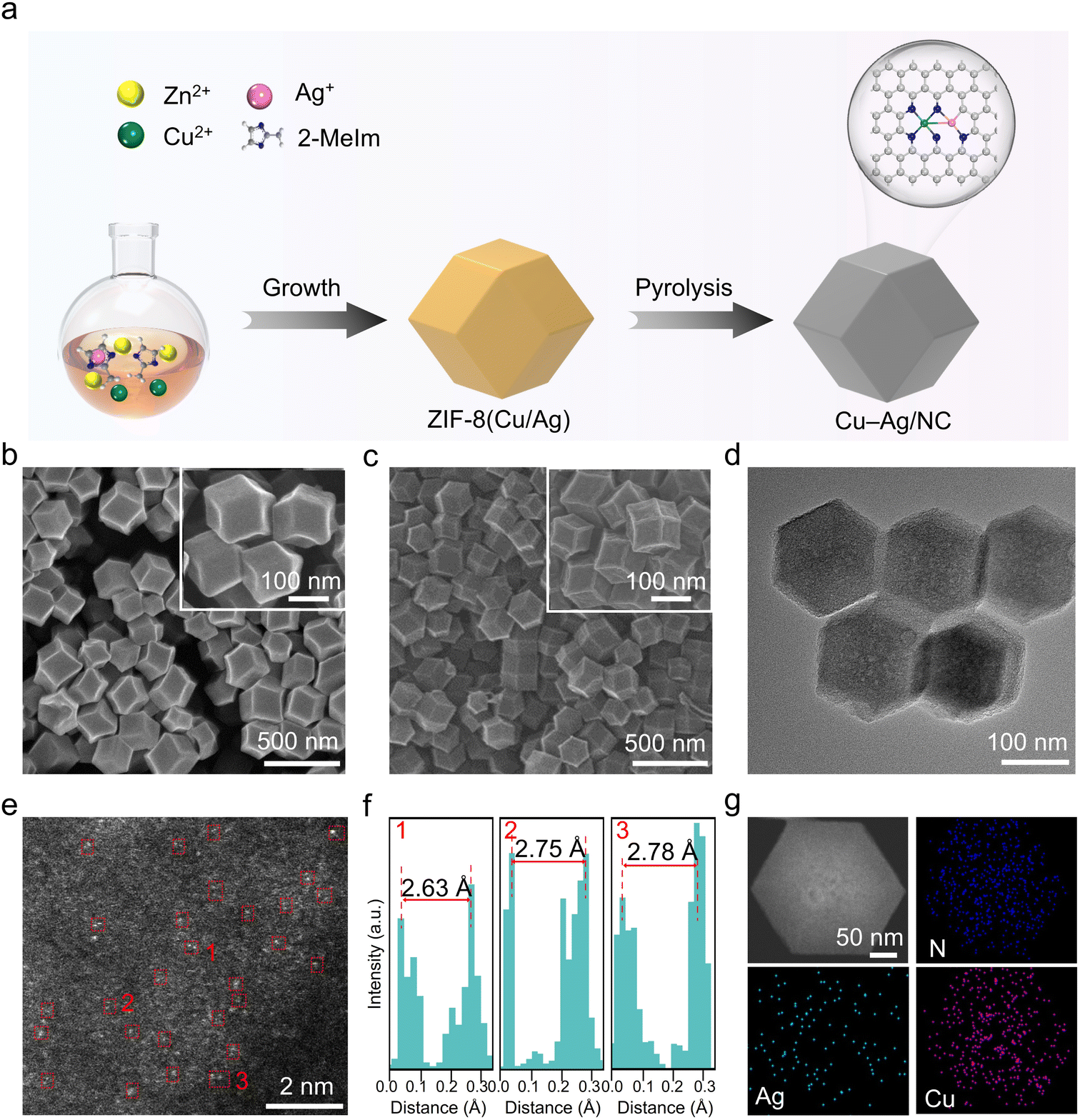 | ||
| Fig. 1 (a) Schematic illustration of the synthesis of Cu–Ag/NC. (b) SEM images of ZIF-8(Cu/Ag). (c) SEM, (d) TEM, and (e) aberration-corrected HAADF-STEM images of Cu–Ag/NC. (f) Line-scanning intensity profiles obtained from regions 1–3 highlighted in (e). (g) HAADF-STEM image and corresponding EDS elemental mapping images of Cu–Ag/NC. | ||
ZIF-8(Cu/Ag) was then calcined at 950 °C under an Ar atmosphere to yield Cu–Ag/NC. SEM images show that Cu–Ag/NC retains the dodecahedral morphology of ZIF-8(Cu/Ag) (Fig. 1c). PXRD patterns of Cu–Ag/NC show two peaks at approximately 28.4° and 42.3°, corresponding to the (002) and (100) planes of graphite carbon (Fig. S2†). Aggregated metal nanoparticles are not observed in the transmission electron microscopy (TEM) image (Fig. 1d). The aberration-corrected high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) image shows neighboring dual dots, suggesting the existence of atomic pairs on carbon substrates (Fig. 1e). The average distance between the dimer dots is measured to be 2.6–2.8 Å (Fig. 1f). Energy-dispersive X-ray spectroscopy (EDS) images show that Cu, Ag, and N are uniformly distributed throughout the structure (Fig. 1g). The contents of Cu and Ag in the Cu–Ag/NC are 0.69 and 0.42 wt%, respectively (Table S1†).
As control samples for the catalytic performance comparison, Cu/NC, Ag/NC, and NC were synthesized using the same procedure with ZIF-8(Cu), ZIF-8(Ag), and ZIF-8 as precursors, respectively (Fig. S3 and S4†). PXRD patterns of Cu/NC, Ag/NC, and NC only exhibit two peaks assigned to carbon, indicating no obvious metal aggregates (Fig. S2†). TEM images of Cu/NC, Ag/NC, and NC also confirm the absence of metal aggregates (Fig. S5–S7†). EDS mapping images further demonstrate the uniform distribution of Cu or Ag on the N-doped carbon matrix.
X-ray photoelectron spectroscopy (XPS) was used to analyze the surface compositions and valence states of Cu–Ag/NC, Cu/NC, Ag/NC, and NC. The Cu 2p XPS spectrum of Cu–Ag/NC displays peaks at 954.4 and 934.2 eV, corresponding to Cu 2p1/2 and Cu 2p3/2 of Cu+. The binding energy of Cu–Ag/NC shifts toward lower values compared to Cu/NC (Fig. 2a). The Ag 3d XPS spectrum of Cu–Ag/NC shifts to higher binding energies compared with Ag/NC (Fig. 2b), indicating electrons transfer from Ag to Cu. Cu–Ag/NC samples with different Ag/Cu atomic ratios also show binding energy shifts in Cu and Ag, further confirming electron transfer from Ag to Cu atoms (Fig. S8 and S9†). The high-resolution N 1s XPS spectra reveal pyridinic (398.5 eV), pyrrolic (400.3 eV), graphitic (401.3 eV), and oxidized N (402.6 eV) species in all samples (Fig. 2c and S10†). The metal–N peak at 399.4 eV indicates the coordination of Ag and Cu atoms to N on the carbon matrix.36
 | ||
| Fig. 2 (a) Cu 2p XPS spectra of Cu–Ag/NC and Cu/NC. (b) Ag 3d XPS spectra of Cu–Ag/NC and Ag/NC. (c) N 1s XPS spectra of Cu–Ag/NC. (d) XANES spectra of the Cu K-edge of Cu foil, CuO, CuPc, and Cu–Ag/NC. (e) Fourier-transform EXAFS spectra of Cu foil, CuO, CuPc, and Cu–Ag/NC. (f) Experimental and fitting EXAFS curves of Cu in Cu–Ag/NC in R space. (g) XANES spectra of the Ag K-edge of Ag foil, Ag2O, and Cu–Ag/NC. (h) Fourier-transform EXAFS spectra of Ag foil, Ag2O, and Cu–Ag/NC. (i) Experimental and fitting EXAFS curves of Ag in Cu–Ag/NC in R space. (j) WT plots of Cu foil, Cu in Cu–Ag/NC, Ag foil, and Ag in Cu–Ag/NC. | ||
X-ray absorption fine structure (XAFS) spectroscopy was employed to investigate the electronic state and local coordination structure of Cu/Ag sites. Cu K-edge X-ray absorption near edge structure (XANES) spectra reveal that the absorption edge position of Cu in Cu–Ag/NC and Cu/NC is located between Cu foil and CuO, indicating that the average valence states of Cu are between 0 and +2 (Fig. 2d and S5†). Compared with Cu/NC, the absorption edge position of Cu in Cu–Ag/NC shifts to lower energy, indicating the electronic donation effect of Ag for the formation of electron-rich Cu+, which is consistent with the XPS results. The Fourier transform (FT) of the extended X-ray absorption fine structure (EXAFS) spectra of Cu–Ag/NC shows two peaks at 1.47 and 2.11 Å, corresponding to Cu–N and Cu–Ag coordination, respectively (Fig. 2e). The Cu–Cu peak is not detected in both Cu–Ag/NC and Cu/NC, confirming that Cu is in atomic form. Similarly, the Ag K-edge XANES spectra indicate that the valence state of Ag is between 0 and +1 (Fig. 2g). The EXAFS spectra of the Ag K-edge show a major peak at 1.61 Å and a second peak at 2.49 Å (Fig. 2h), assigned to Ag–N coordination and Cu–Ag bonding, respectively.
EXAFS wavelet transform (WT) analysis was performed in both k and R space to further discriminate the backscattering atoms (Fig. 2j, S11, and S12†). Compared to the spectra of Cu foil and CuPc, the spectrum of Cu–Ag/NC shows two obvious maximum intensities at 5.08 and 10.74 Å−1, ascribed to the Cu–N and Cu–Ag interaction, respectively. The Cu–Cu path is not detected in the WT-EXAFS of Cu–Ag/NC, suggesting the absence of Cu-containing nanoparticles. In the Ag WT-EXAFS spectrum of Cu–Ag/NC, the Cu–Ag path is not as obvious as that in Cu K-edge wavelet-transform EXAFS, which may be due to the different signal intensities generated by different modes of atomic vibration.37 The quantitative least squares EXAFS curves in R and k space fitting analysis for Cu–Ag/NC indicate that the coordination numbers of Cu–N, Ag–N, and Cu–Ag are 3.8, 1.8, and 1.1, respectively (Fig. 2f, i, S13, and Table S2†). These results confirm the presence of Cu–Ag pairs composed of CuN4 and AgN2 sites, with an electronic interaction between Cu and Ag atoms.
The CO2RR performance of the synthesized catalysts was evaluated using an H-cell with a CO2-saturated 0.1 M KHCO3. Linear sweep voltammetry (LSV) was conducted with a three-electrode system in an H-type cell. The current densities of all catalysts in CO2-saturated electrolytes are higher than those in Ar-saturated electrolytes, indicating a preference for the CO2RR over the HER (Fig. S14†). Within the potential range of 0 to −1.3 V (vs. RHE, the same below if not mentioned), Cu–Ag/NC exhibits higher current densities compared to Cu/NC, Ag/NC, and NC (Fig. S15†). Notably, at −1.25 V, the current densities of Cu–Ag/NC, Cu/NC, Ag/NC, and NC are 29.9, 26.0, 24.1, and 22.1 mA cm−2, respectively. These results demonstrate the superior catalytic activity of dual-atom catalysts to single-atom catalysts.
Gas chromatography was used to quantify gas products, while 1H nuclear magnetic resonance was employed to identify liquid products (Fig. S16†). The distribution of CO2RR products across different potentials was measured for Cu–Ag/NC, Cu/NC, and Ag/NC catalysts (Fig. 3a–c). Cu–Ag/NC produces H2, CO, ethanol, and acetate. The total FEs did not reach 100%, which should be due to the different analytical techniques used for the detection of gas and liquid products. Across the entire potential range, Cu–Ag/NC achieves a maximum FE of 72% for C2 products, with acetate as the dominant product (Fig. 3a). The highest FE of acetate for the Cu–Ag/NC catalyst is 50% at −0.5 V. It is worth noting that Cu–Ag/NC is among the most selective catalysts for acetate (Table S3†). In contrast, Cu/NC reaches only a FE of 10% for acetate at −0.5 V (Fig. 3b), while Ag/NC produces no liquid products and primarily generates CO, with a maximum FE of 67% for CO at −0.6 V (Fig. 3c). The introduction of non-acetate-selective Ag can improve the selectivity of Cu sites toward acetate production. Additionally, Cu–Ag/NC shows lower FE for H2 and CO compared to Cu/NC, indicating that Ag doping significantly suppresses the HER and promotes CO conversion to C2 products (Fig. 3d).
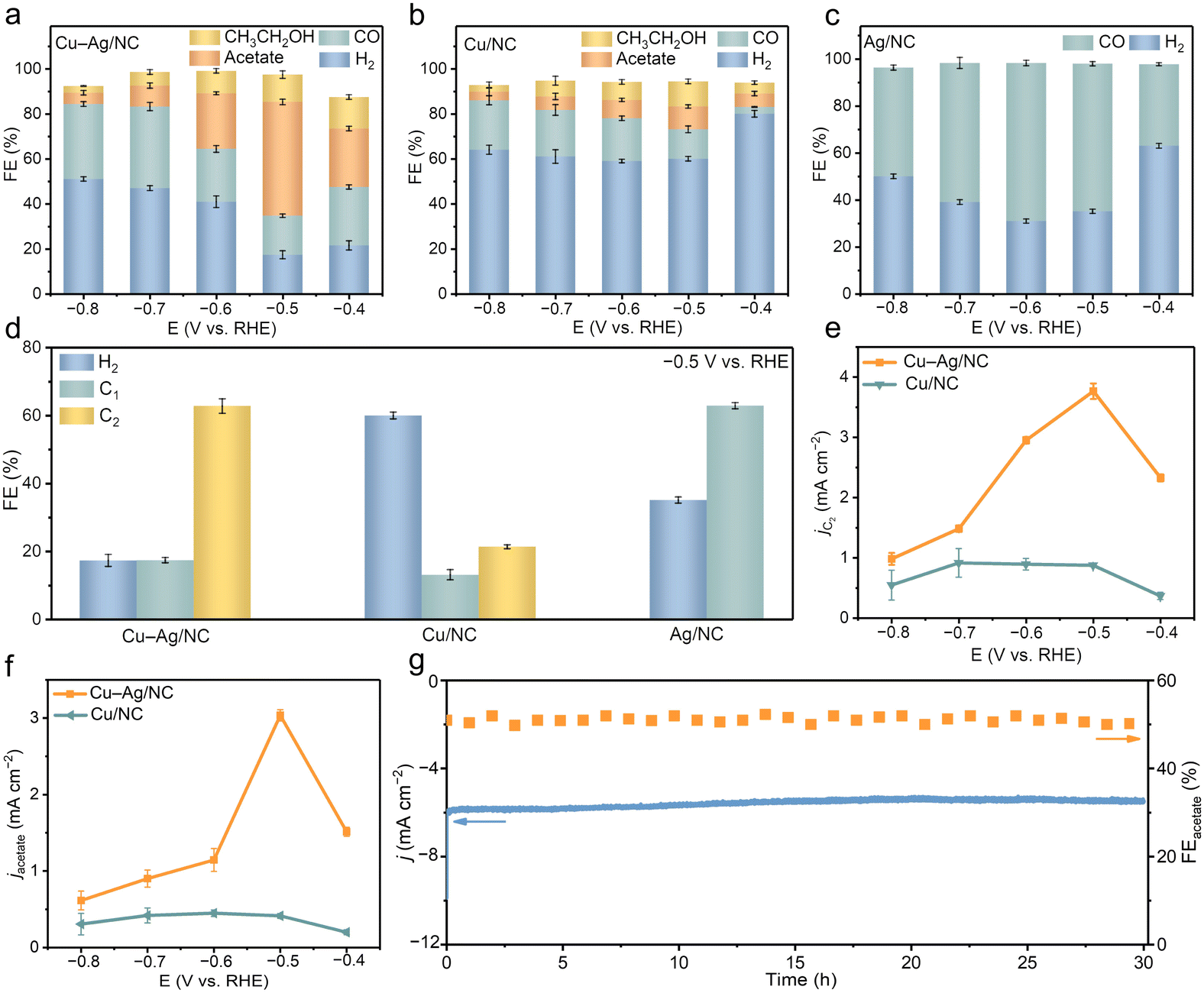 | ||
| Fig. 3 (a–c) FE of all the products obtained on (a) Cu–Ag/NC, (b) Cu/NC, and (c) Ag/NC at different potentials. (d) FE of H2, C1, and C2 on different catalysts. (e) jC2 and (f) jacetate for the CO2RR on Cu–Ag/NC and Cu/NC. (g) The long-term stability of Cu–Ag/NC at −0.5 V vs. RHE. | ||
At −0.5 V, the current densities of C2 products and acetate for Cu–Ag/NC reach 3.77 and 3.01 mA cm−2, respectively, outperforming Cu/NC (Fig. 3e and f). This improvement in acetate yield is attributed to the presence of Ag. Furthermore, we also regulate the proportions of Cu and Ag in Cu–Ag/NC. Cu–Ag/NC catalysts with different Cu-to-Ag ratios show atomically dispersed sites (Fig. S17†). By adjusting the Cu-to-Ag ratio in Cu–Ag/NC, a “volcano” trend in acetate selectivity is observed (Fig. S18 and S19†). Therefore, a suitable Ag content provides an appropriate amount of *CO to further react on the Cu site to produce acetate.
To understand the origin of the high activity of Cu–Ag/NC, we assessed the electrochemically active surface area (ECSA) using cyclic voltammetry (CV) in a non-Faraday region (Fig. S20†). The double-layer capacitance (Cdl) of Cu–Ag/NC is 78.3 mF cm−2, similar to that of Ag/NC (71.5 mF cm−2) and Cu/NC (70.2 mF cm−2) (Fig. S21†), suggesting their similar ECSA. N2 adsorption/desorption measurements show that Cu–Ag/NC has a similar surface area to the other catalysts (Fig. S22–25†). Therefore, the improved performance of Cu–Ag/NC should be attributed to the enhanced intrinsic activity and selectivity due to the presence of Ag. Electrochemical impedance spectroscopy measurement reveals that Cu–Ag/NC exhibits the smallest semicircle in Nyquist plots under open circuit potential (Fig. S26†), indicating its smallest charge transfer resistance during the CO2RR.
The catalytic stability of Cu–Ag/NC was further evaluated through long-term CO2 electrolysis at −0.5 V. The current density remains stable for nearly 30 h (Fig. 3g). XRD patterns of Cu–Ag/NC show no significant changes after the reaction (Fig. S27a†). SEM and TEM images show the maintenance of the structure without aggregation of metal nanoparticles (Fig. S27b and c†). The aberration-corrected HAADF-STEM image reveals the preservation of the atomic pairs on carbon substrates (Fig. S27d†). The Fourier transform (FT) of the XAFS spectrum confirms that metal sites remain in the atomic form after the reaction (Fig. S27e and f†). These results demonstrate the high catalytic activity, selectivity, and stability of Cu–Ag/NC for the CO2RR to acetate.
To investigate the reaction pathways for acetate formation over Cu–Ag/NC, in situ Fourier-transform infrared spectroscopy (FTIR) was used to monitor intermediates during the CO2RR (Fig. 4). At −0.3 V, the FTIR spectra for Cu–Ag/NC and Cu/NC show peaks at 1660, 1420, and 1293 cm−2, assigned to *COOH (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching), *CH3 (C–H bending), and *OOCCH3 (C–O stretching), respectively.38,39 The detected *CH3 and *OOCCH3 intermediates suggest that acetate may be formed from the coupling of *CH3 and CO2. The CO2 partial pressure experiment demonstrates that the FE of acetate decreases as CO2 partial pressure is reduced (Fig. S28†). In addition, a comparative CO reduction experiment shows no formation of acetate (Fig. S29†). These results confirm that CO2 participation is essential for *CH3 coupling. Compared with Cu/NC, the intensities of *COOH and *OOCCH3 on the Cu–Ag/NC are relatively higher, indicating that the introduction of Ag in Cu–Ag/NC is beneficial for the generation of intermediates necessary for acetate production. In addition, the peaks at 2097 cm−1 of Cu–Ag/NC are attributed to the atop configuration of *CO adsorption. However, no obvious *CO adsorption peak is detected for Cu/NC, which is likely due to its rapid conversion into *CH3 upon formation. The observed *CO in the spectra of Cu–Ag/NC likely originates from Ag, where it forms but has not yet transferred to Cu, suggesting that the introduction of Ag accelerates the formation of *CO. Based on the above results, the possible pathway for electrocatalytic CO2 to acetate is proposed. First, CO2 is reduced to *COOH and then to *CO via proton-coupled electron transfer. Second, *CO is hydrogenated to *CHO and then to *CH3. Finally, *CH3 couples with CO2 to form *OOCCH3 and further hydrogenation to acetate.40,41 We can reasonably infer that the single-atomic Ag-modified Cu structure is more favorable for increasing the conversion rate of CO2 to CO and further producing acetate than Cu/NC, which should be the reason of its excellent yield of the acetate products.
O stretching), *CH3 (C–H bending), and *OOCCH3 (C–O stretching), respectively.38,39 The detected *CH3 and *OOCCH3 intermediates suggest that acetate may be formed from the coupling of *CH3 and CO2. The CO2 partial pressure experiment demonstrates that the FE of acetate decreases as CO2 partial pressure is reduced (Fig. S28†). In addition, a comparative CO reduction experiment shows no formation of acetate (Fig. S29†). These results confirm that CO2 participation is essential for *CH3 coupling. Compared with Cu/NC, the intensities of *COOH and *OOCCH3 on the Cu–Ag/NC are relatively higher, indicating that the introduction of Ag in Cu–Ag/NC is beneficial for the generation of intermediates necessary for acetate production. In addition, the peaks at 2097 cm−1 of Cu–Ag/NC are attributed to the atop configuration of *CO adsorption. However, no obvious *CO adsorption peak is detected for Cu/NC, which is likely due to its rapid conversion into *CH3 upon formation. The observed *CO in the spectra of Cu–Ag/NC likely originates from Ag, where it forms but has not yet transferred to Cu, suggesting that the introduction of Ag accelerates the formation of *CO. Based on the above results, the possible pathway for electrocatalytic CO2 to acetate is proposed. First, CO2 is reduced to *COOH and then to *CO via proton-coupled electron transfer. Second, *CO is hydrogenated to *CHO and then to *CH3. Finally, *CH3 couples with CO2 to form *OOCCH3 and further hydrogenation to acetate.40,41 We can reasonably infer that the single-atomic Ag-modified Cu structure is more favorable for increasing the conversion rate of CO2 to CO and further producing acetate than Cu/NC, which should be the reason of its excellent yield of the acetate products.
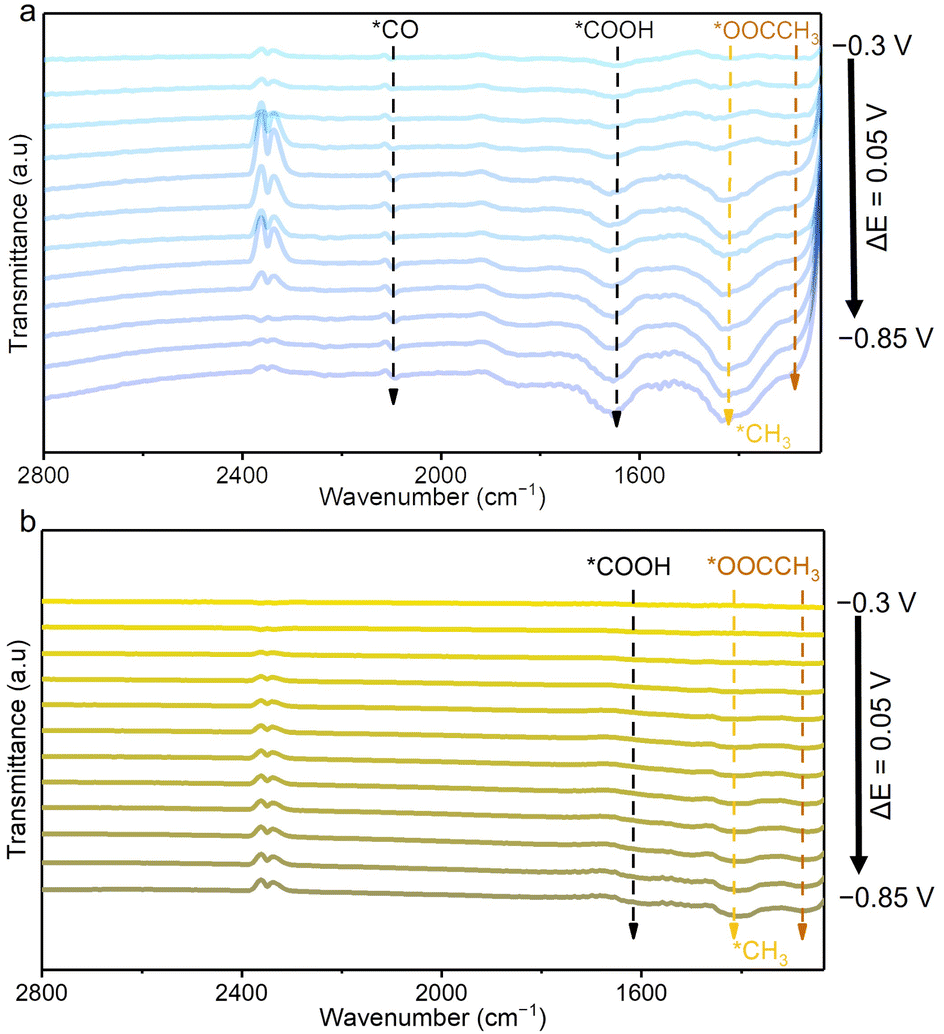 | ||
| Fig. 4 In situ ATR-FTIR spectra of (a) Cu–Ag/NC and (b) Cu/NC under CO2RR conditions. | ||
DFT calculations were performed to elucidate the cooperative role of Cu and Ag sites in the CO2RR to acetate. The catalyst models of Cu–Ag/NC and Cu/NC were built based on the XAFS results (Fig. S30†). Ag/NC is not considered as it produces no acetate under the investigation condition. The Gibbs free energy for intermediate formation on the metal sites of each model was computed. The generation of *CO via proton-coupled electron transfer of CO2 is first calculated (Fig. S31–33†). The free energy change (ΔG) of *COOH formation over the Ag site is much lower than that over Cu sites, indicating that the Ag site in Cu–Ag/NC acts as the active site for the generation of *CO (Fig. 5a). The subsequent protonation of *CO and C–C coupling to afford acetate was calculated (Fig. 5c). The protonation of *CO to *CHO on Cu sites in Cu/NC is exothermic by 0.62 eV. Interestingly, the free energy difference for Cu sites in Cu–Ag/NC is significantly lower than that of Cu/NC, indicating the introduction of Ag can promote the protonation of *CO and C–C coupling to afford acetate. As Cu sites should serve as the active centers for the C–C coupling, the produced *CO may transfer from Ag to Cu sites. We calculated the intrinsic binding strength between Ag and Cu surfaces to CO. The binding energy of CO on the Ag site is weaker than that on Cu (Fig. 5b), indicating that the transfer of CO from Ag to Cu site is thermodynamically favorable. Therefore, CO2 is first reduced to CO over Ag in Cu–Ag/NC and then CO is desorbed from Ag and re-adsorbed to Cu in Cu–Ag/NC to form *CO, which is reduced to *CH3 and coupled with CO2 to form the *OOCCH3 intermediate and finally form acetate (Fig. S34†).
 | ||
| Fig. 5 (a) Calculated free energy of CO2 to CO over Cu in Cu–Ag/NC, Ag in Cu–Ag/NC, and Cu/NC. (b) Calculated binding energy of CO on Ag and Cu sites of Cu–Ag/NC. (c) Calculated free energy of CO to acetate over Cu in Cu–Ag/NC, Ag in Cu–Ag/NC, and Cu/NC. (d) The charge density difference of Cu–Ag/NC and Cu/NC models. The iso-surfaces in yellow and cyan represent electron accumulation and repulsion, respectively. (e and f) Calculated partial density of states for (e) Cu–Ag/NC and (f) Cu/NC. | ||
We also investigated the free energy diagram of the HER using DFT calculations. The Gibbs free energies of *H on the metal atom sites of Cu/NC, Cu in Cu–Ag/NC, and Ag in Cu–Ag/NC are calculated to be 1.60, 1.72, and 2.01 eV, respectively (Fig. S35†). Cu in Cu–Ag/NC shows a higher free energy for *H formation compared with Cu/NC, indicating that the introduction of Ag can suppress the HER. The limiting potential difference (ΔU) between the CO2RR and HER (calculated as UL (CO2) − UL (H2), where UL is the applied potential that all reaction steps are downhill) is used as the indicator of the CO2RR selectivity (Fig. S36†). Cu–Ag/NC shows a more positive ΔU value, further confirming the role of Ag in suppressing the HER and improving CO2RR selectivity.
To elucidate the promoting effect of introduced Ag atoms on the catalytic performance of Cu–Ag/NC sites, the charge density difference and projected density of states were calculated (Fig. 5d). The electron density of Cu sites in Cu–Ag/NC is higher than that of Cu/NC, indicating that electrons transfer from Ag to the Cu center. Bader charge analysis reveals that the oxidation state of Cu for Cu/NC and Cu–Ag/NC is 1.17 and 1.27 e−, respectively. Therefore, the electronic modulation effect offered by the adjacent Ag atom leads to the reduced oxidation state of Cu in Cu–Ag/NC. The partial density of state (PDOS) analysis reveals that the d-band center in Cu–Ag/NC (−3.409 eV) was much closer to the Fermi level than that of Cu/NC (−3.616 eV) (Fig. 5e and f). The upshift of the d-band center in Cu–Ag/NC indicates that Ag enhances the bonding strength of the adsorbate at the Cu surface and lowers reaction barriers for acetate production. These results align with experimental observations, illustrating the synergistic effect of Cu and Ag in promoting the CO2RR to acetate.
3 Conclusion
In summary, we report the construction of dual-atomic Cu–Ag pairs for selective CO2RR to acetate. The neighboring Ag sites provide *CO for Cu sites to promote acetate production. The Cu–Ag/NC catalyst shows excellent CO2RR performance toward C2 products, affording a high FE of 50% for acetate and 72% for C2 products, which are significantly higher than that over Cu/NC. Moreover, Cu–Ag/NC maintains its activity and selectivity during extended operation. DFT calculation and in situ characterization reveal that the Cu–Ag bonding not only promotes the CO spillover, but also modifies the electronic structure of Cu to reduce the reaction barrier of CO to acetate. This work provides valuable insights into the rational design of efficient catalysts for selective CO2RR.Data availability
The data supporting this manuscript have been included as part of the ESI.†Author contributions
L. C. and Y. L. conceived the idea and designed the work. Z. F. performed material synthesis, characterization and catalytic tests. C. H., H. T., and K. S. helped with the characterization. Z. F., L. C., and Y. L. co-wrote the paper. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the National Natural Science Foundation of China (No. 22138003, 22422806, 22378136, and 22108083), the Guangdong Pearl River Talents Program (No. 2021QN02C847 and 2021ZT09Z109), the Natural Science Foundation of Guangdong Province (No. 2024A1515011196 and 2023B1515040005), the Fundamental Research Funds for the Central Universities (2024ZYGXZR011), the Science and Technology Program of Guangzhou (2025A04J5244), the State Key Laboratory of Pulp and Paper Engineering (No. 2023PY06 and 2024ZD09), and the TCL Young Talent Program.Notes and references
- D. D. Zhu, J. L. Liu and S. Z. Qiao, Adv. Mater., 2016, 28, 3423–3452 CrossRef CAS PubMed.
- Y. Wu, S. Cao, J. Hou, Z. Li, B. Zhang, P. Zhai, Y. Zhang and L. Sun, Adv. Energy Mater., 2020, 10, 2000588 CrossRef CAS.
- C. Costentin, M. Robert and J.-M. Saveant, Chem. Soc. Rev., 2013, 42, 2423–2436 RSC.
- O. S. Bushuyev, P. D. Luna, C. T. Dinh, L. Tao, G. Saur, J. V. D. Lagemaat, S. O. Kelley and E. H. Sargent, Joule, 2018, 2, 825–832 CrossRef CAS.
- S. Chen, C. Ye, Z. Wang, P. Li, W. Jiang, Z. Zhuang, J. Zhu, X. Zheng, S. Zaman, H. Ou, L. Lv, L. Tan, Y. Su, J. Ouyang and D. Wang, Angew. Chem., Int. Ed., 2023, 62, e202315621 CrossRef CAS PubMed.
- T. Wang, X. Sang, W. Zheng, B. Yang, S. Yao, C. Lei, Z. Li, Q. He, J. Lu, L. Lei, L. Dai and Y. Hou, Adv. Mater., 2020, 32, 2002430 CrossRef CAS PubMed.
- N. S. Spinner, J. A. Vega and W. E. Mustain, Catal. Sci. Technol., 2012, 2, 19–28 RSC.
- H. Liu, J. Xia, N. Zhang, H. Cheng, W. Bi, X. Zu, W. Chu, H. Wu, C. Wu and Y. Xie, Nat. Catal., 2021, 4, 202–211 CrossRef CAS.
- C. Hu, Y. Zhang, A. Hu, Y. Wang, X. Wei, K. Shen, L. Chen and Y. Li, Adv. Mater., 2023, 35, 2209298 CrossRef CAS PubMed.
- Z. Fan, R. Luo, Y. Zhang, B. Zhang, P. Zhai, Y. Zhang, C. Wang, J. Gao, W. Zhou, L. Sun and J. Hou, Angew. Chem., Int. Ed., 2023, 62, e202216326 CrossRef CAS PubMed.
- S. Dey, F. Masero, E. Brack, M. Fontecave and V. Mougel, Nature, 2022, 607, 499–506 CrossRef CAS PubMed.
- Z. Guo, P. Zhou, L. Jiang, S. Liu, Y. Yang, Z. Li, P. Wu, Z. Zhang and H. Li, Adv. Mater., 2023, 35, 2202298 Search PubMed.
- Y. Qiao, W. Lai, K. Huang, T. Yu, Q. Wang, L. Gao, Z. Yang, Z. Ma, T. Sun, M. Liu, C. Lian and H. Huang, ACS Catal., 2022, 12, 2357–2364 CrossRef CAS.
- S.-H. Zhou, W. Wei, X. Cai, D.-D. Ma, S.-M. Wang, X. Li and Q.-L. Zhu, Adv. Funct. Mater., 2024, 34, 2311422 CrossRef CAS.
- Y. Zheng, A. Vasileff, X. Zhou, Y. Jiao, M. Jaroniec and S.-Z. Qiao, J. Am. Chem. Soc., 2019, 141, 7646–7659 CrossRef CAS PubMed.
- B. Yin, C. Wang, S. Xie, J. Gu, H. Sheng, D.-X. Wang, J. Yao and C. Zhang, Angew. Chem., Int. Ed., 2024, 63, e202405873 CrossRef CAS PubMed.
- C. Xia, X. Wang, C. He, R. Qi, D. Zhu, R. Lu, F.-M. Li, Y. Chen, S. Chen, B. You, T. Yao, W. Guo, F. Song, Z. Wang and B. Y. Xia, J. Am. Chem. Soc., 2024, 146, 20530–50538 CrossRef CAS PubMed.
- J.-X. Wu, S.-Z. Hou, X.-D. Zhang, M. Xu, H.-F. Yang, P.-S. Cao and Z.-Y. Gu, Chem. Sci., 2019, 10, 2199–2205 RSC.
- X. Yan, M. Zhang, Y. Chen, Y. Wu, R. Wu, Q. Wan, C. Liu, T. Zheng, R. Feng, J. Zhang, C. Chen, C. Xia, Q. Zhu, X. Sun, Q. Qian and B. Han, Angew. Chem., Int. Ed., 2023, 62, e202301507 CrossRef CAS PubMed.
- Q. Qian, J. Zhang, M. Cui and B. Han, Nat. Commun., 2016, 7, 11481 CrossRef PubMed.
- K. P. Kuhl, E. R. Cave, D. N. Abram and T. F. Jaramillo, Energy Environ. Sci., 2012, 5, 7050–7059 RSC.
- P. Zhu, C. Xia, C.-Y. Liu, K. Jiang, G. Gao, X. Zhang, Y. Xia, Y. Lei, H. N. Alshareef, T. P. Senftle and H. Wang, Proc. Natl. Acad. Sci. U.S.A., 2021, 118, e2010868118 CrossRef CAS PubMed.
- Y. Ji, Z. Chen, R. Wei, C. Yang, Y. Wang, J. Xu, H. Zhang, A. Guan, J. Chen, T.-K. Sham, J. Luo, Y. Yang, X. Xu and G. Zheng, Nat. Catal., 2022, 5, 251–258 CrossRef CAS.
- P. Zhu and H. Wang, Nat. Catal., 2021, 4, 943–951 CrossRef CAS.
- Y. Guo, H. Yang, X. Zhou, K. Liu, C. Zhang, Z. Zhou, C. Wang and W. Lin, J. Mater. Chem. A, 2017, 5, 24867–24873 RSC.
- B. Yang, L. Chen, S. Xue, H. Sun, K. Feng, Y. Chen, X. Zhang, L. Xiao, Y. Qin, J. Zhong, Z. Deng, Y. Jiao and Y. Peng, Nat. Commun., 2022, 13, 5122 CrossRef CAS PubMed.
- Y. Zhou, F. Che, M. Liu, C. Zou, Z. Liang, P. D. Luna, H. Yuan, J. Li, Z. Wang, H. Xie, H. Li, P. Chen, E. Bladt, R. Quintero-Bermudez, T.-K. Sham, S. Bals, J. Hofkens, D. Sinton, G. Chen and E. H. Sargent, Nat. Chem., 2018, 10, 974–980 CrossRef CAS PubMed.
- A. Bagger, W. Ju, A. S. Varela, P. Strasser and J. Rossmeisl, ACS Catal., 2019, 9, 7894–7899 CrossRef CAS.
- J. Hussain, H. Jónsson and E. Skulason, ACS Catal., 2018, 8, 5240–5249 CrossRef CAS.
- Y. Y. Birdja, E. Pérez-Gallent, M. C. Figueiredo, A. J. Göttle, F. Calle-Vallejo and M. T. M. Koper, Nat. Energy, 2019, 4, 732–745 CrossRef CAS.
- X. J. She, L. Zhai, Y. Wang, P. Xiong, M. M.-J. Li, T.-S. Wu, M. C. Wong, X. Guo, Z. Xu, H. Li, H. Xu, Y. Zhu, S. C. E. Tsang and S. P. Lau, Nat. Energy, 2024, 9, 81–89 CrossRef CAS.
- Z. Wei and P. Sautet, Angew. Chem., Int. Ed., 2022, 61, e202210060 CrossRef CAS PubMed.
- D. Zang, Q. Li, G. Dai, M. Zeng, Y. Huang and Y. Wei, Appl. Catal., B, 2021, 281, 119426 CrossRef CAS.
- X.-F. Qiu, J.-R. Huang, C. Yu, Z.-H. Zhao, H.-L. Zhu, Z. Ke, P.-Q. Liao and X.-M. Chen, Angew. Chem., Int. Ed., 2022, 61, e202206470 CrossRef CAS PubMed.
- H.-L. Zhu, J.-R. Huang, M.-D. Zhang, C. Yu, P.-Q. Liao and X.-M. Chen, J. Am. Chem. Soc., 2024, 146, 1144–1152 CrossRef CAS PubMed.
- C. Hu, Y. Wang, J. Chen, H.-F. Wang, K. Shen, K. Tang, L. Chen and Y. Li, Small, 2022, 18, 2201391 CrossRef CAS PubMed.
- Y. Li, B. Wei, M. Zhu, J. Chen, Q. Jiang, B. Yang, Y. Hou, L. Lei, Z. Li, R. Zhang and Y. Lu, Adv. Mater., 2021, 33, 2102212 CrossRef CAS PubMed.
- P. Zhao, H. Jiang, H. Shen, S. Yang, R. Gao, Y. Guo, Q. Zhang and H. Zhang, Angew. Chem., Int. Ed., 2023, 62, e202314121 CrossRef CAS PubMed.
- M.-D. Zhang, D.-H. Si, J.-D. Yi, S.-S. Zhao, Y.-B. Huang and R. Cao, Small, 2020, 16, 2005254 CrossRef CAS PubMed.
- H. Cheng, X. Wu, M. Feng, X. Li, G. Lei, Z. Fan, D. Pan, F. Cui and G. He, ACS Catal., 2021, 11, 12673–12681 CrossRef CAS.
- M. Feng, X. Wu, H. Cheng, Z. Fan, X. Li, F. Cui, S. Fan, Y. Dai, G. Lei and G. He, J. Mater. Chem. A, 2021, 9, 23817–23827 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc07772e |
| This journal is © The Royal Society of Chemistry 2025 |