
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCustomizing circularly polarized afterglow by stepwise chiral amplification in BINAPs/BINAPOs†
Bo
Yang
a,
Suqiong
Yan
a,
Shirong
Ban
a,
Hui
Ma
a,
Yuan
Zhang
a,
Fanda
Feng
a and
Wei
Huang
 *ab
*ab
aState Key Laboratory of Coordination Chemistry, School of Chemistry and Chemical Engineering, Nanjing University, Nanjing 210023, P. R. China. E-mail: whuang@nju.edu.cn
bShenzhen Research Institute of Nanjing University, Shenzhen 518057, P. R. China
First published on 21st April 2025
Abstract
Overcoming spin-forbidden radiation in chiral phosphors has attracted enormous attention because of their capacity to exhibit circularly polarized organic ultra-long room temperature phosphorescence (CP-OURTP). However, their development has been hindered by the short lifetimes and low dissymmetry factors, which are attributed to the differing parity selection rules that govern the electric and magnetic dipole moments in chiral molecules and poor triplet populations via intersystem crossing (ISC). Considering stepwise chiral amplification at molecular and supramolecular aspects, herein, we first reported donor-decorated BINAPs/BINAPOs with tunable D–A character and triplet incubation, which could enable hybridized local and charge-transfer (HLCT) characteristics, heavy atoms, and p–π* effects. These emitters could serve as guests in the polymer matrix. The doped phosphorescent polymer exhibits unimolecular circularly polarized luminescence (C) with high quantum efficiency, impressive CP-OURTP lifetimes (up to 1.02 s), and decent dissymmetry factors (10−3 level). Comprehensive studies unveil that the impressive CP-OURTP from monomer emission is ascribed to the 1HLCT-controlled ISC, long-lived 3LE-governing triplet radiation, and superior electric–magnetic dipole moment environments. Moreover, given the high RTP activity of rigid polymerization, we demonstrate their potential application in CP-OURTP amplification. Using in situ chiral liquid crystal polymerization, RM257 liquid crystals doped with 0.1–1.0 wt% PO1 guests demonstrate a secondary helical assembly, showing an amplified gCP-RTP factor (±0.11) and a long lifetime (0.83 s) after photopolymerization. The current materials' excellent performance in CP-OURTP and structural dependence could lead to their use in afterglow patterns for multiple optical encryption.
Introduction
The exploitation of organic chiral materials with optimized luminous efficiencies and multiple exciton harvest channels is a stepping stone for light-emitting materials, such as circularly polarized organic light-emitting diodes (CP-OLEDs)1–3 and chirality-induced spin electronics.4 In contrast, the development of CP-OURTP materials has been extremely slow.5–10 However, CP-OURTP unlocks opportunities for promising applications in circularly polarized phosphorescent OLEDs,11,12 3D imaging,13 biological imaging,14 and photodynamic therapy.15 Up to now, the gCP-RTP factor of reported CP-OURTP emitters has struggled to exceed ±0.1 and they have synchronously suffered from poor quantum efficiency and inferior phosphorescence lifetimes (τphos).16 Although some progress has been made through rational molecular design and dual-emissive material design,17–23 efficient unimolecular CP-OURTP is still scarce for simple small molecules.24–30 Therefore, exploiting high-performance CP-OURTP backbones with |gCP-RTP| > 0.1 and τphos > 0.5 s is urgent for material applications.In principle, two aspects might be thoroughly considered in the design of RTP emitters. For instance, heteroatom/heavy insertion and self-assembly strategies have been proposed to accelerate spin–orbit coupling (SOC) and spin-flipping rates based on the El-Sayed rule.27–30 Meanwhile, the facile modulation of charge transfer by structural tailoring is important for differing parity selection rules that govern the electric and magnetic dipole moments in an ideal circularly polarized light pump.22,31,32 Recently, the HLCT tactic has become a novel design landscape in hot-exciton emitters, which has benefited from the conspicuous ISC or high-lying reversal ISC (hRISC) processes and triplet populations via hybrid LE and CT transitions.33–35 Their selective ISC/RISC avenues could be adjusted by optimizing energy levels and CT components at molecular levels, leading to phosphorescent afterglow or thermally active delay fluorescent (TADF) afterglow.36,37
Recently, many researchers have been inspired by p–π*/n–π* effects and have presented new design protocols to control RTP emergence in phosphorus compounds. For instance, some donor-substituted phosphines and their oxides/phosphoniums have demonstrated crystalline RTP emission, where resonance-induced ISC strengthens and mixed p–π* & π–π* transitions and intermolecular electron coupling are responsible for better triplet populations.38–47 Although some rigid crystals show CP-OURTP with low g factors (Fig. 1a, |gCP-RTP| = 10−3 level),48,49 their emission is annihilated in solution and doped polymers because of structural motion, where the emission is driven by rigid electronic interactions from both intra- and intermolecular components, as opposed to those derived solely from unimolecular entities.48,50 This limits the possibility of desired CP-OURTP amplification by simple doping dispersion and complex chiral self-assembly.
 | ||
| Fig. 1 (a) The well-known design of phosphorus compounds for RTP/CP-OURTP. (b) TD-DFT guided molecular design and chemical structures of CP-OURTP active BINAPs. Schematic CP-RTP enhancement by stepwise chiral induction and the amplification mechanism. | ||
For a typical BINAP matrix, the orthotropic orientation and structural motion of two naphthyls create inefficient π-extension, weak light absorbance, and limited triplet populations,50,51 which prevent the visible light radiation of phosphorescence and fluorescence in the unimolecular state. As demonstrated by Wu et al., racemic BINAP crystals possess p–π* & π–π* transitions and weak RTP in the microsecond range (Fig. 1a).50 In addition, Kono et al. reported that BINAP doped polymethyl methacrylate (PMMA) films have circularly polarized fluorescence (CPF) in the UV region (λem = 355 nm, ϕPL = 3%, and gCPF ≈ 10−4). Similarly, their oxides (BINAPOs) display low CPF performances (λem = 354 nm, ϕPL = 7%, and gCPF ≈ 1.2 × 10−3). However, the phosphorescence has not been mentioned in their report due to weak emissive status.51 Based on this context and quantum chemistry computation-guided molecular design, we suppose that the chiral electronic transition could be partly switched from π–π* to HLCT by integrating the weaker D–A moiety with significant steric hindrance (Fig. 1b). The presence of HLCT and multiple p–π* effects stemming from the lone-pair electrons residing on the N and heavy P atoms can facilitate the acceleration of ISC, which successfully produces stable triplet populations. Thus, these emitter-doped PVP or PMMA could emit CP-OURTP with decent fluorescence and phosphorescence glum metrics (gCPF = 6.4 × 10−3 and gCP-RTP = 3.0 × 10−3) and long RTP lifetimes (up to 1.02 s), which are significantly higher than those of previous chiral phosphines and BINAP. In addition, the multiple SOC channels and high-lying reversal ISC (RISC) processes can be altered by manipulating the P-atom valence state or donor, which subsequently enables efficient fluorescence or RTP in solution, solid, and polymer states, transcending the limitations of phosphine crystals for flexible device manufacturing. Importantly, assisted by the chiral induction between the PO1 phosphor and polymerization precursor (RM257), chiral liquid crystal structures and rigid environments are obtained synchronously via chiral photopolymerization (Fig. 1b). Finally, these helical films exhibit an amplified gCP-RTP factor of ±0.11 and a lifetime of 0.83 s.
Results and discussion
Molecular design, synthesis, and characterization
Conventional phosphines and their derivatives (Fig. 1a) typically suffer from a low quantum efficiency (ϕPL) and small π-conjugation, making them unsuitable as CP-RTP emitters. For example, commercial BINAPs only show weak CPF in the UV region.51 At first, we have explored their triplet situation in a unimolecular state according to low-temperature phosphorescence measurements. As shown in Fig. 2, the (R)-BINAP in 2-methyltetrahydrofuran can emit both fluorescence and phosphorescence at 77 K (Fig. 2c). The fluorescent and phosphorescent bands are centered at 367 and 500 nm, respectively. The phosphorescence lifetime reaches up to 1.2 s (Fig. S1c†). Moreover, the (R)-BINAP-doped PVP film facilitates weak fluorescence and green RTP emission (τphos = 0.4 s, Fig. 2d and f), which is identical to that in cryogenic solutions. For this long-lived triplet decay, time-dependent density functional theory (TD-DFT) calculations reveal hybrid p–π* & π–π* transitions for S0 → S1/Tn (Fig. 2b), which cause limited ISC because of large single-triplet splitting energy (1.07 eV) and a small SOC element (0.40 cm−1, Fig. 2e and S2†). Unfortunately, its ϕPL is too low (2.4%). This discovery motivates us to further optimize the molecular structure, especially for energy level optimization and quantum efficiency improvement. | ||
| Fig. 2 (a) Target chemical structures with two TPA groups at different positions. Their θe-m and gabs factors are obtained using TD-DFT calculations. (b) Electron and hole distribution analysis of S0 → S1, T1, and T8 transitions. (c) PL and delay PL (DPL) emission of (R)-BINAP in 2-Me-THF at 77 K (delay 0.1 s, λex = 320 nm). (d) PL and DPL emission of (R)-BINAP@PVP at 298 K (0.5 wt%, delay 0.1 s, λex = 320 nm). (e) The energy-level diagrams and SOC coefficients (ξsoc) for (R)-BINAP. (f) Lifetime decay profiles of (R)-BINAP@PVP at 298 K. (g and h) Single crystal structures of BINAP and (Rac)-P3, respectively. The H-atoms are omitted for clarity. (i) Crystal packing of (Rac)-P3. | ||
Based on the inspiration of similar valence electrons and electronegativity of N and P elements, extended electron-rich arylamines in BINAP are beneficial for incubating extra CT characteristics and enhancing quantum efficiencies. However, the best chiral transition environment during the emission is also required. At first, gabs is predicted using the formula according to the density functional theory (DFT):52
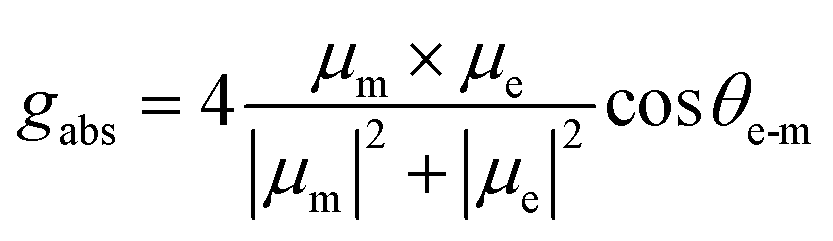
BINAP and five BINAP derivatives have been simulated using TD-DFT calculations. Interestingly, BINAP possesses a moderate θe-m of 31.3° and |gabs| of 0.01. However, the θe-m values are increased for 3,3′/4,4′-substituted BINAPs, in which the μm and μe are orthometric, resulting in worse chiral conditions. The θe-m values are decreased from 4,4′ to 7,7′-substituted BINAPs, while the gabs factors are gradually increased (Fig. 2a and S3†). The 7,7′-substituted BINAPs could employ an outstanding S0 → S1 transition (|gabs| = ∼0.01), which is very close to that of the BINAP matrix. Following this theoretical guidance, we introduce donors of N-phenylcarbazole (CZ) or triphenylamine derivatives (TPAs) at 7,7′-positions to build rigid and weak D−A systems.53,54 Moreover, by oxidation of tervalent phosphines, pentavalent D–A emitters are facilely established to modulate the electronic structure and luminescence (Fig. 1b).
The synthesis and separation of target racemic P1–P3 are achieved according to the protocol in our recent work (Fig. S4†).55,56P1 and P2 could be resolved into their pure enantiomers by high-performance liquid chromatography (HPLC) on chiral stationary phase columns of CHIRAL IE or CHIRAL IB-N5 (>99% ee, Fig. S5†). Employing oxidation by hydrogen peroxide, extra PO1–PO3 emitters are obtained in equivalent yields. The structures of products are confirmed by nuclear magnetic resonance (NMR), high-resolution mass spectrometry (HRMS), and single-crystal X-ray diffraction (SCXRD, see the ESI† for details).
X-ray diffraction analyses
The SCXRD analysis of (Rac)-P3 (CCDC 2344550) reveals a C2-symmetrical conformation with an axially chiral dihedral angle of 75.7°, a large N⋯P isolation of 9.40 Å, and a P⋯P distance of 3.88 Å (Fig. 2h and S6†), implying intramolecular charge transfer potential within the compound. The periodic (R) and (S)-isomers of P3 are packed in cells without obvious π–π interactions by virtue of nonplanar geometry (Fig. 2i and S6a–f†), which is well conducive to suppressing the π–π aggregation and fluorescence quenching. DFT-optimized P3 has N⋯P and P⋯P lengths of 8.66 and 4.18 Å, respectively. After the oxidation of P-atoms to afford PO3, a shorter N⋯P distance (8.58 Å) but longer P⋯P separation (4.31 Å) is established according to the density functional theory (DFT) simulation (Fig. S6g†). Due to the repulsion of two P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O units, the dihedral angle of naphthyl exhibits a more splayed configuration with a larger degree of 80.6°, which could influence the electron transfer process and optical properties. In contrast to the precursor, the chiral dihedral angle of P3 (75.7°) is smaller than that of BINAP (88.5°), while the P⋯P distance also follows this tendency (Fig. 2g and h).
O units, the dihedral angle of naphthyl exhibits a more splayed configuration with a larger degree of 80.6°, which could influence the electron transfer process and optical properties. In contrast to the precursor, the chiral dihedral angle of P3 (75.7°) is smaller than that of BINAP (88.5°), while the P⋯P distance also follows this tendency (Fig. 2g and h).
Theoretical studies
We performed DFT theoretical calculations at the B3LYP/6-31G(d) basis57 set for all compounds to understand the frontier orbitals and excited-state properties (Fig. S7–S23†). Both compounds adopted similar scissor-like configurations with the BINAP core, consistent with the single-crystal structure (Fig. 3a). The highest occupied molecular orbital (HOMO) electrons are equally distributed across carbazole and binaphthyl cores but the lowest occupied molecular orbital (LUMO) electrons are predominantly localized on binaphthyl moieties in P1 (Fig. 3a), which facilitate typical HLCT in the electron excitation process.34,58 In addition, P1 displays a small tindex value (0.034 Å) and hole–electron centroid distance (Dindex = 1.82 Å), coupled with a substantial Sr function of 0.61 a.u. (denotes overlap integral of electrons and holes over 61%; the definitions of tindex, Dindex, and Sr functions are given in the DFT calculation section in the ESI†),59,60 indicating partial CT contributions in this weak D–A structure. Notably, as the donor character increases from P1/PO1 to P3/PO3, a gradual separation between the HOMO and LUMO is observed, resulting in more efficient CT and an orbital gap (ΔEH-L) decrease from 3.77 to 3.40 eV. The HOMO level is elevated, while the LUMO is decreased. Moreover, the calculated oscillator strengths (f) of S1 are increased with donor strength. | ||
| Fig. 3 Theoretical calculation. (a) The analysis of HOMO and LUMO distributions and their calculated energy gaps of ΔEH–L (isovalue = 0.02). (b) Luminescence mechanism of the HLCT-governed CP-OURTP emission for the doped films based on weak D–A–D emitters. (c) The energy-level diagrams, molecular orbital characters, and SOC coefficients (ξsoc) for P1, PO2, and PO3. (d) Electron and hole distribution analysis of S0 → S1 and S0 → T1/T5 transitions at the S0 geometry (isovalue: 0.002). | ||
The singlet and triplet energy levels and key transition configurations calculated using TD-DFT are illustrated in Fig. 3b, c, and S10–S23.† Both P(III) and P(V) compounds (S1-geometry) have distinct S1–T1 energy gaps (0.85 to 0.20 eV), where the gaps are smaller for TPA substitutes because of significant CT components. The lowest triplet level of T1 is close to S1 in PO3, with a very narrow splitting energy of 0.20 eV. Importantly, the CT-type structures of P3 and PO3 display small energy gaps of S1-T1 (0.21 and 0.20 eV), which implies possible RISC at room temperature in these D–A architectures, where the corresponding S1 and T1 states belong to 1CT and 3LE.35,61 In contrast, P1 has great S1–T2/T1 gaps as high as 0.67 and 0.85 eV. The S1 states of P1 and PO1 possessed similar HLCT transition characteristics, i.e., CZ to BINAP and π–π* transitions of BINAP. The T1 state is governed by 3LE (π–π*) in binaphthyl itself, and the higher T3/4 transition originates from the delocalized CZ donors and binaphthyl acceptor, together with low ΔE(S1–T3/4) (Fig. 3d, S11 and S12†). The P and N atom orbitals also participate in these above-mentioned charge transfers, i.e. the N-atoms contribute larger HOMO–LUMO transition components (−14.81 to −22.91%) than P-atoms (4.26 to 0.68%) for six compounds (Fig. S7–S9†). These features could promote spin-flipping via the SOC process between the p–π* and π–π* configurations, and the current systems could follow the analogous El-Sayed rule and CT effect.9,15 Interestingly, the donor effect gradually overwhelms the valence state for orbital level modulation (Fig. 3a). For example, the change of the energy levels and SOC values is negligible between P3 and PO3, while the diversities between P1 and PO1 are large (Fig. S10 and S18†). This fact is mainly caused by fewer orbital transitions for P-atoms in the stronger D–A structures (Fig. S7–S9†).
As shown in Fig. 3c and S18–S24,† spin–orbit mean-field (SOMF)62 allocated TD-DFT profiles reveal that P1 (S1-geometry) has a small spin–orbit coupling matrix <S|ĤSOC|T > of S1–T1 spin–flip transitions (ξ{S1–T1} = 0.29 cm−1), but larger magnitudes of ∑ξ{S1–T3/4} = 1.07 cm−1 and ξ{S1–T2} (1.00 cm−1), suggesting preponderance channels of S1–T2/3/4 for the ISC. The following IC and phosphorescence radiation is slow because of a decent ξ{S0–T1} value (3.23 cm−1). In comparison, the stronger D–A emitters possess distinct CT characteristics for S0–S1 and ISC assignment (Fig. S19–S24†). For instance, the PO2 and PO3 exhibit limited ISC from S1 to the lowest T1/2 (0.42/0.02 and 0.41/0.19 cm−1), which is lower than that of P1/PO1 (Fig. 3c and S18a†). This outcome demonstrates more efficient ISC in carbazole-fused emitters.34,37 Switching to BINAPs with a more robust CT characteristic, the SOC matrix elements and ΔES1–T1 gaps are mostly governed by electron donors rather than the chemical valence state of TPA-fused compounds.
Furthermore, the S0-geometric calculations also afford extra evolution tendency about the phosphorescence superiority of HLCT-governed P1 and PO1 (Fig. S10–S17†). This speculation is confirmed in the following experimental observations. Thus, the theoretical calculations demonstrate that the double incorporation of weak donor units substantially altered the triplet arrangement and excited-state properties of BINAPs. Moreover, this D–A construction change would influence CPL performance, where the asymmetry factors are negatively correlated with CT metrology (vide infra).
Photophysical properties
The photophysical properties of the six compounds are studied by spectral measurements at solution, doping polymer, and solid states (Fig. 4, S25–S36, Tables S3 and S4†). P1–P3 show shifted UV-vis absorption curves from 300 to 430 nm in dichloromethane (DCM, Fig. 4a), where the ICT bands are located at ∼325 and 360 nm for P1 and P2/P3, respectively. TPA-decorated derivatives P2 and P3 display enhanced CT bands because of stronger donor strength, which agrees with higher oscillator strengths (f, S0 → S1) from TD-DFT simulations (0.177 for P1 to 0.441 for P3). Compared to P1–P2, their oxides PO1–PO2 exhibit a slightly blue-shifted absorption but significant emission efficiency in DCM (Fig. 4a and d). For instance, the primitive P1 and P2 reveal a structureless fluorescence emission at 427 and 482 nm (ϕPL = 0.12, 33.7%) with Commission Internationale del’Éclairage 1931(CIE 1931) coordinates of (0.154, 0.043) and (0.149, 0.258), but PO1 and PO2 emit slight blue-shift light at 418 and 474 nm with significant ϕPL values of 63.1% and 72.8%, respectively (Fig. 4a, d and Table S3†). Moreover, PO3 shows red-shifted emission with a broader band, suggesting pronounced structure dynamics in the excited state in the D–A emitters. Notably, the enhanced CT effect of methoxyls on TPAs contributes a near-unity PLQY of 95.3% for PO3 in degassed dichloromethane (DCM). Importantly, O-atom insertion not only improves ϕPL for P1–P3 but also the photochemical stability in solvents (Fig. 4d and S37†), which could benefit from the ICT effect and bonding saturation of P atoms. The trivalent phosphines would undergo oxidation and slight decomposition in solutions by oxygen corrosion and photoactivation.56 These processes are confirmed by corresponding spectral measurements (Fig. S38†). | ||
| Fig. 4 Photophysical properties. (a) UV-vis and PL spectra of six emitters in DCM (298 K, λex = 350 nm, 10−5 M). (b) PL and delay PL spectra of P1 in DCM (77 K, λex = 350 nm, 10−5 M). (c) Lifetime decay profiles of all compounds in DCM at 77 K. (d) PLQY values of the six emitters in DCM and PMMA (0.5 wt%) at 298 K. (e) PL mapping spectra and intensity profiles of the (R)-1@PVP film with different excitation wavelengths (298 K, 0.5 wt%). (f) Delay PL mapping spectra of the (R)-1@PVP film with different excitation wavelengths (298 K, 0.5 wt%, delay 0.1 s). (g and h) PL and delay PL spectra of phosphines or their oxides in PVP (298 K, 0.5 wt%, delay 0.1 s); inserted pictures are of the emissive polymer. (i) Lifetime decay profiles for all doped PVP films (monitoring the delay PL peak). (j) Representative photographs of afterglow images for cryogenic DCM solutions at 77 K and polymers at 298 K. (k and l) CIE 1931 chromaticity coordinates for emitter doped PVP films (298 K). | ||
As shown in Fig. S39,† the UV-vis spectrum of PO2 is almost insensitive to the solvent type but the solvatochromic emission changes from 428 (toluene) to 503 nm (MeCN), while the fluorescence of all oxidative emitters has moderate positive solvatochromism behaviors (Fig. S25†). The general Stokes shifts of D–A pairs in polar solvents imply structural relaxation of CT excited states. Stronger solvatochromic fluorescence is further observed in P3/PO3 compared to P1/PO1, suggesting that the P1/PO1 emission is a HLCT-directorial pathway, whereas the fluorescence of P3/PO3 is better described as having better CT-dominant features (Fig. S25†). In addition, P3 and PO3 have similar oscillator strength for the S1 → S0 transitions and SOC matrix for S1 → T1 (P3: 0.084, 0.43 cm−1 and PO3: 0.081, 0.41 cm−1), which contradicts the higher PLQY in PO3. Given the larger structural deformation and photostability, the lower PLQY is likely due to the structural motion-induced nonradiative decay and light degradation in the P(III) emitter. A recent report has revealed that the spectra of cyclization degradation products are red-shifted and the quantum efficiency is low.56
In consideration of the HLCT, p–π* and heavy effects in these phosphine emitters, the phosphorescence spectra are measured in DCM and 2-Me-THF at 77 K (Fig. S26 and S29†). Expectantly, the intense afterglow of P1–P3 and their PO1–PO3 oxides is observed in DCM at 77 K, while their impressive low-temperature phosphorescence (LTP) lifetimes range from 783 to 898 ms and this afterglow could retain ∼10 s in P1 (Fig. 4b and c), suggesting a phosphorescence radiation process. The LTP peaks of the six emitters range from 518 to 543 nm. The enhanced donors cause lower triplet energy levels in BINAPs, which are smaller than those of the BINAP matrix (500 nm). Taking P1 as a typical sample, it shows ultraviolet color in DCM at 77 K (Video S1†) and displays dual emission at ∼400 nm (τfluo = 4.85 ns, Fig. S28†) and 518 nm with structured features (Fig. 4b). The delayed PL spectra display the same phosphorescence band at 77 K, with a long average lifetime of 857 ms via biexponential decay (Fig. 4c), demonstrating hybrid singlet (Sn) and triplet (Tn) radiation characters under cryogenic conditions. Based on the fluorescence and phosphorescence spectra onset of P1 at 77 K, the moderate energy gap between the lowest S1 and T1 excited states (ΔES1–1) is determined as a large value of 0.83 eV (Fig. 4b), which is well matched to the TD-DFT simulative value (0.86 eV, Fig. 3c). Nevertheless, the significant CT enhancement from the push–pull effect can dramatically separate the HOMO and LUMO spatial distribution (Fig. 3a), resulting in the experimental gap decrease of ΔES1–T1 from 0.83 to 0.45 eV (Table S3 and Fig. S30†). Together with the donor and P-atom valence alteration of conjugated compounds, a significant wavelength adjustment and quantum efficiency enhancement could be easily achieved in diphosphines for the first time. In addition, the phosphorescence composition Iphos./Ifluo. (the ratio of the phosphorescence emission area to the fluorescence spectral area) and the RTP lifetimes are changed in these architectures. As a result, the ratio of Iphos./Ifluo. decreases from 0.39 in P1/PO1 to 0.05 in P3/PO3 at 77 K (Fig. S26†). In contrast to DCM, all emitters in a more rigid 2-Me-THF medium also easily facilitate LTP with similar emission bands but longer lifetimes (1.69–2.23 s, Fig. S31–S32†).
Subsequently, all emitters are doped into polymethyl methacrylate (PMMA) or polyvinyl pyrrolidone (PVP) polymers to restrict molecular motion and isolate molecular oxygen (Fig. S33–S35†).63 As shown in Fig. 4e and h, the doped PVP films of P1@PVP and PO1@PVP emit a near ultraviolet fluorescence peak at ∼410 nm with CIE coordinates of (0.188, 0.157) and (0.162, 0.059), respectively (Fig. 4k), while a short decay at nanosecond levels and an RTP tail at ∼520 nm with an ultralong attenuation dynamic of τavg = 1.02 s (τ1 = 345 ms × 27%; τ2 = 1267 ms × 73%) are determined in P1@PVP (Fig. 4i, details are listed in Table S4†). Obviously, this RTP band overlaps well with their LTP spectra (∼520 nm), demonstrating the same decay channel from the unimolecular emission. This RTP activity is also presented in PMMA and mixed polymer films (Fig. S33–S35, Videos S2 and S3†), which is in contrast to the stereotypical views of previous nonluminous diphosphine monomers.48,50,51 Similarly, RTP with faster decay and lower phosphorescence ratios has been observed in PO1, P2, and P3 doped polymers, and their RTP lifetimes are gradually decreased to 0.88, 0.78, and 0.22 s, respectively (Fig. 4g–i and S32c†). Interestingly, PO2@PVP displays a new mixed RTP-TADF band with a broad feature at 298 K, but the exclusive phosphorescence peak of 543 nm is recovered (τavg = 0.60 s) at 77 K due to suppressed thermal activation for TADF (Fig. 4h and i). Furthermore, PL and delayed PL spectra of PO3@PVP share the same structureless band from 400 nm to 650 nm (Fig. 4h), where the RTP band disappears and the delayed emission lifetime is determined to be 18 ms (Fig. 4i). Taking PO3@PVP as a typical object, the temperature-dependent PL decay of the fluorescence peak demonstrates that the τavg component accelerates as the temperature increases, while the phosphorescence band is suppressed upon heating, suggesting possible TADF characteristics in these strong D–A emitters (Fig. S36b†). Thus, a trace of the fluorescence band is observed in the short-wavelength region for all emitters, showing TADF lifetimes of 14.7–20.1 ms (Fig. 4i and S36a†). By managing the ΔES–T and moderate RISC, the previous TADF-type afterglow emitters of β-diketone chelated difluoroborons can also facilitate improved afterglow quantum yields and maintain long afterglow emission lifetimes up to 187 ms.36,37 Also, the long-lived TADF observation of films is in accordance with experimental larger ΔEST; the slow RISC process would occur via thermal activation at room temperature.64–66 This finding of the RTP → TADF process has also been studied in other achiral weak D–A emitters.8,35,67 In TPA substituted BINAPs/BINAPOs, the small SOC of S1–T1 and S0–T1 limit the ISC process. On the other side, phosphorescence radiation is destroyed due to nonradiative dissipation of the metastable triplet, in line with the larger root of the mean of squared displacement (RMSD) between S0 and S1, i.e. the RMSD value of PO3 (2.0783 Å) is significantly greater than those of PO1 (0.9090 Å, Fig. S18b†). Thus, in addition to the essential triplet populations and structural rigidity, the RTP performance is sensitive to polymer matrices and the environments, such as shorter RTP lifetimes in PMMA (Fig. S35h and Table S4†). These might be related to the rigidity differences of polymer networks.
Chiroptical properties
To evaluate the chiroptical properties of P1–P2 and PO1–PO2 enantiomers, their electronic circular dichroism (ECD) and CPL spectra are collected in solvents, polymers, and pure solid states (Fig. S39 and S40†). At first, the stereo-conformation for chiral-HPLC separated isomers is determined by comparing both experimental ECD and CPL spectra with the ones calculated using the TD-DFT results (Fig. 5a and S41†). The first elution of chiral HPCL belongs to the (R)-isomer and the second is the (S)-isomer. Similar to solvent-insensitive UV-vis absorption, all ECD curves are almost identical in variational solvents from toluene to MeCN (Fig. S39a and b†). In dichloromethane, the trivalent phosphines (R)/(S)-P1 and (R)/(S)-P2 exhibit mirrored ECD curves from 250–450 nm except for red-shifted absorption (Fig. 5a and b). The oxides of PO1 and PO2 have slightly hypochromatic CD signals and higher absorption dissymmetry factors (gabs, Fig. S42†). These |gabs| values manifest a maximum at the lowest-energy absorption band, with a range of 3.4 to 9.6 × 10−3 in eight investigated compounds, suggesting effective chiral induction and expression in this axially chiral D–A skeleton. The oxides have slightly larger gabs values, which are mainly promoted by larger electric-magnetic dipole moment angles (Fig. 5i and S43†). The stereochemical durabilities of (R)-PO1 and (R)-PO2 are elucidated according to chiral conformation analysis using variable temperature ECD spectra. The ECD spectra of these stable phosphine oxides are unchanged after 180 °C heating for several hours, showing ultrahigh stereo conformation persistence by 7,7′-steric hindrance (Fig. S44†). | ||
| Fig. 5 Chiral optical properties. (a and b) ECD spectra of P1 and P2 enantiomers in DCM (10−4 M, fractions 1 and 2 are the (R)-isomer and (S)-isomer, respectively). (c–f) CPL and glum spectra of P1, PO1, P2, and PO2 enantiomers in DCM (10−4 M, λex = 340 nm). (g−h) Solvatochromic CPL and glum spectra of PO2 enantiomers in solvents (inserted picture shows TD-DFT simulated transition electric-magnetic dipole moment and its θe-m angle during the S0 → S1 transition for (R)-PO2). (i) Calculated electric (blue arrow, μe) and magnetic (red arrow, μm) transition dipole moments at the B3LYP/6-31G(d) level for S0 → S1. (j) Chiroptical property performances of four emitters in this work (in DCM or polymers). | ||
Enantiomeric P1, P2, PO1, and PO2 (Fig. 5c–h) emitters show attractive CPL with decent luminescence dissymmetry factors (glum) ranging from ∼5.0 × 10−3 (P1) to 2.1 × 10−3 (P2) in toluene (Fig. 5g and S45†), which is the highest record in already existing phosphine emitters.48,50,51 Furthermore, solvatochromic CPL has also been observed for enantiomeric PO1 and PO2 emitters (Fig. 5g, h and S45†). With increasing solvent polarity, the CPL emission becomes broadband. Specifically, the case in point is that (R)/(S)-PO1 emits near-ultraviolet CPL with negative and positive signals at 416 nm in nonpolar toluene but 440 nm in polar MeCN (ΔλCPL = ∼25 nm), and its considerable glum factors are maintained in all solvents. In comparison to (R)/(S)-PO1, (R)/(S)-PO2 has stronger D–A character, providing a more significant solvatochromic CPL contrast (ΔλCPL = 75 nm).
Notably, the emitter-doped PVP or PMMA polymers produce high |g(CPF)| = 6.4 × 10−3 (P1@PVP) and |g(CPF)| = 1.0 × 10−3 (P2@PVP) for CPF emission (Fig. 6a, b and S46–S48). After complete deoxygenation by UV photoactivation for 5 minutes, the enantiomeric P1@PVP films show a CP-OURTP band beyond 480 nm, with g(CP-RTP) values of ±3.0 × 10−3 at 520 nm. However, the PO1, P2, and PO2 doped PVP films exhibit lower g(CP-RTP) values due to weak phosphorescence and the inferior electro-magnetic transition environment. In addition, the doped PMMA films show similar CPF but fewer phosphorescence components (Fig. S46 and S47†). The afterglow of the P1@PMMA polymer is much brighter at 77 K because of the enhanced rigidity of excited state conformation, which displays an improved circularly polarized low-temperature phosphorescence (CP-LTP) proportion and |g(CP-LTP)| values of ca. 2.0 × 10−2 (Fig. S49†).68,69
 | ||
| Fig. 6 CP-OURTP properties. (a and b) CPL and glum spectra of P1 and P2 enantiomers in doped PVP (0.5 wt%) at 298 K. Continuous UV irradiation (5 min, 15 W light source) was attempted to remove the molecular oxygen residue (λex = 330 nm). (c) POM image showing fingerprint texture for the induced chiral N* phase at 298 K (5CB@(R)-PO1). (d) CPL and glum spectra of chiral liquid crystal films at 298 K (λex = 330 nm). (e) Chemical structures of RM257, Irg651, and emitters. (f) Schematic preparation and assembly of the chiral LC polymer by in situ photopolymerization. (g) Fingerprint texture of the PLC@(R)-PO1 polymer film at 298 K (scale bar is 50 μm). (h) Yellow CP-RTP photographs of transparent PLC@(R)/(S)-PO1 polymer films excited using a 365 nm lamp (polarized glasses are placed between films and the camera). (i) Concentration-dependent PL and delayed PL spectra of PLC@(R)/(S)-PO1 polymer films. (j) Lifetime decay profiles of PLC@(R)/(S)-PO1 polymer films. (k andl) CPL and glum spectra of PLC@(R)/(S)-PO1 polymer films with a 1.0 wt% doping proportion. (m) Changes in gCP-RTP factors and total PLQY values of PLC@(R)/(S)-PO1 films at different doping proportions. | ||
TD-DFT calculations provide further physical insights into the chiral transition processes (Fig. S43 and S50–S54†). For (R)-P1, the S0 → S1 transition processes balanced electron transition dipole moment (μe = 3.76 × 10−18 esu cm), magnetic transition dipole moment (μm = 8.05 × 10−21 erg/Gauss), the large μe–μm angle (θe-m = 159.1°), and a high g(abs,cal) of −8.0 × 10−3, which are in agreement with the experimental results g(abs,exp) = −6.5 × 10−3 (Fig. 5i and S42†). Besides, other TPA-decorated compounds exhibit slightly higher θe-m but lower μm values, producing degressive gabs values for the S0 → S1 transition (Fig. S43†). According to TD-DFT calculated fluorescence emission based on Kasha's rule, the reduced glum (S1 → S0) values of all emitters appeared due to degenerated cosθe-m (θe-m = 147.2 → 91.2°, Fig. S55†). Hence, glum factors are lower than those of g(abs,cal), which is in accord with CD/CPL measurement tendency in solutions (Table S3†). By comparison, with the enhancement rate of CT and higher ϕPL, both g value and RTP lifetime will deteriorate. This result is caused by depressed θe-m magnitude but increased μe (Fig. 5i). Thus, PO1 provides superior ϕPL, CP-OURTP performance, and structural stability, including photostability and exciton stability. Overall, the experimental and theoretical studies revealed that the facile P-functionalities have remarkable impacts on the CPL and RTP of BINAPs/BINAPOs via altering the charge transfer states and p–π* electronic effects.
Chiral photopolymerization induced CP-OURTP amplification
Moreover, stable PO1/PO2 are candidates for emissive chiral inducers for chiral liquid crystal (LCs, 5CB) fabrication, as evidenced by the increased CPL metrics with optimized glum values up to ±0.2 in the assembled superchiral cholesteric mesophase (Fig. 6c, d, S56 and S57†). However, this flow and soft LCs phase cannot offer a rigid excited state for RTP. To overcome this drawback in the 5CB matrix, we further utilize ternary liquid crystal coassembly and sequential chiral polymerization to stabilize the triplet exciton (Fig. 6e and f). The achiral LC precursor RM257 (host), Irg651 (photoinitiator), and chiral phosphorescent inducer PO1 are selected and coassembled via mixing and liquation treatments (for details see the ESI†). The twist supramolecular chirality is induced successfully, where the fingerprint textures are presented (Fig. S58a–c†), suggesting helical architectures of the chiral nematic phase. Before the UV-triggered photopolymerization (Fig. S59†), only the fluorescence phenomenon could be noticed by the naked eye, in line with the flow PO1@5CB system. Remarkably, intense RTP with long decay is observed after in situ UV irradiation (365 nm light, Fig. 6h). The mobility of the transparent films is quenched but the chiral LC textures are still reserved (Fig. 6g and S58d–f†), manifesting expectant rigid networks and helical constructions. No structural color is found in these transparent thin films (Fig. S59†). CD and CPL measurements reveal that these polymeric LC films have increased g factors of CD absorption and CP-RTP emission (Fig. S61†). After optimization of the doping concentration, the PLC@(R)/(S)-PO1 films display increased RTP proportions, lifetimes, and gCP-RTP (Fig. 6i–m). Finally, the high gCP-RTP (±0.11), ϕPL (52.4%), and a long lifetime (τavg = 0.83 s) are obtained in a 1.0 wt% concentration (Fig. 6m). Importantly, the calculated FM (FM = ϕPL × gCP-RTP) value of CP-OURTP reaches up to 0.058. This represents the best values among small molecules and dual-layer reflection devices (Fig. 7a).16,70,71 The high dissymmetry of RTP could be distinguished using homemade polarized glasses (Fig. 6h and S62e†), where one isomer is brighter than the other. This discrimination is impossible for traditional CP-RTP materials due to low gCP-RTP, demonstrating the significance of stepwise chiral amplification.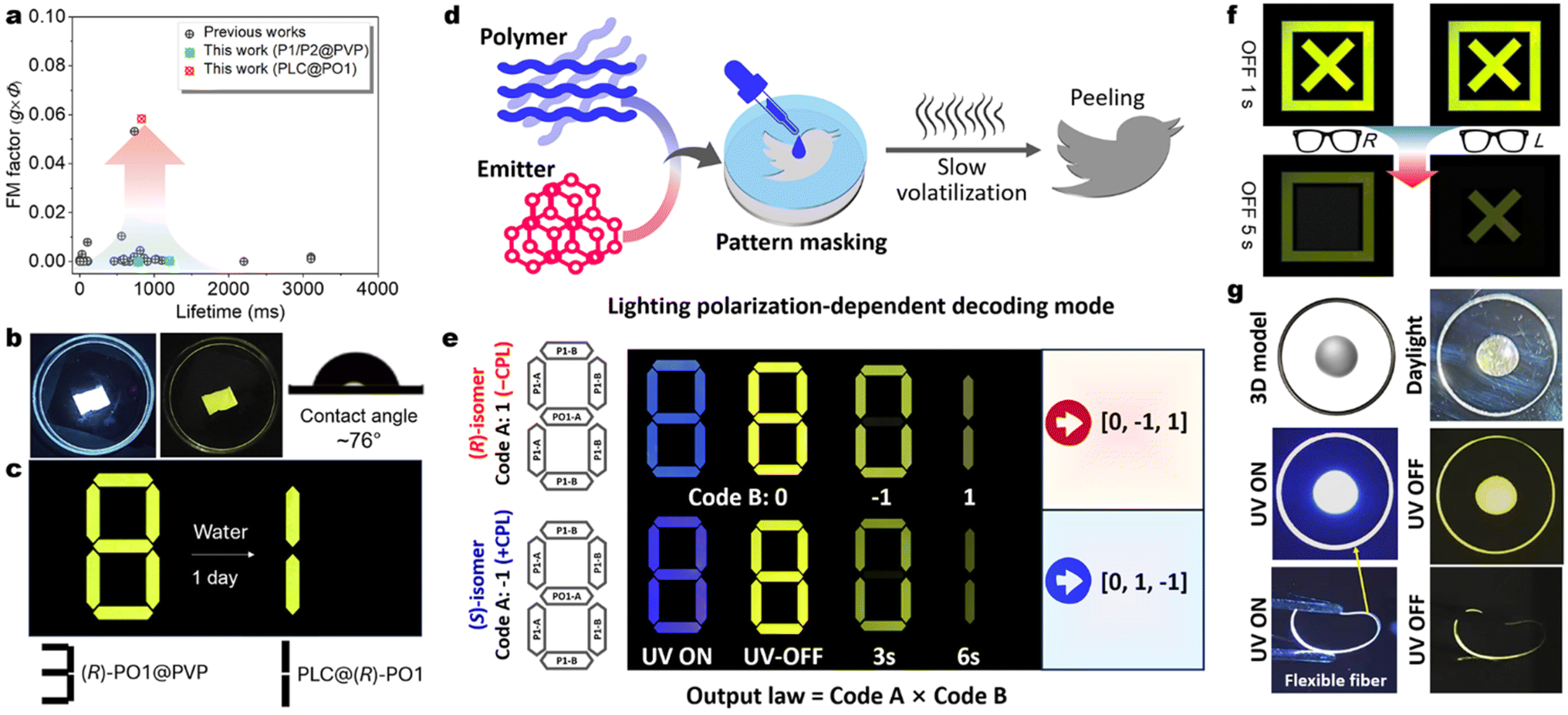 | ||
| Fig. 7 CP-OURTP stability and applications. (a) CP-RTP performance comparison between this work and previous reports (see Table 1 in ref. 16). (b) Circularly polarized fluorescence and afterglow photographs of a PLC@(R)-PO1 film immersed in water environments (soaking for 1 day); the left picture showing the water contact angle. (c) The information is decoded by water-soaking. (d) Schematic diagram of the pattern masking operation for diverse light-emitting pattern preparation and time and polarization-involved information encryption. (e) Illustration of chiral information coding based on Arabic number patterns (A: doped PMMA & PVP and B: doped PMMA film). (f) The information decoding process with L/R polarized glasses. (g) Photographs of various lighting-emitting patterns and polymer fibers (doped PMMA). | ||
Chiral photopolymerization induced stability enhancement
Most conventional CP-OURTP polymers are unendurable to moisture and water, especially for water-soluble polymer matrices, e.g. PVP and poly(vinyl alcohol) (PVA), which can break the hydrogen bonding networks within the polymers and accelerate the triplet non-radiative decay and RTP quenching.72–76 The polymer network of PO1@PVP is gradually digested in water environments and the RTP disappears (Fig. S63†). In contrast, the photopolymeric PLC@PO1 films can suppress water penetration because of their hydrophobic character. This polymeric LC network is dense and electroneutral and exhibits a larger water contact angle (∼76°) compared to the PO1@PVP film (Fig. 7b). This indicates that the combination of the hydrophobic RM257 polymer could prevent water from diffusing into active phosphors and offer the desired helical network for CP-OURTP reinforcement (Fig. 7b).Multiple optical encryption and display applications
The significant CP-OURTP performance of the P1/PO1 doped polymer is suitable for flexible optical encryption devices through pattern masking (Fig. 7c–f). Masking films are prepared by evaporating a DCM solution comprising 0.5 wt% emitters and 99.5 wt% PMMA & PVP (mass ratio = 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and device type A: P1-A and PO1-A) or PMMA (P1-B) in the cell (Fig. 7e). Various patterns are obtained by using different masking cells (Fig. 7f and g), showing high luminous contrast and resolution between blue CPF and yellow CP-OURTP, based on the different RTP lifetimes and polarization differences of enantiomeric P1 and PO1 guests and very close emission color but diverse afterglow visuality in the air. Thereby, decay time-dependent information encryption could be realized. Fig. 7e displays the prepared Arabic numeral “8” with distinct polymer building blocks. The (R)/(S)-polymer systems emit the same color but in the inverse direction of polarization (Fig. 6a). After removing the UV flashlight, the pattern was changed from “8” to “0” and finally “1” by extending the observation time. By linking the code with the essential output law, two distinct sets of information, namely 0, −1, 1 and 0, 1, −1, can be deciphered from (R) and (S)-systems. However, only pseudocode can be obtained when the decoding law is unclear due to the same visual color of patterns.
:1 and device type A: P1-A and PO1-A) or PMMA (P1-B) in the cell (Fig. 7e). Various patterns are obtained by using different masking cells (Fig. 7f and g), showing high luminous contrast and resolution between blue CPF and yellow CP-OURTP, based on the different RTP lifetimes and polarization differences of enantiomeric P1 and PO1 guests and very close emission color but diverse afterglow visuality in the air. Thereby, decay time-dependent information encryption could be realized. Fig. 7e displays the prepared Arabic numeral “8” with distinct polymer building blocks. The (R)/(S)-polymer systems emit the same color but in the inverse direction of polarization (Fig. 6a). After removing the UV flashlight, the pattern was changed from “8” to “0” and finally “1” by extending the observation time. By linking the code with the essential output law, two distinct sets of information, namely 0, −1, 1 and 0, 1, −1, can be deciphered from (R) and (S)-systems. However, only pseudocode can be obtained when the decoding law is unclear due to the same visual color of patterns.
Benefiting from the high gCP-RTP factor and polarization discrimination of PLC@(R)/(S)-PO1, the encryption and decoding procedures by the naked eye with polarized glasses have also been realized. As illustrated in Fig. 7f, one combined pattern (✗ and □) contains PLC@(R)PO1 (✗) and PLC@(S)-PO1 (□). After turning off the UV excitation for 5 s, the afterglow pictures are collected to guarantee adequate afterglow attenuation and contrast. The symbol of  is observed when the polarizer is absent, but the □ symbol is authorized when the polarizer is present. Using the opposite left polarizer glasses, the reversal ✗ sign is decoded.
is observed when the polarizer is absent, but the □ symbol is authorized when the polarizer is present. Using the opposite left polarizer glasses, the reversal ✗ sign is decoded.
Furthermore, using similar RTP emission but distinct water resistance for PLC@(R)-PO1 and (R)-PO1@PVP, a Snapchat decode has also been established (Fig. 7c). Overall, this stepwise chiral amplification strategy underscores the additional accessibility and superiority offered by the introduction of CP-OURTP in the process of multiple anti-counterfeiting.
Conclusion
In summary, we have first validated new tactics for customizing effective CP-OURTP luminescence in unimolecular BINAPs via stepwise chiral amplification. The synchronously enhanced luminescence in solution, polymer, solid, and liquid crystal states go beyond typical phosphines. These weak D–A architectures not only enable fine regulation of HLCT excited states for ISC optimization but also a good chiral transition environment, facilitating record-breaking FM metrics and stability in CP-OURTP. Mechanism studies reveal that their low-lying triplet populations and decay channels highly depend on the LE/CT proportions, 1HLCT/1CT→3LE processes, and singlet-triplet split energy. These properties could be easily optimized by altering the donor strength, together with the associated valency of the P-atom. This stepwise regulation of molecular and supermolecular chirality could break the physical constraint of CP-OURTP, thereby offering a high RTP lifetime (0.83 s) and gCP-RTP factor (±0.11). Moreover, the persistent afterglow of circularly polarized light up to ∼10 s allows multiple encryption applications. Given the diversity and commercial accessibility of phosphine ligand libraries, we believe that this strategy would provide a new future for designing high-performance CP-OURTP materials.Data availability
Detailed experimental details and characterization are listed in the ESI.† The data that support the findings of this study are available from the corresponding author on reasonable request.Author contributions
All authors have given approval to the final version of the manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This research was supported by the National Natural Science Foundation of China (No. 21871133), the Natural Science Foundation of Jiangsu Province (No. SBK20250201551), and the Science, Technology, and Innovation Commission of Shenzhen Municipality (No. JCYJ20180307153251975).References
- F. Furlan, J. M. Moreno-Naranjo, N. Gasparini, S. Feldmann, J. Wade and M. J. Fuchter, Nat. Photonics, 2024, 18, 658–668 CrossRef CAS.
- L. Wan, Y. Liu, M. J. Fuchter and B. Yan, Nat. Photonics, 2023, 17, 193–199 Search PubMed.
- J. R. Brandt, F. Salerno and M. J. Fuchter, Nat. Rev. Chem, 2017, 1, 0045 Search PubMed.
- H. Lu, Z. V. Vardeny and M. C. Beard, Nat. Rev. Chem, 2022, 6, 470–485 Search PubMed.
- G. Chen, B. Huang and W. Zhang, Nat. Commun., 2023, 14, 1514 CrossRef PubMed.
- Z. Huang, Z. He, B. Ding, H. Tian and X. Ma, Nat. Commun., 2022, 13, 7841 CrossRef CAS PubMed.
- H. Li, J. Gu, Z. Wang, J. Wang, F. He, P. Li, Y. Tao, H. Li, G. Xie, W. Huang, C. Zheng and R. Chen, Nat. Commun., 2022, 13, 429 CrossRef CAS PubMed.
- S. Xu, R. Chen, C. Zheng and W. Huang, Adv. Mater., 2016, 28, 9920–9940 Search PubMed.
- W. Zhao, Z. He and B. Z. Tang, Nat. Rev. Mater., 2020, 5, 869–885 CrossRef CAS.
- S. Hirata and M. Vacha, J. Phys. Chem. Lett., 2016, 7, 1539–1545 CrossRef CAS PubMed.
- J. Song, H. Xiao, L. Fang, L. Qu, X. Zhou, Z. X. Xu, C. Yang and H. Xiang, J. Am. Chem. Soc., 2022, 144, 2233–2244 CrossRef CAS PubMed.
- D. W. Zhang, M. Li and C. F. Chen, Chem. Soc. Rev., 2020, 49, 1331–1343 RSC.
- M. Zhang, Q. Guo, Z. Li, Y. Zhou, S. Zhao, Z. Tong, Y. Wang, G. Li, S. Jin, M. Zhu, T. Zhuang and S. H. Yu, Sci. Adv., 2023, 9, eadi9944 CrossRef CAS PubMed.
- Y. Zhao, J. Yang, C. Liang, Z. Wang, Y. Zhang, G. Li, J. Qu, X. Wang, Y. Zhang, P. Sun, J. Shi, B. Tong, H. Y. Xie, Z. Cai and Y. Dong, Angew. Chem., Int. Ed., 2024, 63, e202317431 CrossRef CAS PubMed.
- D. Liu, W. J. Wang, P. Alam, Z. Yang, K. Wu, L. Zhu, Y. Xiong, S. Chang, Y. Liu, B. Wu, Q. Wu, Z. Qiu, Z. Zhao and B. Z. Tang, Nat. Photonics, 2024, 18, 1276–1284 CrossRef CAS.
- X. Zou, N. Gan, Y. Gao, L. Gu and W. Huang, Angew. Chem., Int. Ed., 2025, e202417906 CAS.
- X. Y. Dai, M. Huo and Y. Liu, Nat. Rev. Chem, 2023, 7, 854–874 CrossRef CAS PubMed.
- T. Itoh, Chem. Rev., 2012, 112, 4541–4568 CrossRef CAS PubMed.
- R. Kabe and C. Adachi, Nature, 2017, 550, 384–387 CrossRef CAS PubMed.
- X. Ma, J. Wang and H. Tian, Acc. Chem. Res., 2019, 52, 738–748 CrossRef CAS PubMed.
- R. Kabe, N. Notsuka, K. Yoshida and C. Adachi, Adv. Mater., 2016, 28, 655–660 CrossRef CAS PubMed.
- Kenry, C. Chen and B. Liu, Nat. Commun., 2019, 10, 2111 CrossRef CAS PubMed.
- G. Zhang, G. M. Palmer, M. W. Dewhirst and C. L. Fraser, Nat. Mater., 2009, 8, 747–751 CrossRef CAS PubMed.
- F. Nie and D. Yan, Nat. Commun., 2024, 15, 9491 CrossRef CAS PubMed.
- G. Guo, H. Li, Y. Yan, W. Zhao, Z. Gao, H. Cao, X. Yan, H. Li, G. Xie, R. Chen, Y. Tao and W. Huang, Adv. Mater., 2024, 2412100 CrossRef CAS PubMed.
- M. Zeng, W. Wang, S. Zhang, Z. Gao, Y. Yan, Y. Liu, Y. Qi, X. Yan, W. Zhao, X. Zhang, N. Guo, H. Li, H. Li, G. Xie, Y. Tao, R. Chen and W. Huang, Nat. Commun., 2024, 15, 3053 CrossRef CAS PubMed.
- J. Liu, X. Zhou, X. Tang, Y. Tang, J. Wu, Z. Song, H. Jiang, Y. Ma, B. Li, Y. Lu and Q. Li, Adv. Funct. Mater., 2024, 2414086 Search PubMed.
- H. Gao and X. Ma, Aggregate, 2021, 2, e38 CrossRef CAS.
- Y. Patil, C. Demangeat and L. Favereau, Chirality, 2023, 35, 390–410 CrossRef CAS PubMed.
- A. Forni, E. Lucenti and E. Cariati, J. Mater. Chem. C, 2018, 6, 4603–4626 RSC.
- W. Zhao, Z. He, J. W. Y. Lam, Q. Peng, H. Ma, Z. Shuai, G. Bai, J. Hao and B. Z. Tang, Chem, 2016, 1, 592–602 CAS.
- Q. Zhou, C. Yang and Y. Zhao, Chem, 2023, 9, 2446–2480 CAS.
- G. Li, B. Li, H. Zhang, X. Guo, C. Lin, K. Chen, Z. Wang, D. Ma and B. Z. Tang, ACS Appl. Mater. Interfaces, 2022, 14, 10627–10636 CrossRef CAS PubMed.
- G. Li, K. Xu, J. Zheng, X. Fang, W. Lou, F. Zhan, C. Deng, Y. F. Yang, Q. Zhang and Y. She, J. Am. Chem. Soc., 2024, 146, 1667–1680 CrossRef CAS PubMed.
- D. Cui, L. Zhang, J. Zhang, W. Li, J. Chen, Z. Guo, C. Sun, Y. Wang, W. Wang, S. Li, W. Huang, C. Zheng and R. Chen, Angew. Chem., Int. Ed., 2024, e202411588 CAS.
- X. Wang, Y. Sun, G. Wang, J. Li, X. Li and K. Zhang, Angew. Chem., Int. Ed., 2021, 60, 17138–17147 CrossRef CAS PubMed.
- G. Wang, X. Chen, Y. Zeng, X. Li, X. Wang and K. Zhang, J. Am. Chem. Soc., 2024, 146, 24871–24883 CrossRef CAS PubMed.
- S. Jena, A. T. M. Munthasir and P. Thilagar, J. Mater. Chem. C, 2022, 10, 9124–9131 RSC.
- P. She, J. Duan, J. Lu, Y. Qin, F. Li, C. Liu, S. Liu, Y. Ma and Q. Zhao, Adv. Opt. Mater., 2022, 10, 2102706 CrossRef CAS.
- G. Lu, J. Tan, H. Wang, Y. Man, S. Chen, J. Zhang, C. Duan, C. Han and H. Xu, Nat. Commun., 2024, 15, 3705 Search PubMed.
- J. Wei, M. Zhu, T. Du, J. Li, P. Dai, C. Liu, J. Duan, S. Liu, X. Zhou, S. Zhang, L. Guo, H. Wang, Y. Ma, W. Huang and Q. Zhao, Nat. Commun., 2023, 14, 4839 Search PubMed.
- X. Song, G. Lu, Y. Man, J. Zhang, S. Chen, C. Han and H. Xu, Angew. Chem., Int. Ed., 2023, 62, e202300980 CrossRef CAS PubMed.
- H. Li, X. Li, H. Su, S. Zhang, C. Tan, C. Chen, X. Zhang, J. Huang, J. Gu, H. Li, G. Xie, H. Dong, R. Chen and Y. Tao, Chem. Sci., 2024, 15, 14880–14887 RSC.
- P. Alam, T. S. Cheung, N. L. C. Leung, J. Zhang, J. Guo, L. Du, R. T. K. Kwok, J. W. Y. Lam, Z. Zeng, D. L. Phillips, H. H. Y. Sung, I. D. Williams and B. Z. Tang, J. Am. Chem. Soc., 2022, 144, 3050–3062 Search PubMed.
- M. Tang, J. Wen, Y. Sun, Q. Hou, X. Cai, W. He, X. Xie, H. Ding, F. Li, L. Zheng, Y. Shi and Q. Cao, Adv. Funct. Mater., 2024, 34, 1–10 Search PubMed.
- A. W. K. Law, T. S. Cheung, J. Zhang, N. L. C. Leung, R. T. K. Kwok, Z. Zhao, H. H. Y. Sung, I. D. Williams, Z. Qiu, P. Alam, J. W. Y. Lam and B. Z. Tang, Adv. Mater., 2024, 2410739 CrossRef CAS PubMed.
- Y. Tao, R. Chen, H. Li, J. Yuan, Y. Wan, H. Jiang, C. Chen, Y. Si, C. Zheng, B. Yang, G. Xing and W. Huang, Adv. Mater., 2018, 30, 1803856 Search PubMed.
- B. Yang, S. Yan, Y. Zhang, S. Ban, H. Ma, F. Feng and W. Huang, J. Am. Chem. Soc., 2024, 146, 7668–7678 CrossRef CAS PubMed.
- S. Jena, J. Eyyathiyil, S. K. Behera, M. Kitahara, Y. Imai and P. Thilagar, Chem. Sci., 2022, 13, 5893–5901 RSC.
- X. Wu, C. Y. Huang, D. G. Chen, D. Liu, C. Wu, K. J. Chou, B. Zhang, Y. Wang, Y. Liu, E. Y. Li, W. Zhu and P. T. Chou, Nat. Commun., 2020, 11, 2145 CrossRef CAS PubMed.
- Y. Kono, K. Nakabayashi, S. Kitamura, R. Kuroda, M. Fujiki and Y. Imai, Tetrahedron, 2015, 71, 3985–3989 Search PubMed.
- D. Zhang, M. Li and C. Chen, Chem. Soc. Rev., 2020, 49, 1331–1343 RSC.
- C. Chen, Z. Chi, K. C. Chong, A. S. Batsanov, Z. Yang, Z. Mao, Z. Yang and B. Liu, Nat. Mater., 2021, 20, 175–180 CrossRef CAS PubMed.
- K. Jinnai, R. Kabe, Z. Lin and C. Adachi, Nat. Mater., 2022, 21, 338–344 CrossRef CAS PubMed.
- B. Yang, S. Yan, C. Li, H. Ma, F. Feng, Y. Zhang and W. Huang, Chem. Sci., 2023, 14, 10446–10457 RSC.
- B. Yang, S. Yan, S. Ban, Y. Zhang, H. Ma and W. Huang, Chem. Mater., 2024, 36, 7940–7952 CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- D. Cui, L. Zhang, J. Zhang, W. Li, J. Chen, Z. Guo, C. Sun, Y. Wang, W. Wang, S. Li, W. Huang, C. Zheng and R. Chen, Angew. Chem., Int. Ed., 2024, e202411588 CAS.
- T. Lu, J. Chem. Phys., 2024, 161, 082503 CrossRef CAS PubMed.
- T. Le Bahers, C. Adamo and I. Ciofini, J. Chem. Theory Comput., 2011, 7, 2498–2506 CrossRef CAS PubMed.
- W. Qiu, D. Liu, Z. Chen, Y. Gan, S. Xiao, X. Peng, D. Zhang, X. Cai, M. Li, W. Xie, G. Sun, Y. Jiao, Q. Gu, D. Ma and S. J. Su, Matter, 2023, 6, 1231–1248 CrossRef CAS.
- B. A. Heß, C. M. Marian, U. Wahlgren and O. Gropen, Chem. Phys. Lett., 1996, 251, 365–371 CrossRef.
- H. Thomas, D. L. Pastoetter, M. Gmelch, T. Achenbach, A. Schlögl, M. Louis, X. Feng and S. Reineke, Adv. Mater., 2020, 32, 2000880 Search PubMed.
- Q. Zhang, J. Li, K. Shizu, S. Huang, S. Hirata, H. Miyazaki and C. Adachi, J. Am. Chem. Soc., 2012, 134, 14706–14709 Search PubMed.
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234–238 CrossRef CAS PubMed.
- Q. Zhang, B. Li, S. Huang, H. Nomura, H. Tanaka and C. Adachi, Nat. Photonics, 2014, 8, 326–332 CrossRef CAS.
- R. Chen, R. Feng, Z. Huang, D. Feng, Y. B. Long, J. Zhang, Y. Yang, Z. Ma, Z. Yuan, S. Lu, Z. Zhao and X. Chen, Adv. Funct. Mater., 2024, 34, 2404602 CrossRef CAS.
- S. Ishikawa, D. Sakamaki, M. Gon, K. Tanaka and H. Fujiwara, Chem. Commun., 2024, 60, 4946–4949 Search PubMed.
- W. Huang, C. Fu, Z. Liang, K. Zhou and Z. He, Angew. Chem., Int. Ed., 2022, 61, e202202977 CrossRef CAS PubMed.
- J. Liu, J. J. Wu, J. Wei, Z. J. Huang, X. Y. Zhou, J. Y. Bao, R. C. Lan, Y. Ma, B. X. Li, H. Yang, Y. Q. Lu and Q. Zhao, Angew. Chem., Int. Ed., 2024, 63, e202319536 CrossRef CAS PubMed.
- J. Liu, Z. P. Song, J. Wei, J. J. Wu, M. Z. Wang, J. G. Li, Y. Ma, B. X. Li, Y. Q. Lu and Q. Zhao, Adv. Mater., 2024, 36, 2306834 CrossRef CAS PubMed.
- B. Yang, S. Yan, S. Ban and W. Huang, Inorg. Chem., 2025, 64, 5801–5810 Search PubMed.
- S. Kong, H. Wang, J. Liao, Y. Xiao, T. Yu and W. Huang, Adv. Mater., 2024, 36, 2412468 CrossRef CAS PubMed.
- G. Guo, H. Li, Y. Yan, W. Zhao, Z. Gao, H. Cao, X. Yan, H. Li, G. Xie, R. Chen, Y. Tao and W. Huang, Adv. Mater., 2024, 36, 2412100 Search PubMed.
- W. Huang, C. Fu, Z. Liang, K. Zhou and Z. He, Angew. Chem., Int. Ed., 2022, 61, e202202977 Search PubMed.
- X. Zhao, X. He, Y. Gao, S. Liang, G. Wen, B. Wang, K. Gong, W. Feng and D. Ji, Adv. Opt. Mater., 2025, 2402452 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section; NMR and SCXRD identification of compounds; theoretical calculations; additional figures/charts/tables (PDF); videos of the CP-OURTP phenomenon for materials (ZIP). CCDC 2344550. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc08710k |
| This journal is © The Royal Society of Chemistry 2025 |