
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMultifunctional quantum dot-decorated drug-loaded magnetite nanosystem for dual optical-magnetic resonance imaging and drug delivery
Thi Thu Trang Maia,
Ke Son Phana,
Bich Thuy Doan b,
Thi Thu Huong Lec,
Thi Dieu Thuy Unga,
Quynh Bao Ngoc Nguyena,
Thi Hong Thinh Trana,
Ngoc Quyen Trand,
Le Hang Dangd and
Phuong Thu Ha*a
b,
Thi Thu Huong Lec,
Thi Dieu Thuy Unga,
Quynh Bao Ngoc Nguyena,
Thi Hong Thinh Trana,
Ngoc Quyen Trand,
Le Hang Dangd and
Phuong Thu Ha*a
aInstitute of Materials Science, Vietnam Academy of Science and Technology, 18 Hoang Quoc Viet, Cau Giay, Hanoi, Vietnam. E-mail: thuhp@ims.vast.ac.vn
bThe Institute I-CLeHS Institute of Chemistry for Life and Health Sciences, ENSCP Chimie ParisTech, PSL Université, CNRS UMR, Paris, 8060, France
cFaculty of Natural Resources and Environment, Vietnam National University of Agriculture, Hanoi, Vietnam
dInstitute of Advanced Technology, Vietnam Academy of Science and Technology, Ho Chi Minh City, Vietnam
First published on 23rd June 2025
Abstract
Early detection of cancer plays a significant role in the effective prognosis, treatment regimen planning, and patient recovery. The aim of this study was to combine a near-infrared (NIR) contrast agent, a magnetic resonance imaging (MRI) contrast agent and a therapeutic agent in an all-in-one nanosystem. In particular, owing to their excellent properties, Fe3O4 magnetic nanoparticles (an MRI contrast agent), CdTe quantum dots (an NIR optical fluorescence agent), and curcumin (an anticancer drug) were incorporated in the nanosystem using carboxymethyl chitosan as the coating polymer. The nanosystem was characterized using DLS, zeta potential analysis, Fourier transform infrared spectroscopy, fluorescence spectroscopy, EDX analysis and TEM analysis. The nanoparticles exhibited average sizes ranging from 100 nm to 170 nm in DI water, PBS and DMEM, with zeta potential in the range of −40 to −64 mV, indicating good stability. The nanoplatform demonstrated potential as a positive MRI contrast agent, exhibiting an r2 relaxivity of 78.33 mM−1 s−1 and an r2/r1 ratio of 470.95 ± 15.11 at 7 T, attributed to second-sphere water interactions. The multifunctional nanoplatform showed great in vitro stability and drug encapsulation over two-month period. The MR and NIR optical imaging efficiencies were evaluated in vitro and in vivo on CT26-bearing mice. The ex vivo signal acquisition was also studied for the tumor, liver, spleen, lung and kidney in the mouse model. Results demonstrated the potential of the prepared multifunctional nanosystem in dual optical MR imaging and passive drug delivery applications.
1. Introduction
Cancer remains one of the leading causes of death in the world.1 Early detection of cancer is a keystone for effective patient prognosis and faster recovery. Precise determination of the location and size of tumors can facilitate the treatment regimen and minimize damage to healthy tissues.2 The current imaging techniques for cancer detection include magnetic resonance imaging (MRI), computed tomography (CT), ultrasonography, positron emission tomography, and near-infrared (NIR) fluorescence.3 Each imaging modality possesses its own unique strengths and limitations; consequently, research on nanotheranostics is focused on integrating multiple nanoprobes to capitalize on their complementary advantages, aiming to achieve more accurate and reliable diagnostic outcomes.4 Dual-modal optical-magnetic imaging, which synergistically combines the high sensitivity of optical imaging with the superior spatial resolution of magnetic resonance imaging (MRI), enables precise early detection of tumors and facilitates comprehensive quantitative analysis of pathophysiological processes in vivo.5–7Optical imaging has emerged as a powerful tool in biological research, providing valuable insights both in vitro and in vivo.8,9 Its primary advantages include high sensitivity, rapid image acquisition, relatively low cost, portability, absence of ionizing radiation, and the capability to provide multiplex fluorescent signals simultaneously.10 Nonetheless, in vivo fluorescence monitoring of biomaterials often produces diffuse signals, making it challenging to accurately determine the implant volume or precise anatomical localization. Fluorescence imaging (FLI) provides important information regarding the distribution and bioresorption of the labeled degradation products that are released from the implant. In contrast, MRI enables pinpointing the precise in vivo anatomical location, including its 3D shape and volume. In recent years, MRI has become one of the most versatile techniques for detailed visualization of internal body organs. The incorporation of contrast agents for MRI provides spatial resolution comparable to that of computed tomography (CT) while yielding superior contrast resolution.11 Furthermore, recent studies have demonstrated the utility of bimodal imaging, combining MRI and fluorescent labeling, to effectively track cells and proteins during oncology diagnosis.12–16 Yang et al. reported the synthesis of Fe3O4 and PdS/CdS quantum dots in a multifunctional therapeutic and diagnostic nanoplatform for deep-tissue bimodal imaging.17
Quantum dots (QDs) are a family of semiconductor fluorescent nanocrystals that possess high quantum yields and demonstrate excellent photo- and bio-stability compared with organic fluorescent dyes, making them promising imaging agents [6–8]. Owing to their bright emission, adjustable wavelengths, and biocompatibility, quantum dots are particularly effective for near-infrared (NIR) optical imaging. Their small size and long-lasting fluorescence make them valuable for pre-clinical imaging and clinical applications. Additionally, surface modifications allow QDs to be targeted toward specific tissues or cells, providing detailed visualization of biological processes.
Magnetic nanoparticles (MNP) have been extensively utilized in biomedical applications, notably as contrast agents in magnetic resonance imaging (MRI) and magnetic hyperthermia therapy.18,19 They possess numerous favorable characteristics, including biocompatibility, biodegradability, and versatility for functionalization.20 In MRI, MNP enhances the image contrast by substantially decreasing the transverse relaxation time (T2) of water protons within tissues in the tumors, liver, or spleen, and has been implemented in clinical practices.21 However, conventional MNP are constrained in oncology-related applications due to their relatively large hydrodynamic size (typically exceeding 100 nm) and tendency for magnetic aggregation, leading to rapid removal by macrophages in the liver and spleen.22 To overcome these limitations, various surface modification strategies have been developed to prolong systemic circulation time and enhance contrast in MRI.
In this study, our focus was on preparing a stable nanosystem containing curcumin as a therapeutic agent and not particularly on developing novel nanomaterials; MNP as the MRI contrast agent and QDs as the NIR optical fluorescence agent. The bimodal imaging applications, namely MR imaging and NIR optical imaging, as well as drug release, will be evaluated. Fe3O4 magnetic nanoparticles and CdTe quantum dots were incorporated into an ultimate system. Carboxymethyl chitosan (CMCS) was used as the coating polymer on account of its biocompatibility, and aqueous solubility. Being non-toxic, anti-cancerous, anti-bacterial, and anti-inflammatory,23 curcumin (Cur) was chosen as a therapeutic agent in the multimodal nanoconjugate. By integrating these components, the study aims to enhance the efficiency of passive targeted delivery, MR and NIR optical imaging for biodistribution and early tumor detection, as well as to overcome current challenges in nanoparticle drug delivery systems.
2. Materials and method
2.1 Materials
Ferric chloride hexahydrate (FeCl3·6H2O, 97%), ferrous chloride tetrahydrate (FeCl2·4H2O, 98%), ammonia (NH3), hydrochloric acid (HCl), curcumin (Cur), cadmium bromide (CdBr2, 99%), and mercaptosuccinic acid (MSA, 99%) were purchased from Aldrich. Carboxymethyl chitosan (CMCS) with 80% decarboxylation, sodium borohydride (NaBH4, 99%), tellurium powder (Te, 99.8%), and phosphate buffered saline (PBS, pH 7.4), Dulbecco's modified Eagle medium (DMEM), fetal bovine serum (FBS) were purchased from Merck. Other chemicals were of HPLC grade. Distilled water was used throughout all experiments.2.2 Methods
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio, were dissolved in a 2 M HCl solution. The solution was transferred into a nitrogen-purged flask. Subsequently, a 2 M ammonium hydroxide solution was added dropwise under continuous magnetic stirring until the pH reached 11. The reaction mixture was then allowed to proceed for 30 minutes at 70 °C. The resulting MNP nanoparticles were washed three times with water and collected by magnetic separation.
:1 molar ratio, were dissolved in a 2 M HCl solution. The solution was transferred into a nitrogen-purged flask. Subsequently, a 2 M ammonium hydroxide solution was added dropwise under continuous magnetic stirring until the pH reached 11. The reaction mixture was then allowed to proceed for 30 minutes at 70 °C. The resulting MNP nanoparticles were washed three times with water and collected by magnetic separation.CdTe quantum dots (QDs) were synthesized according to a previously published procedure.25 Briefly, Te powder and NaBH4 (with a determined amount) were mixed and degassed under a nitrogen atmosphere in 30 minutes. Next, 2 mL of degassed distilled water was slowly added, and the mixture was ultrasonicated for 30 minutes. The temperature was maintained at 120 °C throughout the reaction, resulting in the formation of a NaHTe solution. The freshly prepared 0.625 M NaHTe solution was then rapidly added to a mixture of CdBr2 and MSA (molar ratio of 1:1.5) under a nitrogen atmosphere at room temperature. The formation of CdTe QDs was evidenced by the color transition from red to golden yellow.
The as-synthesized MNP and CdTe (at the molar ratio of 10 : 1) were added dropwise to 4 mg mL−1 of a CMCS solution. The mixture was then ultrasonicated for 1 h and stirred for 24 h to form coated magnetic-fluorescent nanoparticles denoted as CMCS-MNP-QDs (C1). The emulsion solvent evaporation method was used to encapsulate Cur onto the CMCS-MNP-QDs nanoparticles as reported in our previous studies.26 First, a curcumin solution was prepared in ethanol. The obtained Cur solution was then slowly added to the CMCS-MNP-QDs suspension under vigorous stirring. The reaction was allowed to complete in 24 h in a closed flask. Ethanol was then evaporated by vacuum evaporation and the final product of CMCS-MNP-QDs-Cur (C3) was achieved. For comparison purposes, two additional samples were prepared, containing two components, namely CMCS-MNP-Cur (C2) and CMCS-QDs-Cur (C4). The concentrations of each component were kept constant in all samples: specifically, 2 mg mL−1 for MNP, 1 mg mg−1 for curcumin and 0.2 mg mL−1 for QDs.
000 rpm for 15 min, the supernatant of the NPs solution was collected and analyzed using HPLC (UV detector of DAD wavelength 425 nm). The test was performed three times, followed by calculating the EE according to the equation below: 000×g for 10 min. The release profile of curcumin from free curcumin or C3 NPs was calculated according to the formula:
000×g for 10 min. The release profile of curcumin from free curcumin or C3 NPs was calculated according to the formula: 000 ms and TE = 44 ms. For both T1- and T2-mappings, the matrix size = 128 × 64, the field of view = 3.17 × 3.0 cm2, flip angle = 180°, and slice thickness = 1.5 mm were used. The mean and standard deviations for T1 and T2 of each phantom were calculated over a region of interest (ROI) placed in the centre of each phantom. The r1 and r2 relaxivity values were calculated from the slope of the inverse of the longitudinal relaxation time (1/T1) and the transversal relaxation time (1/T2), respectively, versus concentration plots of Fe concentration.
000 ms and TE = 44 ms. For both T1- and T2-mappings, the matrix size = 128 × 64, the field of view = 3.17 × 3.0 cm2, flip angle = 180°, and slice thickness = 1.5 mm were used. The mean and standard deviations for T1 and T2 of each phantom were calculated over a region of interest (ROI) placed in the centre of each phantom. The r1 and r2 relaxivity values were calculated from the slope of the inverse of the longitudinal relaxation time (1/T1) and the transversal relaxation time (1/T2), respectively, versus concentration plots of Fe concentration.MRI acquisitions were conducted on a 7 T vertical spectrometer equipped with an ultra-shielded refrigerated magnet (300 WB, Bruker, Avance II, Wissembourg, France) and an RF birdcage coil with a 40 mm inner diameter (Bruker), along with a nominal 200 mT m−1 actively shielded gradient coil. Mice were anaesthetised with gaseous isoflurane at 1.5% in an air/O2 mixture at 0.5 L min−1 and 0.2 L min−1, respectively. The respiration rate was monitored throughout the procedure, along with the temperature in the cradle. To monitor the NPs' passive targeting, T2*-weighted MRI images were obtained using a fast low-angle shot (FLASH) sequence 4 hours post-injection (dosage of NPs: 15 mg kg−1). Whole-body imaging of the mice was performed using the following parameters: FLASH images employing a Hermitian pulse, echo time (TE) = 5 ms, repetition time (TR) = 350 ms, and a flip angle of 40°, triggered by respiration. A field of view of 3 × 3 cm2, matrix size of 256 × 256 corresponding to 117 μm × 117 μm in-plane resolution, and 23 to 26 slices with a thickness of 1 mm were utilised, resulting in an acquisition time of approximately 6 minutes. All MRI data were transferred to and analysed on a computer equipped with MRI-specific software (ParaVision version 5.1). All images were captured at the same depth to facilitate the comparison of NPs-mediated darkening. The animals were then sacrificed.
For data processing, the MRI image of each slice was opened in ImageJ software (National Institutes of Health, Bethesda) using the BrukerOpener plugin, and the ROI corresponding to the tumor was drawn. The pixel intensity distributions were obtained for each slice of the tumor and then compiled using MATLAB software (R2023a, Natick, Massachusetts, United States) for the whole tumor. To assess the effectiveness of the treatment and compare the different groups of mice, the percentage below the 25th percentile and above the 75th percentile, respectively, %p25 and %Ap75, a post-treatment methodology was used.6 Briefly, the pixel intensity distribution of each tumor was obtained from T2-weighted images, and the percentage of pixels under the value I0.25 = 0.25 × (Intensitymax − Intensitymin) and the percentage of pixels above the value I0.75 = 0.75 × (Intensitymax − Intensitymin) were calculated and written as %p25, %Ap75 using the Matlab software. The two-tailed Student's t-test with unequal variance was used to compare the two groups, and the values p are obtained.
Then, all mice were sacrificed, and the tumors, livers, spleens, lungs, and kidneys were collected for ex vivo signal acquisition.
2.3 Statistical analysis
One-way ANOVA and Student's t-test were used for statistical analysis (p < 0.05) using the Sigma Plot 15 software. The experiments were performed three times and the data are expressed as the mean ± SD of three independent measurements.3. Results and discussion
3.1 Properties of nanoparticles
FTIR spectra of CMCS-MNP-QDs-Cur and their constituents are shown in Fig. 1. It can be seen that some characteristic peaks were shifted in the spectrum of C3 NPs compared to those of pure components. Particularly, the characteristic peak of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond of chitosan and curcumin at 1629 cm−1 was observed at 1564 cm−1, which overlapped with the mixed vibrations at 1512 cm−1 of curcumin. The symmetrical COO- stretching of CMCS (1422 cm−1) overlapped with the CC aromatic vibration of curcumin (1431 cm−1), which shifted to 1402 cm−1 in the C3 NPs spectrum.27,28 The peak at 596 cm−1 attributed to the Fe–O bond vibration also appeared in the spectrum of C3 NPs.6 Fig. 1B presents the fluorescence spectra of the four nanosystems. A high-intensity peak at 684 nm was observed in the CMCS-MNP-QDs (C1) spectrum, corresponding to the fluorescence of CdTe QDs. Curcumin exhibited a broad emission peak between 460 and 550 nm, which was recorded at 526 nm in the spectrum of C2 NPs. These two peaks were also observed in the fluorescence spectra of C3 and C4 NPs with a slight shift. The emission peak of QDs was shifted to 690 nm, while that of Cur was blue-shifted to 518 nm.
O bond of chitosan and curcumin at 1629 cm−1 was observed at 1564 cm−1, which overlapped with the mixed vibrations at 1512 cm−1 of curcumin. The symmetrical COO- stretching of CMCS (1422 cm−1) overlapped with the CC aromatic vibration of curcumin (1431 cm−1), which shifted to 1402 cm−1 in the C3 NPs spectrum.27,28 The peak at 596 cm−1 attributed to the Fe–O bond vibration also appeared in the spectrum of C3 NPs.6 Fig. 1B presents the fluorescence spectra of the four nanosystems. A high-intensity peak at 684 nm was observed in the CMCS-MNP-QDs (C1) spectrum, corresponding to the fluorescence of CdTe QDs. Curcumin exhibited a broad emission peak between 460 and 550 nm, which was recorded at 526 nm in the spectrum of C2 NPs. These two peaks were also observed in the fluorescence spectra of C3 and C4 NPs with a slight shift. The emission peak of QDs was shifted to 690 nm, while that of Cur was blue-shifted to 518 nm.
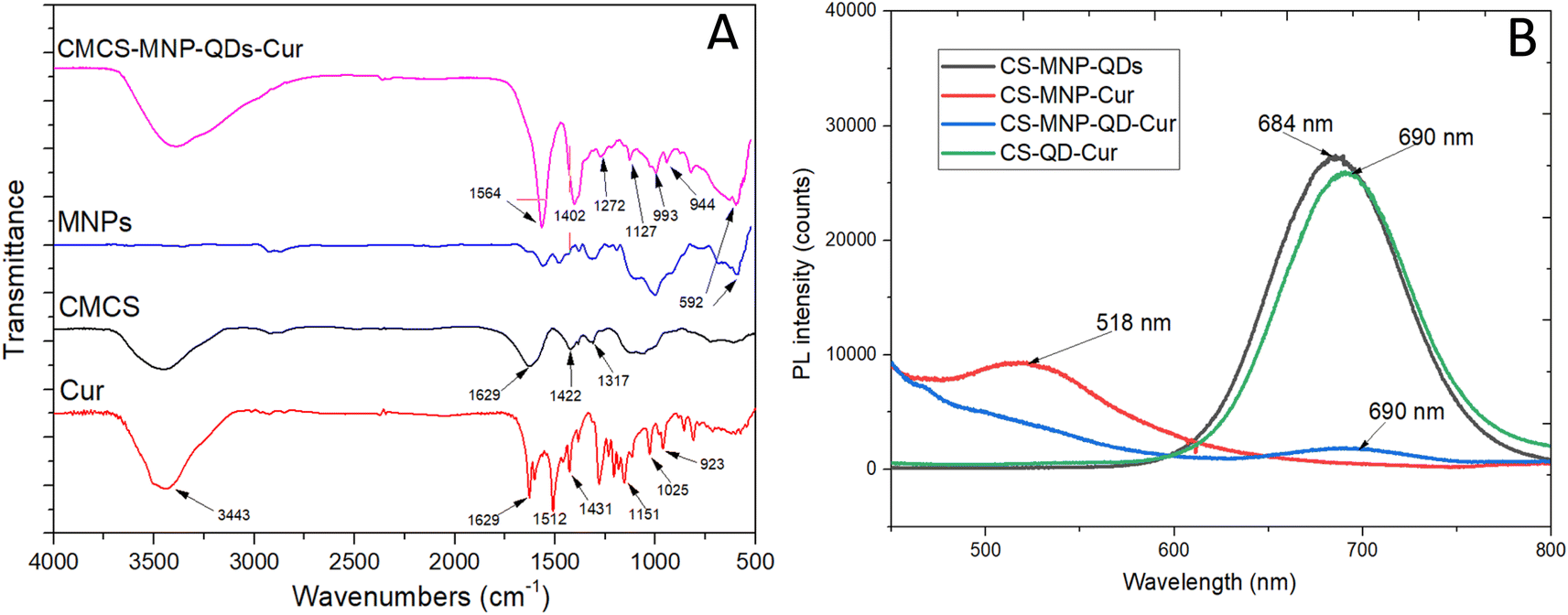 | ||
| Fig. 1 Fourier transform infrared spectra of CMCS-MNP-QDs-Cur (C3) nanoparticles and their constituents (A), and photoluminescence (B) spectra of the four NPs. | ||
Table 1 presents the hydrodynamic size and zeta-potential results of the nanoparticle systems. The hydrodynamic sizes of the nanosystems were measured in DI water, PBS and DMEM and l ranged from 96 nm to 167 nm. These values are within the recommended size range for various biomedical applications that allow for better biodistribution, enhanced cellular uptake, and controlled release of therapeutic agents.29 In PBS and DMEM media, the nanoparticle sizes increased slightly compared to those in DI water. This could be due to protein adsorption, particle aggregation, or the presence of ions in these media.30 The PDI values in all cases were found to be in the range of 0.1–0.3, indicating the monodispersity of the prepared nanoparticles. Besides, the zeta potentials in DI water of the four nanoparticles range from −63.7 to −40.0 mV, suggesting excellent stability of all four nanosystems. The presence of Fe3O4 and CdTe QDs nanoparticles was also confirmed in the EDX spectra of C3 NPs (Fig. 2A). TEM images of C3 NPs showed Fe3O4 particles with a diameter of approximately 15 nm and CdTe QDs particles with a smaller size of 2–5 nm decorated on the surface of Fe3O4. These results are consistent with earlier reports on MNPs and CdTe QDs.31,32
| Sample | In water | In PBS | In DMEM | Zeta potential (mV) | |||
|---|---|---|---|---|---|---|---|
| Z-Average (nm) | PDI | Z-Average (nm) | PDI | Z-Average (nm) | PDI | ||
| CMCS-MNP-QDs (C1) | 96.2 | 0.236 | 110 | 0.146 | 105 | 0.117 | −53.5 |
| CMCS-MNP-Cur (C2) | 167.1 | 0.257 | 176 | 0.202 | 170 | 0.136 | −63.7 |
| CMCS-MNP-QDs-Cur (C3) | 128.1 | 0.352 | 122 | 0.104 | 125 | 0.084 | −50.7 |
| CMCS-QDs-Cur (C4) | 135.9 | 0.356 | 148 | 0.154 | 150 | 0.162 | −40.9 |
 | ||
| Fig. 2 EDX spectrum (A) and TEM image (B) of C3 NPs. | ||
3.2 In vitro stability
The storage stability of four NPs was evaluated by monitoring changes in particle size and encapsulation efficiency of curcumin over a 60 days period at 25 °C. The average particle diameters of all formulations remained stable for 60 days (Fig. 3A), with only slight fluctuations in the PBS and DMEM media (p > 0.05), indicating that they were able to maintain their size for a relatively long time. This indicates no particle aggregation and excellent stability for biological applications. Compared to C2 and C4 NPs, the reduction in size of QDs-decorated curcumin-loaded nanosystem (C3) may be attributed to two primary factors: the enhanced water dispersibility of the encapsulating CMCS polymer and the robust hydrophobic magnetite core.33,34 The Fe3O4 molecules that have been modified on their surface offer the NH2 group to create a hydrogen bond with the –OH groups of curcumin and CMCS, and they can also generate a positive surface charge to attract the negatively charged curcumin through electrostatic interaction.35,36 Compared with the particle size of the other NPs, C3 NPs with an average size of 120 nm in PBS and DMEM solutions were still suitable for better curative effects through the EPR effect. Regarding the encapsulation of curcumin, it was observed that the formulation showed a non-significant change in encapsulation efficiency during 60 days, and no drug degradation was detected (Fig. 3B). These observations indicate that curcumin encapsulated into the polymeric core remained stable for two months of storage. | ||
| Fig. 3 Changes of particle sizes of the four NPs incubated in PBS and DMEM (A), and encapsulation efficiency (B) over 60 days. | ||
3.3 In vitro curcumin release
The release of free curcumin was used as a control. Free curcumin showed a burst release within 12 h at a pH of 7.4; meanwhile, only about 35% and 52% of curcumin were released at pH 5.5 and 6.5, respectively (Fig. 4A). As free curcumin has low solubility in acidic conditions, the release was lower at pH 5.0 than at pH 7.4. On the other hand, the opposite occurs in the C3 NPs. Curcumin was released from C3 NPs in a gradual and continuous manner since the polymer was swollen but not broken at pH 5.5 and 37 °C. The final cumulative drug release was 83.33% at pH 5.5 within 96 h. Compared to pH 7.4 in PBS, the enhanced release was observed at pH 6.5. The cumulative release profile of curcumin from C3 NPs at pH 5.5 was the largest, which stemmed from the protonation of amino groups and the disruption of hydrogen bonding between the carboxyl group of CMCS and the hydroxyl group of curcumin in acidic environments.34 Acidic conditions enhance curcumin delivery to tumor cells due to lower pH; therefore, they mitigate harm to normal cells. Similar curcumin release behavior has been observed in other nanocarriers, such as pH-responsive polymeric nanoparticles,37 hollow mesoporous silica NPs,38 and pectin/gelatin hydrogel,39 primarily due to the hydrophobic properties of curcumin. In line with our findings, the study by Zhai et al. showed the sustained release and maximum curcumin release at pH 5.5 from curcumin-loaded pH-sensitive tri-block copolymer of PCL-PDEA-PSBMA micelles, while a relatively lower release occurred at the physiological pH of 7.4.40 The enhanced release at pH 5.5 is attributed to the PDEA block, which could be protonated, leading to a faster drug release in an acidic environment.40 Similar results have been reported in the literature, where curcumin was covalently conjugated to the hydrophilic terminals of Poloxam 188 chains, which would be broken down and result in the faster release of curcumin in pH 6.4 and pH 5.0, compared to that in the physiological pH 7.4.41 The cancer cells were then cultured and the supernatant of the conditioned medium was collected and measured for the release profile of curcumin. The curcumin release from C3 NPs exposed to CM-CT26 for 96 h was significantly higher than those exposed to DMEM and FBS, 60.42% compared to 27.81 and 25.32%, respectively (p < 0.0001) (Fig. 4B). Thus, an enhanced release of curcumin from C3 NPs was observed after being exposed to low pH and CM-CT26, mimicking the tumor pH conditions. | ||
| Fig. 4 (A) Drug release at pH 7.4, 6.5, and 5.5 and (B) kinetics of the release of curcumin from C3 NPs in the presence of CM-CT26, DMEM and FBS. | ||
The MRI contrast enhancement capabilities of C1, C2, and C3 were evaluated using in vitro MR images captured at varying iron concentrations under both T1-weighted and T2-weighted modes. Fig. 5A shows the T2-weighted images of the nanoconjugates. As the Fe concentration increases, the T2-weighted MR images appear darker. This could be due to a decrease in the spin–spin relaxation time (T2) because the protons of water molecules are affected by the dipole moment of the nanosystems. Additionally, the carboxyl groups of CMCS promote the adsorption of water molecules on the nanosystem surface, thereby altering the longitudinal relaxation time (T1).42 Fig. 5B illustrates the linear relationship between relaxation rate (R1 = 1/T1 and R2 = 1/T2) and Fe concentration. The slopes of the plots represent longitudinal relaxivity (r1) and transverse relaxivity (r2) (Table 2). While C2 exhibited the highest r2 value, the highest r2/r1 value was obtained for C3 NPs. Previous studies have also indicated that the incorporation of drugs such as Dox or curcumin can improve the r2 value.43,44 Therefore, C2 and C3 exhibit the potential to be T2-contrast enhancement agents.
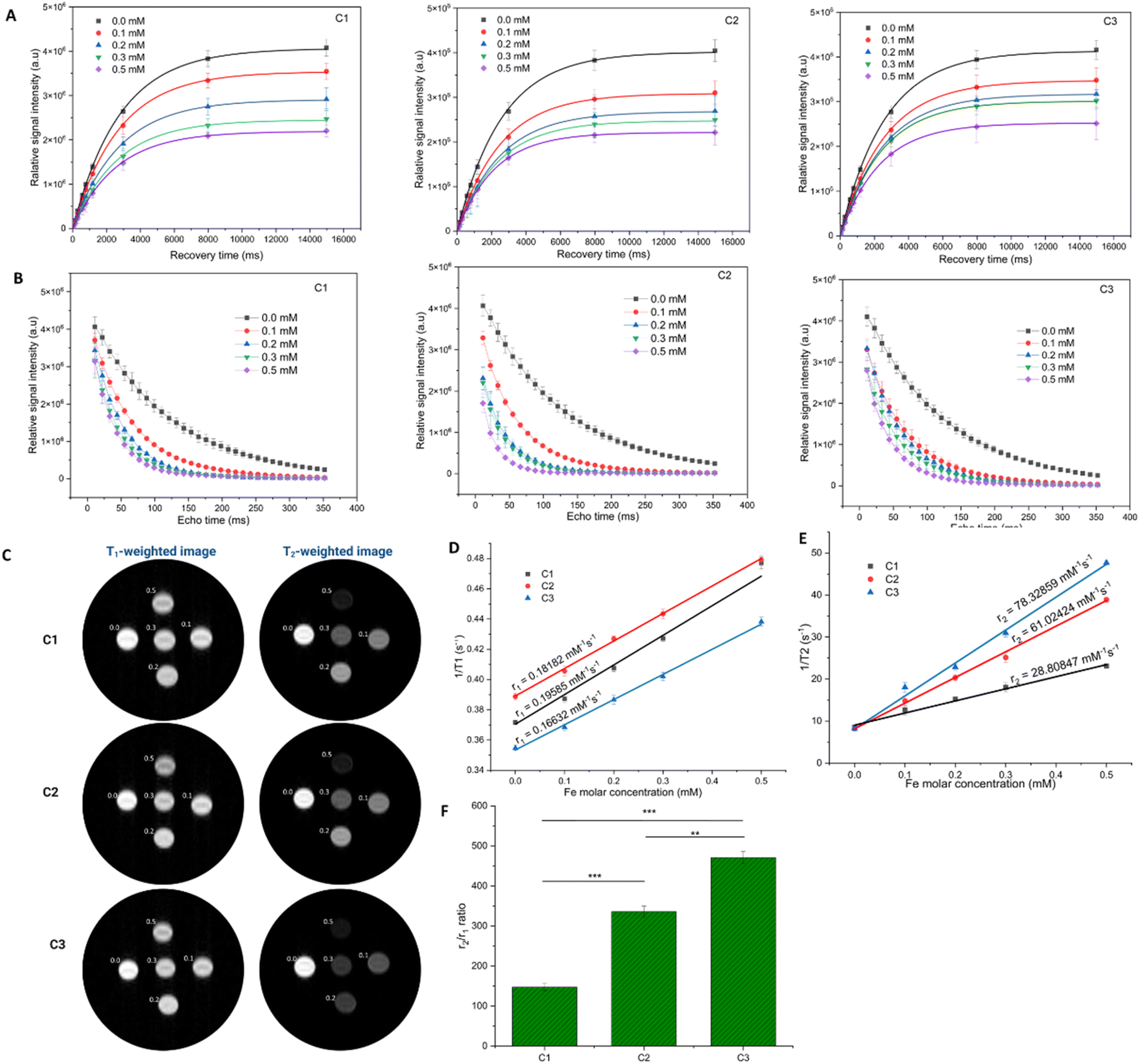 | ||
| Fig. 5 The relative signal intensity (SI) in a partial saturation experiment versus (A) recovery time (TR) and (B) echo time (TE) of NPs. Recovery time (TR) is the repetition time between two 90° pulses. The signal recovery is 63% after a period of T1. The signal decay is 37% after a period of T2. (C) T1-weighted and T2-weighted MR images of NPs. Plots of relaxation rates T1-1 (D) and T2-1 (E) versus concentration and relaxivity ratio r2/r1 (F) of C3 NPs. Statistical analysis was performed with a two-tailed Student's t-test with unequal variance, *p value < 0.05, **p value < 0.01, ***p value < 0.001. | ||
| No. | Wavelength corresponding to curcumin (nm) | Radiance enhancement ratio of NPs [C0]/agarose [0] | Signal-to-noise ratio (SNR) | Wavelength corresponding to QDs (nm) | Radiance enhancement ratio of NPs [C0]/agarose [0] | Signal-to-noise ratio (SNR) | ||
|---|---|---|---|---|---|---|---|---|
| λexcitation | λemission | λexcitation | λemission | |||||
| I | C1 NPs | |||||||
| 1 | 412 | 497 | 0.35 | 8.58 | 487 | 672 | 2.96 | 89.29 |
| 2 | 522 | 0.43 | 12.41 | 697 | 3.41 | 92.69 | ||
| 3 | 547 | 0.22 | 15.48 | 722 | 2.83 | 78.35 | ||
| 4 | 572 | 0.63 | 19.36 | 747 | 2.19 | 54.32 | ||
| 5 | 437 | 497 | 6.06 | 10.47 | 512 | 672 | 5.99 | 128.78 |
| 6 | 522 | 0.34 | 12.87 | 697 | 6.88 | 162.73 | ||
| 7 | 547 | 0.52 | 16.33 | 722 | 5.46 | 112.40 | ||
| 8 | 572 | 0.61 | 20.21 | 747 | 4.0 | 72.82 | ||
| 9 | 462 | 497 | 0.40 | 14.35 | 537 | 672 | 6.04 | 127.01 |
| 10 | 522 | 0.45 | 15.97 | 697 | 7.66 | 170.72 | ||
| 11 | 547 | 0.53 | 17.99 | 722 | 6.43 | 154.48 | ||
| 12 | 572 | 0.61 | 19.48 | 747 | 4.40 | 80.37 | ||
|
||||||||
| II | C2 NPs | |||||||
| 13 | 412 | 497 | 0.09 | 1.94 | 487 | 672 | 0.28 | 6.83 |
| 14 | 522 | 0.015 | 2.58 | 697 | 0.47 | 7.11 | ||
| 15 | 547 | 0.126 | 3.89 | 722 | 0.63 | 8.82 | ||
| 16 | 572 | 0.18 | 7.72 | 747 | 0.72 | 8.27 | ||
| 17 | 437 | 497 | 0.09 | 1.94 | 512 | 672 | 0.34 | 9.54 |
| 18 | 522 | 0.09 | 2.31 | 697 | 0.60 | 16.11 | ||
| 19 | 547 | 0.13 | 3.69 | 722 | 0.84 | 19.04 | ||
| 20 | 572 | 0.18 | 5.73 | 747 | 0.94 | 19.85 | ||
| 21 | 462 | 497 | 0.17 | 3.89 | 537 | 672 | 0.33 | 9.29 |
| 22 | 522 | 0.10 | 2.63 | 697 | 0.60 | 16.79 | ||
| 23 | 547 | 0.13 | 3.07 | 722 | 0.84 | 21.44 | ||
| 24 | 572 | 0.19 | 5.32 | 747 | 0.93 | 21.72 | ||
|
||||||||
| III | C3 NPs | |||||||
| 25 | 412 | 497 | 0.19 | 8.29 | 487 | 672 | 6.79 | 197.62 |
| 26 | 522 | 0.38 | 10.35 | 697 | 15.03 | 279.31 | ||
| 27 | 547 | 0.73 | 20.41 | 722 | 8.73 | 271.10 | ||
| 28 | 572 | 0.92 | 27.43 | 747 | 6.89 | 174.99 | ||
| 29 | 437 | 497 | 0.19 | 6.08 | 512 | 672 | 8.42 | 123.86 |
| 30 | 522 | 0.40 | 12.07 | 697 | 12.83 | 169.89 | ||
| 31 | 547 | 0.74 | 23.23 | 722 | 11.87 | 145.37 | ||
| 32 | 572 | 0.91 | 25.93 | 747 | 8.89 | 100.78 | ||
| 33 | 462 | 497 | 0.27 | 7.35 | 537 | 672 | 7.55 | 202.71 |
| 34 | 522 | 0.43 | 12.63 | 697 | 11.39 | 277.53 | ||
| 35 | 547 | 0.79 | 21.16 | 722 | 10.92 | 265.19 | ||
| 36 | 572 | 0.95 | 25.68 | 747 | 8.2 | 166.81 | ||
|
||||||||
| IV | C4 NPs | |||||||
| 37 | 412 | 497 | 0.25 | 5.00 | 487 | 672 | 10.00 | 288.02 |
| 38 | 522 | 0.44 | 9.60 | 697 | 20.44 | 388.40 | ||
| 39 | 547 | 1.19 | 26.52 | 722 | 20.97 | 337.89 | ||
| 40 | 572 | 0.17 | 46.01 | 747 | 14.00 | 225.63 | ||
| 41 | 437 | 497 | 0.26 | 6.55 | 512 | 672 | 28.98 | 541.21 |
| 42 | 522 | 0.47 | 11.08 | 697 | 40.17 | 668.42 | ||
| 43 | 547 | 1.09 | 27.86 | 722 | 36.23 | 583.84 | ||
| 44 | 572 | 1.76 | 43.61 | 747 | 26.27 | 391.76 | ||
| 45 | 462 | 497 | 0.37 | 8.33 | 537 | 672 | 22.44 | 488.79 |
| 46 | 522 | 0.54 | 11.92 | 697 | 29.30 | 533.27 | ||
| 47 | 547 | 1.29 | 29.18 | 722 | 37.47 | 705.64 | ||
| 48 | 572 | 2.10 | 47.34 | 747 | 27.61 | 441.48 | ||
3.4 In vitro and in vivo MRI measurements
To unveil the potential of NPs as a contrast agent for MRI, the dual-mode T1- and T2-weighted MR images, along with the corresponding longitudinal and transverse relaxation rates at various concentrations, were evaluated using a Bruker 300 WB magnet at 25 °C in 25 mM of phosphate buffer (Fig. 5A and B). Because of the practical applicability to in vivo systems, all the experiments were done in PBS. The phantoms were prepared in a microtube with different concentrations of Fe (0, 0.1, 0.2, 0.3, and 0.5 mM). As shown in Fig. 5C, both T1- and T2-weighted MR images exhibited evident concentration-dependent signals of brightness and darkness. The r1 relaxivity was obtained for five different concentrations of Fe using an inversion recovery pulse. The plot of the longitudinal relaxation rate 1/T1 versus Fe concentration is linear and shows r1 relaxivities of 0.19585, 0.18182 and 0.16632 mM−1 s−1 for C1, C2 and C3 NPs, respectively (Fig. 5D). Also, the r2 relaxivities of these NPs were obtained using a saturation transfer pulse at 7 T as 28.80847, 61.02424 and 78.32859 mM−1 s−1, respectively. These values were obtained from the slope of the linear plot of transverse relaxation rate 1/T2 versus the concentration of Fe in PBS (Fig. 5E). The relaxivity must be derived from the second sphere water relaxation, which is the hydrogen bonding interaction of the bulk water molecule with oxygen atoms in the structure of the Fe3O4 core of NPs. Fig. 5F indicates that the relaxivity ratio r2/r1 of C3 NPs was 470.95 ± 15.11, which is 1.4 times higher than that of C2 NPs (335.63 ± 13.90) and 3.2 times higher than that of C1 NPs (147.09 ± 8.99). This difference (p < 0.001) was also evident in T2-weighted MR imaging (Fig. 5D), highlighting the superior performance of C3 NPs as a contrast agent for MRI imaging. This indicated the ability of curcumin and QDs to enhance the signal in vitro of C3 NPs. In a previous study, we demonstrated that the chemical structures of the drug (i.e., doxorubicin) and dye agent (i.e., cyanine 5.5) significantly contribute to the enhancement of the T2 relaxivities of Fe3O4 NPs through π–π and ρ–π conjugation.45 Remarkably, the r2/r1 value of C3 NPs was much higher than that of doxorubicin-QDs-Fe3O4-alginate NPs at 7 T, also presented in our previous study.46 Another study by Ghorbaanee et al. also showed that the presence of curcumin increased the negative contrast in MRI images as compared to curcumin-free NPs.44 The r2 of curcumin-loaded Fe3O4-PEI-CUR NPs increased by about 20% compared to the non-curcumin-loaded Fe3O4 nanoparticles.The effectiveness and passive targeting of C3 NPs by MRI imaging was assessed using a post-processing methodology previously described with a new parameter.47 The pixel intensity distribution on the T2*-weighted images was quantified to generate %p25 and %Ap25, which were used as reference parameters to compare the various groups (Fig. 6A and B). The proportion of pixels below the threshold was computed and recorded as %p25 (0.25(Intensitymax − Intensitymin)). %Ap0.75 was defined as % above the 75th percentile computing bright intensity pixels attributed to inflammation by MRI. There was a substantial difference (p < 0.0001) in %p25 between the injected mice and non-injected mice (35.78% and 5.77%, respectively). This indicated the passive accumulation of C3 NPs in these tumors. This finding could be explained by the particle size range of C3 NPs, which was within the range of 100-200 nm for the enhanced permeability and retention effect (EPR).48 Also, the CT26 tumor showed the ability to increase blood vessels around it, forming these highly vascularized tumors, which can improve permeability and retention of C3 NPs.49,50 Moreover, the %Ap0.75 in the treated tumor was significantly higher compared to the control groups, p < 0.001, with the %Ap75 increasing from 3.83% for the control to 18.67% for C3 NPs, a factor of 4.9 (Fig. 6E). Six hours after injecting the C3 NPs, the %p0.25 and %Ap0.75 remarkably increased compared to those in the control mice, reflecting necrosis and inflammation, respectively, which are the expected consequences of the NP treatment.
 | ||
| Fig. 6 MRI images of control mice (A) and C3 NPs (B); histogram pixel number from MRI images using a homemade Matlab programme: control mice (C) and C3 NPs (D); and (E) percentile %p25 and %Ap75 graphs. | ||
3.5 In vitro NIR optical imaging
To optimize the excitation and emission wavelengths of each NPs, the radiance enhancement ratio and signal-to-noise ratio (SNR) were determined by fluorescence imaging in solution (Table 2). The C1 NPs exhibited the best signal quality and most trustworthy data for fluorescence imaging when excited at 487 nm with a 697 nm emission (Fig. 7A). These wavelengths were close to the excitation and emission wavelengths of QDs. Besides, the radiance enhancement ratio of C2 NPs was under 1.0, and the signal-to-noise ratio was also relatively low at all wavelength conditions (Fig. 7B). At an excitation wavelength of 487 nm and an emission wavelength of 697 nm, the contrast between the C3 NPs and the background was the highest (Fig. 7C), with a radiance enhancement ratio of 15.03 and an SNR of 279.31. Without Fe3O4 NPs, meanwhile, fluorescence optical images of C4 NPs showed the optimal excitation and emission wavelengths at 512 nm and 697 nm, respectively (Fig. 7D). This phenomenon could be explained by the interaction between Fe3O4 NPs and curcumin/QDs encapsulated in the CMCS polymer of C3 NPs. It was demonstrated that the interaction between the drug and Fe3O4 NPs caused both blue-shift and quenching effects in the radiance enhancement ratio and signal-to-noise ratio of fluorescence optical imaging.43,45 Thus, the ideal parameters for in vivo fluorescence imaging for biodistribution of C3 NPs are the highest radiance enhancement ratio and SNR at the excitation/emission wavelength of 487/697 nm. | ||
| Fig. 7 Fluorescence imaging in solutions at different excitation and emission wavelengths of (A) C1 NPs, (B) C2 NPs, (C) C3 NPs, and (D) C4 NPs. | ||
3.6 In vivo and ex vivo NIR optical imaging
The inclusion of different therapeutic modalities, as well as the clinical need for visual diagnosis and monitoring treatment progress, have become more important in the development of “all-in-one” nanotheranostics. Herein, to demonstrate the ability of C3 NPs to undergo passive targeting and treatment by the quantitative MRI signal, the in vivo behavior of the C3 NPs was investigated by measuring the average radiance of optical imaging in the mice model (Fig. 8A). After intravenous administration of C3 NPs (13.5 mg QDs per kg body), the tissue biodistribution was determined at seven time points (5 min, 15 min, 30 min, 1 h, 3 h, 6 h, and 24 h) (Fig. 8B). Fig. 8C–F illustrate the average radiance (ph s−1 cm−2 sr−1) in fluorescence intensity of the tumors, liver, kidneys, and spleen, respectively. As shown in Fig. 8G, at 5 min post-injection, a weak fluorescence signal was detected at the tumor site, indicating gradual accumulation of the C3 NPs due to permeability and retention (EPR) effect. It has been reported that nanomaterials within the 20–200 nm range can extravasate and accumulate in tumors via the EPR effect, allowing for the passive accumulation of C3 NPs in the tumor areas.51,52 Also, impaired lymphatic drainage hinders the elimination of NPs from the tumor tissue, resulting in increased NP accumulation.53 Meanwhile, the fluorescence intensity progressively increased over time, and the maximum fluorescence was obtained after 6 h post-injection, which reaffirmed the in vivo tumor passive targeting effects of C3 NPs. Subsequently, with the passage of time, the fluorescence intensity in the tumor progressively decreases, with only a weak fluorescence observed after 24 h, suggesting that C3 NPs could be metabolized gradually over time. In addition, according to the distribution pattern shown in Fig. 8G, the C3 NPs accumulated faster in the spleen within the first 30 min after injection, and at 1 h post-injection, the accumulation in the liver increased up to the highest signal of the injected dose. The accumulation of nanoparticles in the spleen, liver, and kidneys gradually decreased from 1 h to 24 h, which may be related to the elimination process of C3 NPs. Furthermore, the FLI intensity ratios of the tumors/liver at all time points were higher than 1.0, suggesting greater accumulation in tumor cells than in the liver (Fig. 8H). On the other hand, the FLI intensity of the liver/kidneys was approximately 0.5 at all seven time points. Compared to the liver, the rate of accumulation in the kidneys during the studied time interval was remarkably higher. These results affirm that C3 NPs can passively accumulate at the tumor efficiently and facilitate the monitoring of treatment progress by NIR fluorescence optical imaging. In another report, Tamarov et al. have shown that the PEG-BPSi-Cy7.5 NPs exhibited high fluorescence and were observed in the stomach, intestine, and kidney.54 This finding demonstrated that NPs underwent the hepatobiliary clearance pathway, where the NPs were transported from the liver to the stomach and small intestine and could be finally removed from the body by fecal excretion. Zheng et al. have also shown that the intravenously administered curcumin@IR783 nanocolloid accumulated in the tumor site via the EPR effect, and the fluorescence signal at the tumor site peaked at 24 h.55 Notably, the liver exhibited a robust fluorescent signal, which was attributed to the reticuloendothelial system absorption and drug hepatorenal metabolism as reported in other studies.43,56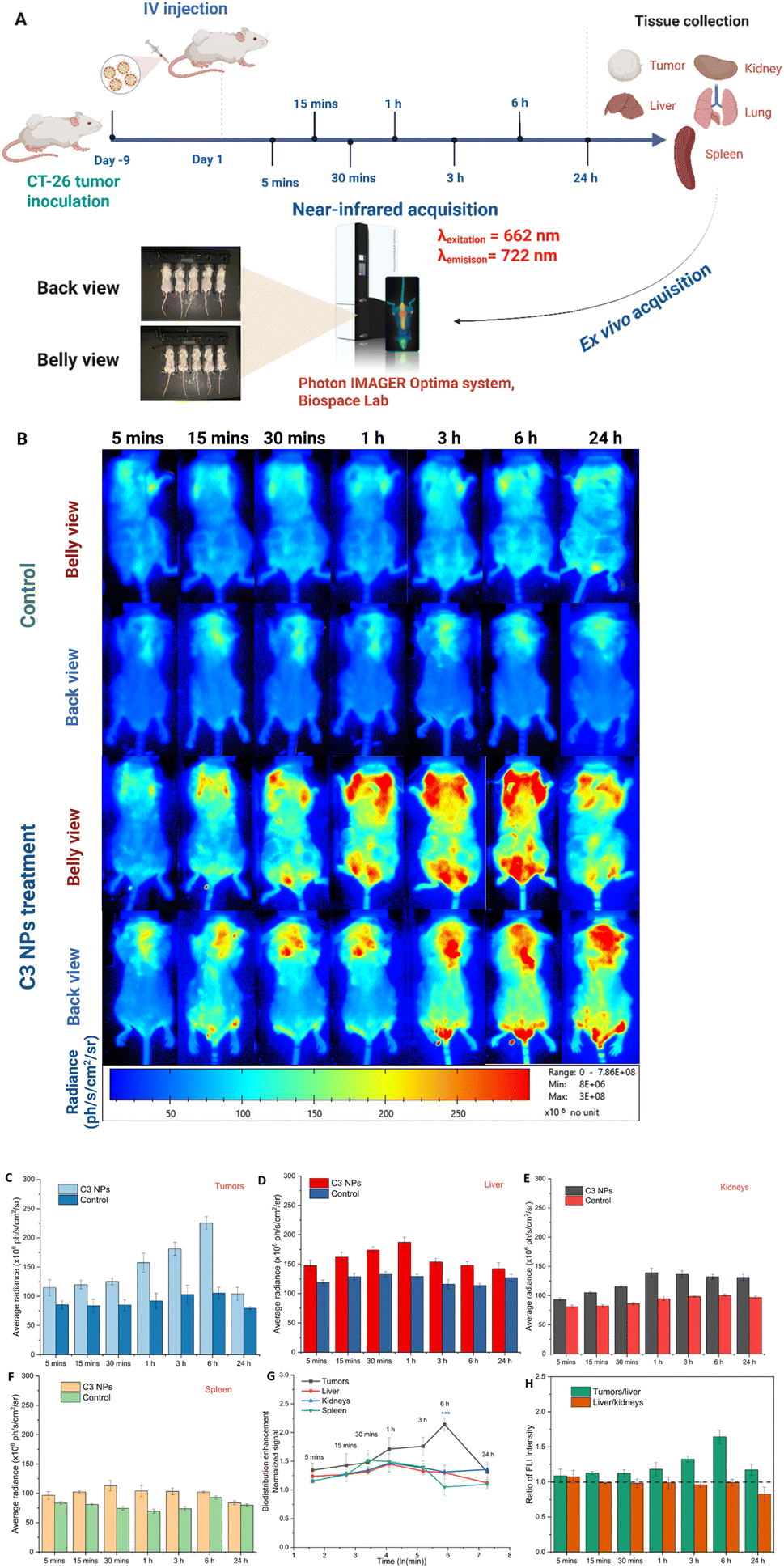 | ||
| Fig. 8 (A) Schematic illustration showing the treatment regimen: CT26 cell inoculation in the two mouse flanks. The mice (n = 5) received C3 NPs treatments; (B) the FL images of mice treated with various time points; (C–F) average FL intensity in tumors, liver, kidney, spleen, respectively; (G) quantitative analysis of the biodistribution in (B); (H) FLI ratios of tumors/liver and liver/kidneys. Statistical analysis was performed with a two-tailed Student's t-test with unequal variance, *p value < 0.05, **p value < 0.01, ***p value < 0.001. | ||
After 24 h post injection, the mice were sacrificed, and major organs including the liver, spleen, lungs, and kidneys, as well as the tumor, were resected to evaluate the tissue distribution of C3 NPs (Fig. 9A and B). From the qualitative analysis of the images, a higher average radiance intensity was observed from the tumors of mice injected with the nanoparticles compared to control mice (Fig. 9C). This also indicated that the fluorescence emission from the tumor occurred as a result of the accumulation of nanoparticles via the EPR effect. In mice treated with C3 NPs, the liver/kidneys ratio was generally lower than 1.0, and in particular, the detected tumors/liver ratio was lower than 0.5 (Fig. 9D). Clearly, part of the NPs eventually accumulated in the liver, spleen, and kidneys, which are the organs responsible for eliminating alien bodies from systemic circulation. Interestingly, a 4.09-fold higher fluorescence was observed from the lungs of C3-NPs-injected mice than that of the control mice. The signals from these tissues may be a result of some free QDs dissociated from the C3 NPs. Gabuzenko et al. have observed that QDs accumulated in the lungs and spleen 1 hour after intravenous administration.57 Yaghini et al. also found that bio CFQD® nanoparticles were predominantly accumulated in the liver and spleen till 90 days after IV injection in rats.58 The accumulation of QDs in the liver and spleen after systemic injection has been frequently reported and is associated with the clearance of NPs from the circulatory system by mononuclear phagocytic system cells.59,60
 | ||
| Fig. 9 (A) Images of liver, kidneys, spleen, tumor, and lungs, collected from tumor mice at 24 h after nanoparticle injection; (B) FL imaging of major organs and tumor; (C) average FL intensity of each organ; (D) ratios of ex vivo FLI intensity. | ||
4. Conclusion
In a nutshell, we report an ultimate system CMCS-MNP-QDs-Cur composed of the NIR dye QDs, consisting of the natural compound curcumin as the therapeutic agent and Fe3O4 magnetic nanoparticles as the contrast agent. The nanosystems exhibited small particle sizes within the 100–200 nm range with excellent stability in both DI water, PBS, and DMEM media. Moreover, the encapsulated curcumin remained stable for more than two months of storage. The MRI results of C3 NPs demonstrated their passive accumulation in tumor areas 6 hours post-injection. The nanosystem shows promise as a T2-weighted MRI contrast agent, with an r2 relaxivity of 78.33 mM−1 s−1 and an r2/r1 ratio of 470.95 ± 15.11 at 7 T, primarily due to the second-sphere water interactions. The in vivo treatment results evidenced an increase in necrosis and inflammation at the tumor site, correlated with the proportion of pixels falling below the 25th and above the 75th percentiles in intensity on the T2*-weighted images. The NIR optical imaging of the incorporated CdTe QDs effectively assessed the in vivo biodistribution of the nanoparticles. These interesting results indicate the potential application of the multimodal nanosystem for in vivo MRI and NIR optical imaging as well as passive targeting.Data availability
All the data that support the findings of this study are included in the manuscript.Author contributions
T. T. T. M.: investigation, writing – original draft, review and editing; K. S. P.: investigation, data analysis, review and editing; B. T. D., T. D. T. U., Q. B. N. N., T. H. T. T., D. L. H.: investigation; T. T. H. L.: investigation, review and editing; N. Q. T.: investigation, funding acquisition; P. T. H.: conceptualization, funding acquisition, review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the Vietnam Academy of Science and Technology (VAST) under Grant Number NCXS.01.01/23-25.References
- F. Bray, M. Laversanne, H. Sung, J. Ferlay, R. L. Siegel, I. Soerjomataram and A. Jemal, CA Cancer J. Clin., 2024, 74, 229–263 CrossRef PubMed.
- P. Repetowski, M. Warszynska and J. M. Dabrowski, Adv. Colloid Interface Sci., 2025, 336, 103356 CrossRef CAS PubMed.
- M. Jiang, Y. Liu, K. Wang, Y. Dong, X. Hu and Y. Yuan, Cell Biomater., 2025, 1, 100044 CrossRef.
- A. N. Al-Thani, A. G. Jan, M. Abbas, M. Geetha and K. K. Sadasivuni, Life Sci., 2024, 352, 122899 CrossRef CAS PubMed.
- C. Zhang, R. Cha, K. Long, Y. Liu, Y. Dong, Y. Zhang, X. Wang and X. Jiang, ACS Appl. Mater. Interfaces, 2024, 16, 68905–68917 CrossRef CAS PubMed.
- T. T. H. Le, K. S. Phan, B. T. Doan, T. T. T. Mai, H. B. H. Bui, H. N. Pham, T. D. T. Ung, Y. Wang, N. Q. Tran, L. H. Dang and P. T. Ha, RSC Adv., 2025, 15, 9644–9656 RSC.
- I. Vermeulen, E. M. Isin, P. Barton, B. Cillero-Pastor and R. M. A. Heeren, Drug Discov. Today, 2022, 27, 2086–2099 CrossRef CAS PubMed.
- E. K. Um, S. Wang, J. Qu and L. Liu, Cancer Med., 2024, 13, e70155 CrossRef PubMed.
- T. Sun, H. Zhao, L. Hu, X. Shao, Z. Lu, Y. Wang, P. Ling, Y. Li, K. Zeng and Q. Chen, Acta Pharm. Sin. B, 2024, 14, 2428–2446 CrossRef CAS PubMed.
- N. Sakurai, K. Ishigaki, K. Terai, T. Heishima, K. Okada, O. Yoshida, Y. Kagawa and K. Asano, BMC Vet. Res., 2022, 18, 374 CrossRef CAS PubMed.
- J. Lohrke, T. Frenzel, J. Endrikat, F. C. Alves, T. M. Grist, M. Law, J. M. Lee, T. Leiner, K. C. Li, K. Nikolaou, M. R. Prince, H. H. Schild, J. C. Weinreb, K. Yoshikawa and H. Pietsch, Adv. Ther., 2016, 33, 1–28 CrossRef PubMed.
- H. Xu, P. Yu, R. P. Bandari, C. J. Smith, M. R. Aro, A. Singh and L. Ma, Nanomaterials, 2024, 14, 1177 CrossRef CAS PubMed.
- A. Berdichevski, H. Simaan Yameen, H. Dafni, M. Neeman and D. Seliktar, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 5147–5152 CrossRef CAS PubMed.
- C. Li, C. Li, J. Zhou, Y. Wang, H. Wu, L. Xu, Y. Li, X. Sui, G. Jiang, Y. Li, Z. Hu, J. Tian and F. Yang, Mol. Pharm., 2025, 22, 1198–1209 CrossRef CAS PubMed.
- M. Duraiyarasu, S. S. Kumaran and R. Mayilmurugan, ACS Biomater. Sci. Eng., 2023, 9, 3570–3580 CrossRef CAS PubMed.
- N. Zhang, G. Shu, E. Qiao, X. Xu, L. Shen, C. Lu, W. Chen, S. Fang, Y. Yang, J. Song, Z. Zhao, J. Tu, M. Xu, M. Chen, Y. Du and J. Ji, ACS Appl. Mater. Interfaces, 2022, 14, 20603–20615 CrossRef CAS PubMed.
- F. Yang, A. Skripka, M. S. Tabatabaei, S. H. Hong, F. Ren, A. Benayas, J. K. Oh, S. Martel, X. Liu, F. Vetrone and D. Ma, ACS Nano, 2019, 13, 408–420 CrossRef CAS PubMed.
- D. Pouliquen, J. J. Le Jeune, R. Perdrisot, A. Ermias and P. Jallet, Magn. Reson. Imaging, 1991, 9, 275–283 CrossRef CAS PubMed.
- M. Bañobre-López, A. Teijeiro and J. Rivas, Rep. Pract. Oncol. Radiother., 2013, 18, 397–400 CrossRef PubMed.
- T. L. Doane and C. Burda, Chem. Soc. Rev., 2012, 41, 2885–2911 RSC.
- M. Rahman, Nanotheranostics, 2023, 7, 424–449 CrossRef PubMed.
- J. Xie, G. Liu, H. S. Eden, H. Ai and X. Chen, Acc. Chem. Res., 2011, 44, 883–892 CrossRef CAS PubMed.
- K. Kaur, A. K. Al-Khazaleh, D. J. Bhuyan, F. Li and C. G. Li, Antioxidants, 2024, 13, 1092 CrossRef CAS PubMed.
- L. T. T. Huong, L. M. Huong, T. T. H. Ha, P. H. Nam and H. P. Thu, J. Sci. Technol., 2018, 61–68 Search PubMed.
- T. D. T. Ung, T. K. C. Tran, T. N. Pham, D. N. Nguyen, D. K. Dinh and Q. L. Nguyen, Adv. Nat. Sci. Nanosci. Nanotechnol., 2012, 3, 043001 CrossRef CAS.
- Q. T. Phan, M. H. Le, T. T. Le, T. H. Tran, P. N. Xuan and P. T. Ha, Int. J. Pharm., 2016, 507, 32–40 CrossRef CAS PubMed.
- Z. Chen, S. Cao, H. Wang, Y. Li, A. Kishen, X. Deng, X. Yang, Y. Wang, C. Cong, H. Wang and X. Zhang, PLoS One, 2015, 10, e0116553 CrossRef PubMed.
- M. M. H. Khalil, H. Mahdy, D. Y. Sabry and E. H. Ismail, J. Sci. Res., 2014, 6, 509–519 CrossRef.
- J. Wu, J. Pers. Med., 2021, 11, 771 CrossRef PubMed.
- M. Yuan, T. H. Yan, J. Li, Z. Xiao, Y. Fang, Y. Wang, H. C. Zhou and J. P. Pellois, Nanomedicine, 2021, 35, 102392 CrossRef CAS PubMed.
- T. T. H. Le, T. Q. Bui, T. M. T. Ha, M. H. Le, H. N. Pham and P. T. Ha, J. Mater. Sci., 2018, 53, 13826–13842 CrossRef CAS.
- S. T. Pham and Q. L. Nguyen, Commun. Phys., 2010, 20, 377–384 Search PubMed.
- B. Xie, Y. Hou, C. Liu and Y. Li, Eur. Polym. J., 2022, 168, 111090 CrossRef CAS.
- X.-G. Chen and H.-J. Park, Carbohydr. Polym., 2003, 53, 355–359 CrossRef CAS.
- P. T. Ha, M. H. Le, T. M. N. Hoang, T. T. Huong Le, T. Q. Duong, T. H. Ha Tran, D. L. Tran and X. P. Nguyen, Adv. Nat. Sci.: Nanosci. Nanotechnol., 2012, 3, 035002 Search PubMed.
- T. Bashkeran, A. Harun Kamaruddin, H. Umakoshi, N. M. Watanabe and M. J. J. Mohd Nadzir, Heliyon, 2024, e18710 Search PubMed.
- X. Yang, M. Dongdong, J. Ning, W. Chaoxing, Z. Jinbo, H. Yanqiu, L. Jiaming, J. Rui, Z. Xueyun and W. Sun, J. Biomater. Sci. Polym. Ed., 2024, 35, 628–656 CrossRef CAS PubMed.
- F. R. Wibowo, O. A. Saputra, W. W. Lestari, M. Koketsu, R. R. Mukti and R. Martien, ACS Omega, 2020, 5, 4261–4269 CrossRef CAS PubMed.
- N. S. Bostancı, S. Büyüksungur, N. Hasirci and A. Tezcaner, Biomater. Adv., 2022, 134, 112717 CrossRef PubMed.
- M. Cai, M. Leng, A. Lu, L. He, X. Xie, L. Huang, Y. Ma, J. Cao, Y. Chen and X. Luo, Colloids Surf., B, 2015, 126, 1–9 CrossRef CAS PubMed.
- X.-B. Fang, J.-M. Zhang, X. Xie, D. Liu, C.-W. He, J.-B. Wan and M.-W. Chen, Int. J. Pharm., 2016, 502, 28–37 CrossRef CAS PubMed.
- N. Torkashvand and N. Sarlak, Eur. Polym. J., 2019, 118, 128–136 CrossRef CAS.
- K. Son Phan, B. Thuy Doan, T. Thu Huong Le, T. Thu Trang Mai, T. Dieu Thuy Ung, T. Quang Bui, S. Boumati and P. Thu Ha, ChemistrySelect, 2023, 8, e202303580 CrossRef CAS.
- M. Ghorbaanee, A. A. Salarian and V. Saba, J. Inorg. Organomet. Polym. Mater., 2018, 28, 2169–2178 CrossRef CAS.
- K. Son Phan, H. Nghi Do, B. Thuy Doan, T. Thu Huong Le, T. Thu Trang Mai, Q. Bao Ngoc Nguyen, T. Nham Dong, B. Hung Bui Ha, V. Dung Dang, L. H. Dang, N. Quyen Tran and P. Thu Ha, ChemMedChem, 2025, 20, e202400586 CrossRef CAS PubMed.
- P. T. Ha, T. T. H. Le, T. D. T. Ung, H. D. Do, B. T. Doan, T. T. T. Mai, H. N. Pham, T. M. N. Hoang, K. S. Phan and T. Q. Bui, New J. Chem., 2020, 44, 17277–17288 RSC.
- S. Boumati, A. Sour, V. Heitz, J. Seguin, G. Beitz, Y. Kaga, M. Jakubaszek, J. Karges, G. Gasser, N. Mignet and B.-t. Doan, ACS Appl. Bio Mater., 2023, 6, 4791–4804 CrossRef CAS PubMed.
- M. M. Rahman, M. R. Islam, S. Akash, M. Harun-Or-Rashid, T. K. Ray, M. S. Rahaman, M. Islam, F. Anika, M. K. Hosain, F. I. Aovi, H. A. Hemeg, A. Rauf and P. Wilairatana, Biomed. Pharmacother., 2022, 153, 113305 CrossRef CAS PubMed.
- C. J. Thébault, G. Ramniceanu, S. Boumati, A. Michel, J. Seguin, B. Larrat, N. Mignet, C. Ménager and B.-T. Doan, J. Controlled Release, 2020, 322, 137–148 CrossRef PubMed.
- J. Seguin, C. Nicolazzi, N. Mignet, D. Scherman and G. G. Chabot, Tumour Biol., 2012, 33, 1709–1717 CrossRef CAS PubMed.
- V. Lenders, X. Koutsoumpou, P. Phan, S. J. Soenen, K. Allegaert, S. de Vleeschouwer, J. Toelen, Z. Zhao and B. B. Manshian, Chem. Soc. Rev., 2023, 52, 4672–4724 RSC.
- Q. Wang and Y. Bao, Free Radic. Biol. Med., 2021, 167, 125–140 CrossRef CAS PubMed.
- I. B. Belyaev, O. Y. Griaznova, A. V. Yaremenko, S. M. Deyev and I. V. Zelepukin, Adv. Drug Del. Rev., 2025, 219, 115550 CrossRef CAS PubMed.
- K. Tamarov, J. T.-W. Wang, J. Kari, E. Happonen, I. Vesavaara, M. Niemelä, P. Perämäki, K. T. Al-Jamal, W. Xu and V.-P. Lehto, ACS Appl. Mater. Interfaces, 2021, 13, 40392–40400 CrossRef CAS PubMed.
- M. Zheng, J. Zhang, C. Deng, L. Chen, H. Zhang, J. Xin, O. Aras, M. Zhou, F. An and Y. Ren, Mater. Des., 2024, 241, 112900 CrossRef CAS.
- E. García de Jalón, K. Kleinmanns, V. Fosse, B. Davidson, L. Bjørge, B. E. Haug and E. McCormack, Mol. Imaging Biol., 2023, 25, 144–155 CrossRef PubMed.
- O. B. Garbuzenko, G. Mainelis, O. Taratula and T. Minko, Cancer Biol. Med., 2014, 11, 44–55 CAS.
- E. Yaghini, H. Turner, A. Pilling, I. Naasani and A. J. MacRobert, Nanomed.: Nanotechnol. Biol. Med., 2018, 14, 2644–2655 CrossRef CAS PubMed.
- H. H. Gustafson, D. Holt-Casper, D. W. Grainger and H. Ghandehari, Nano Today, 2015, 10, 487–510 CrossRef CAS PubMed.
- K. C. Nguyen, Y. Zhang, J. Todd, K. Kittle, D. Patry, D. Caldwell, M. Lalande, S. Smith, D. Parks, M. Navarro, A. Massarsky, T. W. Moon, W. G. Willmore and A. F. Tayabali, Chem. Res. Toxicol., 2019, 32, 1491–1503 Search PubMed.
| This journal is © The Royal Society of Chemistry 2025 |