
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEfficient synthesis of tributyl citrate plasticizer via esterification reaction using SO42−/ZrO2–TiO2 as catalysts†
Heyuan Song*a,
Shuangtai Lei ab,
Weiguo Fangb,
Fuxiang Jinb,
Meirong Kangb,
Jing Chenb and
Hailong Liu*b
ab,
Weiguo Fangb,
Fuxiang Jinb,
Meirong Kangb,
Jing Chenb and
Hailong Liu*b
aSchool of Chemistry and Chemical Engineering, Lanzhou Jiaotong University, Lanzhou, 730070, China. E-mail: heyuansong@mail.lzjtu.cn; Tel: +86-931-4938755
bState Key Laboratory for Oxo Synthesis and Selective Oxidation, Lanzhou Institute of Chemical Physics, Chinese Academy of Sciences, Lanzhou 730000, China. E-mail: hlliu@licp.cas.cn; Tel: +86-931-4968070
First published on 9th June 2025
Abstract
Tributyl citrate, an environment-friendly and nontoxic plasticizer, is commonly synthesized via the esterification of citric acid and n-butanol. Highly effective SO42−/ZrO2–TiO2 catalysts were prepared by coprecipitation and impregnation for tributyl citrate production. The excellent conversion of 95.1% for citric acid and selectivity of 98.8% for tributyl citrate were achieved over a 3SO42−/ZrO2–TiO2 (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) catalyst with Zr/Ti mole ratio ∼4/1 under optimal reaction conditions of 120 °C and acid-to-alcohol ratio of 1/5. The textural and surface properties of the catalysts were characterized by nitrogen adsorption–desorption, XRD, pyridine-FTIR, etc. The characterization results confirmed that the addition of TiO2 promoted the formation of ZrO2 tetragonal phase to a certain extent, increased the specific surface area of the catalyst, stabilized the combination of SO42− and ZrO2, and reduced the loss of active sites. At the same time, the 3SO42−/ZrO2–TiO2 (4:1) catalyst has stronger acidity and higher acid content, which greatly improves the activity and stability of the catalyst. No significant decrease in the catalytic activity and selectivity were found during 10 runs. A promising solid acid catalyst with an excellent performance was developed for esterification reactions.
:1) catalyst with Zr/Ti mole ratio ∼4/1 under optimal reaction conditions of 120 °C and acid-to-alcohol ratio of 1/5. The textural and surface properties of the catalysts were characterized by nitrogen adsorption–desorption, XRD, pyridine-FTIR, etc. The characterization results confirmed that the addition of TiO2 promoted the formation of ZrO2 tetragonal phase to a certain extent, increased the specific surface area of the catalyst, stabilized the combination of SO42− and ZrO2, and reduced the loss of active sites. At the same time, the 3SO42−/ZrO2–TiO2 (4:1) catalyst has stronger acidity and higher acid content, which greatly improves the activity and stability of the catalyst. No significant decrease in the catalytic activity and selectivity were found during 10 runs. A promising solid acid catalyst with an excellent performance was developed for esterification reactions.
Introduction
Recently, a variety of plasticizers such as phthalates, citrates, and phosphates have been developed to change the properties of different polymers for the various operating conditions. However, plasticizer compounds containing aromatic rings are typically toxic to humans and the environment.1 Tributyl citrate (TBC) and acetyl-tributyl citrate (A-TBC), which have high thermal stabilities, are new environment-friendly plasticizers that have been gradually used in many polymer industries. In 2019, the global TBC market size was approximately 14500 tons, valued at approximately $63 million. TBC is utilized as a solvent in the production of poly-vinyl chloride (PVC) and its copolymers, which are applied in the creation of food packaging films.1,2 In addition, TBC is extensively used as a safe and nontoxic plasticizer in various sectors, including toy manufacturing, medical products (such as enteric coatings for controlled drug release systems), printing ink coatings, biodegradable polymers, cosmetics, and food additives.3,4 Nowadays, citric acid (CA) is largely produced via biological fermentation in industry. Esterification of CA derived from renewable resources with n-butanol has created a fully bio-renewable and eco-friendly approach for the efficient production of the nontoxic TBC plasticizer in the presence of various acidic catalysts (Scheme 1).5 Classical esterification reactions are generally catalyzed using concentrated protic acids, such as inexpensive H2SO4, hydrochloric acid, etc. Although protic acids exhibited high catalytic activity in the reactions, serious corrosion of equipment occurred and increasing byproducts existed during the total process. Additionally, a stoichiometric base was employed to neutralize the acidic solution, and a large amount of waste was generated. The utilization of efficient and recyclable solid acid catalysts can avoid the insurmountable shortcomings of protonic acids.6
 | ||
| Scheme 1 The esterification of CA with n-butyl CA to form TBC. | ||
In recent years, a variety of solid catalysts have been developed for the synthesis of TBC, including solid superacids, molecular sieves, supported heteropolyacids, poly-ionic liquid and so on.7–9 Based on their strong acidity and high catalytic activity, the solid acids for SO42− supported over MxOy oxides have been paid much attention for the esterification of CA and n-butanol. Among them, SO42−/ZrO2 catalysts, with high activity, has been widely used in esterification.10 Xu et al.11 prepared a solid superacid catalyst M-S2O82−/ZrO2 for esterification of TBC,the conversion rate decreased to 85.5% after repeated use for four times; Yang et al.1 prepared a ZS/HMS solid acid catalyst loaded with zirconium sulfate (ZS) on HMS molecular sieves. In the esterification reaction of CA and n-butanol, the conversion rate of CA was 93%. Nandiwale et al.12 loaded phosphorus onto USY molecular sieves and prepared a P/USY catalyst with 2% phosphorus content, which could be reused for 5 times in esterification reaction of CA and n-butanol. Wang et al.3 prepared a supported PTA/PANI catalyst with high activity and stability, and the selectivity of TBC was 95% at 170 °C. However, the industrial application of SO42−/MxOy catalysts is frequently limited by their instability in aqueous environments, mainly due to the leaching of SO42− ions resulting in poor catalyst reuse.13 Therefore, with the increasing market demand for TBC, it is imperative to develop more efficient and stable solid acid catalysts to promote the esterification of CA and n-butanol in industry.
This study presents a novel approach to prepare highly active and stable solid acid catalysts (SO42−/ZrO2–TiO2) for selectively producing nontoxic TBC plasticizers through the esterification of renewable CA and n-butanol. The SO42−/ZrO2–TiO2 catalyst with different SO42− contents and Zr/Ti mole ratios prepared by the precipitation impregnation method showed the best catalytic performance for TBC synthesis among the various solid acid catalysts investigated. Additionally, a range of characterization methods, such as X-ray diffraction (XRD), Fourier-transform infrared spectroscopy (FT-IR), ammonia temperature programmed desorption (NH3-TPD), nitrogen adsorption–desorption isotherms (BET), pyridine Fourier-transform infrared spectroscopy (Py-FTIR), scanning electron microscopy (SEM), transmission electron microscopy (TEM), and elementary analysis were utilized to determine the physical and chemical characteristics of the catalysts. Finally, the reaction conditions were optimized, and the structure–activity relationship and reusability of the catalyst were studied.
Experimental section
Materials
Zirconium nitrate (Zr(NO3)4·5H2O, AR) and tributyl citrate (C18H32O7, AR) were purchased from Shanghai Macklin Biochemical Co., Ltd. Citric acid (C6H10O8, AR), n-butanol (CH3(CH2)3OH, AR), ammonium hydroxide (NH3·H2O, 25–28 wt%), and concentrated sulfuric acid (H2SO4, ∼98%) were purchased from Tianjin Fuchen Chemical Reagent Co., Ltd. Titanium(IV) tetrachloride (TiCl4, AR) was obtained from Tianjin Kemi Ou Chemical Reagent Co., Ltd. All chemicals and reagents were used directly without any additional purification.Catalyst preparation
The SO42−/ZrO2–TiO2 catalyst was prepared by coprecipitation–impregnation method. In general, 0.5 M Zr(NO3)4 and TiCl4 solutions are prepared respectively according to different Zr/Ti ratios, and then they are mixed. Under violent stirring, the excess ammonia water (25%) was added into the mixed solution until pH ∼9 at room temperature. After aging for 3 h, the precipitate is then filtered and wash with distilled water until no Cl− ions (detected with a 1 mol per L AgNO3 solution). After drying at 110 °C for 12 h, the obtained powder samples were ground and impregnated in varying concentrations (1.0, 2.0, 3.0, 4.0, and 5.0 M) of H2SO4 solution for 1 h. After filtration, washing, and drying, the sample was calcined at 650 °C for 4 h. The resulting catalysts, xSO42−/ZrO2–TiO2 (m:n), where “x” denotes the concentration of H2SO4 used, where m:n stand for the ratios of Zr/Ti.
Catalyst characterization
XRD patterns were acquired with a RigakuD/MAX-2400 diffractometer, utilizing Cu Kα radiation, and operated at voltages and currents of 40 kV and 30 mA respectively.FT-IR spectroscopy was performed using a Nicolet 5700 infrared spectrometer. The spectra were captured from 4000–400 cm−1 at room temperature.
SEM was performed using an SU8020 (Hitachi, Japan) electron microscope. The microscope was operated at acceleration and landing voltages of 15 kV and 1.0 kV, respectively. Prior to the test, the powdered sample was uniformly glued to a sample table coated with a conductive adhesive.
TEM was performed using an FEI JEM2010 electron microscope operated at an accelerating voltage of 200 kV. Prior to the analysis, the catalysts were finely ground into a powder, dispersed in anhydrous ethanol using ultrasonic waves, and then applied to a carbon-coated copper grid.
The specific surface area and pore structure of the catalyst samples were measured using a Micromeritics Tristar II 3020 analyzer at liquid-nitrogen temperature. Before the test, the samples were degassed at 90 °C for 1 h and at 300 °C for 4 h to remove the physically adsorbed water and impurities. The Brunauer–Emmett–Teller (BET) equation was used to calculate the specific surface area, and the pore volume and size of the samples were calculated based on P/P0 = 0.99 single point adsorption.
NH3-TPD spectra were collected using a DAX-7000 automatic chemisorption analyzer (Hunan Huasi Company). Before each measurement, the samples were pretreated at 200 °C for 1 h under 40 mL min−1 of He stream. The pretreated samples were then exposed to 40 mL min−1 of 10% NH3–N2 gas stream at 100 °C for 1 h, and then heated to 800 °C at a heating rate of 10 °C min−1 in 40 mL min−1 of He stream. The NH3 desorption signal was collected using a thermal conductivity detector (TCD), and the desorption peak of NH3 was quantitatively analyzed using the pulse method.
The presence of Lewis and Brønsted acid sites on the catalyst surface was determined using Bruker's Tensor 27 Fourier Transform Infrared Spectrometer (Py-FTIR). Samples were pre-treated at 400 °C and 10−3 Pa for 1 h before testing, and a blank experiment was conducted by collecting the spectrum of the sample disk. Pyridine adsorption was performed at room temperature, followed by desorption at 150–400 °C for 1 h. After cooling to room temperature, the infrared spectrum in the range of 1700–1400 cm−1 was recorded.
The sulfur (S) element content of the catalyst was measured using an Agilent 725-ES Inductively Coupled Plasma Emission Spectrometer (ICP-OES). Prior to testing, the samples were neutralized with nitric acid to slightly acidify the solution by microwave ablation.
Catalytic tests
The esterification reaction of citric acid (CA) and n-butanol was performed in a three-necked flask with a condenser and a water konckout vessel with stirring at 500 rpm. Typically, 0.1 mol of citric acid, 0.45 mol of n-butanol, and 1 wt% catalyst were added into the reactor. The mixture was heated to at 120 °C and maintained for 4 h. The resulting liquid products were qualitatively and quantitatively analyzed using an Agilent 7890A/5975C Gas Chromatography-Mass Spectrometry (GC-MS) system with an HP-5MS column,and an Agilent GC-7820N instrument with an HP-5 capillary column (30 m × 0.32 mm × 0.25 μm) and FID, respectively. The citric acid conversion and product selectivity were calculated as follows:(1) The all liquid after reaction were separated with the catalyst and weighed as W.
(2) The reaction solution with mass W1 was calibrated with a KOH solution using phenolphthalein as an indicator.9
All products were weighed to calculate the CA mass in the remaining products and marked as n2.


where W is the total mass of the reaction mixture, W1 is the calibrated mass of the reaction mixture, C is the KOH concentration, V is the volume of KOH utilized, n1 is the initial molar quantity of citric acid, n2 is the post-reaction molar quantity of citric acid, m1 is the concentration of TBC determined by GC, m2 is the concentration of MBC identified by GC, m3 is the concentration of DBC quantified by GC, M1 is the molecular weight of TBC, M2 is the molecular weight of MBC, and M3 is the molecular weight of DBC.

Results and discussion
Catalysts characterization
The XRD patterns of the calcined 3SO42−/ZrO2–TiO2 catalysts with different Zr/Ti molar ratios are shown in Fig. 1. In all catalysts, a pronounced and well-defined peak is visible at 2θ = 30°, indicative of high crystallinity. In addition, in Fig. 1(d–f), a moderate peak was observed near 50°, along with several smaller peaks at 35°, 60°, and 63°.14,15 These peaks are indicative of ZrO2's tetragonal phase, which is known to boost catalytic performance.16–18 The 2θ peaks at 25.1° is characteristic of the anatase phase of TiO2, whereas peaks at 2θ values of 28.3°, 53.5°, and 54.7° correspond to the monoclinic phase of ZrO2.13,19 Evidently, with an increase of Zr/Ti molar ratios, the samples 3SO42−/ZrO2–TiO2 (4:1) and 3SO42−/ZrO2–TiO2 (2:1) predominantly crystallize into the tetragonal structure, as indicated by the broad peaks at 2θ = 30°. Notably, higher crystallinity is observed in the 3SO42−/ZrO2–TiO2 (4:1) sample when the Zr/Ti molar ratio reaches 4:1, as shown in Fig. 1(e). No other sulfur-related peaks were detected apart from those of TiO2 and ZrO2, suggesting a high dispersion of SO42− ions within the catalyst.
 | ||
| Fig. 1 XRD patterns of 3SO42−/ZrO2–TiO2 catalyst, (a) 3SO42−/ZrO2–TiO2 (1:4), (b) 3SO42−/ZrO2–TiO2 (1:2), (c) 3SO42−/ZrO2–TiO2 (1:1), (d) 3SO42−/ZrO2–TiO2 (2:1), (e) 3SO42−/ZrO2–TiO2 (4:1), (f) 3SO42−/ZrO2. | ||
ICP-OES was used to analyze the elemental content of the synthesized catalyst (Table 1). It was found that the elemental content ratio was consistent with that of the designed catalyst, indicating that the designed catalyst was successfully prepared.
| Catalysts | Zr contentsa (wt%) | Ti contentsa (wt%) | S contentsa (wt%) | Surface areab (m2 g−1) | Pore volumeb (cm2 g−1) | Pore sizeb (nm) |
|---|---|---|---|---|---|---|
| a Determined by ICP-OES analysis.b Calculated from the BET method. | ||||||
| 3SO42−/ZrO2 | 70.55 | — | 0.44 | 25.8 | 0.06 | 8.7 |
| 3SO42−/ZrO2–TiO2 (1:4) |
14.91 | 59.84 | 0.46 | 30.8 | 0.12 | 16.1 |
| 3SO42−/ZrO2–TiO2 (1:2) |
20.92 | 42.41 | 0.60 | 33.1 | 0.20 | 24.6 |
| 3SO42−/ZrO2–TiO2 (1:1) |
32.67 | 32.25 | 0.38 | 34.8 | 0.13 | 15.0 |
| 3SO42−/ZrO2–TiO2 (2:1) |
43.86 | 22.70 | 0.71 | 47.1 | 0.19 | 16.1 |
| 3SO42−/ZrO2–TiO2 (4:1) |
55.58 | 13.85 | 0.55 | 58.9 | 0.18 | 12.0 |
Table 1 summarizes the results of physical N2 adsorption and desorption isotherm analyses. As the mole ratios of Zr to Ti in the 3SO42−/ZrO2–TiO2 catalyst increase, the BET surface area of the catalyst continuously increasing from 30.8 to 58.9 m2 g−1. However, the pore volume and average pore diameter initially increased and then decreased, which reached the maximum value for the catalyst with Zr/Ti mole ratios ∼1/2. For the 3SO42−/ZrO2–TiO2 catalyst, the degree of aggregation of the metal oxide precursors was significantly affected by the free hydroxide content on the surface of the particles.20 The 3SO42−/ZrO2–TiO2 (4:1) catalyst with the largest Zr/Ti ratio had the largest specific surface area, which is significantly high compared with other catalysts, but the void volume and average pore size are not the largest. This change was caused by the addition of a certain amount of TiO2, which ultimately improved the catalytic activity.20 The type IV isotherms of 3SO42−/ZrO2–TiO2 catalysts all indicate the presence of typical mesoporous structural features, and H2 type hysteresis loop indicates that the catalyst is irregularly close-packed spherical pore (Fig. S1†). The high specific surface area and moderate average pore size are conducive for enhancing the exposure of active sites and the diffusion of substrates and products, thus improving the catalytic performance.
As shown in Fig. 2, FT-IR spectroscopy was employed to analyze the skeletal structure of the 3SO42−/ZrO2–TiO2 series catalysts with different Ti/Zr mole ratios. All the catalyst samples displayed similar structural characteristics, as evidenced by the comparable absorption bands across the spectra. In the 3SO42−/ZrO2–TiO2 series catalysts, distinct bands at 3427 and 1635 cm−1 were identified, indicative of the stretching vibration absorptions of surface hydroxyl groups and chemisorbed water molecules, respectively.14 The bands at 1139 cm−1 and 1046 cm−1 relate to the stretching vibrations of the O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) SO and O–S–O bonds, whereas the 1245 cm−1 band is linked to the bidentate chelation of SO42− ions with the metal oxide, implying that sulfate groups were introduced into the ZrO2 framework.21–24 And the covalent double bond properties can induce acid properties. The 510 cm−1 frequency band corresponds to the vibrations in the Ti–O–Ti, Zr–O–Zr, or Ti–O–Zr networks within the catalyst.
SO and O–S–O bonds, whereas the 1245 cm−1 band is linked to the bidentate chelation of SO42− ions with the metal oxide, implying that sulfate groups were introduced into the ZrO2 framework.21–24 And the covalent double bond properties can induce acid properties. The 510 cm−1 frequency band corresponds to the vibrations in the Ti–O–Ti, Zr–O–Zr, or Ti–O–Zr networks within the catalyst.
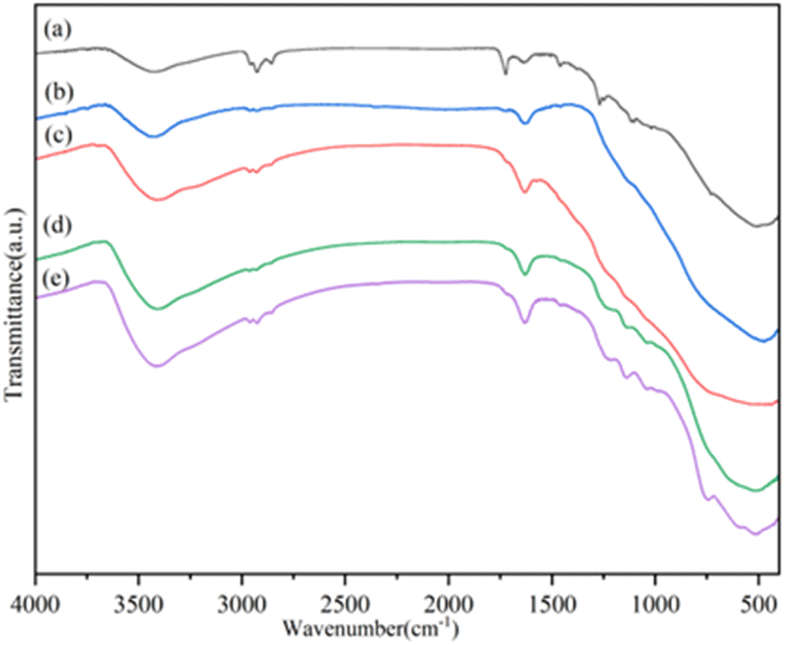 | ||
| Fig. 2 FI-IR spectra of SO42−/ZrO2–TiO2 catalyst series. (a) SO42−/ZrO2–TiO2 (1:1), (b) SO42−/ZrO2–TiO2 (1:4), (c) SO42−/ZrO2–TiO2 (1:2), (d) SO42−/ZrO2–TiO2 (2:1), (e) SO42−/ZrO2–TiO2 (4:1). | ||
Fig. 3 presents SEM images of the samples with different Zr/Ti mole ratios. Fig. 3(a)–(d) show significant aggregation of the nanoparticles. These aggregations led to fewer interparticle voids, resulting in lower BET surface areas ranging from 30.8 to 47.1 m2 g−1, as indicated in Table 2. Additionally, Fig. 4(e) shows some irregular spherical crystal particles. They are closely packed, forming an abundance of intercrystalline pores within the materials. This structural arrangement likely contributed to the larger BET surface areas observed in these samples.25 With increasing Zr/Ti ratios, the catalyst particle size decreased, thereby increasing the specific surface area and catalytic activity.
 | ||
| Fig. 3 SEM images of 3SO42−/ZrO2–TiO2 catalysts, (a) SO42−/ZrO2–TiO2 (1:1), (b) SO42−/ZrO2–TiO2 (1:4), (c) SO42−/ZrO2–TiO2 (1:2), (d) SO42−/ZrO2–TiO2 (2:1), (e) SO42−/ZrO2–TiO2 (4:1). | ||
| Catalyst | Acitidya (μmol g−1) | Sum | ||
|---|---|---|---|---|
| Weak | Moderate | Strong | ||
| a Calculated from the NH3-TPD method. | ||||
| 3SO42−/ZrO2–TiO2 (1:4) |
82.1 | 82.8 | 80.7 | 245.6 |
| 3SO42−/ZrO2–TiO2 (1:2) |
65.3 | 72.0 | 70.5 | 207.8 |
| 3SO42−/ZrO2–TiO2 (1:1) |
62.8 | 61.4 | 52.3 | 176.5 |
| 3SO42−/ZrO2–TiO2 (2:1) |
82.1 | 87.6 | 93.6 | 263.3 |
| 3SO42−/ZrO2–TiO2 (4:1) |
69.1 | 80.6 | 153.5 | 303.2 |
 | ||
| Fig. 4 TEM images of 3SO42−/ZrO2–TiO2 catalysts, (a) SO42−/ZrO2–TiO2 (1:1), (b) SO42−/ZrO2–TiO2 (1:4), (c) SO42−/ZrO2–TiO2 (1:2), (d) SO42−/ZrO2–TiO2 (2:1), (e) SO42−/ZrO2–TiO2 (4:1). | ||
Fig. 4 shows TEM images of samples with different Zr/Ti ratios. They all have similar crystal structures. As the Zr/Ti ratio increases, the grain arrangement becomes more compact, forming a smaller grain spacing. In addition, placing titanium oxide in zirconium crystals and sulfate ions caused the zirconium crystals to decompose, inhibiting the tetragonal monoclinic phase transition that occurs in coarser grain sizes.26 Moreover, the monotonic structure of the titanium oxide crystal affected the zirconia crystal, thereby improving its catalytic activity of the zirconia crystal.27,28
Fig. 5 shows a comparison of the NH3-TPD curves of all five catalysts. The ammonia desorption curve of the catalyst showed desorption peaks at 200, 400 and 600 °C, which are ascribed as weak, moderate and strong acid sites, respectively. With the increase of the Zr/Ti ratio, the acid site of the catalyst decreases first and then increases. When the Zr/Ti ratio is 1, the acid site of the catalyst is the least and the total acid content is 176.5 μmol g−1. When the Zr/Ti ratio continues to increase to 4, the catalyst has a strong peak at 600 °C, which is the strong acid site, and the total acid content also increases to 303.2 μmol g−1 (Table 2). Generally, in esterification reactions catalyzed by solid acids, the more strongly acidic the sites are, the more conducive the catalytic reactions and the more significant the effect of the catalyst.29
 | ||
| Fig. 5 NH3-TPD profiles of prepared catalysts. | ||
Fig. 6 displays the Py-FTIR spectra of pyridine adsorbed on 3SO42−/ZrO2–TiO2 at varying temperatures, with the desorption profiles of pyridine at 150, 300, and 400 °C corresponding to weak, medium, and strong acid sites, respectively. The characteristic peak of pyridine ion was formed at the Lewis acid site at 1446 cm−1, the coordination bond between pyridine and Brønsted and Lewis acid site was formed at 1490 cm−1, and the formation of pyridine at the Brønsted acid site occurred at 1540 cm−1.30,31 Fig. 6(a) shows the pyridine characteristic peaks of 3SO42−/ZrO2–TiO2 catalysts with different Zr/Ti ratios. As shown in Table 3, with increasing Zr/Ti ratios, the number of acidic sites gradually increased and 3SO42−/ZrO2–TiO2 (4:1) catalyst contained the most acid sites. As observed in Fig. 6(b), there is a noticeable decrease in the number of both Brønsted and Brønsted + Lewis acid sites with an increase in the pyridine desorption temperature across each spectrum. At the desorption temperature of 300 °C, the Brønsted acid site disappeared. When the desorption temperature increased to 400 °C, the Lewis acid site was not completely desorbed, but the peak of the Brønsted + Lewis acid site became much weaker. All the strong acid sites in 3SO42−/ZrO2–TiO2(4:1) catalysts were Lewis acid sites. This is primarily because of the transformation of Brønsted acid sites into Lewis acid sites through dehydroxylation, leading to the scenario where peaks associated with Brønsted + Lewis acid sites predominantly represent Lewis acid sites.
 | ||
| Fig. 6 Py-FTIR spectra of SO42−/ZrO2–TiO2 catalyst, (a) desorption spectra of different catalysts at 200 °C, (b) desorption spectra of SO42−/ZrO2–TiO2 (4:1) catalyst at different temperatures. | ||
| Catalyst | Conversion/% | Selectivity/% | ||
|---|---|---|---|---|
| MBC | DBC | TBC | ||
| a Reaction conditions: n-butyl alcohol/CA = 4.5, 120 °C, 1 wt% catalyst, 4 h reaction time. | ||||
| 1SO42−/ZrO2 | 64.8 | 25.5 | 30.5 | 44.0 |
| 2SO42−/ZrO2 | 68.9 | 24.8 | 31.3 | 43.9 |
| 3SO42−/ZrO2 | 78.9 | 21.4 | 29.3 | 49.3 |
| 4SO42−/ZrO2 | 73.7 | 22.6 | 29.6 | 47.8 |
| 5SO42−/ZrO2 | 72.7 | 26.5 | 30.4 | 43.1 |
Catalytic performance
The catalytic activity of SO42−/ZrO2 catalyst at different sulfuric acid concentrations of 1–5 M was investigated (Table 3). As the amount of H2SO4 impregnation increased from 1–3 M, the catalytic effect has also been significantly enhanced. In the esterification reaction, the conversion rate of CA and selectivity of TBC continuously improved, the conversion rate increased from 64.8% to 78.9%, and the selectivity increased from 44.0% to 49.3%. At this time, the selectivity of the by-products monobutyl citrate (MBC) and dibutyl citrate (DBC) decreased to 21.4% and 29.3%, respectively. When the impregnation amount increased from 3 M to 5 M, both the conversion rate of CA and the selectivity for TBC started declining. This was mainly because the SO42− bound to the catalyst was saturated. According to the characterization results of NH3-TPD, the amount of acid on 3 SO42−/ZrO2 catalyst was the largest (Fig. S2 and Table S1†). Therefore, 3SO42−/ZrO2 catalyst showed the best catalytic performance with CA conversion of 78.9% and TBC selectivity of 49.3%. The influence of reaction parameters was also studied.The effect of different metals on SO42−/ZrO2 catalyst was investigated under the condition of 3 M H2SO4 concentration. The catalytic activity of different oxides on the modification of 3SO42−/ZrO2 catalyst was examined. The 3SO42−/ZrO2 catalyst was modified by adding Al2O3, MgO, Fe2O3, and TiO2. The activity of the modified catalyst was investigated, and it was found that the conversion of CA was not significantly improved, but the selectivity for TBC was significantly improved after TiO2 modification (Table 4). The TBC selectivity increased from 49.3% to 57.2%. Therefore, 3SO42−/ZrO2–TiO2 catalyst was selected as the catalyst for esterification. Combining the BET and XRD results, the 3SO42−/ZrO2–Al2O3 and 3SO42−/ZrO2–TiO2 catalysts were found to have larger specific surface areas than the 3SO42−/ZrO2–Fe2O3 and 3SO42−/ZrO2–MgO catalysts (Fig. S3 and Table S2†). However, 3SO42−/ZrO2–Al2O3 is an amorphous material, and its catalytic activity is not as good as that of 3SO42−/ZrO2–TiO2. Therefore, 3SO42−/ZrO2–TiO2 was selected as the catalyst to study the catalytic performance.
| Catalyst | Conversion/% | Selectivity/% | ||
|---|---|---|---|---|
| MBC | DBC | TBC | ||
| a Reaction conditions: n-butyl alcohol/CA = 4.5, 120 °C, 1 wt% catalyst, 4 h reaction time. | ||||
| 3SO42−/ZrO2–Al2O3 | 74.3 | 28.9 | 26.4 | 44.7 |
| 3SO42−/ZrO2–MgO | 71.8 | 30.4 | 26.5 | 43.1 |
| 3SO42−/ZrO2–Fe2O3 | 73.1 | 27.1 | 27.2 | 45.7 |
| 3SO42−/ZrO2–TiO2 | 79.5 | 18.5 | 24.2 | 57.2 |
The influence of different Zr/Ti ratios on 3SO42−/ZrO2–TiO2 catalyst was investigated under the condition of 3 M H2SO4 concentration. The catalytic activity of 3SO42−/ZrO2–TiO2 composite catalysts with different ratios was examined. Table 5 presents the results of CA esterification catalyzed by 3SO42−/ZrO2–TiO2 complex catalysts with different Zr/Ti ratios. With an increase in the TiO2 content, the conversion of CA and the selectivity for the TBC both decreased, and the addition of a small amount of TiO2 improved the catalytic activity. When the Zr/Ti ratio of the catalyst was 4:1, the CA conversion and TBC selectivity were the highest at 84.8% and 73.9%, respectively. According to BET and NH3-TPD characterization results, 3SO42−/ZrO2–TiO2 (4:1) catalyst had the largest specific surface area and most acid sites.
| Catalyst | Conversion/% | Selectivity/% | ||
|---|---|---|---|---|
| MBC | DBC | TBC | ||
| a Reaction conditions: n-butyl alcohol/CA = 4.5, 120 °C, 1 wt% catalyst, 4 h reaction time. | ||||
| 3SO42−/ZrO2–TiO2 (1:4) |
79.8 | 16.8 | 17.4 | 65.9 |
| 3SO42−/ZrO2–TiO2 (1:2) |
76.8 | 20.6 | 24.2 | 55.3 |
| 3SO42−/ZrO2–TiO2 (1:1) |
79.5 | 18.5 | 24.2 | 57.2 |
| 3SO42−/ZrO2–TiO2 (2:1) |
81.3 | 14.3 | 19.3 | 66.4 |
| 3SO42−/ZrO2–TiO2 (4:1) |
84.8 | 9.8 | 16.3 | 73.9 |
The conversion rate of CA and selectivity of the TBC were different under different reaction conditions. Thus, the effects of reaction parameters such as the alcohol–acid ratio, catalyst dosage, reaction temperature, and reaction time on TBC production were optimized over the most active 3SO42−/ZrO2–TiO2 (4:1) to achieve a high CA conversion rate and TBC selectivity.
Optimizing the n-butyl alcohol to CA molar ratio is crucial, as a lower ratio may prevent chemical equilibrium, whereas a higher ratio could dilute and reduce catalytic efficiency. This effect was examined by varying the concentration of CA from 3:1 to 7:1 ratio. At lower ratios of 3:1 and 4:1, the CA conversions were 75.4% and 79.4%, respectively (Fig. 7). Increasing the n-butyl alcohol concentration to 5:1 improved the CA conversion to 86.0%. However, at higher molar ratios (6:1 and 7:1), a slight decrease in CA conversion was observed, likely due to dilution effects. Therefore, the most effective reactant molar ratio was determined to be 5:1, which achieved the highest CA conversion of 86.0% and TBC selectivity of 81.1%. At this time, the selectivity of the by-products MBC and DBC decreased to 8.2% and 10.7%, respectively.
 | ||
| Fig. 7 Effect of reactant mole ratio on the esterification of CA with n-butyl alcohol, reaction conditions: 120 °C, 6 wt% catalyst, 4 h reaction time. | ||
The effect of temperature on the reaction was evaluated using the 3SO42−/ZrO2–TiO2 (4:1) catalyst across a range of 110–150 °C (Fig. 8). An increase in temperature from 110 to 140 °C resulted in an increase in CA conversion from 62.5 to 93.8%. At 110 °C, the CA conversion and TBC selectivity were relatively low, at 62.5 and 29.8%, respectively. Increasing the temperature to 120 °C increased both CA conversion and TBC selectivity to 86.0% and 81.1%, respectively. Further temperature increasing up to 140 °C enhanced CA conversion to 93.8% and TBC selectivity to 96.6%. However, a slight decrease in both CA conversion and TBC selectivity was noted at 150 °C. Thus, 140 °C was identified as the optimum temperature for the reaction with the 3SO42−/ZrO2–TiO2 (4:1) catalyst.
 | ||
| Fig. 8 Effect of temperature on the esterification of CA with n-butyl alcohol, reaction conditions: n-butyl alcohol/CA = 5, 6 wt% catalyst, 4 h reaction time. | ||
The effect of varying the catalyst dosage of 3SO42−/ZrO2–TiO2(4:1) from 2 wt% to 8 wt% relative to the total reactant mass was explored. The results showed that increasing the catalyst dosage from 2 wt% to 6 wt% led to an increase in the CA conversion from 89.0% to 93.9%, respectively; however, beyond this, the conversion decreased to 92.5% (as shown in Fig. 9). The TBC selectivity improved from 82.9% to 96.4%. However, with 8 wt% catalyst, the TBC selectivity decreased from 96.4% to 91.2%.
 | ||
| Fig. 9 Effect of catalyst dosage on the esterification of CA with n-butyl alcohol, reaction conditions: n-butyl alcohol/CA = 5 and 120 °C, 4 h reaction time. | ||
The performance of the 3SO42−/ZrO2–TiO2 (4:1) catalyst was assessed over a reaction time ranging from 2 h to 10 h (Fig. 10). After 2 h, CA conversion and TBC selectivity stood at 84.9% and 63.3%, respectively. Extending the reaction time to 8 h resulted in a CA conversion of 95.1% and TBC selectivity of 98.8%. When the reaction time was increased to 10 h, the CA conversion reached 96.8%, with a TBC selectivity of 100%. The minimal change in the CA conversion and attainment of 100% TBC selectivity between 8 and 10 h suggests that 8 h is the optimal reaction time.
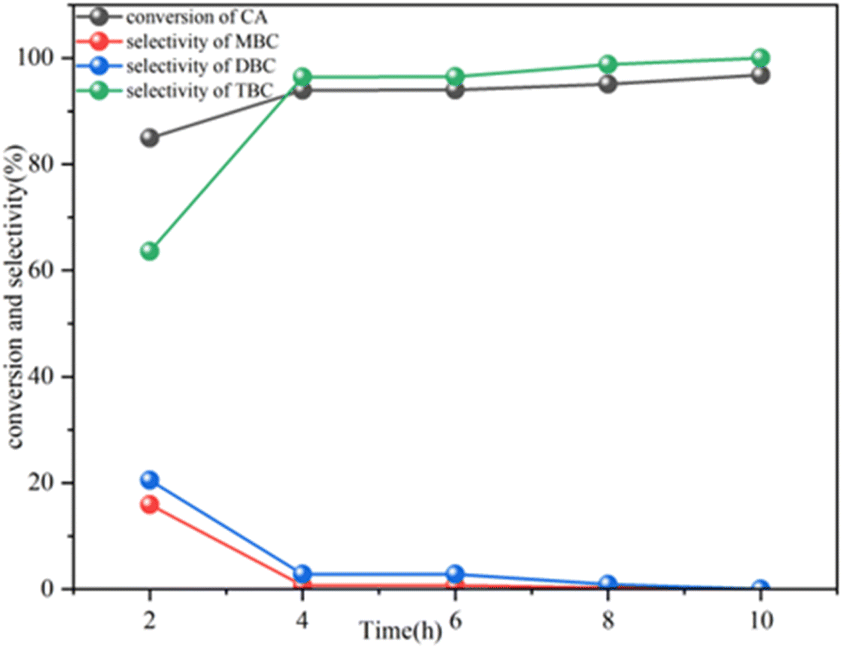 | ||
| Fig. 10 Effect of reaction time on the esterification of CA with n-butyl alcohol. Reaction conditions: n-butyl alcohol/CA = 5 and 120 °C, 6 wt% catalyst. | ||
Catalyst reuse cycle
Recycle test of the catalyst was conducted under optimal conditions (n-butyl alcohol/CA = 5:1, catalyst amount: 6 wt%, and 120 °C), with a run time of 8 h for each test (Fig. 11). In our study, the effect of catalyst reuse on catalytic activity was the key focus, particularly in terms of CA conversion and TBC selectivity. After each reaction, the catalyst was removed from the reaction mixture, cleaned with methanol, and dried for subsequent use. This process is exemplified using the 3SO42−/ZrO2–TiO2 (4:1) catalyst in esterification reactions. As observed from Fig. 11, both the conversion of CA and the selectivity of TBC using the 3SO42−/ZrO2–TiO2 (4:1) catalyst exhibited a slight decline with each successive reuse of the catalyst. After ten cycles, there was a noticeable decrease in performance: the conversion rate of CA decreased from 95.1% to 87.3%, and the selectivity of TBC decreased from 98.8% to 91.6%. This trend highlights the need to consider the durability and longevity of catalysts in practical applications, especially when they are reused multiple times. By collecting the catalyst after the reaction, the physical loss was found to be 6.3%. The catalysts were characterized before and after the reaction. As presented in Table S3,† the specific surface area of the catalyst after use was reduced from 58.9 m2 g−1 to 44.5 m2 g−1, which was caused by the collapse of the catalyst pore. Simultaneously, the S content in the catalyst decreased from 0.48% to 0.22%, indicating that the SO42− supported by the catalyst was lost, but the decrease in catalytic activity was not significant.
 | ||
| Fig. 11 Reusability of 3SO42−/ZrO2–TiO2 (4:1) in the TBC production process. | ||
Catalyst mechanism
The esterification of CA with n-butanol can be explained by the interaction between the adsorbed CA and free n-butanol, which adheres to the Eley–Rideal (E–R) model, as outlined in ref. 32–35. Scheme 2 present the E–R mechanism for the esterification process under the influence of the 3SO42−/ZrO2–TiO2 (4:1) catalyst based on the proposed model.36,37 Typically, the CA carboxyl group is first adsorbed and activated by a catalyst, leading to the formation of a carbonyl ion and its subsequent reaction with a hydroxyl group. This step is followed by hydrogen transfer and dehydration, resulting in the formation of an intermediate that ultimately yields the TBC via a desorption process. Notably, the mass transfer resistance of large molecular substances, such as CA and its ester, can affect the efficiency of the catalyst, as indicated in ref. 38. Thus, the catalyst performance is influenced not only by the number of active sites but also by its textural properties.
 | ||
| Scheme 2 Mechanistic representation of esterification of CA monohydrate with n-butanol to synthesize TBC on Lewis acid sites. | ||
Conclusions
This study demonstrated that the incorporation of TiO2 into the 3SO42−/ZrO2 catalyst markedly improved its specific surface area and pore size. All samples tested in this study exhibited the formation of only a tetragonal ZrO2 crystal phase. The results showed that while synthesizing TBC through the esterification of CA and n-butanol, modifying 3SO42−/ZrO2 with TiO2 substantially enhanced the activity and stability of the catalyst. The 3SO42−/ZrO2–TiO2 catalyst demonstrated optimal catalytic performance, achieving a CA conversion rate of 93.9% and TBC selectivity of 96.4% under specific conditions of a 5:1 molar ratio of n-butanol to CA, 6.0 wt% catalyst dosage, 4 h reaction time, and a temperature of 120 °C. Notably, the catalytic activity of the 3SO42−/ZrO2–TiO2 (4:1) catalyst remained robust after 10 cycles. This underscores its potential as an efficient and environment-friendly catalyst for the esterification of CA and n-butanol.
Data availability
All relevant date are within the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (22102198), Natural Science Foundation of Gansu Province (22JR5RA349) and Gansu Province Key Research and Development Program – Industrial Projects (23YFGA0041).References
- H. Yang, H. Song, H. Zhang, P. Chen and Z. Zhao, J. Mol. Catal. A: Chem., 2014, 381, 54–60 CrossRef CAS
.
- A. Chaos, A. Sangroniz, A. Gonzalez, M. Iriarte and J. R. Sarasua, Polym. Int., 2018, 68(1), 125–133 CrossRef
- L. Wang, B. Ding and M. Zhang, RSC Adv., 2019, 9(57), 33124–33129 RSC
- J. Han, M. Zhang, H. Zhang, H. Liu and S. Xu, J. Vinyl Addit. Technol., 2022, 28(4), 751–761 CrossRef CAS
- K. Y. Nandiwale, S. P. Borikar and V. V. Bokade, Clean: Soil, Air, Water, 2015, 43(6), 927–931 CAS
- P. Gupta and S. Paul, Catal. Today, 2014, 236, 153–170 CrossRef CAS
- R. Sirsam, D. Hansora and G. A. Usmani, J. Inst. Eng. (India): Ser. E, 2016, 97(2), 167–181 CAS
- V. C. Nguyen, N. Q. Bui, P. Mascunan, T. T. H. Vu, P. Fongarland and N. Essayem, Appl. Catal., A, 2018, 552, 184–191 CrossRef CAS
- X. U. Junming, J. C. Jiang, Z. Y. Zuo and J. Li, Process Saf. Environ., 2010, 88(1), 28–30 CrossRef
- Y. Liao, X. Huang, X. Liao and B. Shi, J. Mol. Catal. A: Chem., 2011, 347(1–2), 46–51 CrossRef CAS
- J. Xu, J. Jiang, L. V. Wei and Y. Gao, Chem. Eng. Commun., 2010, 198(4), 474–482 CrossRef
- K. Y. Nandiwale, P. Gogoi and V. V. Bokade, Chem. Eng. Res. Des., 2015, 98, 212–219 CrossRef CAS
- Z. Ye, H. Chen, X. Cui, J. Zhou and J. Shi, Mater. Lett., 2009, 63(27), 2303–2305 CrossRef CAS
- J. Yan, W. Ni, K. You, T. Duan, R. Deng, Y. Chen, F. Zhao, P. Liu and H. A. Luo, Res. Chem. Intermed., 2021, 47(9), 3569–3582 CrossRef CAS
- S. Wang, X. Meng, N. Liu and L. Shi, Sep. Purif. Technol., 2023, 308, 122731 CrossRef CAS
- B. S. Fu, L. J. Gao, L. Niu, R. Wei and G. M. Xiao, Energy Fuels, 2009, 23, 569–572 CrossRef CAS
- H. Wang, Y. Li, F. Yu, Q. Wang, B. Xing, D. Li and R. Li, Chem. Eng. J., 2019, 364, 111–122 CrossRef CAS
- T. Witoon, T. Numpilai, N. Dolsiririttigul, N. Chanlek, Y. Poo-arporn, C. K. Cheng, B. V. Ayodele, M. Chareonpanich and J. Limtrakul, Int. J. Hydrogen Energy, 2022, 47(98), 41374–41385 CrossRef CAS
- S. Wang, J. Pu, J. Wu, H. Liu, H. Xu, X. Li and H. Wang, ACS Omega, 2020, 5(46), 30139–30147 CrossRef CAS PubMed
- H. Yao, J. Li, J. Li, X. Liang, G. Wang and H. Luo, Chin. J. Chem. Eng., 2023, 61, 24–36 CrossRef
- L. E. Davies, N. A. Bonini and E. E. Gonzo, Proce. Mater. Sci., 2015, 8, 592–598 CrossRef
- X. Li, K. Nagaoka, R. Olindo and J. Lercher, J. Catal., 2006, 238(1), 39–45 CrossRef
- L. Li, B. Yan, H. Li, S. Yu, S. Liu, H. Yu and X. Ge, Fuel, 2018, 226, 190–194 CrossRef
- W. Li, F. Ma, F. Su, L. Ma, S. Zhang and Y. Guo, ChemSusChem, 2011, 4(6), 744–756 CrossRef
- G. Shi, F. Yu, Y. Wang, D. Pan, H. Wang and R. Li, Renewable Energy, 2016, 92, 22–29 CrossRef
- Y. Liu, Z. Li, J. J. Leahy and W. Kwapinski, Energy Fuels, 2015, 29(6), 3691–3698 CrossRef
- M. Fan, Z. Si, W. Sun and P. Zhang, Fuel, 2019, 252, 254–261 CrossRef
- Z. Li, R. Wnetrzak, W. Kwapinski and J. J. Leahy, ACS Appl. Mater. Interfaces, 2012, 4(9), 4499–4505 CrossRef
- K. Saravanan, B. Tyagi, R. S. Shukla and H. C. Bajaj, Fuel, 2016, 165, 298–305 CrossRef
- H. Yan, Y. Yang, D. Tong, X. Xiang and C. Hu, Catal. Commun., 2009, 10(11), 1558–1563 CrossRef CAS
- Z. Ali Saleh Bairq, H. Gao, Y. Huang, H. Zhang and Z. Liang, Appl. Energy, 2019, 252, 113440 CrossRef CAS
- S. K. Das and S. A. El-Safty, ChemCatChem, 2013, 5(10), 3050–3059 CrossRef CAS
- K. Saravanan, B. Tyagi and H. C. Bajaj, Appl. Catal., B, 2016, 192, 161–170 CrossRef CAS
- L. F. Isernia, Microporous Mesoporous Mater., 2014, 200, 19–26 CrossRef CAS
- S. Wang, L. Xu, C. Tian and Y. Guan, Int. J. Polym. Sci., 2018, 2018, 1–9 Search PubMed
- H. Li, S. Zhao, W. Zhang, H. Du, X. Yang, Y. Peng, D. Han, B. Wang and Z. Li, Fuel, 2023, 342, 127786 CrossRef
- L. Jiang, L. Zhou, J. Chao, H. Zhao, T. Lu, Y. Su, X. Yang and J. Xu, Appl. Catal., B, 2018, 220, 589–596 CrossRef
-
(a) J. Fu, Z. Cai, Y. Gong, S. E. O'Reilly, X. Hao and D. Zhao, Colloids Surf., A, 2015, 484, 1–8 CrossRef
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ra02407b |
| This journal is © The Royal Society of Chemistry 2025 |