
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOn the impact of aromatic core fluorination in hydrogen-bonded liquid crystals†
Ahmed F. Darweesh *a,
Christian Andersb,
B. S. Ranjithac,
G. Shankerc and
Mohamed Alaasar*ab
*a,
Christian Andersb,
B. S. Ranjithac,
G. Shankerc and
Mohamed Alaasar*ab
aDepartment of Chemistry, Faculty of Science, Cairo University, 12613 Giza, Egypt. E-mail: darweesh@sci.cu.edu.eg
bInstitute of Chemistry, Martin Luther University Halle-Wittenberg, 06120 Halle, Germany. E-mail: mohamed.alaasar@chemie.uni-halle.de
cDepartment of Chemistry, Bangalore University, Jnana Bharathi Campus, Bengaluru, 560056, India
First published on 3rd March 2025
Abstract
Herein we report the impact of fluorine substitution on the liquid crystalline (LC) self-assembly of supramolecular hydrogen-bonded rod-like architectures. This was systematically investigated by introducing fluorine atom at different positions or with different numbers on the investigated supramolecules. Therefore, eight different groups of hydrogen-bonded LCs (HBLCs) were designed and synthesized in which four 4-hexyloxybenzoic acid derivatives without or with fluorine substitution were used as proton donors. The proton acceptors are fluorinated or nonfluorinated alkyloxyazopyridine derivatives. The hydrogen bond formation between the complementary components was proved using FTIR and 1H NMR spectroscopy. All HBLCs were investigated for their mesomorphic behaviour using various tools such as differential scanning calorimetry (DSC), polarized optical microscopy (POM) and X-ray diffraction (XRD). Depending on the position and number of fluorine atoms different LC phases were observed including nematic, orthogonal non-tilted smectic A (SmA) or tilted smectic C (SmC) phases in addition to an unknown X phase. Depending on the position of the fluorine substitution, it was proved from the XRD investigations that different types of cybotactic nematic phases (NCyb) are exhibited by the reported HBLCs. Because of cis–trans photoisomerization under light irradiation of the reported HBLCs, their photo responsivity was investigated in solutions as well as between the different LC phases. This report provides key insights into the structure–property relationships of HBLCs, which might be of interest for optical storage device applications.
1. Introduction
Non-covalent interactions are distinct from covalent ones and ensure functionality of biological systems such as proteins or DNA.1 Among them, vital hydrogen bonding plays a crucial role in numerous instances in nature and biological systems,2–5 driving the field of supramolecular chemistry and gives inspiration for material scientists to design interesting functional materials such as liquid crystals (LCs).6Decisively, shape anisotropy, molecular interaction direction, and its strength support the achievement of LC phases and mesophase formation.7,8 The advantage of using non-covalent interactions between two mesogenic or non-mesogenic components to produce LC materials is to avoid the many steps required for organic synthesis, thus reducing the total cost and environmental pollution.9 The field of hydrogen-bonded liquid crystals (HBLCs) was initiated by Kato's group,10 when first examples of HBLCs were reported between the 4-alkoxybenzoic acids as the proton donors and pyridine derivatives as the proton acceptors. Later various classes of HBLCs have been designed and prepared. These encompass low and high molecular weight calamitics,11,12 bent-core molecules,13–15 LC polymers,16 and polycatenars,17–19 with a broad application potential in technological fields.
Fluorine substitution in organic compounds exhibits captivating and unusual properties due to a combination of polar and steric effects, in addition to the strong covalent bond between fluorine and carbon, which imparts greater stability to the compounds.20 The introduction of fluorine into distinct classes of LCs, including calamitics,21,22 discotics,23,24 bent-core,25–27 LC polymers,28,29 and other non-conventional LCs,30 does not compromise the material's liquid crystal nature. As a result of its larger size compared to hydrogen, fluorine provides more steric hindrance and greater polarity, which affects the melting point, mesophase characteristics, and phase transition temperatures. To modify the liquid crystalline behaviour, fluorine could be added at various positions in the molecular structure of a given mesogen. Therefore, fluorine was used in perfluorinated terminal chains,31–33 in the aromatic backbone as an ending group34,35 or as a lateral group at different positions.22,36–38 These distinct forms of molecular tailoring play a crucial role in unravelling fundamental structure–property relationships and developing materials tailored for various applications. Recently, fluorinated rod-like molecules were found to exhibit the ferroelectric nematic phase in addition to other polar LC phases.39–41
Most of the HBLCs incorporate azopyridine derivatives as proton acceptors.42–47 Azopyridines demonstrate appealing characteristics, including their ability to act as promesogenic material and undergoing trans–cis photoisomerization upon light irradiation. These traits render them extremely valuable in the domain of photonics applications.48–52
Previously, we have reported the effect of mono aromatic core fluorination in the azopyridine derivatives used as hydrogen-bond acceptor on the LC behaviour of rod-like molecules.53 However, fluorination of the proton donor i.e. the benzoic acid derivatives with different degrees is not investigated yet. Therefore, herein we investigate systematically the impact of aromatic core fluorination with different number and positions on the liquid crystalline behaviour of HBLCs. For this purpose, two different types of azopyridine derivatives (AHn and AFn, Fig. 1) were used as the proton acceptors, while the proton donors are 4-hexyloxybenzoic acid (B6H) and its fluorinated derivatives (B6F2, B6F3 and B6F23 Fig. 1).
 | ||
| Fig. 1 Model of HBLCs and the chemical structures of hydrogen-bond donors and hydrogen-bond acceptors under discussion. | ||
Therefore, eight different combinations between these components were achieved resulting in total of 32 HBLCs (HHHn–FFFn, Scheme 1). The effect of varying the terminal chain length attached to the proton acceptor (AHn or AFn) on the LC behaviour was also investigated (n = 8, 10, 12, 14). Moreover, the trans–cis photoisomerization of the reported materials were studied in solution as well as between different LC phases. This wide variety of molecular building blocks allows a systematic study of structure–property relationships and provide photo-switchable functional materials which could be of interest for applications in thermo-responsive optical filters and sensors.54
 | ||
| Scheme 1 Synthesis of the new HBLCs HHHn–FFFn. Reagents and conditions: (i) NaNO2/HCl/H2O, 0 °C, 5 h; (ii) NaOH, NaHCO3; (iii) BrCnH2n+1, K2CO3, DMF, 80 °C, 18 h; (iv) BrC6H13, K2CO3, n-Bu4NI, butanone, reflux, 5 h; (v) KOH, ethanol, reflux, HCl; (vi) resorcinol, t-butanol, NaOCl2, NaH2PO4·2H2O, stirring, r.t., HCl. | ||
2. Synthesis
The synthesis of the target materials was performed as illustrated in Scheme 1. The proton acceptors i.e. the azopyridine derivatives with or without fluorine substitution (AHn and AFn) were synthesized using previously reported protocols.19,43,55 The proton donors with different degree of fluorination were synthesized starting from different key intermediates using standard reported procedures,56,57 while the non-fluorinated 4-hexyloxybenzoic acid B6H was commercially available. The general synthetic procedures and analytical data for the synthesized intermediates and target molecules are provided in ESI.†3. Results and discussion
3.1. Analysis of the hydrogen-bond formation
To identify the hydrogen bond formation between the complementary components – the benzoic acid derivatives and the azopyridines – we conducted both FTIR measurements using the conventional KBr method and NMR analysis for some selected examples. As a representative example the FTIR spectra of the supramolecule HHH12 (green curve) and its components B6H (blue curve) and AH12 (red curve) are presented in Fig. 2a and b (for the complete spectra refer to Fig. S12a†).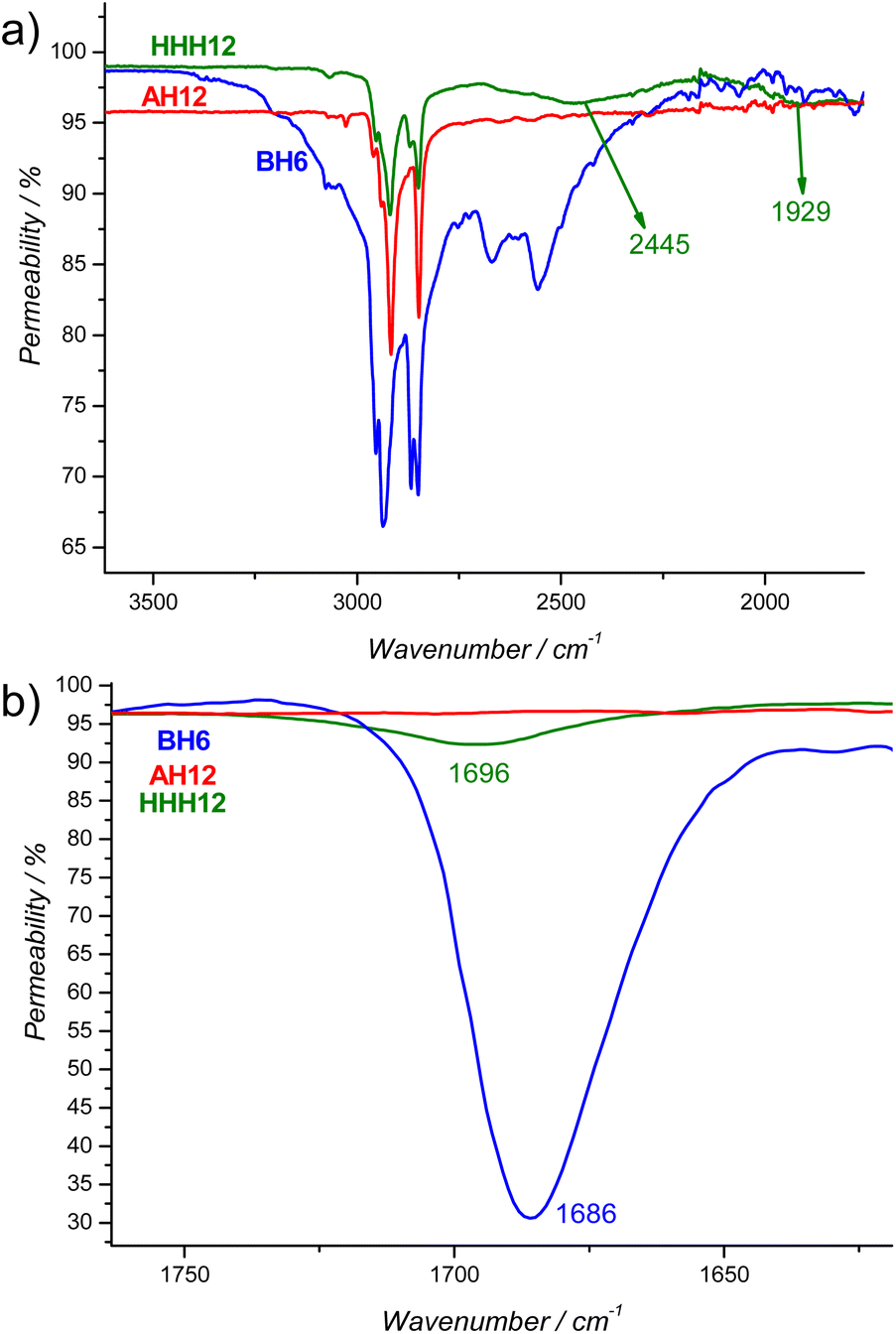 | ||
| Fig. 2 FTIR spectra of the supramolecule HHH12 (green) and its complementary components B6H (blue), AH12 (red) in the crystalline state at room temperature enlarged area (a) between 1750 cm−1 and 3600 cm−1 and (b) between 1620 cm−1 and 1760 cm−1. | ||
As is evident from Fig. 2a, the FTIR bands of the supramolecule HHH12 differ significantly from those of the pure components. The broad band observed in the range of 2500–3300 cm−1 for B6H, due to the formation of carboxylic dimer, is diminished in HHH12. Instead, two new broadened bands centred at approximately 2445 and 1929 cm−1 appear in HHH12, which is ascribed to Fermi resonance overtone.53 Additionally, the band at 1686 cm−1 corresponding to νC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration of the pure acid (BH6), broadens, decreases in intensity, and shifts to a higher wavenumber (∼1696 cm−1) upon complexation with the azopyridine AH12 (Fig. 2b).
O stretching vibration of the pure acid (BH6), broadens, decreases in intensity, and shifts to a higher wavenumber (∼1696 cm−1) upon complexation with the azopyridine AH12 (Fig. 2b).
These observations align with previously reported hydrogen-bonded systems involving pyridine derivatives and benzoic acids,53,58,59 confirming the successful hydrogen-bond formation between the azopyridine derivative AH12 and the acid BH6.
Next, the FTIR spectra for HHH12 and its related analogues with varying numbers and positions of fluorine substitutions were also recorded, and the results are given in Fig. S13–S18.† This investigation aimed to study how the strength of the hydrogen bond could be influenced by the position and degree of core fluorination. Regardless of the number or position of the fluorine atoms, the formation of H-bonds between each azopyridine derivative and the benzoic acids was successfully confirmed. In all cases, the two bands resulting from the Fermi resonance overtone were observed, along with changes in the intensity and wavenumber of the carbonyl band (νCO) of the pure acids compared to their complexed form (Fig. S13–S18†).
Table 1 summarizes the wavenumber shifts (ΔνCO) calculated between the νCO band for the pure acids and their corresponding supramolecules. As shown in Table 1, the lowest ΔνCO value is calculated for the nonfluorinated supramolecule HHH12. Introducing one fluorine atom at the ortho position with respect to the alkoxy chain in the azopyridine derivative AH12 in case of HHF12 results in no change in ΔνCO compared to HHH12 (Fig. S13†). This is because the fluorine atom is positioned far from the interacting carboxylic group, thus having almost no effect on the strength of the formed hydrogen bond.
| Pure acid | νCO cm−1 |
Supramolecule | νCO cm−1 |
ΔνCO |
|---|---|---|---|---|
| a ΔνCO represents the change in the carbonyl band of the pure acid component upon complexation with the azopyridine derivative. |
||||
| B6H | 1686 | HHH12 | 1696 | 10 |
| B6H | 1686 | HHF12 | 1696 | 10 |
| B6F3 | 1667, 1681 | FHH12 | 1691, 1703 | 24, 22 |
| B6F3 | 1667, 1681 | FHF12 | 1691, 1703 | 24, 22 |
| B6F2 | 1676 | HFH12 | 1693 | 17 |
| B6F23 | 1681 | FFH12 | 1698 | 17 |
| B6F2 | 1676 | HFF12 | 1693 | 17 |
On the other hand, shifting the F atom from the azopyridine derivative to the benzoic acid derivative, while maintaining its ortho position relative to the terminal alkoxy chain in case of FHH12, resulted in a more pronounced effect (Fig. S14†). The ΔνCO value for FHH12 is ∼24 and 22, indicating that the formed H-bond in this case is stronger compared to those in HHH12 or HHF12. This can be attributed to the electron-withdrawing effect of the fluorine atom, which increases the acidic character of the proton donor (B6F3). Similar ΔνCO values were also recorded for FHF12, further confirming that the fluorine atom on the azopyridine derivative has no significant effect on the strength of hydrogen bond.
Finally, introducing a fluorine atom adjacent to the carboxylic group (HFH12 or HFF12) or using two F atoms (FFH12) resulted in similar ΔνCO values in all cases ∼17. This suggests that for all fluorinated supramolecules constructed using any of the fluorinated benzoic acid derivatives (B6F3, B6F2 or B6F23) higher ΔνCO values could be calculated compared to the supramolecules derived from the nonfluorinated acid BH6 or the fluorinated azopyridine derivative AF12. This indicates that fluorine substitution on the benzoic acid side results in a more stabilized hydrogen bond formation.
To confirm the formation of the hydrogen bond in solution, we have also investigated some selected supramolecules using 1H NMR spectroscopy. Fig. 3 represents the 1H NMR of HHH12 (black) without fluorine substitution, alongside the spectra of its precursor acid B6H (blue) and the azopyridine derivative AH12 (red) in the aromatic region. The results reveal distinct signal differences between the pure components and their mixed state. In the supramolecule HHH12, the 1H NMR spectrum shows an increase of the chemical shift of the pyridine ring protons of AH12, as well as the signals of the two protons next to the azo group in the benzene ring. Additionally, a slight shift in the chemical shift values for B6H protons upon complexation with AH12 could be observed. These spectral changes confirm the successful formation of hydrogen bond between the complementary components in chloroform.
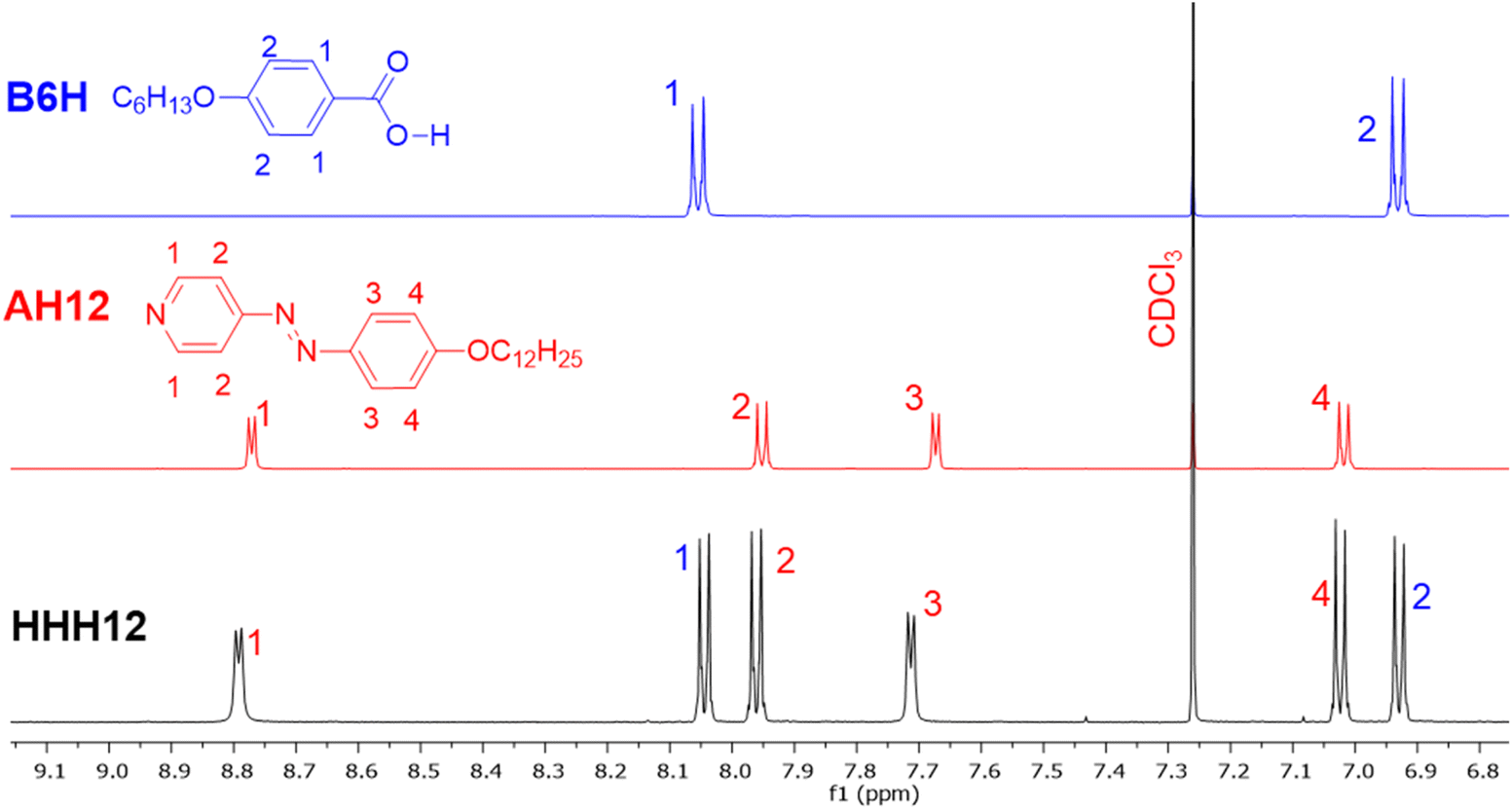 | ||
| Fig. 3 1H NMR spectra (500 MHz, CDCl3) of the supramolecule HHH12 (black) and its complementary components the azopyridine derivative AH12 (red) and the benzoic acid derivative B6H (blue) in the aromatic region (for the whole range see Fig. S19† in the ESI). | ||
To further investigate the influence of fluorine on the strength of the hydrogen bond, we examined FFH12, which features double fluorine substitution on the benzoic acid derivative (Fig. 4), and FHH12, which has single fluorine atom instead of two (Fig. S21† in the ESI). As can be seen from Fig. 4, the increase of the chemical shift values of all AH12 protons after hydrogen formation (FFH12, black spectrum) is more pronounced compared to that observed in the nonfluorinated supramolecule (HHH12, Fig. 3). This is due to the stronger interaction between the double fluorinated acid B6F23 and AH12. Additionally, the change in the shape of the signals for the two protons of the B6F23 after complexation further supports this conclusion.
 | ||
| Fig. 4 1H NMR spectra (500 MHz, CDCl3) of the supramolecule FFH12 (black) and its complementary components the azopyridine derivative AH12 (red) and the benzoic acid derivative B6F23 (blue) in the aromatic region (for the whole range see Fig. S20† in the ESI). | ||
Similar trends were also observed in the 1H NMR spectra of FHH12 when compared to its pure components, B6F3 and AH12 (Fig. S21† in the ESI). These findings lead to the conclusion that fluorination of the proton donor i.e. the benzoic acid derivative enhances the interaction between the two components, resulting in stronger hydrogen bond formation. Moreover, it confirms the stability of the hydrogen bond formation in solution.
3.2. Mesomorphic behaviour of synthesized HBLCs
Before discussing the LC behaviour of the target HBLCs we should refer to the fact that all azopyridine derivatives used in this work with or without fluorine are nonmesomorphic materials and melts directly to the isotropic liquid state.19,53 On the other hand, the benzoic acid derivatives are either nematogenic or crystalline materials as indicated by POM investigations. The transition temperatures of the proton donors as well as for the proton acceptors are summarized in Tables S1 and S2,† respectively. Therefore, the observation of different mesophases in the HBLCs under POM could be considered as a further indication of the formation of the hydrogen-bonded supramolecular complexes.The synthesized complementary components i.e. the benzoic acid derivatives and the azopyridine derivatives were used to prepare eight different groups of supramolecular HBLC complexes, which differ from each other's in the degree and position of fluorine substitution (Scheme 1). The formation of the hydrogen-bonded supramolecular complex between the complementary components was further confirmed by DSC investigations. For example, Fig. 5 shows the DSC heating and cooling cycles for the supramolecular complex FHH8 and its complementary components the 3-fluoro-4-n-hexyloxybenzoic acid (B6F3) and the 4-(4-octyloxyphenylazo)pyridine (AH8). Upon mixing the two components the individual transition temperatures recoded for each of them before mixing totally disappeared (red and blue curves in Fig. 2) and new transition temperatures are exhibited by the formed HBLC (green curves in Fig. 2). This clearly indicates the formation of the hydrogen bond between AH8 and B6F3 yielding the desired HBLC FHH8. Similar investigations were performed for all synthesized HBLCs (Tables S3–S10 and Fig. S22–S29† in ESI for additional DSC traces) and the results confirm the successful formation of the supramolecular complexes in all cases.
 | ||
| Fig. 5 DSC Heating and cooling cycles of the HBLC FHH8 (green curves) and its individual components, the proton donor B6F3 (blue curves) and the proton acceptor AH8 (red curves) at a scanning rate of 10 K min−1. | ||
The phase behaviour of all prepared supramolecules is represented graphically in Fig. 6 and the numerical data for transition temperatures and the corresponding enthalpy values are given in Table S3–S10† in the ESI.
 | ||
| Fig. 6 Schematic illustration of combined DSC and POM data on cooling at a rate of 10 K min−1 for the synthesized HBLCs. N = nematic phase, SmA = smectic A, SmC = smectic C, X = unknown LC phase; Nx = nematic x phase. | ||
 | ||
| Fig. 7 POM textures observed for: (a) HHH8 in the nematic phase at 128 °C, (b) HHH14 in the SmA phase at 120 °C and (c) HHH12 in the synclinic SmC phase at 105 °C. | ||
The N phase is retained for the next homologue HHH10 but with shorter range followed by wide range of SmA phase. Notably, for all members with n ≥ 10–14 both melting and crystallization temperatures are lowered compared to HHH8 resulting in wider LC range. For the supramolecules with n = 12 and 14 the N phase totally disappeared and the SmA phase is retained. Interestingly, an additional LC phase is observed below the SmA for HHH12 and HHH14, which cannot be detected by DSC investigations (Table S3†) indicating a second order phase transition. This phase is characterized by its high birefringence in all areas and the appearance of Schlieren texture with four-brush disclinations (indicated by white circle in Fig. 7c). These observations are characteristic for the synclinic tilted smectic C phase.
Changing the F position from the azopyridine derivative side to be on the other terminal of the supramolecule i.e. at ortho position in the benzoic acid derivative (B6F3) leads to the HBLCs FHHn (Fig. 6c and Table S5†). Interestingly, this modification results in different phase sequences compared to their related HBLCs HHFn, where the N phase is totally removed and instead a SmA phase is observed for all members of FHHn (compare Fig. 6b and c). On the other hand, the SmC is observed for a small range in case of FHH8 and its range is increasing upon chain elongation. The loss of N phase and appearance of smectic phases in HHFn is an indication for favouring layer structure in such HBLCs, which is a result of increasing core–core interaction. This indicates that the strength of the hydrogen bond between the complementary components in these HBLCs plays an important role in determining the type of LC phases. Introducing F in the benzoic acid derivative increases its character as proton donor, meaning stronger H-bonding interaction and more core–core interaction leading to the observed lamellar phases (SmA and SmC phases). In contrast, introducing the F at the azopyridine derivative (HHFn) does not affect the H-bonding strength to the same extent compared to that in case of FHHn because it is far from the two interacting positions. Instead, because of its steric effect the lamellar structure is only favoured with chain elongation and the nematic phase is retained for homologues with n ≤ 12 (Fig. 6b). This hypothesis is supported by the observed phase sequences in case of the next mono fluorinated HBLCs i.e. HFHn. The later have the F atom in inner position instead of outer one in case of FHHn, which results in more steric interaction leading to the formation of N phases in all HFHn homologues (Fig. 6e and 8). The N phase is the only observed mesophase for the shortest homologue and on chain elongation its range is decreasing and become very short for the longest chain homologue (HFH14). More interesting for all HFHn with n ≥ 10 a different LC phase characterized by its homeotropic texture with bright lines (Fig. 8c) is observed below the N phase. The range of this phase is increasing with chain elongation (Table S7†). Based on XRD results (Section 3.3), the exact type of this LC phase cannot be solved and therefore it is assigned as X phase. From these observations, it could be concluded that changing the position of the F atom not only alters the LC range or sequence but also changes the type of LC phases.
 | ||
| Fig. 8 POM textures observed for: (a) nematic droplets of HHF10 at the transition from the isotropic liquid phase at 115 °C; (b) nematic phase of HHF12 at 116 °C; (c) unknown phase of HFH10 at 90 °C. | ||
Changing the position of the F atom in FHFn from the ortho position in the benzoic acid derivative to be in a meta position i.e. using B6F2 as the proton donor leads to the HFFn series (Fig. 6f). This results in more steric interactions, leading to disfavouring of layer structures. Therefore, the N phase is observed for all members with n ≥ 8–12 and for the longest homologue the N phase is totally removed and SmC–SmA phase sequence appeared. More interesting, for HFF12 two different types of N phases are observed as indicated from the POM investigations (Fig. 9). Both N phases exhibit Schlieren textures, which flashes on applying shearing stress confirming the nematic nature. Unfortunately, it was not possible to further investigate the lower temperature N phase with XRD due its monotropic nature and therefore it is assigned as Nx phase.
 | ||
| Fig. 9 POM textures observed on cooling for: (a) N phase of HFF12 at 95 °C, (b) Nx phase of HFF12 at 70 °C and (c) the SmC phase of HFF14 at 96 °C. | ||
These results give insights about the structure–property relationship in these HBCLs and emphasizes the role of aromatic core fluorination, its degree and position as key factors in determining and controlling the LC phase types and ranges. This strategy could be used as a successful tool for advancing new materials for certain type of applications.
3.3. X-ray diffraction (XRD) investigations
To prove the different types of LC phases observed for the synthesized HBLCs we have performed XRD investigations for some selected examples. All experiments were performed using a surface-aligned sample, prepared by slow cooling of a small drop of the sample on a glass substrate from the isotropic liquid state.The first example is the nonfluorinated supramolecule HHH14, which display two LC phases as confirmed from POM investigations. In the wide-angle region (WAXS) a broad diffuse scattering in temperature range of the lower temperature LC phase with d value ∼ 0.44 nm is observed at T ∼ 100 °C (see the inset in Fig. 10a). This indicates a liquid crystalline phase with no fixed positions of the supramolecules. In the small-angle region (SAXS) at the same temperature a sharp peak is observed at 2θ ∼ 2.19° indicating a layer structure with a second order peak at 2θ ∼ 4.34° (Fig. 10a). The calculated d value is ∼4.04 nm which is smaller than the molecular length of HHH14 calculated with Materials Studio for all-trans conformation of the alkyl chains (Lmol = 4.44 nm). This indicates the presence of a tilted SmC phase with a tilt angle β of ∼24.5° calculated according to cos![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) β = d/Lmol. In the higher temperature LC phase (Fig. 10b), the WAXS region still shows a wide diffuse scattering confirming the transition from the SmC phase to another LC phase. In the SAXS region the first and second order reflections could be also seen proving the presence of another lamellar LC phase with a d value ∼ 4.13 nm, which is larger than that calculated for the SmC phase. These results confirm the presence of SmA phase with partial intercalation of the aromatic cores at higher temperature for HHH14, which agrees with the homeotropic, and fan-shaped textures observed under POM (Fig. 7b).
β = d/Lmol. In the higher temperature LC phase (Fig. 10b), the WAXS region still shows a wide diffuse scattering confirming the transition from the SmC phase to another LC phase. In the SAXS region the first and second order reflections could be also seen proving the presence of another lamellar LC phase with a d value ∼ 4.13 nm, which is larger than that calculated for the SmC phase. These results confirm the presence of SmA phase with partial intercalation of the aromatic cores at higher temperature for HHH14, which agrees with the homeotropic, and fan-shaped textures observed under POM (Fig. 7b).
 | ||
| Fig. 10 Plot of the integrated scattered intensity as a function of 2θ in the small-angle region (SAXS) for HHH14: (a) the SmC phase at T = 100 °C and (b) the SmA phase at T = 128 °C. The insets in (a) and (b) show the WAXS region in both phases. | ||
The next example is the monofluorinated HFH10 supramolecule. On cooling HFH10 from the isotropic liquid and at T ∼ 110 °C a diffuse wide-angle scattering (WAXS) reflex at d = 0.44 nm is observed confirming its LC nature (Fig. 11a). The SAXS pattern recorded at the same temperature shows one scattering peak at 2θ ∼ 2.36°. The peak is relatively sharp, and its intensity increased with decreasing temperature in the N phase. The appearance of such sharp peak in the N phase is not common for rod-like molecules, which usually display a broad halo scattering.60 This peak corresponds to d value of ∼3.74 nm, which is lower the molecular length of the compound calculated with Materials Studio for all-trans conformation of the alkyl chains Lmol = 4.09 nm. The presence of this peak and the d-value suggest the presence of cybotactic clusters of Sm-like structure (Cyb) in the nematic phase of HFH10 and therefore the nematic phase could be assigned as NCyb one. Such NCyb phase was reported recently for few examples of rod-like LC phases.33,61–65 The NCyb phase was also proved for the supramolecule HFF8 from XRD investigations (see Section 2 in the ESI†).
 | ||
| Fig. 11 Plot of the integrated scattered intensity as a function of 2θ in the small-angle region (SAXS) for HFH10 on cooling: (a) the N phase at T = 110 °C, (b) the X phase at T = 98 °C. The insets in (a) and (b) show the WAXS region in both phases. | ||
At the transition to the next LC phase on further cooling of HFH10 the single peak recorded in SAXS at T ∼ 98 °C become sharper with d ∼ 3.88 nm and a second reflex at 2θ ∼ 2.67° (Fig. 10b). The calculated d-value is larger than that observed in the NCyb phase and less than the molecular length (Lmol = 4.04 nm). This inline was the presence of titled lamellar phase. However, based on the observed texture under POM given in Fig. 8c and the second small reflection at 2θ ∼ 2.67°, which are not typical for known SmC phases, the exact structure of this lower temperature LC phase cannot be solved at the current stage and therefore it is assigned as X phase.
It should be noted that the observation of NCyb phase in the investigated supramolecules as confirmed from XRD investigations, indicates that different types of nematic phases are exhibited by the reported supramolecules. Therefore, depending on the adjacent LC phase the nematic phase of series HHHn could be assigned as NCybA phase formed by cybotactic clusters of the SmA type, while that of HHFn series is NCybC formed by SmC clusters. The third type is the NCybX phase adjacent to the unknown X phase in HFHn supramolecules. This proves that the position of the fluorine substitution plays a very important role in determining the type of the cybotactic clusters present in such nematic phases.
3.4. Photo-responsive behaviour
The focus of the current work was not only on the structure–property relationships of HBLCs by correlating the results discussed in the previous sections from DSC, POM, FTIR, 1H NMR and XRD but also to investigate the photo-responsive behaviour of the reported HBLCs. We designed the HBLCs to be photo-responsive because of the trans–cis photoisomerization of the azopyridine unit upon light irradiation. Therefore, to investigate the effect of fluorine substitution on the photoisomerization in solution we have selected the HBLCs with n = 8 from all series for such study (Fig. 12a–h). | ||
| Fig. 12 Spectral changes observed under UV light irradiation for: (a) HHH8, (b) HHF8, (c) FHH8, (d) FHF8, (e) HFH8, (f) HFF8, (g) FFH8, (h) FFF8 dissolved in chloroform. | ||
All selected HBLCs were dissolved in chloroform ∼2 mg/10 mL and measured with UV-vis spectrometry before and after UV illumination with time intervals of 5 seconds. Before UV irradiation all materials exhibit a strong absorption band between 355–358 nm, resulting from the π–π* transition. This indicates the presence of the stable trans isomer. After irradiation with UV light different behaviour is observed for the HBLCs, which depends strongly on the degree and position of fluorine atoms. Therefore, for the nonfluorinated complex HHH8 there is a slow transformation to the cis isomer with time, indicated by the decreasing of the initial absorption band at 355 nm and the appearance of another weak band around 448 nm. The latter is due to the n–π* transition, which corresponding to the cis isomer. These observations confirm the trans–cis photoisomerization of HHH8 under UV irradiation. Introducing a fluorine atom at ortho position with respect to the terminal chain in the azopyridine derivative in case of HHF8, results in faster trans–cis photoisomerization as indicated by the less time required for reducing the band at 357 nm (Fig. 12b). Changing the F atom to the other end at the complex in case of FHH8 suppress the photoisomerization completely as almost no change in the absorption band at 357 nm is observed. Using two F atoms at both terminals (X = Z = F, FHF8) results in a very similar behaviour to that of HHF8.
This process is more pronounced when the F atom is used next to the carboxylic group in the benzoic acid derivative (HFH8), which shows the fastest photoisomerization among all investigated HBLCs. For the remaining two complexes having two fluorines X, Y = F (FFH8) or three fluorines X, Y, Z = F (FFF8), no significant difference could be observed, where a slow photoisomerization process is observed in both cases. Therefore, from this investigation it could be concluded that compared to the nonfluorinated supramolecule (Fig. 12a) the trans–cis photoisomerization of mono fluorinated HBLCs strongly depend on the position of the F atom. Therefore, to reduce the trans–cis photoisomerization time the F atom should be introduced at outer position on the azopyridine side (Fig. 12b) or inner position on the benzoic acid side (Fig. 12e) instead of using it on outer position on the acid side (Fig. 12c). If the supramolecule contain two F atoms then at least one of them should be positioned at the azopyridine side (Fig. 12d and f) instead of having both of them at the acid side (Fig. 12g). In the latter case the trans–cis photoisomerization needs longer time, which is comparable to the triple fluorinated supramolecule (Fig. 12h). These different behaviours could be attributed to the steric effects resulting from using fluorine atoms at different positions. Keeping the measured solutions for all supramolecules in dark overnight and measuring them again results in almost identical spectra observed for the freshly prepared ones before light irradiation. This confirms that the trans–cis photoisomerization is completely a reversible process.
We have selected the supramolecules HHH8 and FFH8 as representative examples to investigate the photoisomerization between the different types of LC phases (Fig. 13). Isothermal UV light irradiation (365 nm) of the N phase of HHH8 at T = 125 °C results in a rapid transition to the isotropic liquid phase within three seconds (Fig. 13a and b). On removing the light source, the N phase texture is retained immediately, indicating a fast and reversible photo switching between N and isotropic liquid phase. This is a result of trans–cis photoisomerization, whereby the bent-shaped cis-isomer tends to destabilize the N phase, thus inducing a phase transition to the isotropic state. Similar observations were also observed between N and SmA (Fig. 13c and d) as well as between SmC and SmA (Fig. 13e, f) phases of FFH8. However, these transitions are slower (∼7 seconds) compared to that between N and isotropic states in case of HHH8. This might be attributed to the higher order of the SmA and SmC phases, resulting in lower mobility of the mesogens in these LC phases compared to the less order found in the N phase. These observations could be of interest for applications such as optical information storage device applications and non-linear optics.48,65
 | ||
| Fig. 13 Reversible isothermal photo-off–on switching between the different LC phases on cooling for: (a and b) supramolecule HHH8 between N and isotropic phases at 125 °C, (c and d) supramolecule FFH8 between N and SmA phases at 125 °C and (e and f) supramolecule FFH8 between SmC and SmA at 110 °C. | ||
4. Conclusion
This study provides a comprehensive exploration of the structure–property relationships in hydrogen-bonded liquid crystals (HBLCs), with a particular focus on the impact of aromatic core fluorination on their self-assembly and phase behaviour. By systematically varying the number and positions of fluorine substitutions in both proton donors (benzoic acid derivatives) and proton acceptors (azopyridine derivatives), we designed and synthesized eight distinct groups of HBLCs. These supramolecular complexes were thoroughly characterized using FTIR, NMR, DSC, POM, and XRD techniques, revealing a rich diversity of LC phases, including nematic, SmA, SmC, and an unidentified X phase.An interesting finding is the identification of cybotactic nematic phases (NCyb), which are rare in rod-like LCs. The type of cybotactic clusters (NCybA, NCybC, or NCybX) strongly depends on the adjacent LC phase, which could be modified by the degree and position of the fluorine substitution. This highlights the critical role of fluorine substitution in modulating molecular packing and intermolecular interactions. Specifically, fluorination of the benzoic acid derivatives was shown to enhance hydrogen bond strength as proved from FTIR and NMR investigations, leading to more stabilized and ordered LC phases, while fluorination of the azopyridine derivatives had a negligible effect on hydrogen bond strength but influenced phase transitions and mesophase stability.
Additionally, the photo-responsive behaviour of these HBLCs was investigated in soultion, demonstrating that the position and degree of fluorination significantly impact the rate of trans–cis photoisomerization. For instance, mono fluorination near the carboxylic group in the benzoic acid derivatives or at the outer side of the azopyridine derivative accelerated the photoisomerization, while double fluorination requires certain distribution of the fluorine atoms. On the other hand, the photoisomerization behaviour of triple fluorinated supramolecules was found to be comparable to the nonfluorinated ones. Regardless the degree of fluorination reversible relaxation from cis to trans isomer was found in all cases. Reversible photo-switching between LC phases (e.g., SmC–SmA or N–Iso) was also achieved, showcasing the potential of these materials for applications in optical storage devices and non-linear optics.
In summary, this work not only advances our understanding of the role of aromatic core fluorination in tuning the properties of HBLCs but also provides a versatile platform for designing functional materials with tailored mesomorphic and photo-responsive behaviours avoiding many steps of organic synthesis.
Data availability
Experimental data, tables of transition temperatures and additional XRD data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the German Research Foundation (DFG) (AL2378/1-2, 424355983, RTG 2670, 436494874). A. F. Darweesh acknowledges the support by the Alexander von Humboldt Foundation for the research fellowship at Martin Luther University Halle-Wittenberg, Germany.References
- J. Lehn, Supramolecular Chemistry, Wiley, 1995 Search PubMed.
- J. D. Watson and F. H. C. Crick, Nature, 1953, 171, 737–738 CrossRef CAS PubMed.
- J. Buchs, L. Vogel, D. Janietz, M. Prehm and C. Tschierske, Angew. Chem., 2017, 129, 286–290 CrossRef.
- F. Malotke, M. Saccone, C. Wölper, R. Y. Dong, C. A. Michal and M. Giese, Mol. Syst. Des. Eng., 2020, 5, 1299–1306 RSC.
- D. L. Nelson and M. M. Cox, Lehninger Principles of Biochemistry, W. H. Freeman and Company, New York, 5th edn, 2008 Search PubMed.
- X.-H. Cheng and H.-F. Gao, in Hydrogen Bonded Supramolecular Materials, Springer, Berlin, Heidelberg, 2015, pp. 133–183 Search PubMed.
- G. V. Oshovsky, D. N. Reinhoudt and W. Verboom, Angew. Chem., Int. Ed., 2007, 46, 2366–2370 CrossRef CAS PubMed.
- T. Kato, N. Mizoshita and K. Kanie, Macromol. Rapid Commun., 2001, 22, 797–814 CrossRef CAS.
- C. M. Paleos and D. Tsiourvas, Liq. Cryst., 2001, 28, 1127–1161 CrossRef CAS.
- T. Kato and J. M. J. Fréchet, J. Am. Chem. Soc., 1989, 111, 8533–8534 CrossRef CAS.
- B. Friot, D. Boyd, K. Willis, B. Donnio, G. Ungar and D. W. Bruce, Liq. Cryst., 2000, 27, 605–611 CrossRef CAS.
- L. Vogel, D. Janietz, M. Prehm and C. Tschierske, Soft Matter, 2018, 14, 806–816 RSC.
- N. Gimeno, M. B. Ros, J. L. Serrano and M. R. De La Fuente, Chem. Mater., 2008, 20, 1262–1271 CrossRef CAS.
- J. Wang, Y. Shi, K. Yang, J. Wei and J. Guo, RSC Adv., 2015, 5, 67357–67364 RSC.
- M. Alaasar, C. Tschierske and M. Prehm, Liq. Cryst., 2011, 38, 925–934 CrossRef CAS.
- D. Stewart and C. T. Imrie, Macromolecules, 1997, 30, 877–884 CrossRef CAS.
- M. Alaasar, X. Cai, F. Kraus, M. Giese, F. Liu and C. Tschierske, J. Mol. Liq., 2022, 351, 118597 CrossRef CAS.
- M. Alaasar, J. C. Schmidt, X. Cai, F. Liu and C. Tschierske, J. Mol. Liq., 2021, 332, 115870 CrossRef CAS.
- M. Alaasar, S. Poppe, Q. Dong, F. Liu and C. Tschierske, Chem. Commun., 2016, 52, 13869–13872 RSC.
- M. Hird, J. W. Goodby, R. A. Lewis and K. J. Toyne, Mol. Cryst. Liq. Cryst., 2003, 401, 1–18 CrossRef.
- D. Demus and H. Zaschke, Flüssige Kristalle in Tabellen II, veb Deutscher verlag für grundstoffindustrie, 1984 Search PubMed.
- T. H. Aldahri, M. Alaasar and H. A. Ahmed, Liq. Cryst., 2023, 50, 1059–1068 CrossRef CAS.
- S. Sergeyev, W. Pisula and Y. H. Geerts, Chem. Soc. Rev., 2007, 36, 1902–1929 RSC.
- M. Powers, R. J. Twieg, J. Portman and B. Ellman, J. Chem. Phys., 2022, 157, 134901 CrossRef CAS PubMed.
- M. Alaasar, M. Prehm, M. G. Tamba, N. Sebastián, A. Eremin and C. Tschierske, ChemPhysChem, 2016, 17, 278–287 CrossRef CAS PubMed.
- B. N. Sunil, M. K. Srinatha, G. Shanker, G. Hegde, M. Alaasar and C. Tschierske, J. Mol. Liq., 2020, 304, 112719 CrossRef CAS.
- H. N. S. Murthy and B. K. Sadashiva, J. Mater. Chem., 2004, 14, 2813–2821 RSC.
- L. Caillier, E. Taffin De Givenchy, S. Géribaldi and F. Guittard, J. Mater. Chem., 2008, 18, 5382–5389 RSC.
- S. Iamsaard, E. Anger, S. J. Aßhoff, A. Depauw, S. P. Fletcher and N. Katsonis, Angew. Chem., 2016, 128, 10062–10066 CrossRef.
- J. L. Serrano, Metallomesogens – Synthesis, Properties, and Applications, VCH, Weinheim, Germany, 1996 Search PubMed.
- M. M. Zhou, J. He, H. M. Pan, Q. Zeng, H. Lin, K. Q. Zhao, P. Hu, B. Q. Wang and B. Donnio, Chem.–Eur. J., 2023, 29, e202301829 CrossRef CAS PubMed.
- F. B. Meng, X. Z. He, X. D. Zhang, Y. Ma, H. L. Han and H. Lu, Colloid Polym. Sci., 2011, 289, 955–965 CrossRef CAS.
- M. Alaasar and S. Poppe, Liq. Cryst., 2020, 47, 939–949 CrossRef CAS.
- M. A. El-Atawy, A. Z. Omar, M. L. Alazmi, M. S. Alsubaie, E. A. Hamed and H. A. Ahmed, Heliyon, 2023, 9, e14871 CrossRef CAS PubMed.
- P. Kirsch, M. Bremer, M. Heckmeier and K. Tarumi, Angew. Chem., Int. Ed., 1999, 38, 1989–1992 CrossRef CAS PubMed.
- M. Poppe, C. Chen, F. Liu, S. Poppe and C. Tschierske, Chem.–Eur. J., 2017, 23, 7196–7200 CrossRef CAS PubMed.
- M. Spengler, R. Y. Dong, C. A. Michal, M. Pfletscher and M. Giese, J. Mater. Chem. C, 2017, 5, 2235–2239 RSC.
- M. Hird, Chem. Soc. Rev., 2007, 36, 2070–2095 RSC.
- C. J. Gibb, J. Hobbs, D. I. Nikolova, T. Raistrick, S. R. Berrow, A. Mertelj, N. Osterman, N. Sebastián, H. F. Gleeson and R. J. Mandle, Nat. Commun., 2024, 15, 5845 CrossRef CAS PubMed.
- S. Brown, E. Cruickshank, J. M. D. Storey, C. T. Imrie, D. Pociecha, M. Majewska, A. Makal and E. Gorecka, ChemPhysChem, 2021, 22, 2506–2510 CrossRef CAS PubMed.
- J. Karcz, J. Herman, N. Rychłowicz, P. Kula, E. Górecka, J. Szydlowska, P. W. Majewski and D. Pociecha, Science, 2024, 384, 1096–1099 CrossRef CAS PubMed.
- H. Ren, P. Yang and H. Yu, Molec, 2022, 27, 3977 CrossRef CAS PubMed.
- Y. Chen, H. Yu, L. Zhang, H. Yang and Y. Lu, Chem. Commun., 2014, 50, 9647–9649 RSC.
- M. Alaasar and C. Tschierske, Liq. Cryst., 2019, 46, 124–130 CrossRef CAS.
- M. Hagar, H. A. Ahmed and O. A. Alhaddad, Liq. Cryst., 2019, 46, 1440–1451 CrossRef CAS.
- L. J. McAllister, J. Taylor, N. E. Pridmore, A. J. McEllin, A. C. Whitwood, P. B. Karadakov and D. W. Bruce, CrystEngComm, 2023, 25, 1683–1692 RSC.
- M. Alaasar, C. Anders, R. Pashameah and A. F. Darweesh, Liq. Cryst., 2023, 50, 2397–2412 CrossRef CAS.
- B. S. Ranjitha, D. Sandhya Kumari, A. Shetty, G. Shanker, M. Alaasar, R. Pashameah and G. Hegde, J. Mol. Liq., 2023, 383, 121985 CrossRef CAS.
- D. S. Kumari, A. Shetty, B. S. Ranjitha, M. Vandana, G. Shanker, M. Alaasar and G. Hegde, Heliyon, 2024, 10, e37455 CrossRef PubMed.
- Y. Ni, X. Li, J. Hu, S. Huang and H. Yu, Chem. Mater., 2019, 31, 3388–3394 CrossRef CAS.
- H. Yu, H. Liu and T. Kobayashi, ACS Appl. Mater. Interfaces, 2011, 3, 1333–1340 CrossRef CAS PubMed.
- H. Liu, T. Kobayashi and H. Yu, Macromol. Rapid Commun., 2011, 32, 378–383 CrossRef CAS PubMed.
- M. Alaasar, J. C. Schmidt, A. F. Darweesh and C. Tschierske, J. Mol. Liq., 2020, 310, 113252 CrossRef CAS.
- M. Giese, T. Krappitz, R. Y. Dong, C. A. Michal, W. Y. Hamad, B. O. Patrick and M. J. MacLachlan, J. Mater. Chem. C, 2015, 3, 1537–1545 RSC.
- M. Spengler, R. Y. Dong, C. A. Michal, W. Y. Hamad, M. J. MacLachlan and M. Giese, Adv. Funct. Mater., 2018, 28, 1800207 CrossRef.
- B. O. Lindgren and T. Nilsson, Acta Chem. Scand., 1973, 27, 888 CrossRef CAS.
- K. Schwetlick, Organikum, Wiley-VCH, Weinheim, Germany, 21st edn, 2001, p. 500 Search PubMed.
- T. Kato, J. M. J. Frechet, P. G. Wilson, T. Saito, T. Uryu, A. Fujishima, C. Jin and F. Kaneuchi, Chem. Mater., 1993, 5, 1094 CrossRef CAS.
- Y. Arakawa, Y. Sasaki and H. Tsuji, J. Mol. Liq., 2019, 280, 153–159 CrossRef CAS.
- B. S. Ranjitha, M. Alaasar and G. Shanker, J. Mol. Struct., 2024, 1305, 137626 CrossRef CAS.
- W. Nishiya, Y. Takanishi, J. Yamamoto and A. Yoshizawa, J. Mater. Chem. C, 2014, 2, 3677–3685 RSC.
- Y. Uchida, T. Akita, K. Hanada, D. Kiyohara and N. Nishiyama, J. Mater. Chem. C, 2022, 10, 6621–6627 RSC.
- Y. Kimoto, A. Nishizawa, Y. Takanishi, A. Yoshizawa and J. Yamamoto, J. Phys. Chem. B, 2013, 117, 6290–6293 CrossRef CAS PubMed.
- Y. Arakawa, Y. Sasaki, K. Igawa and H. Tsuji, New J. Chem., 2017, 41, 6514–6522 RSC.
- M. Alaasar, T. Nirgude and C. Anders, J. Mol. Liq., 2024, 414, 126174 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ra00670h |
| This journal is © The Royal Society of Chemistry 2025 |