
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDual-functional co-crystal of streptavidin and ssDNA: electrostatic assembly with positively charged peptide tags†
Ayasa Nagatani a,
Kosuke Minamihata*a,
Motoyasu Adachibc,
Rie Wakabayashia,
Masahiro Gotoad and
Noriho Kamiyaad
a,
Kosuke Minamihata*a,
Motoyasu Adachibc,
Rie Wakabayashia,
Masahiro Gotoad and
Noriho Kamiyaad
aDepartment of Applied Chemistry, Graduate School of Engineering, Kyushu University, 744 Motooka, Fukuoka 819-0395, Japan
bInstitute for Quantum Life Science, National Institutes for Quantum Science and Technology, 4-9-1 Anagawa, Inage, Chiba 263-8555, Japan
cDepartment of Quantum Life Science, Graduate School of Science, Chiba University, Chiba, 263-8522, Japan
dDivision of Biotechnology, Center for Future Chemistry, Kyushu University, 744 Motooka, Fukuoka 819-0395, Japan
First published on 3rd March 2025
Abstract
We have achieved a novel co-crystal in which the dual functions of the protein and single-stranded DNA are maintained by introducing a charged peptide tag at the C-terminus of the protein. The functionalities allowed the co-crystals to be modified with high selectivity. Additionally, we have confirmed that energy transfer occurs between the two molecules modified within the co-crystal. Therefore, this co-crystal has the potential as a novel biomaterial applicable to biosensors.
Proteins and nucleic acids are the primary biomolecules that constitute living organisms. Proteins are crucial in metabolism1 and immune function,2,3 whereas nucleic acids store and express genetic information,4 with DNA often called the blueprint of life. These biomolecules underpin the overall functioning of biological systems, and they each have their own distinct roles, in addition to co-assembling to form essential biological structures. For example, viruses,5 nucleosomes,6 and ribosomes7 all exhibit high functionality8 that cannot be achieved by individual proteins or nucleic acids. Therefore, research involving the artificial creation of co-assemblies of proteins and nucleic acids, particularly DNA, is being conducted for applications in catalytic reactions,9,10 biosensors,11,12 cancer treatment,13,14 and other fields.
Crystals have a three-dimensional (3D) structure in which the molecules in the assembly are arranged in a regular pattern. The regular arrangement enables crystals to achieve high-efficiency molecular separation15 and energy transfer,16 and thus, these crystals may have medical applications.17,18 These features and functionalities of crystals are derived from the nanosized structural arrangement of the molecules and not from the functionality of the molecules themselves. The crystallization of diverse molecules with distinct functionalities is expected to create highly functional crystals with multiple capabilities. However, existing functional crystals have mainly been constructed using small molecules, which possess limited functionalities of their own, as their framework. Other research has focused on creating crystals of high-functionality biomacromolecules, typically proteins and nucleic acids. The self-assembly of co-crystals is challenging because proteins and nucleic acids are markedly different biopolymers, both chemically and structurally. Although many examples of co-crystals formed by covalently linking these biomacromolecules have been reported, the driving force for co-crystal formation has primarily been complementary base-pairing of DNA.19 Existing methods often result in the loss of the key function of the nucleic acids, causing the functionality of the co-crystal to depend largely on the function of the protein alone. Consequently, the functionality of protein and nucleic acid co-crystals has been nearly identical to that of the protein alone. If it were possible to form co-crystals of proteins and nucleic acids while maintaining the functions of both, their cooperative action could lead to the creation of new functional biomaterials.
In this study, our goal was to create co-crystals composed of proteins and nucleic acids without relying on their intrinsic functions, thereby creating a new biomaterial exhibiting the functionalities of both types of biomolecules.
The rapid co-crystallization of streptavidin (SA) with single-stranded DNA (ssDNA) has been achieved by using electrostatic interactions between a genetically introduced peptide tag and ssDNA.20 Strong electrostatic interactions between proteins and nucleic acids tend to cause the formation of aggregates in which the molecules are randomly arranged and which is often accompanied by the denaturation of the constituent proteins. However, this technique has been used to achieve SA and ssDNA co-crystallization by introducing a positively charged peptide tag, namely, RXY (where X > 3), at the C-terminus of SA by genetic modification and simply mixing the SA containing the peptide tag with ssDNA. Because this method does not depend on the functionalities of either SA or ssDNA, the SA-ssDNA co-crystals are expected to exhibit both their functionalities, although further functional evaluation of the co-crystals has not yet been conducted. Additionally, the effects of the electrostatic interactions between SA and ssDNA on co-crystallization remain unclear. Furthermore, SA's biotin-binding capability enables the incorporation of diverse molecules, making it an ideal model for creating multifunctional protein crystals composed of both ssDNA and proteins.
Herein, we investigated the effect of the ssDNA sequence and the length of the positively charged peptide tag on co-crystal formation. SA modified with R4Y and R6Y (hereafter, referred to as SA-R4Y and SA-R6Y, respectively) were prepared, and co-crystallization with ssDNA of different lengths at different salt concentrations was conducted to reveal the effects of the electrostatic interactions on co-crystal formation. Furthermore, we verified the selective incorporation of biotinylated molecules in the co-crystals and the complementary base-pairing ability of the ssDNA.
First, the effect of the base type of ssDNA on co-crystallization was examined by mixing either SA-R4Y or SA-R6Y with ssDNA of NX (where N = A, T, G, or C and X = 8, 10, 12, or 20). Co-crystal formation was observed when ssDNAs composed of A, T, and C were used (Fig. 1). Fluorescence was observed from co-crystals prepared with SA-R6Y and T8 ssDNA modified with a fluorescent molecule at the 5′-terminus, indicating the presence of ssDNA in the co-crystal (Fig. S1†). Conversely, no crystals were obtained with ssDNA of GX (G-ssDNA) and only aggregates were formed, which was attributed to the tendency of G-rich DNA sequences to adopt higher-order structures, such as triplets and quadruplexes.21 Therefore, circular dichroism spectra were used to confirm whether G-ssDNA formed higher-order structures (Fig. S2†). The spectra of parallel quadruplexes exhibit a positive band at 260 nm.22 A peak similar to that of G-quadruplexes was observed for the GX-ssDNA. Therefore, the G-ssDNA formed a self-assembled structure, disrupting the alignment with either SA-R4Y or SA-R6Y and resulting in the formation of aggregates.
 | ||
| Fig. 1 Microscope images of crystals obtained by co-crystallization of SA-RnY (n = 4 or 6) and ssDNA (NX: N = A, T, G, or C; X = 8, 10, 12, or 20 nt). Co-crystals were prepared with SA-R4Y or SA-R6Y (10 μM) and each ssDNA (10 μM) in 10 mM Tris–HCl (pH 8.0) at 25 °C. Scale bars: 100 μm. | ||
The length of the positively charged peptide tag also affected the co-crystallization. SA-R6Y only crystallized with short ssDNAs (8–12 nt), whereas with longer-length ssDNAs (20 nt), aggregation instead of crystallization was observed. In contrast, SA-R4Y co-crystallized with all the ssDNA lengths. Additionally, SA-R4Y even formed co-crystals with 40-nt-long ssDNA (Fig. S3†). The aggregation of SA-R6Y with long ssDNAs, which had the highest number of negative charges tested here, was attributed to the excessively strong interactions that resulted in the misalignment of the SA and ssDNA molecules. This finding indicates that there is an optimal range for the strength of the electrostatic interactions for co-crystallization.
In assembly formation using protein surface charges, the surface charge can be modulated by electrostatic shielding with the addition of salts.23 To support the existence of an optimal electrostatic interaction strength for co-crystallization, we investigated the effect of NaCl addition on the formation of SA and ssDNA co-crystals. Although SA-R6Y and T20 ssDNA formed aggregates at low salt concentrations, the addition of 50 mM or more NaCl facilitated the formation of co-crystals (Fig. 2A). Moreover, a higher salt concentration was correlated with larger-sized co-crystals. However, SA-R4Y formed no co-crystals with an increase in the NaCl concentration. Furthermore, ssDNA shorter than 20 nt in length, which had previously resulted in co-crystal formation with SA-R6Y, did not form co-crystals at a NaCl concentration of 75 mM or higher (Fig. 2B and S4†). These results indicate that co-crystal formation is facilitated by the establishment of an appropriate electrostatic interaction between the positively charged peptide tag and the ssDNA. Therefore, by regulating the electrostatic interaction between the two, co-crystals can be formed with ssDNAs of any length. This electrostatic interaction can be modulated through the design of the positively charged peptide tag and manipulation of the solution environment.
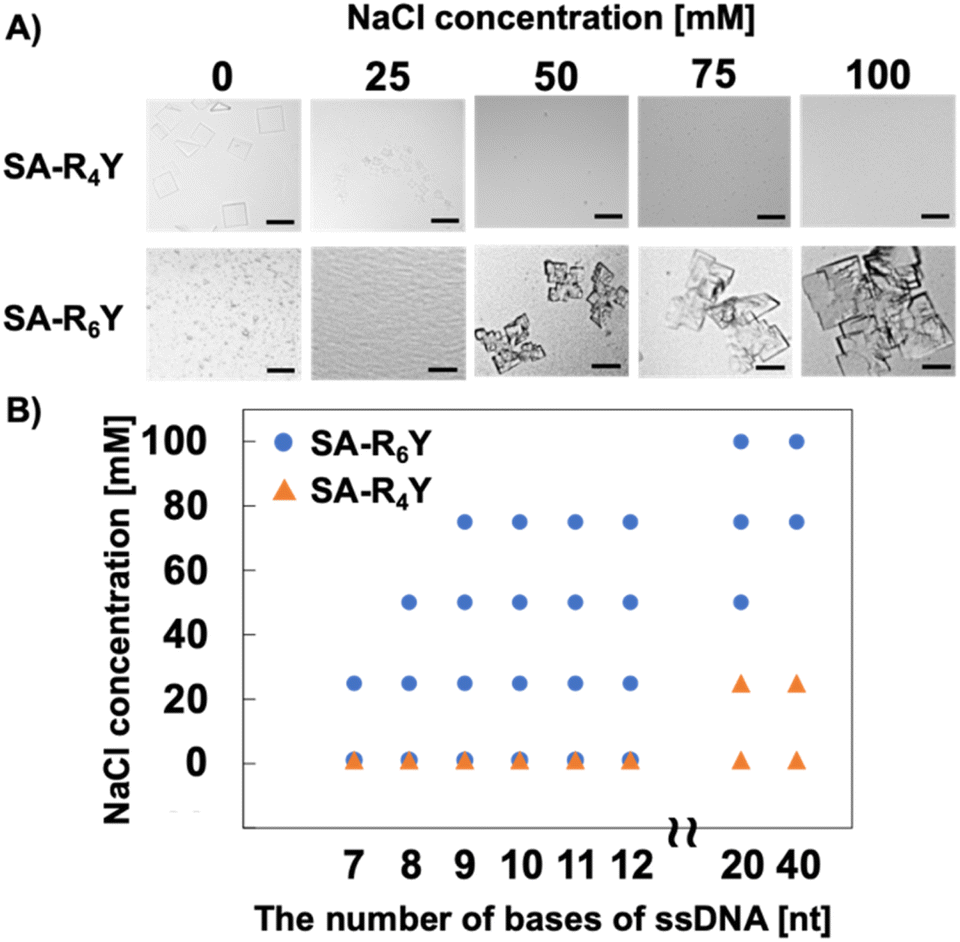 | ||
| Fig. 2 Effect of NaCl on the co-crystallization of SA-RnY and ssDNA. (A) Microscope images of co-crystal formation of SA-R4Y or SA-R6Y with T20 ssDNA. Scale bars: 100 μm. (B) Phase diagrams of SA-R4Y or SA-R6Y with T20 ssDNA. Co-crystallization was conducted in individual wells, and conditions where co-crystals formed in more than two wells are indicated by blue circles and orange triangles respectively. | ||
We determined the crystal structure of SA-R6Y and A9 co-crystals, and the results of the single-crystal X-ray structural analysis are presented in Fig. 3. In addition, we obtained the crystal structure of SA-R6Y and C9 co-crystals (Fig. S5†), which exhibited a different structure from that of SA-R6Y and A9. The co-crystal with A9 crystallized in space group P1,24 whereas the co-crystal with C9 crystallized in space group C2.25 These results indicated that co-crystals of SA and ssDNA can form at least two types of crystal structure. This was supported by the formation of different shapes of SA and ssDNA crystals. Although the co-crystals of SA-R6Y and A8 were mainly plate-like, trapezoidal crystals were also formed (Fig. 1). The crystal (space group and cell constants) of both SA with A9 and C9 obtained were different from those of SA reported in the Protein Data Bank (PDB) and are new SA crystal systems. Both crystal structure analyses showed that the SA molecules formed two-dimensional structures with a spacing of approximately 2.9 nm between the C-termini of each SA molecule (Fig. 3A). This suggests that ssDNA may be positioned within this space, linking the SA molecules via electrostatic interactions. Although the diameter of double-stranded DNA is around 2.4 nm, ssDNA probably occupies a smaller space, and thus fitted between the C-termini of the SA molecules along with the positively charged peptide tags. However, the G-quadruplex structure has a larger diameter of approximately 2.8 nm, which would complicate its accommodation in the same space alongside the peptide tags, and would cause aggregate formation when oligo ssDNAs composed solely of guanine were used.
 | ||
| Fig. 3 Crystal structure of SA-R6Y/A9. (A) Packing of each layer of SAs showing the layers. Enlarged view of the C-terminal region of SAs in the crystal showing the distance between the Ser136 residues. (B) 3D packing of SAs showing the repeating unit of three layers in different colours. Enlarged view of the layer thickness. | ||
Because the pores between the SA molecules in each layer were interconnected and formed penetrating vacant spaces in the 3D co-crystal structure of SA-R6Y and A9, the long ssDNAs that could not fit within a single layer of SA could still form crystals by spanning the penetrating pore spaces. The 20 nt ssDNA, which could not fit within a single layer of SA, can form a packing arrangement similar to that observed in the crystal structure obtained with A9 (Fig. 3B). Additionally, when the crystal was observed under white light in bright-field microscopy (Fig. S6†), it exhibited colour. We expected SA and ssDNA to be aligned separately, and the domains occupied by ssDNA are alternately present relative to the domains occupied by SA molecules. Different molecules possess different diffractive indexes; therefore, the co-crystals of SA and ssDNA behaved like a diffraction grating, exhibiting properties similar to those of a photonic crystal.
Next, we assessed the functionalities of SA and ssDNA in the co-crystals. In these experiments, we used SA-R4Y as SA-RnY because it readily crystallizes with long ssDNA. We used biotin-4-fluorescein (B4F) to investigate the biotin-binding ability of SA in the co-crystals. The co-crystals were not stained with fluorescein, whereas the co-crystals treated with B4F exhibited green fluorescence (Fig. 4A and B). Quantitative analysis of the amount of fluorescein incorporated into the crystals revealed that B4F displayed a significantly higher incorporation ratio than fluorescein (Fig. 4B). These results indicate that the SA in the co-crystals retained its biotin-binding ability. We also evaluated the complementary strand recognition ability of ssDNA in the co-crystals, using the ssDNA shown in Fig. 4C. Co-crystals containing ssDNA complementary to the fluorescein-labelled ssDNA (FL-ssDNA) exhibited strong fluorescein fluorescence, indicating that the complementary base-pairing ability of the ssDNA in the co-crystals was maintained (Fig. 4D and E). Measurements of the supernatant concentration of FL-ssDNA confirmed that complementary ssDNA was incorporated to a greater extent than noncomplementary ssDNA. Thus, these co-crystals exhibited high selectivity for recognizing ssDNA. We treated the co-crystals with both B4F and tetramethyl rhodamine (TAMRA)-labelled ssDNA to confirm that the functionalities of both SA and ssDNA were retained in the co-crystals. The TAMRA-labelled ssDNA was incorporated selectively into the co-crystals (Fig. S7†).
 | ||
| Fig. 4 Functionalities of SA and ssDNA. (A) Fluorescein microscopy images of co-crystals treated with B4F and fluorescein. (B) Ratio of fluorescent molecule uptake. (C) ssDNA sequences studied. (D) Fluorescence microscopy images of co-crystals formed with complementary, ssDNA1, ssDNA2, or noncomplementary ssDNA treated with fluorescein-labelled ssDNA (yellow markers indicate the locations where mutations were introduced). (E) Ratio of ssDNA uptake (N = 3, mean ± SE, *p < 0.05, ***p < 0.001. BFI: bright field image, FI: fluorescent image). | ||
Fig. 5 shows that TAMRA-labelled ssDNA and B4F were co-localized in the co-crystals. The Z-stacking results of the co-crystals incorporating TAMRA-labelled ssDNA and B4F confirmed that the two molecules were present on the crystal surface and were almost identical (Fig. S8†). Therefore, we expected that energy transfer could occur between these two molecules, and we investigated whether fluorescence resonance energy transfer (FRET) occurred. To confirm the FRET results using confocal laser scanning microscopy (CLSM), we observed the fluorescence intensity changes in fluorescein, the donor molecule, upon bleaching of the FRET acceptor molecule, TAMRA (Fig. 5C and D). An increase in the fluorescence intensity from fluorescein was observed in the region where TAMRA was bleached (Fig. 5D). This finding confirms that our approach has the potential to be used to form an energy-exchangeable biomaterial. Moreover, reports have indicated that combining photonic crystals and the FRET system can enhance energy transfer.26 There are no reported examples of a photonic crystal formed from protein and DNA in which energy transfer has been observed. Eventually, this co-crystal may function as a novel biomaterial that promotes highly efficient energy transfer.
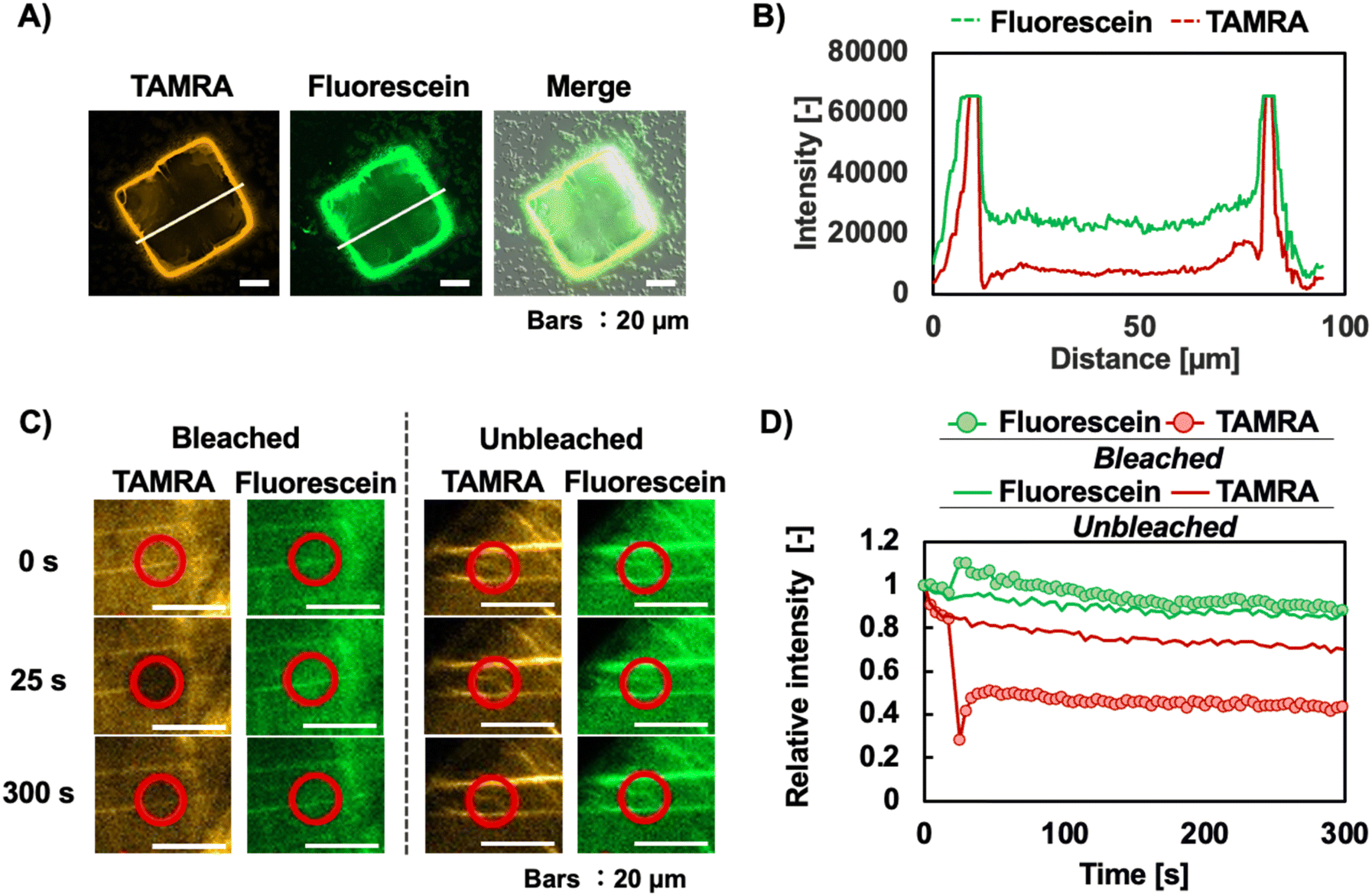 | ||
| Fig. 5 Co-crystallization with dual-molecule modification. (A) Fluorescence microscopy images of SA-ssDNA co-crystals treated with B4F- and TAMRA-labelled ssDNA. (B) Fluorescence intensities of TAMRA and fluorescein along the lines shown in the CLSM images in panel A. (C) Confirmation of FRET between two molecules modified for co-crystallization using CLSM. (D) Relative intensity during FRET observations. Bleached and unbleached measurements were performed in the same crystal. | ||
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
A. N.: writing of the original draft, investigation, and methodology. M. A., R. W., M. G.: investigation, and methodology; K. M.: conceptualization, methodology, validation, writing/reviewing, editing; N. K.: supervision, validation, and editing. All authors contributed to the discussion of the paper and approved the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by JSPS KAKENHI Grant Numbers JP18K14067 (to K. M.), JP22H01884 (to R. W.), JP23H00247 (to N. K.). X-ray diffaction data collection was performed under the approval of the Photon Factory Program Advisory Committee (Proposal No. 2014G645). We thank Amy Capes, PhD from Edanz (https://jp.edanz.com/ac) for editing a draft of this manuscript.Notes and references
- R. J. Argüello, A. J. Combes, R. Char, J. P. Gigan, A. I. Baaziz, E. Bousiquot, V. Camosseto, B. Samad, J. Tsui, P. Yan, S. Boissonneau, D. Figarella-Branger, E. Gatti, E. Tabouret, M. F. Krummel and P. Pierre, Cell Metab., 2020, 32, 1063 CrossRef PubMed.
- F. Hu, X. Yu, H. Wang, D. Zuo, C. Guo, H. Yi, B. Tirosh, J. R. Subjeck, X. Qiu and X. Y. Wang, Eur. J. Immunol., 2011, 41, 1086 CrossRef PubMed.
- C. Hetz, K. Zhang and R. J. Kaufman, Nat. Rev. Mol. Cell Biol., 2020, 21, 421 CrossRef.
- P. Nie, Y. Bai and H. Mei, Molecules, 2020, 25, 3483 CrossRef.
- A. A. T. Naqvi, F. Anjum, A. Shafie, S. Badar, A. M. Elasbali, D. K. Yadav and Md. I. Hassan, PLoS One, 2021, 16, e0261497 CrossRef.
- M. Eitok, L. Sato, T. Senda and M. Horikoshi, Cell. Mol. Life Sci., 2008, 65, 414 CrossRef PubMed.
- J. H. Cate, M. M. Yusupov, G. Z. Yusopova, T. N. Earnest and H. F. Noller, Science, 1999, 285, 2095 CrossRef.
- M. Jinek, K. Chylinski, I. Fonfara, M. Hauer, J. A. Doudna and E. Charpentier, Science, 2012, 337, 816 CrossRef CAS PubMed.
- J. D. Brodin, E. A. Auyeung and C. A. Mirkin, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 4564 CrossRef CAS PubMed.
- T. M. Nguyen, E. Nakata, M. Saimura, H. Dinh and T. Morii, J. Am. Chem. Soc., 2017, 139, 8487 CrossRef CAS PubMed.
- J. Gao, X. Hua, R. Yuan, Q. Li and W. Xu, Talanta, 2021, 234, 122643 CrossRef CAS PubMed.
- L. Li, L. Wang, Q. Xu, L. Xu, W. Liang, Y. Li, M. Ding, A. Aldalbahi, Z. Ge, L. Wang, J. Yan, N. Lu, J. Li, Y. Wen and G. Liu, ACS Appl. Mater. Interfaces, 2018, 10, 6895 CrossRef CAS.
- D. Lee, S. Baek, Y. Y. Kim, Y. Bang, H. N. Jung, H. J. Im and Y. K. Song, ACS Appl. Mater. Interfaces, 2022, 14, 37493 CrossRef CAS PubMed.
- H. V. P. Thelu, S. Atchimnaidu, D. Perumal, K. S. Harikrishnan, S. Vijayan and R. Varghese, ACS Appl. Bio Mater., 2019, 2, 5227 CrossRef CAS PubMed.
- E. Barea, C. Montoro and J. A. R. Navarro, Chem. Soc. Rev., 2014, 43, 5419 RSC.
- H. Sha and B. Yan, Talanta, 2023, 275, 124326 CrossRef.
- Y. Sun, L. Zheng, Y. Yang, X. Qian, T. Fu, X. Li, Z. Yang, H. Yan, C. Cui and W. Tan, Nanomicro Lett., 2020, 12, 103 CAS.
- X. Zou, X. S. Li, Q. Sun and E. q. Gao, CrystEngComm, 2024, 26, 356 Search PubMed.
- J. R. McMillan, O. G. Hayes, P. H. Winegar and C. A. Mirkin, Acc. Chem. Res., 2019, 52, 1939 CrossRef CAS PubMed.
- K. Minamihata, K. Tsukamoto, M. Adachi, R. Shimizu, M. Mishina, R. Kuroki and T. Nagamune, Chem. Commun., 2020, 56, 3891 RSC.
- D. Sen and W. Gilbert, Nature, 1990, 344, 410 CrossRef CAS.
- J. Kypr, I. Kejnovská, D. Renčiuk and M. Vorlíčková, Nucleic Acids Res., 2009, 37, 1713 CrossRef CAS PubMed.
- M. I. Jacobs, P. Bansal, D. Shukla and C. M. Schroeder, ACS Cent. Sci., 2022, 8, 1350 CrossRef CAS.
- The co-crystal structure of SA-R6Y and A9 has been deposited in the PDB under accession number 8ZR1.
- The co-crystal structure of SA-R6Y and C9 has been deposited in the PDB under accession number 8ZR2.
- Y. Li, S. Hu, C. Chen, N. Alifu, X. Zhang, J. Du, C. Li, L. Xu, L. Wang and B. Dong, Talanta, 2023, 258, 124435 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Materials and methods, morphological observation of co-crystal, crystal structure analysis data, and functional evaluation in ssDNA on the co-crystal. See DOI: https://doi.org/10.1039/d4ra08326a |
| This journal is © The Royal Society of Chemistry 2025 |