
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTheoretical and experimental studies on stability, bonding and isolation of elusive bis-(dichloro-aluminium) oxides supported by donor-base ligands†
Maria Francis ,
Kishor Shinde and
Sudipta Roy*
,
Kishor Shinde and
Sudipta Roy*
Department of Chemistry, Indian Institute of Science Education and Research (IISER) Tirupati, Tirupati 517619, Andhra Pradesh, India. E-mail: roy.sudipta@iisertirupati.ac.in
First published on 25th March 2025
Abstract
Herein, we depict the stability, and bonding studies of bis-(dichloro-aluminium) oxides Cl2Al–O–AlCl2 supported by a pair of homo-/hetero-leptic donor base ligands L, L′ [L, L′ = cyclic alkyl(amino) carbene (cAACMe; 1); N-heterocyclic carbene (NHCMe; 2); di-amido carbene (DACMe; 3); L = cAACMe, L′ = NHCMe; (4)] with a general formula (L)Al(Cl)2–O–Al(Cl)2(L′) (1–4) by NBO, QTAIM and EDA-NOCV analyses. Theoretical calculations suggest that 1–4 possess favorable interaction energies (ΔEint), and bond dissociation energies between L/L′, and Al(Cl)2–O–Al(Cl)2 fragments via the formation of two dative L→Al bonds [1 > 4 > 2 > 3]. This trend is rationalized by the σ-donor ability of the ligands L/L′. Moreover, we depict the first successful solid-state isolation of the colorless compound (cAAC)Al(Cl)2–O–Al(Cl)2(cAAC) (1′) by reacting cAAC and AlCl3 in the presence of a controlled amount of H2O, where two equiv. of cAAC is being utilized as the base. 1′ has been structurally characterized by single-crystal X-ray diffraction, and further studied by NMR spectroscopy.
Introduction
The elusive oxo-bridged di-chloro aluminium compounds have been rarely documented in the literature. The low coordinate oxo-bridged Al compounds, known as the aluminoxanes have attracted significant interest among chemists due to their utility as active catalysts in the polymerization processes of epoxides,1,2 aldehydes,3,4 and olefins.5 The discovery of methyl-aluminoxane (MAO) [(Me)AlO]n in 1980 has been marked as the pivotal development in this field introducing a highly active co-catalyst that effectively promotes the polymerization of ethylene, and propylene.6,7 Aluminoxanes with general formula [(R)AlO]n or [R2AlOAlR2]n, can be synthesized through the controlled hydrolysis of organo-aluminium compounds. This process can be initiated using water in the form of the moisture present in hydrated metal salts8 or by reacting those compounds with oxygen-containing entities.9 In recent years, substantial advancements in the structural characterization of aluminoxanes have been documented, despite the inherent complexity of their synthetic routes.8,9 In 2003, Roesky and co-workers unveiled the isolation of the pioneering bimetallic aluminoxane, featuring the terminal hydroxide groups (Fig. 1, A) via hydrolysis of an aluminium compound [LAl(I)2] (L = HC{MeCNDipp}2, Dipp = 2,6-iPr2C6H3) within a biphasic liquid ammonia/toluene system, leading to the isolation of a previously unknown molecule [{LAl(OH)}2(μ-O)].10 In 2005, the same group highlighted the reaction between an Al(I) monomer LAl (L = HC[(CMe)(NAr)]2, Ar = 2,6-iPr2C6H3), and molecular oxygen, leading to the stabilization of a neutral bis-aluminium oxide [LAlO]2 (Fig. 1, B).11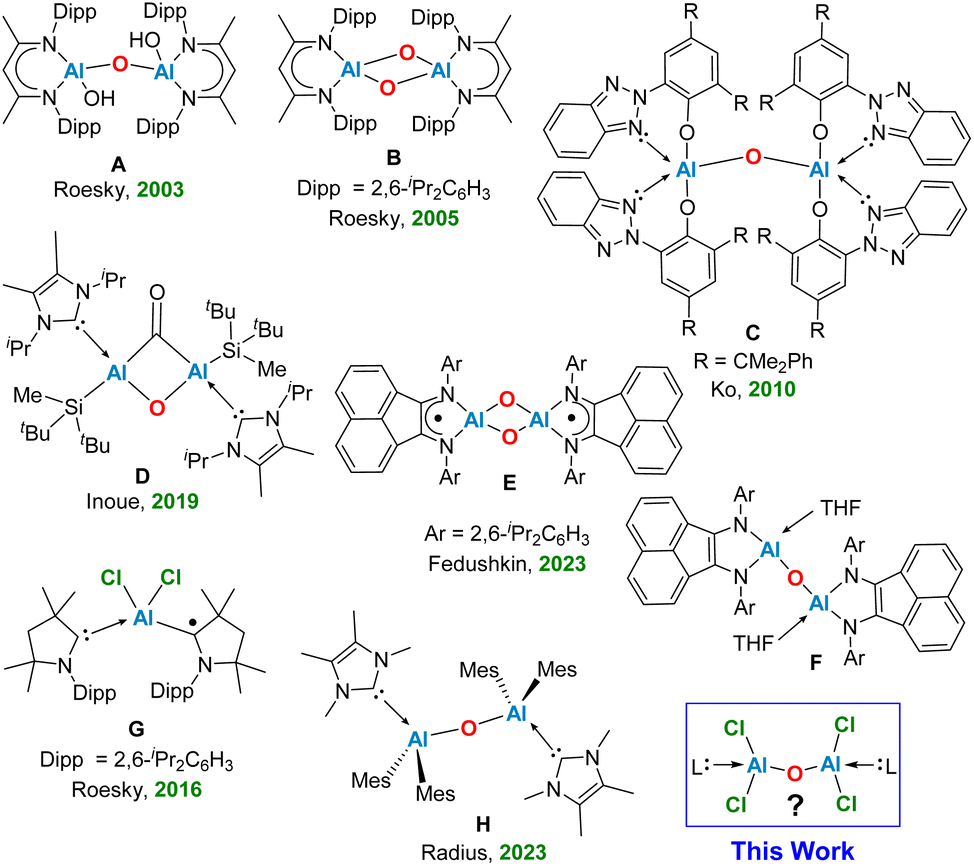 | ||
| Fig. 1 Representative oxo-/di-chloro oxo-aluminium compounds isolated in the laboratory. | ||
In 2010, Ko and colleagues discovered the aluminium-methyl derivative with bulky ligands [(CMe2PhBTP)2AlMe] (CMe2PhBTP = 2-(2H-benzotriazol-2-yl)-4,6-bis(1-methyl-1-phenylethyl)-phenolate), which readily reacted with water to produce the dimeric oxo-aluminium species C (Fig. 1).12 In a ground-breaking study, Inoue and team successfully synthesized the first known di-alumene species ([((NHC)(Si)Al)2] Si = SitBu2Me) stabilized by the donor base ligand NHC (N-heterocyclic carbene) with a formal Al![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Al double bond. The reaction of a toluene solution of this dialumene under an atmosphere of CO2 at −78 °C to 50 °C by a gradual heating, produced the novel oxo-di-aluminacarbonyl compound D, showcasing an unusual central Al(μ-CO)(μ-O)Al bridged motif (Fig. 1).13 Very recently, Fedushkin and colleagues have elucidated a fascinating reaction of a dialane [(dpp-bian)Al–Al(dpp-bian)] as a potential four-electron reducing agent with N2O in toluene at room temperature, leading to the formation of the corresponding oxides [(dpp-bian)Al(μ2-O)2Al(dpp-bian)] (E), and [(dpp-bian)Al(THF)–(μ-O)Al(THF)(dpp-bian)] (F) (dpp-bian = 1,2-bis[(2,6-diisopropylphenyl)imino]-acenaphthene) (Fig. 1).14 In 2016, Roesky and co-workers reported a neutral radical (Me2-cAAC)2AlCl2 (G, Fig. 1) stabilized by cyclic (alkyl)(amino)carbenes (cAACs), synthesized by reduction of the Me2-cAAC→AlCl3 adduct with KC8 in the presence of another equiv. of Me2-cAAC.15 The EDA-NOCV analysis performed on G suggested an interaction between the neutral AlCl2 radical and (cAAC)2 in a singlet electronic state, and (cAAC)2 in a triplet electronic state interacting with AlCl2, featuring the equal probability electron sharing and coordinate bond due to negligible energy difference between ΔEorb values of the two bonding possibilities. The mono-radical (Me2-cAAC)2AlCl2 (G) is a π-type radical with the electron densities on
Al double bond. The reaction of a toluene solution of this dialumene under an atmosphere of CO2 at −78 °C to 50 °C by a gradual heating, produced the novel oxo-di-aluminacarbonyl compound D, showcasing an unusual central Al(μ-CO)(μ-O)Al bridged motif (Fig. 1).13 Very recently, Fedushkin and colleagues have elucidated a fascinating reaction of a dialane [(dpp-bian)Al–Al(dpp-bian)] as a potential four-electron reducing agent with N2O in toluene at room temperature, leading to the formation of the corresponding oxides [(dpp-bian)Al(μ2-O)2Al(dpp-bian)] (E), and [(dpp-bian)Al(THF)–(μ-O)Al(THF)(dpp-bian)] (F) (dpp-bian = 1,2-bis[(2,6-diisopropylphenyl)imino]-acenaphthene) (Fig. 1).14 In 2016, Roesky and co-workers reported a neutral radical (Me2-cAAC)2AlCl2 (G, Fig. 1) stabilized by cyclic (alkyl)(amino)carbenes (cAACs), synthesized by reduction of the Me2-cAAC→AlCl3 adduct with KC8 in the presence of another equiv. of Me2-cAAC.15 The EDA-NOCV analysis performed on G suggested an interaction between the neutral AlCl2 radical and (cAAC)2 in a singlet electronic state, and (cAAC)2 in a triplet electronic state interacting with AlCl2, featuring the equal probability electron sharing and coordinate bond due to negligible energy difference between ΔEorb values of the two bonding possibilities. The mono-radical (Me2-cAAC)2AlCl2 (G) is a π-type radical with the electron densities on 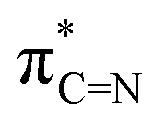 orbital of cAAC ligand, similar to the diradical (Me2-cAAC)2SiCl2.15b Radius and colleagues synthesized dialane compounds stabilized by NHC and examined their reactivity with chalcogenides and oxygen. Using pyridine N-oxide as an oxygen donor, they isolated the compound ({(IMeMe)·AlMes2}2–μ-O) (Mes = 2,4,6-trimethylphenyl) (H, Fig. 1), exhibiting a central linear Al–O–Al bridge positioned on a crystallographic inversion center.16 However, no reports have been documented on the computational and experimental studies on the stabilization of the elusive neutral bis-(dichloro-aluminium) oxides [Cl2Al–O–AlCl2]. In this report, we shed light on the stability and bonding of five neutral monomeric bis-(dichloro-aluminium) oxides, supported by a pair of donor-base ligands with general formula (L)Al(Cl)2OAl(Cl)2(L′) (L, L′ = cAACMe, 1; L, L′ = NHCMe, 2; L, L′ = diamidocarbene (DACMe, 3), L = cAACMe, L′ = NHCMe, 4), and the first successful laboratory isolation of the cAAC-stabilized bis-(dichloro-aluminium) oxide (1′) by the activation of water molecule in presence of cAAC and AlCl3.
orbital of cAAC ligand, similar to the diradical (Me2-cAAC)2SiCl2.15b Radius and colleagues synthesized dialane compounds stabilized by NHC and examined their reactivity with chalcogenides and oxygen. Using pyridine N-oxide as an oxygen donor, they isolated the compound ({(IMeMe)·AlMes2}2–μ-O) (Mes = 2,4,6-trimethylphenyl) (H, Fig. 1), exhibiting a central linear Al–O–Al bridge positioned on a crystallographic inversion center.16 However, no reports have been documented on the computational and experimental studies on the stabilization of the elusive neutral bis-(dichloro-aluminium) oxides [Cl2Al–O–AlCl2]. In this report, we shed light on the stability and bonding of five neutral monomeric bis-(dichloro-aluminium) oxides, supported by a pair of donor-base ligands with general formula (L)Al(Cl)2OAl(Cl)2(L′) (L, L′ = cAACMe, 1; L, L′ = NHCMe, 2; L, L′ = diamidocarbene (DACMe, 3), L = cAACMe, L′ = NHCMe, 4), and the first successful laboratory isolation of the cAAC-stabilized bis-(dichloro-aluminium) oxide (1′) by the activation of water molecule in presence of cAAC and AlCl3.
Computational methods
The geometry optimizations, and frequency calculations for the hypothesized compounds (1–4) have been performed at BP86-D3(BJ)/def2-TZVPP17a–c level in both singlet and triplet electronic states, employing Grimme D3 with Becke–Johnson damping dispersion correction.17d,e The potential energy surface (PES) was found to have a minimum as there was no imaginary frequency present. The Gaussian 09 program package was utilized to perform all the density functional theory (DFT) calculations.18 The natural bond orbital (NBO)19 analysis has been performed using NBO 6.0 program to evaluate the partial charges, Wiberg bond indices (WBI),20 and the natural bond orbitals. All the predicted compounds (1–4) have been found to possess singlet ground states. To elucidate the bonding characteristics within the (L)Al(Cl)2–O–Al(Cl)2(L′) species, Energy Decomposition Analysis (EDA)21 integrated with the Natural Orbital for Chemical Valence (NOCV)22 approach was employed using the ADF 2020.105 software suite. The EDA-NOCV analysis, predicated on geometries optimized at the BP86-D3(BJ)/def2-TZVPP level and conducted at the BP86-D3(BJ)/TZ2P level, facilitates the dissection of the intrinsic interaction energy (ΔEint) between two molecular fragments into four constituent energy components (see ESI†).Results and discussion
We initiated our studies with optimizations of the predicted molecules in both singlet and the triplet states. The optimized geometries of the hypothetical neutral monomeric bis-(dichloroaluminium) oxides, (L)Al(Cl)2–O–Al(Cl)2(L′), where, L, L′ = cAACMe (1); L, L′ = cAACDipp (1′); L, L′ = NHCMe (2); L, L′ = NHCDipp (2′); L, L′ = DACMe (3) and L = cAACMe, L′ = NHCMe (4) in their singlet states are represented in Fig. 2.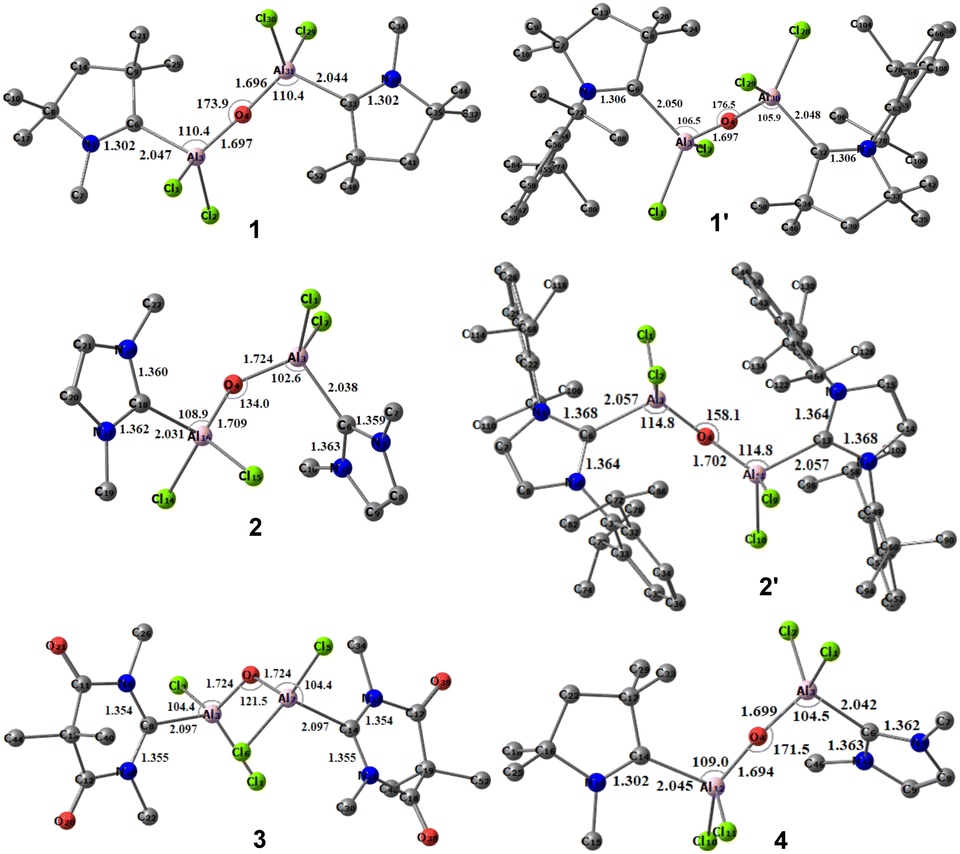 | ||
| Fig. 2 Optimized geometries (at BP86/def2-TZVPP level of theory) of the species (L)Al(Cl)2–O–Al(Cl)2(L) (1–4) in the singlet ground states. L, L′ = cAACMe (1); L, L′ = NHCMe (2), L, L′ = DACMe (3) and L = cAACMe; L′ = NHCMe (4). | ||
The computed CL–Al distances for these compounds are found to be 2.047, 2.031, 2.097 and 2.045 Å (Table 1), respectively, which are comparable to the previously reported carbene donor C→Al bond lengths in (iPr2Im)·AlH3 (2.0405(17) Å),23 (cAAC)2Al(Cl)2 (2.097(2) Å), cAAC:→AlCl3 (2.037(1) Å),15 and (NHC)2Al2H4 (2.0860(13) Å).24 However, they are found to be slightly longer than the covalent Al–C single bond lengths observed in LHAlMe2Cl (1.950(3) and 1.980(2); L = HC[C(Me)N(Ar)]2, Ar = 2,6-iPr2C6H3),25 and Al2[CH(SiMe3)2]4 (1.982(3)–1.985(3) Å).26 In the species 1–4, the Al–O distances remain relatively consistent, hovering around 1.7 Å. In compounds 1 (L, L′ = cAACMe) and 4 (L = cAACMe; L′ = NHCMe), the central Al(Cl)2–O–Al(Cl)2 moiety demonstrates a nearly linear configuration, with an Al–O–Al bond angle of 173.9° and 171.5°. In contrast, compounds 2 and 3 exhibit reduced bond angles of 134.0° and 121.5°, respectively. But when the Me groups of 2 were replaced by the Dipp (2,6-diisopropylphenyl) groups (2′), the Al–O–Al bond angle was increased to 158.1°, which can be attributed to the higher steric effect (see ESI†).
| Compound | Bond | Bond length | Compound | Bond | Bond length |
|---|---|---|---|---|---|
| 1 | CcAAC–Al | 2.047 | 2′ | CNHC–Al | 2.057 |
| Al–O | 1.697 | Al–O | 1.702 | ||
| 1′ | CcAAC–Al | 2.050 | 3 | CDAC–Al | 2.097 |
| Al–O | 1.697 | Al–O | 1.724 | ||
| 2 | CNHC–Al | 2.031, 2.038 | 4 | CcAAC–CNHC | 2.045 |
| 2.042 | |||||
| Al–O | 1.709, 1.724 | Al–O | 1.694 | ||
| 1.699 |
To have a detailed understanding of the distribution of electron densities in the hypothetical species 1–4, we have performed the Natural Bond Orbital (NBO) analyses (Table S9, ESI†). The respective Kohn–Sham orbitals show that the LUMOs for all three species are the π* orbitals of the CN bond of the carbenes, which play a crucial role in their reactivity and interaction with other molecules.15b The lower lying 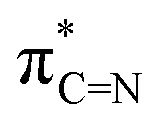 orbital of cAAC, in general, is beneficial for the intra-molecular charge transfer, small molecule activation, enhancement of photoluminescence properties, hosting the extra electron densities from bonded element, and formation of the cAAC-centred radical anion intermediate for reduction of cAAC containing compounds leading to the formation of low-valence cAAC-containing compounds.15b,c For compounds 1, 2, and 4 the HOMO represents the lone pairs of electrons on the central bridging oxygen atom. We observed that the fully occupied frontier orbitals of these molecules do not only represent the π orbitals of the phenyl rings of the Dipp groups, rather, those exhibit through-space interaction of these π orbitals with the non-bonding electrons of the Cl and the O atoms (see ESI† for the respective Hirshfeld plots). Such secondary interactions indirectly play important role in overall stabilization of the respective molecules. For example, the HOMO−1 of 4 represents through space interaction of the lone pair of electrons on bridging O-atom with the π electron cloud of the aromatic ring of the Dipp group. The HOMO of 1′ shows through-space interaction between the lone pair of electrons on Cl atoms with the central O atom and the π electron cloud of the aromatic rings. Similarly, the HOMO of 2′ shows the through-space interaction between the lone pair of electrons on Cl-atoms and the π electron cloud of the aromatic ring.
orbital of cAAC, in general, is beneficial for the intra-molecular charge transfer, small molecule activation, enhancement of photoluminescence properties, hosting the extra electron densities from bonded element, and formation of the cAAC-centred radical anion intermediate for reduction of cAAC containing compounds leading to the formation of low-valence cAAC-containing compounds.15b,c For compounds 1, 2, and 4 the HOMO represents the lone pairs of electrons on the central bridging oxygen atom. We observed that the fully occupied frontier orbitals of these molecules do not only represent the π orbitals of the phenyl rings of the Dipp groups, rather, those exhibit through-space interaction of these π orbitals with the non-bonding electrons of the Cl and the O atoms (see ESI† for the respective Hirshfeld plots). Such secondary interactions indirectly play important role in overall stabilization of the respective molecules. For example, the HOMO−1 of 4 represents through space interaction of the lone pair of electrons on bridging O-atom with the π electron cloud of the aromatic ring of the Dipp group. The HOMO of 1′ shows through-space interaction between the lone pair of electrons on Cl atoms with the central O atom and the π electron cloud of the aromatic rings. Similarly, the HOMO of 2′ shows the through-space interaction between the lone pair of electrons on Cl-atoms and the π electron cloud of the aromatic ring.
Compound 3 demonstrates σ-donation from the DAC moiety to Al. Moreover, for compounds 1, 2, and 4, the σ-donation from cAAC/NHC is represented in the HOMO−1, and HOMO−8 orbitals, respectively; suggesting variations in the electron donation capabilities among the compounds 1–4 (see ESI†). We could observe comparable results when the methyl groups in 1, and 2 are replaced by sterically bulkier Dipp groups (1′–2′). The NBO analyses performed on bulky analogues 1′, and 2′ at BP86/def2-TZVPP level of theory showed the presence of a single occupancy bond between CcAAC/NHC–Al, which is majorly polarised towards CL atom (86–88.3%). The WBI for C–Al bond is approximately 0.4, which implies its single bond nature. The HOMO−3, and HOMO−10 of 1′, and 2′, respectively confirm σ-donation from carbene to Al. The bond between Al–O is polarized exclusively towards the O atom due to its higher electronegativity. The LUMO of 1′ and 2′ corresponds to the π* of CN. HOMO of 1′ and 2′ lies on the π orbitals of the phenyl ring. HOMO of 1′ also represents the lone pairs present on the oxygen atom (Fig. 3).
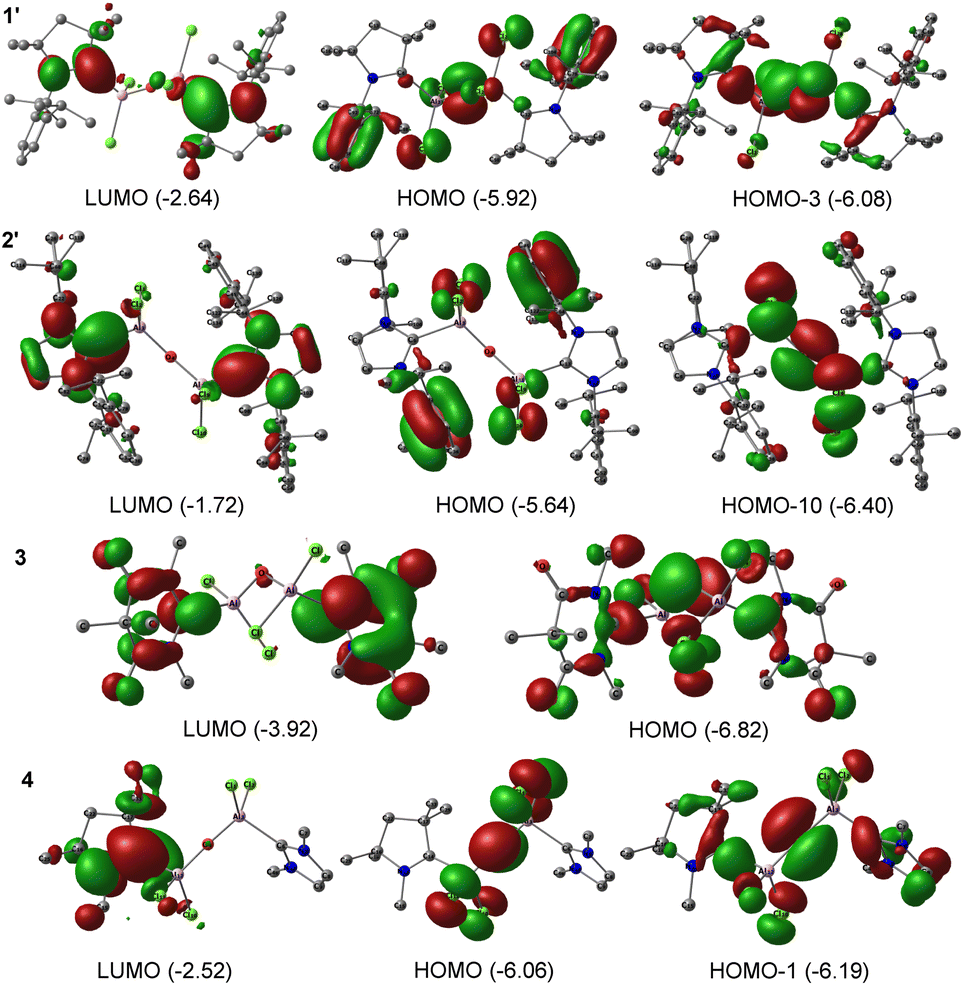 | ||
| Fig. 3 Selected Kohn–Sham orbitals of compounds 1′–2′, and 3 and 4 at BP86/def2-TZVPP level of theory. The values in the parentheses are the energies of the orbitals in eV. | ||
The single bond occupancy was observed along the Ccarbene–Al bond, which is significantly polarized towards the carbene moiety. This polarization is indicative of the unique bonding interactions within 1–4. The Wiberg Bond Indices (WBI) further elaborate on the nature of the C–N bonds within the compounds; for instance, compound 1 possesses a WBI of 1.60, implying a double bond characteristic. Contrastingly, in compound 2, the WBI drops to 1.29, suggesting a diminishing double bond nature, as visualized in the HOMO−4 orbital. The NBO results do not distinguish between a covalent dative bond or a covalent electron-sharing bond, and therefore NBO cannot accurately determine the true nature of the bond. In this regard, the energy decomposition analysis natural orbitals of chemical valence (EDA-NOCV)21,22 approach is the most suitable tool for gaining insight into the nature of the chemical bonds of (L)Al(Cl)2OAl(Cl)2(L′) [L, L′ = cAACMe (1), L, L′ = NHCMe (2), L, L′ = DACMe (3), L = cAACMe, L′ = NHCMe (4)]. The magnitudes of |ΔEorb| from different bonding scenarios were compared, and the bonding scenario with the lowest value of ΔEorb is considered to be the best (i.e., the most feasible) bonding scenario, since it will require the least change in the electronic structure of the fragments to maintain the electronic structure of the final molecule at its equilibrium geometry.15a In the present study, the best bonding description of 1–4 is illustrated considering different bonding possibilities (Scheme 1) by varying the charge, and electronic states of the interacting fragments [(L)(L′)], and Al(Cl)2OAl(Cl)2, specifically: (a) neutral [(L)(L′)] and Al(Cl)2OAl(Cl)2 fragments in their electronic singlet state forming two dative bonds, (b) doubly charged [(L)(L′)]2+ and [Al(Cl)2OAl(Cl)2]2− fragments in their electronic triplet state leading to the formation of two σ electron sharing bonds, and (c) singly charged [(L)(L′)]+ and [Al(Cl)2OAl(Cl)2]− fragments in electronic doublet state, which would interact to form an electron sharing and a dative bond. The EDA-NOCV results depicted from Table 2 show that for carbene-containing species 1–4, the fragmentation scheme involving neutral [(L)(L′)] and [Al(Cl)2OAl(Cl)2] fragments in the electronic singlet state (Scheme 1(a)) forming dative bonds gives the smallest ΔEorb and is therefore the best bonding scenario.
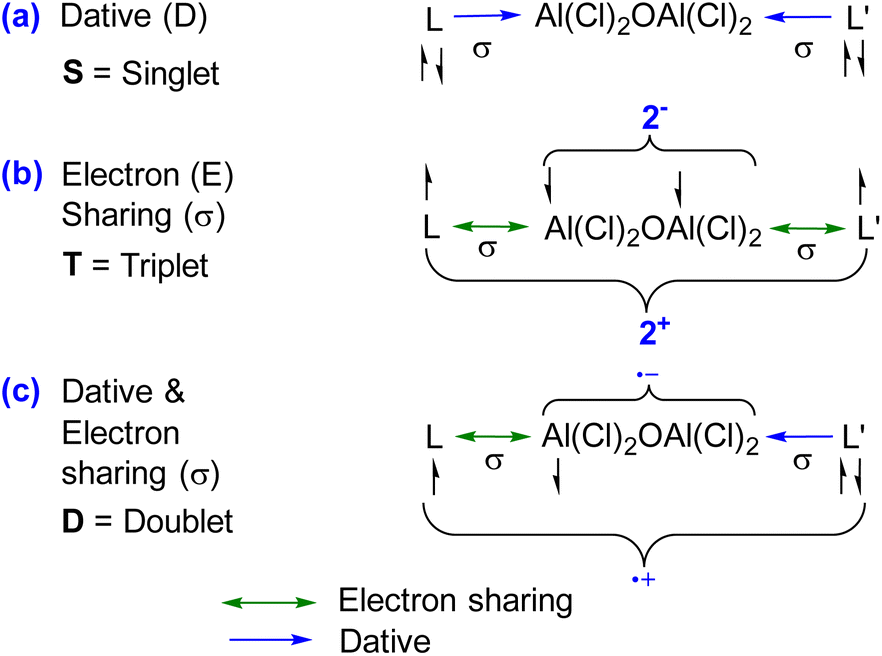 | ||
| Scheme 1 Possible bonding scenarios between interacting fragments [(L)(L′)] and Al(Cl)2OAl(Cl)2 of 1–4. | ||
| Compound | Energy | L–Al(Cl)2OAl(Cl)2–L | LAl(Cl)2–O–Al(Cl)2L |
|---|---|---|---|
| 1 | ΔEint | −157.9 | −770.45 |
| ΔEPauli | 231.9 | 405.59 | |
| ΔEele | −233.6 (59.9%) | −899.60 (76.5%) | |
| ΔEdis | −28.6 (7.3%) | −4.33 (0.4%) | |
| ΔEorb | −127.7 (32.8%) | −272.10 (23.1%) | |
| 2 | ΔEint | −154.0 | −783.38 |
| ΔEPauli | 225.6 | 405.22 | |
| ΔEele | −227.7 (60%) | −905.05 (76.1%) | |
| ΔEdis | −25.6 (6.7%) | −3.91 (0.3%) | |
| ΔEorb | −126.4 (33.3%) | −280.64 (23.6%) | |
| 3 | ΔEint | −136.6 | −795.98 |
| ΔEPauli | 221.3 | 441.01 | |
| ΔEele | −204.7 (57.2%) | −916.54 (74.1%) | |
| ΔEdis | −30.4 (8.5%) | −4.15 (0.3%) | |
| ΔEorb | −122.8 (34.3%) | −316.31 (25.6%) | |
| 4 | ΔEint | −154.1 | −774.1 |
| ΔEPauli | 225.3 | 408.0 | |
| ΔEele | −228.1 (60.1%) | −907.2 (76.7%) | |
| ΔEdis | −25.1 (6.6%) | −4.16 (0.4%) | |
| ΔEorb | −126.2 (33.3%) | −270.7 (22.9%) |
Table 2 encapsulates the results, revealing that in these compounds, the bonding between the aluminium (Al) and the carbene ligand (L/L′) predominantly stems from electrostatic interactions, constituting approximately 57.2–60% of the bond character. The covalent interactions contribute significantly as well, accounting for 32.8–34.3% of the bonding nature, while dispersion forces contribute marginally between 8.5% and 6.6%, which is significant.
A deeper analysis involves dissecting the ΔEorb, the energy associated with the orbital interactions, into pairwise contributions to understand the specifics of bonding. The study identifies two significant orbital contributions, ΔEorb(1) (with same phase; ++) and ΔEorb(2) (with opposite phase; +−) which mainly correspond to the σ donation from the ligand (L) to the aluminium centre (Al) (Fig. 4).
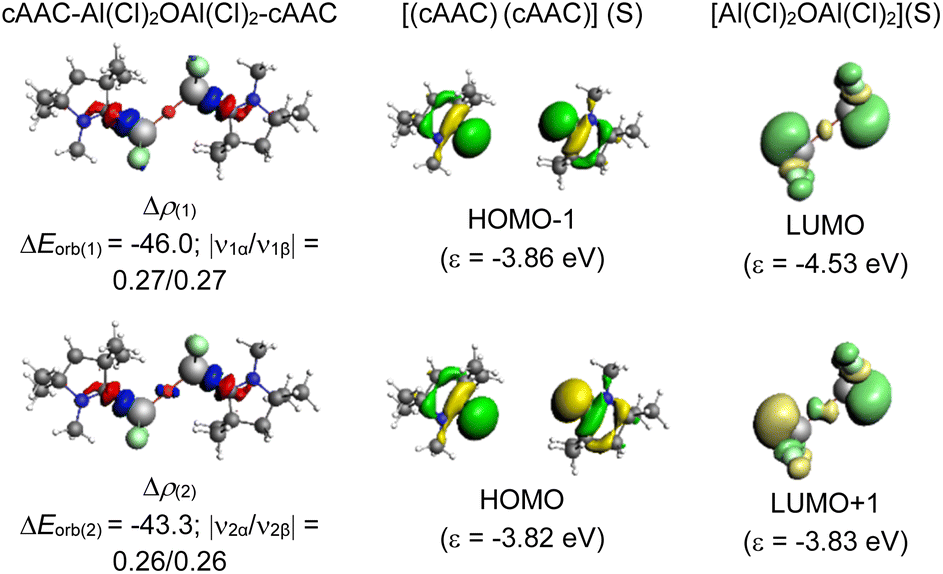 | ||
| Fig. 4 The shape of the deformation densities Δρ(1)–(2) that correspond to ΔEorb(1)–(2), and the associated MOs of cAAC–Al(Cl)2OAl(Cl)2–cAAC (1) and the fragments orbitals of [(cAAC)(cAAC)] and [Al(Cl)2OAl(Cl)2] in the singlet state (S) at the BP86-D3(BJ)/TZ2P level. Isosurface values are 0.003 au for Δρ(1)–(2). The eigenvalues |νn| give the size of the charge migration in e. | ||
The dative in-phase (++) σ donation from HOMO−1 of the ligands [(L)(L)] into LUMO of the [Al(Cl)2OAl(Cl)2] forms the ΔEorb(2) in 2, and 3. The ΔEorb(2) of 4 features the σ donation from HOMO−1 of the ligands [(cAAC)(NHC)] into LUMO+1 of the [Al(Cl)2OAl(Cl)2] in out-of-phase (+−) combination.
Roesky and co-workers studied the bonding of the neutral radical (Me2-cAAC)2AlCl2 (ref. 15) using EDA-NOCV studies, and predicted two bonding scenarios. The first scenario proposed an interaction between an AlCl2 radical and (cAAC)2 in a singlet electronic state, whereas the second scenario treated (cAAC)2 in a triplet electronic state interacting with AlCl2, leading to the formation of a shared electron bond and a coordination bond. The findings indicated that the energy difference for both scenarios (ΔEorb) was nearly identical, suggesting both are similarly probable. Further analysis of the fragments' orbitals revealed the primary contribution to ΔEorb in the original study was the transfer of the unpaired electron from aluminium to the cAAC ligands' LUMO, amounting to −123.9 kcal mol−1. However, in our analysis of compound 1, the primary contribution to ΔEorb, totalling −89.3 kcal mol−1 or 70% of ΔEorb, stemmed from the electron donation from the cAAC2 ligand fragments to the Al(Cl)2OAl(Cl)2 center. Whereas, the stabilization arising from the coordinate bond (CcAAC→Al) in (Me2-cAAC)2AlCl2 is only −59.6 kcal mol−1, i.e., 27.7% of the ΔEorb. The bond dissociation energy for the (cAAC)2 and Al(Cl)2OAl(Cl)2 is 118.9 kcal mol−1 is significantly higher compared to (Me2-cAAC)2AlCl2 (−71.0 kcal mol−1). The computed interaction energies (ΔEint) and bond dissociation energies (BDEs) establish a distinct trend of the ligand strength in the order of 1 > 4 > 2 > 3, which directly correlates with the π-acceptor and σ-donor properties of the respective carbenes. The BDE values follow a similar pattern with 1 showing 118.9 kcal mol−1, while 2 and 3 exhibit 113.9 kcal mol−1 and 89.6 kcal mol−1, respectively. The heteroleptic system, 4 falls in between at 113.1 kcal mol−1 for the L–(Al(Cl)2OAl(Cl)2)–L′ bonds. The stronger bonding interaction in 1 and 4 is attributed to the enhanced π-acceptor ability of cAAC, which enables greater orbital overlap with the electron-deficient aluminium centre, leading to better charge delocalization and bond stabilization. On the other hand, the DAC-stabilized compound 3 exhibits the weakest interaction, as reflected in both its lower BDE (89.6 kcal mol−1) and interaction energy (−136.6 kcal mol−1). This can be attributed to its diminished donor–acceptor interactions and reduced charge transfer efficiency, making it inherently less stable compared to cAAC and NHC. The WBI analysis also supports these findings, with the C–N bond in 1 exhibiting a WBI of 1.60, indicative of substantial double-bond character, while 2 shows a lower value of 1.29, signifying weaker π-conjugation.
To study the nature of the central Al–O bonds, we performed EDA-NOCV calculations, cleaving Al–O bonds in 1–4. Three bonding possibilities were generated by changing the charge and the multiplicity of the fragments. The first possibility considered the interaction of neutral triplet fragments leading to the formation of electron-sharing bonds (Scheme 2(a)). The second bonding possibility considered the interaction of a combination of dative and electron-sharing bonds from the interaction of singly charged doublet fragments (Scheme 2(b)). The third and last considers the interaction of doubly charged singlet fragments [(O2−) and ((L)Al(Cl2))(Al(Cl)2(L′))2+] forming dative bonds (O→Al). It was found the ΔEorb was found the least for the third possibility and hence considered the best bonding scenario (Scheme 2(c)).
 | ||
| Scheme 2 Possible bonding scenarios of the Al–O bonds of 1–4. | ||
The bonding between the Al and O atoms predominantly stems from electrostatic interactions, constituting approximately 74.1–76.7% of the bond character (Fig. 5, see ESI†). The covalent interactions contribute significantly, accounting for 22.9–25.6% of the bonding nature, while dispersion forces contribute marginally between 0.3 and 0.4%, which is significantly lower than that of CL–Al bonds. A deeper bonding analysis involves dissecting the ΔEorb, the energy associated with the orbital interactions, into pairwise contributions to understand the specifics of covalent bonding. The study identifies four significant orbital contributions, ΔEorb(1)–ΔEorb(4), which mainly correspond to the σ and π donation from the O-atom to the aluminium centre (Al). ΔEorb(1) and ΔEorb(2) correspond to the σ donation from O2− to Al centre in which the major contribution is from ΔEorb(1) (42.6–50.3%).
 | ||
| Fig. 5 The shape of the deformation densities Δρ(1)–(4) that correspond to ΔEorb(1)–(4), and the associated MOs of (cAAC)Al(Cl)2–O–Al(Cl)2(cAAC) (1) and the fragments orbitals of [(cAACAl(Cl)2)2]2+ and [O]2− in the singlet state (S) at the BP86-D3(BJ)/TZ2P level. Isosurface values are 0.001 au for Δρ(1)–(4). The eigenvalues |νn| give the size of the charge migration in e. The direction of the charge flow of the deformation densities is red → blue. | ||
In 1 and 3 it is the dative out-phase (+−) σ donation from HOMO−1 of O into LUMO of the ((L)Al(Cl2))(Al(Cl)2(L′))2+ whereas in 2 and 4 it is the dative in-phase (++) σ donation from HOMO−1 of O into LUMO of the [((L)Al(Cl2))(Al(Cl)2(L′))]2+. ΔEorb(3) and ΔEorb(4), which contribute minorly to the corresponds to the ΔEorb, are π donation from O to Al fragments. The dative in-phase (++) π donation from O to Al forms the ΔEorb(3)–(4) in 1 and 4, whereas, dative out-phase (+−) π donation from O to Al form the ΔEorb(3)–(4) in 2 and 3 (Scheme 3).
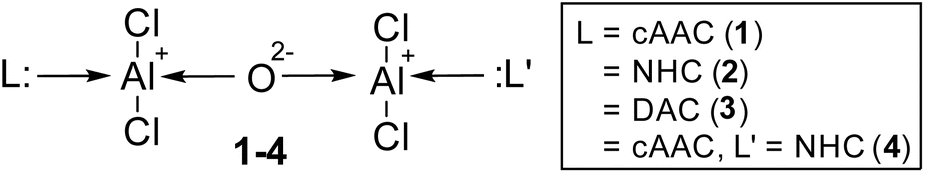 | ||
| Scheme 3 Predicted best bonding scenario between O and Al centres of 1–4. | ||
Synthesis
To a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar mixture of Me2-cAAC:→LiOTf (Me2-cAAC::C(N-2,6-iPr2C6H3)(CMe2)2(CH2)) and AlCl3, toluene (10 mL) was added at −5 °C (ice/water mixture) to which 1 equiv. of water was added using a micro-syringe and the reaction mixture was slowly raised to room temperature (rt), The resulting reaction solution was stirred at rt for 12 h. Afterwards, the white precipitate was filtered, and the pale-yellow filtrate was concentrated under reduced pressure, and stored for crystallization at ambient temperature to obtain the colorless cubes of compound 1′ after 1–2 days in 42% yield (Scheme 4).
:1 molar mixture of Me2-cAAC:→LiOTf (Me2-cAAC::C(N-2,6-iPr2C6H3)(CMe2)2(CH2)) and AlCl3, toluene (10 mL) was added at −5 °C (ice/water mixture) to which 1 equiv. of water was added using a micro-syringe and the reaction mixture was slowly raised to room temperature (rt), The resulting reaction solution was stirred at rt for 12 h. Afterwards, the white precipitate was filtered, and the pale-yellow filtrate was concentrated under reduced pressure, and stored for crystallization at ambient temperature to obtain the colorless cubes of compound 1′ after 1–2 days in 42% yield (Scheme 4).
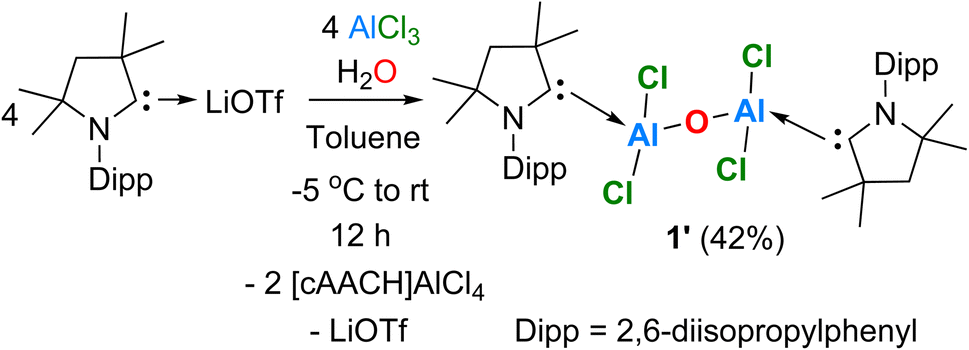 | ||
| Scheme 4 Synthesis of [(cAACDipp)Al(Cl2)–O–Al(Cl2)(cAACDipp)] (1′). | ||
Compound 1′ was structurally characterized by single-crystal X-ray diffraction, and further studied by NMR spectroscopy, and elemental analyses. The white residue obtained was also characterized by the NMR spectroscopy, which revealed the formation of the salt [cAACH]AlCl4 (see ESI†). The powder of 1′ was found to be thermally stable up to 200 °C under an inert atmosphere, and then decomposed to a black liquid in the temperature range of 201–204 °C.
The crystals of 1′ were found to be soluble in organic solvents, such as, toluene, THF, etc. The toluene solution of 1′ was found to be stable for over one month under an argon atmosphere. The 13C NMR spectrum of a deuterated benzene solution of 1′ exhibited the Ccarbene peak at 206.6 ppm at 298 K, which is upfield shifted compared to that of the free carbene,27 and downfield shifted when compared to that of ({(IMeMe)·AlMes2}2–μ-O)(172.4 ppm).16 The molecular structure of 1′ is shown in Fig. 6.
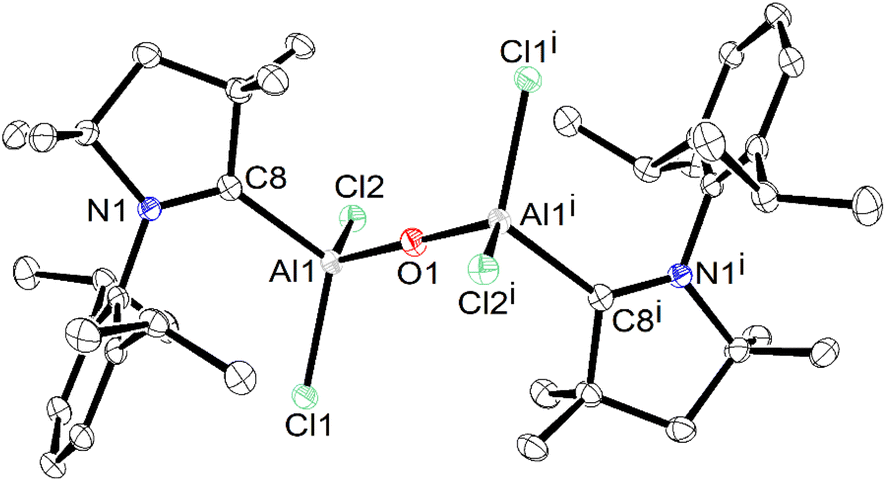 | ||
| Fig. 6 Molecular structure of isolated (cAACDipp)Al(Cl2)–O–Al(Cl2)(cAACDipp) (1′). The anisotropic displacement parameters are depicted at the 50% probability level. Hydrogen atoms, and solvent (toluene) are omitted for clarity. Selected experimental [calculated at BP86/def2-TZVPP] bond lengths [Å] and bond angles [°]: C8–Al1 2.063(2) [2.050], Al1–Cl2 2.1659(7) [2.168], Al1–Cl1 2.1494(8) [2.175], Al1–O1 1.6780(6) [1.697], C8–N1 1.297(2) [1.306]; N1–C8–Al1 129.73(14) [128.5], Al1–O1–Al1i 180.00 [176.5]. | ||
Compound 1′ crystallizes in the P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) triclinic space group, and features a linear (Cl)2AlOAl(Cl)2 units with an inversion centre passing through O1. The aluminium atoms in 1′ display a distorted tetrahedral geometry and form bonds with two chlorine atoms, a cAAC ligand, and a μ-O unit. The Al–Cl bond lengths in 1′ measure 2.1659(7) and 2.1494(8) Å, which are comparable to those observed in cAAC2AlCl2 (2.1612(7)–2.1638(7) Å), and cAAC:→AlCl3 adduct (2.1315(5)–2.1439(5) Å).15 The Al–CcAAC bond length in 1′ is found to be 2.063(2) Å, which lies in-between to those observed in Me2-cAAC:→AlCl3 adduct (2.037(1) Å), featuring the σ-donating CcAAC:→Al bond, and in (Me2-cAAC)2AlCl2 (2.097(2) Å), featuring one shorter CcAAC:–Al electron-sharing covalent bond (1.967(2) Å) and a relatively longer CcAAC:→Al σ-donating bond (2.097(2) Å).15 The Al–CcAAC bond in 1′ seems to be longer as expected than those present in compounds stabilized by bulky non-carbene ligands, e.g., LHAlMe2Cl (1.967(2) Å) (L = HC[C(Me)N(Ar)]2, Ar = 2,6-iPr2C6H3) and Al2[CH(SiMe3)2]4 (1.982(3)–1.985(3) Å), where the Al–CcAAC bonds are covalent single bonds.25,26 The Al–O bond length observed in 1′ is 1.6780(6) Å, which is significantly shorter than that (1.7252(4) Å) present in ({(IMeMe)·AlMes2}2–μ-O) (H), possibility due to the bulky Mes group on Al.16
triclinic space group, and features a linear (Cl)2AlOAl(Cl)2 units with an inversion centre passing through O1. The aluminium atoms in 1′ display a distorted tetrahedral geometry and form bonds with two chlorine atoms, a cAAC ligand, and a μ-O unit. The Al–Cl bond lengths in 1′ measure 2.1659(7) and 2.1494(8) Å, which are comparable to those observed in cAAC2AlCl2 (2.1612(7)–2.1638(7) Å), and cAAC:→AlCl3 adduct (2.1315(5)–2.1439(5) Å).15 The Al–CcAAC bond length in 1′ is found to be 2.063(2) Å, which lies in-between to those observed in Me2-cAAC:→AlCl3 adduct (2.037(1) Å), featuring the σ-donating CcAAC:→Al bond, and in (Me2-cAAC)2AlCl2 (2.097(2) Å), featuring one shorter CcAAC:–Al electron-sharing covalent bond (1.967(2) Å) and a relatively longer CcAAC:→Al σ-donating bond (2.097(2) Å).15 The Al–CcAAC bond in 1′ seems to be longer as expected than those present in compounds stabilized by bulky non-carbene ligands, e.g., LHAlMe2Cl (1.967(2) Å) (L = HC[C(Me)N(Ar)]2, Ar = 2,6-iPr2C6H3) and Al2[CH(SiMe3)2]4 (1.982(3)–1.985(3) Å), where the Al–CcAAC bonds are covalent single bonds.25,26 The Al–O bond length observed in 1′ is 1.6780(6) Å, which is significantly shorter than that (1.7252(4) Å) present in ({(IMeMe)·AlMes2}2–μ-O) (H), possibility due to the bulky Mes group on Al.16
The QTAIM28 analyses on 1′ at the BP86/def2-TZVPP level of theory depicted the electron densities (ρ(r)) of 0.068–0.109 and positive Laplacian (∇2ρ(r)) at the bond critical point (BCP) of the C–Al and Al–O bonds in 1′ suggested the closed-shell interactions as expected (Fig. 7).
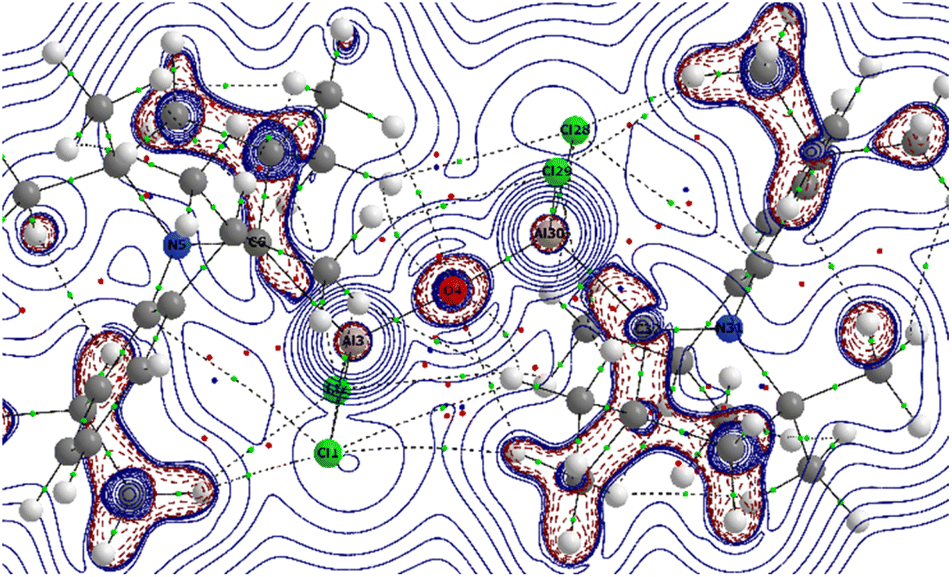 | ||
| Fig. 7 Contour plot of Laplacian distribution [∇2ρ(r)] in the Al3–O4–Al30 plane of (cAACDipp)Al(Cl2)–O–Al(Cl2)(cAACDipp) (1′). Solid blue lines indicate the areas of charge concentration (∇2ρ(r) < 0) while dotted purple lines denotes charge depletion (∇2ρ(r) > 0). Solid lines connecting atomic nuclei (black) are the bond paths and green spheres on bond path indicates the BCP. | ||
Conclusions
In summary, we have hypothesized the possible stabilization of the elusive neutral bis-(dichloro-aluminium) oxides in presence of donor-base ligands [(L)Al(Cl)2OAl(Cl)2(L)] [(L, L′ = donor base ligands; e.g., cAAC, NHC, and DAC); 1–4], and predicted their electron densities distribution and bonding scenarios by quantum chemical calculations. The NBO analysis revealed that the LUMOs comprise of the π* orbitals of the CN bond, a finding that portends significant repercussions for their reactivity and potential interaction abilities. The HOMOs in compounds 1, 2, and 4 are the lone pairs on the oxygen atoms. σ-Donation from Ccarbene to the aluminium centre in 1, and 4 is represented by the HOMO−1, and in 2 by HOMO−8 orbital. Moreover, the analysis revealed a noteworthy polarization of the Ccarbene–Al bond towards the carbene, thereby introducing an additional layer of complexity to the bonding interactions. The EDA-NOCV analysis revealed that the bond between the ligands (L, L′), and the Al(Cl)2OAl(Cl)2 core is mediated through a purely dative mechanism involving neutral singlet fragments. Intriguingly, the bond between the [((L)Al(Cl2))(Al(Cl)2(L′))] units and oxygen was identified as a dative bond, albeit with singlet doubly charged fragments, thus delineating a complex interaction framework within these molecular systems. The cAACDipp analogue (cAACDipp)Al(Cl2)–O–Al(Cl2)(cAACDipp) (1′) has been successfully synthesized, isolated, and structurally characterized by single-crystal X-ray diffraction, and studied further by NMR spectroscopy.
Data availability
Deposition numbers 2374368 (for 1′), 2376845 (for [cAACH]AlCl4) contain the supplementary crystallographic data for this paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
SR gratefully acknowledges ANRF-SERB, New Delhi for the POWER grant (SPG/2021/003237), and IISER Tirupati for the central HPC facility. MF and KS thank CSIR and IISER Tirupati for SRF, and JRF, respectively. We thank BBS for the initial results.References
- R. O. Colclough, G. Gee, W. C. E. Higginson, J. B. Jackson and M. Litt, The polymerization of epoxides by metal halide catalysts, J. Polym. Sci., 1959, 34, 171–179 CrossRef CAS.
- E. J. Vandenberg, Organometallic catalysts for polymerizing monosubstituted epoxides, J. Polym. Sci., 1960, 47, 486–489 CrossRef CAS.
- S. Ishida, Stereospecific polymerization of acetaldehyde. Part I, J. Polym. Sci., 1962, 62, 1–14 CrossRef.
- T. Saegusa, Y. Fujii, H. Fujii and J. Furukawa, Polymerization of acetaldehyde by triethylaluminum/water system, Makromol. Chem., 1962, 55, 232–235 CrossRef.
- C. Longiave and R. Castelli, Quelques types particuliers de catalyseurs au cobalt dans la polymérisation du butadiéne, J. Polym. Sci., Polym. Symp., 1963, 4, 387–398 CrossRef.
- H. Sinn, W. Kaminsky, H. Vollmer and R. Woldt, “Living polymers” on polymerization with extremely productive Ziegler catalysts, Angew Chem. Int. Ed. Engl., 1980, 19, 390–392 CrossRef.
- H. Sinn and W. Kaminsky, Advances in Organometallic Chemistry, ed. F. G. A. Stone and R. West, Academic Press, 1980, vol. 18, pp. 99–149 Search PubMed.
- M. R. Mason, J. M. Smith, S. G. Bott and A. R. Barron, Hydrolysis of tri-tert-butylaluminum: the first structural characterization of alkylalumoxanes [(R2Al)2O]n and (RAlO)n, J. Am. Chem. Soc., 1993, 115, 4971–4984 CrossRef CAS.
- R. J. Wehmschulte and P. P. Power, A new synthetic route to organoalumoxanes (RAlO)n: synthesis of (Mes*AlO)4 (Mes* = –C6H2-2,4,6-tertBu3) and its reactions with AlR3 (R = Me or Et), J. Am. Chem. Soc., 1997, 119, 8387–8388 CrossRef CAS.
- G. Bai, H. W. Roesky, J. Li, M. Noltemeyer and H. Schmidt, Synthesis, structural characterization, and reaction of the first terminal hydroxide-containing alumoxane with an [{Al(OH)}2(μ-O)] core, Angew. Chem., Int. Ed., 2003, 42, 5502–5506 CrossRef CAS PubMed.
- H. Zhu, J. Chai, V. Jancik, H. W. Roesky, W. A. Merrill and P. P. Power, The selective preparation of an aluminum oxide and its isomeric C–H activated hydroxide, J. Am. Chem. Soc., 2005, 127, 10170–10171 CrossRef CAS PubMed.
- C. Li, C. Tsai, C. Lin and B. Ko, Synthesis, structural characterization and reactivity of aluminium complexes supported by benzotriazole phenoxide ligands: air-stable alumoxane as an efficient catalyst for ring-opening polymerization of L-lactide, Dalton Trans., 2011, 40, 1880–1887 RSC.
- C. Weetman, P. Bag, T. Szilvási, C. Jandl and S. Inoue, CO2 fixation and catalytic reduction by a neutral aluminum double bond, Angew. Chem., Int. Ed., 2019, 58, 10961–10965 CrossRef CAS PubMed.
- T. S. Koptseva, M. V. Moskalev, E. V. Baranov and I. L. Fedushkin, Reduction of nitrous oxide and binding of carbon dioxide by acenaphthene-1,2-diimine aluminum compound, Organometallics, 2023, 42, 965–970 CrossRef CAS.
- (a) B. Li, S. Kundu, S. A. Claudia, H. Zhu, H. Keil, R. HerbstIrmer, D. Stalke, B. Schwederski, W. Kaim, D. M. Andrada, G. Frenking and H. W. Roesky, A stable neutral radical in the coordination sphere of aluminum, Angew. Chem., Int. Ed., 2017, 56, 397–400 CrossRef CAS PubMed; (b) S. Roy, K. C. Mondal and H. W. Roesky, Cyclic alkyl(amino) carbene stabilized low coordinate metal complexes of enduring nature, Acc. Chem. Res., 2016, 49, 357–369 CrossRef CAS PubMed; (c) S. K. Kushvaha, A. Mishra, H. W. Roesky and K. C. Mondal, Recent advances in the domain of cyclic (alkyl)(amino) carbenes, Chem.–Asian J., 2022, 17, e202101301 CrossRef PubMed.
- L. Werner, J. Hagn and U. Radius, NHC-stabilized dialanes(4) of Al2Mes4, Chem.–Eur. J., 2023, 29, e202303111 CrossRef CAS PubMed.
- (a) A. D. Becke, Density-functional exchange-energy approximation with correct asymptotic behavior, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS PubMed; (b) J. P. Perdew, Density-functional approximation for the correlation energy of the inhomogeneous electron gas, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 8822–8824 CrossRef PubMed; (c) F. Weigend and R. Ahlrichs, Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: design and assessment of accuracy, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 CAS; (d) S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed; (e) S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu, J. Chem. Phys., 2010, 132, 154104 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision C.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- (a) A. E. Reed, R. B. Weinstock and F. Weinhold, Natural population analysis, J. Chem. Phys., 1985, 83, 735–746 CrossRef CAS; (b) A. E. Reed, L. A. Curtiss and F. Weinhold, Intermolecular interactions from a natural bond orbital, donor–acceptor viewpoint, Chem. Rev., 1988, 88, 899–926 CAS; (c) E. D. Glendening, C. R. Landis and F. Weinhold, NBO 6.0: natural bond orbital analysis program, J. Comput. Chem., 2013, 34, 1429–1437 CAS.
- K. B. Wiberg, Application of the pople-santry-segal CNDO method to the cyclopropylcarbinyl and cyclobutyl cation and to bicyclobutane, Tetrahedron, 1968, 24, 1083–1096 CAS.
- (a) K. Morokuma, Molecular Orbital Studies of Hydrogen Bonds. III. C=O⋯H–O Hydrogen Bond in H2CO⋯H2O and H2CO⋯2H2O, J. Chem. Phys., 1971, 55, 1236–1244 CAS; (b) T. Zeigler and A. Rauk, On the calculation of bonding energies by the Hartree Fock Slater method, Theor. Chim. Acta, 1977, 46, 1–10 Search PubMed.
- (a) M. Mitoraj and A. Michalak, Donor–acceptor properties of ligands from the natural orbitals for chemical valence, Organometallics, 2007, 26, 6576–6580 CAS; (b) M. Mitoraj and A. Michalak, Applications of natural orbitals for chemical valence in a description of bonding in conjugated molecules, J. Mol. Model., 2008, 14, 681–687 CAS; (c) M. P. Mitoraj and A. Michalak, σ-Donor and π-acceptor properties of phosphorus ligands: an insight from the natural orbitals for chemical valence, Inorg. Chem., 2010, 49, 578–582 CAS; (d) M. P. Mitoraj, A. Michalak and T. Ziegler, A combined charge and energy decomposition scheme for bond analysis, J. Chem. Theory Comput., 2009, 5, 962–975 CAS.
- H. Schneider, A. Hock, R. Bertermann and U. Radius, Reactivity of NHC-alane adducts towards N-heterocyclic carbenes and cyclic (alkyl)(amino)carbenes: ring expansion, ring opening, and Al–H bond activation, Chem.–Eur. J., 2017, 23, 12387–12398 CrossRef CAS PubMed.
- S. J. Bonyhady, D. Collis, G. Frenking, N. Holzmann, C. Jones and A. Stasch, Synthesis of a stable adduct of dialane(4) (Al2H4) via hydrogenation of a magnesium(I) dimer, Nat. Chem., 2010, 2, 865–869 CrossRef CAS PubMed.
- Y. Yang, H. Li, C. Wang and H. W. Roesky, Studies of the ligand effect on the synthesis of dialuminoxanes by various β-diketiminato ligands, Inorg. Chem., 2012, 51, 2204–2211 CrossRef CAS PubMed.
- W. Uhl, Tetrakis[bis(trimethylsilyl)methyl]dialan(4), eine Verbindung mit Aluminium–Aluminium-Bindung, Z. Naturforsch., B: J. Chem. Sci., 1988, 43, 1113–1118 CrossRef CAS.
- V. Lavallo, Y. Canac, C. Präsang, B. Donnadieu and G. Bertrand, Stable cyclic (alkyl)(amino)carbenes as rigid or flexible, bulky, electro-rich ligands for transition-metal catalysts: a quaternary carbon atom makes the difference, Angew. Chem., Int. Ed., 2005, 44, 5705–5709 CrossRef CAS PubMed.
- (a) R. F. W. Bader, A quantum theory of molecular structure and its applications, Chem. Rev., 1991, 91, 893–928 CrossRef CAS; (b) R. F. W. Bader, Atoms in Molecules: A Quantum Theory, Clarendon Press, 1994 Search PubMed; (c) R. F. W. Bader, Atoms in molecules, Acc. Chem. Res., 1985, 18, 9–15 CrossRef CAS; (d) C. F. Matta and R. J. Boyd, The Quantum Theory of Atoms in Molecules: from Solid State to DNA and Drug Design, Wiley-VCH, Weinheim, 2007 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Computational details, synthesis and single-crystal X-ray diffraction data of 1′, NMR spectra, elemental analysis (PDF). CCDC 2374368 (1′) and 2376845 ([cAACH]AlCl4). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5ra00211g |
| This journal is © The Royal Society of Chemistry 2025 |