
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThermally activated delayed fluorescence materials: innovative design and advanced application in biomedicine, catalysis and electronics
Ehsan Ullah Mughal
 *a,
Syeda Fariha Kainat
a,
Abdulaziz M. Almohyawib,
Nafeesa Naeem
a,
Essam M. Husseinbc,
Amina Sadiq
d,
Ahmad Abd-El-Azize,
Ning Mae,
Alaa S. Abd-El-Aziz
e,
A. Timoumif,
Ziad Moussag,
Nermeen Saeed Abbashi and
Saleh A. Ahmed
*b
*a,
Syeda Fariha Kainat
a,
Abdulaziz M. Almohyawib,
Nafeesa Naeem
a,
Essam M. Husseinbc,
Amina Sadiq
d,
Ahmad Abd-El-Azize,
Ning Mae,
Alaa S. Abd-El-Aziz
e,
A. Timoumif,
Ziad Moussag,
Nermeen Saeed Abbashi and
Saleh A. Ahmed
*b
aDepartment of Chemistry, University of Gujrat, Gujrat-50700, Pakistan. E-mail: ehsan.ullah@uog.edu.pk
bDepartment of Chemistry, Faculty of Science, Umm Al-Qura University, 21955 Makkah, Saudi Arabia. E-mail: saahmed@uqu.edu.sa
cDepartment of Chemistry, Faculty of Science, Assiut University, 71516, Assiut, Egypt
dDepartment of Chemistry, Govt College Women University, Sialkot-51300, Pakistan
eQingdao Innovation and Development Center, Harbin Engineering University, Qingdao, China 266400
fDepartment of Physics, Faculty of Science, Umm Al-Qura University, Makkah, 24382, Saudi Arabia
gDepartment of Chemistry, College of Science, United Arab Emirates University, P.O. Box 15551, Al Ain, United Arab Emirates
hDepartment of Chemistry, Faculty of Science, Taibah University, Medina, Saudi Arabia
iDepartment of Chemistry, Faculty of Science, Helwan University, Cairo, A. R., Egypt
First published on 7th March 2025
Abstract
Thermally Activated Delayed Fluorescence (TADF) materials have emerged as a revolutionary class of functional compounds, driven by their unique ability to utilize excitons from both singlet and triplet states for efficient fluorescence emission. This manuscript provides an overview of recent innovations in TADF material design, focusing on molecular strategies to achieve optimal TADF properties, including small singlet–triplet energy gaps (ΔEST) and high photoluminescence quantum yields. We explore the diverse applications of TADF materials, spanning OLEDs, biomedical imaging, photosensitizers, photocatalysis, UV photodetectors (UVOPDs), electrogenerated chemiluminescence, triplet–triplet annihilation (TTA) sensitizers, organic hybrid microwire radial heterojunctions, multicolor luminescent micelles, mechano-luminescence (ML), light-emitting electrochemical cells (LEECs), and fluorescent probes. The integration of TADF materials in these technologies highlights their potential to enhance performance and efficiency. Through this review, we aim to elucidate the fundamental principles governing TADF behavior and present a forward-looking perspective on the synthetic methodologies and new, versatile applications of materials.
 Ehsan Ullah Mughal | Ehsan Ullah Mughal obtained his PhD degree in Organic Chemistry from Bielefeld University Germany in 2013. Currently, he is working as Tenured Associate Professor in Department of Chemistry, University of Gujrat, Pakistan. He was a post-doc fellow at Max-Planck Institute for Polymer Research, Mainz, Germany from 2013 to 2014. His current research interests include the design and synthesis of bioactive heterocycles, synthetic flavonoids, transition metal-based terpyridine complexes and their uses in the fabrication of DSSCs and as efficient photocatalysts. His research in the field has resulted in more than 100 published articles (research papers and review papers) in reputable high impact journals. His publications have made a great impact in the field by receiving more than 2330 citations with h-index of 29. |
 Syeda Fariha Kainat | Syeda Fariha Kainat earned her BS in 2021 and MPhil in 2023 from the University of Gujrat, Pakistan. She has published two research articles and 1 review article in high-impact journals. Her research focuses on the synthesis and photocatalytic applications of terpyridine-based metal complexes. |
 Nafeesa Naeem | Nafeesa Naeem received her BS and MPhil degrees from the University of Gujrat, Pakistan, in 2018 and 2020, respectively. She is currently pursuing a PhD in Chemistry under the supervision of Dr Ehsan Ullah Mughal at University of Gujrat, Pakistan. She has published over 40 publications (research papers and review papers) in reputable high impact international journals and receiving more than 1230 citations and an h-index of 21. Her current research focuses on the synthesis, biological evaluation, photophysical studies, and photocatalytic applications of various heterocyclic scaffolds. |
 Amina Sadiq | Amina Sadiq is an Associate Professor of Organic Chemistry at the Government College Women University in Sialkot, Pakistan. She earned her PhD in 2013 from Bielefeld University, Germany, under the supervision of Prof. Dr Norbert Sewald, and later conducted postdoctoral research with the same group. She published more than 65 publications in high ranked journals and receiving more than 1580 citations and an h-index of 24. Her research focuses on the design and synthesis of bioactive heterocycles, synthetic flavonoids, and chalcone-based metal complexes, along with evaluating their biological activities. |
 Alaa S. Abd-El-Aziz | Alaa S. Abd-El-Aziz is a Full Professor at Harbin Engineering University in China. He currently building an international research program in the field of polymers and materials and their applications in biomedicine and industry. He has extensive experience in the design and synthesis of organic and organometallic polymers and dendrimers with optical and biological properties. His current research is focused on the synthesis of functional polymeric materials as sensors, biosensors or therapeutic agents. He serves on many international advisory and editorial boards. |
 Saleh A. Ahmed | Prof. Dr Saleh A. Ahmed received his PhD in photochemistry (photochromism) under the supervision of Prof. Heinz Dürr at Saarland University, Germany. He has more than 20 years of experience as a postdoctoral fellow, senior researcher and visiting professor in France, Japan, Germany, Italy and USA. Since 2010 he promoted to full professor of organic photochemistry. He published more than 290 publications in high ranked journals and more than 10 US-patents. His research interests include synthesis and photophysical properties of novel organic compounds, and developments of synthetic methodologies for the synthesis of compounds with unique theoretical and biological applications. |
1. Introduction
Since the first report of luminescent materials over four centuries ago, this important class of materials has continued to attract the attention of many research groups in academia and industry.1–6 Luminescent materials have found a broad range of industrial applications from materials science to biomedical technology.7,8 Over the past few decades, efficient luminescent emitters, including metal–organic frameworks,9 lanthanide metal-based nanoparticles,10 and inorganic quantum dots,11 have been extensively investigated. However, emitters composed of organic molecules featuring high-order π-conjugation and heteroatoms within their structures have been shown to offer high fluorescence quantum yields, good photostability, improved biocompatibility, and low phototoxicity in biological applications.12–14 Additionally, numerous π-conjugated luminescent molecules have been investigated for their fluorescence properties in nanoaggregates and thin films, making them valuable in sensing and imaging applications.15,16 Moreover, organic luminescent materials have gained significant attention due to their tunable luminescence properties, such as the color, duration, and intensity of emissions, which can be adjusted through molecular design. These materials have found advanced applications in fields like biomedicine, electroluminescence, optoelectronics, organic lasers, organic sensors, and organic light-emitting diodes (OLEDs).17–19 Organic molecules exhibiting thermally activated delayed fluorescence (TADF) properties have become a compelling class of functional materials due to their increasing demand and significant design advancements in recent years.20 Before using the term TADF, this phenomenon was referred to as “E-type” delayed fluorescence. In the early 1920s, French Nobel Laureate Jean-Baptiste Perrin (Nobel Prize in Physics, 1926) reported the “E-type” delayed fluorescence in “Lumière et réactions chimiques, rapport au 2e Conseil de Chimie Solvay, Bruxelles”.21 Later, Delorme and Perrin reported delayed fluorescence from uranyl salts and solutions in 1929 in an article from “Le Journal de la Physique et la Radium”.22 In early 2024, Huang and Cole generated a database of TADF molecules and made it available through Figshare.23 The first TADF-based OLED, which was designed using a fluorescein derivative, was described by Adachi et al. in 2012.17 The increased demand for new TADF materials has been clearly shown in the recent literature. Fig. 1 shows the diverse fields where this class of materials has been utilized, including light emitting devices, photocatalysts and photodetectors, as well as their biomedical applications. As an example, TADF applications of mechanoluminescence from simple organic molecules by positional isomerism were first observed by Xiong et al.24 TADF-emitting compounds can be used as probes for conventional fluorescence bioimaging or be used for their sensitivity to temperature or oxygen levels.25 Suleymanova et al. designed a novel liquid-crystal TADF emitter based on substituted carbazoles and terephthalonitrile.26,27 Also, Chen et al. reported an example of a liquid crystal multi-resonance TADF emitter that showed preferential horizontal orientation.28 While these examples highlight the importance of this growing field, this review gives a comprehensive description of the design and applications of various types of TADF materials. This will attract a broad interest from many interdisciplinary fields, including materials science, chemistry, physics, biology, and medicine. | ||
| Fig. 1 Applications of TADF materials. | ||
1.1. Classification of TADF materials: design strategies and mechanistic insights
TADF materials can be broadly classified into three main categories based on their electronic structures and charge-transfer characteristics.Key features:
➢ Faster ISC and RISC rates due to enhanced SOC.
➢ Lower ΔEST, leading to efficient TADF emissions.
➢ Potential dual-emission behavior from mixed triplet and singlet states.
Representative examples: Pt(II) and Au(I) complexes with D–A architectures. Organometallic TADF materials with ligand-field modifications to control emission wavelengths.33,35–37
1.1.2.1 Long-range charge transfer (LRCT) TADF. In LRCT-TADF systems, the donor and acceptor units are spatially separated by extended π-conjugated linkers, resulting in highly polarized charge-transfer states. While this leads to a significantly small ΔEST, the reduced spin–orbit coupling often slows down the RISC process.40–43
Key features:
➢ Extremely small ΔEST (∼0.01–0.05 eV), enhancing TADF efficiency.
➢ Increased excited-state lifetimes due to reduced SOC.
➢ High emission stability with minimized aggregation-induced quenching.
Representative examples: multi-resonance boron–nitrogen (BN) embedded systems. Large π-extended D–A systems with steric hindrance to prevent aggregation.44–48
1.1.2.2 Short-range charge transfer (SRCT) TADF. In contrast, SRCT-TADF materials have donor and acceptor units in proximity, leading to stronger electronic coupling and moderate charge separation. This configuration ensures a balance between ΔEST reduction and efficient spin mixing for faster RISC.49–52
Key features:
➢ Moderate ΔEST (∼0.05–0.1 eV), enabling both efficient ISC and RISC.
➢ Shorter excited-state lifetimes than LRCT-TADF, improving device efficiency.
➢ Strong fluorescence-phosphorescence coupling, leads to high photoluminescence quantum yield (PLQY).
Representative examples: carbazole-based D–A molecules with rigidified linkers.53–55 Polycyclic acceptors paired with nitrogen-rich donors to optimize charge transfer.56,57
Key features:
➢ Non-bonded through-space interactions control charge-transfer dynamics.
➢ Efficient TADF behavior with tunable emission colors.
➢ Improved molecular stability due to minimized intramolecular vibrations.
Representative examples: twisted or V-shaped donor–acceptor systems with spatial charge confinement.59–62 Supramolecular host–guest complexes enabling intermolecular TADF.63,64
2. Mechanism of TADF
The luminescence process involves the absorption of energy to form excitons, followed by radiative decay from the excited state to the ground state.65,66 The excited states can possess divergent spin multiplicities, and returning to the ground state therefore results in either fluorescence (from singlet states) or phosphorescence (from triplet states) as shown in Fig. 2. Transitions between two electronic states possessing the same spin multiplicity are possible according to quantum mechanics, but transitions between states with divergent spin multiplicities, i.e., intersystem crossing (ISC) with spin changes, are not allowed. In fluorescence, a radiative transition from singlet excitons to the lowest singlet excited state occurs before being reduced to S0, a ground state of many types of organic molecules that are generally singlets. This process is extremely rapid, with a life span of nanoseconds. In contrast, the phosphorescence mechanism requires a radiative transition of triplet excitons from the T1 to the ground states, which can be theoretically achievable.67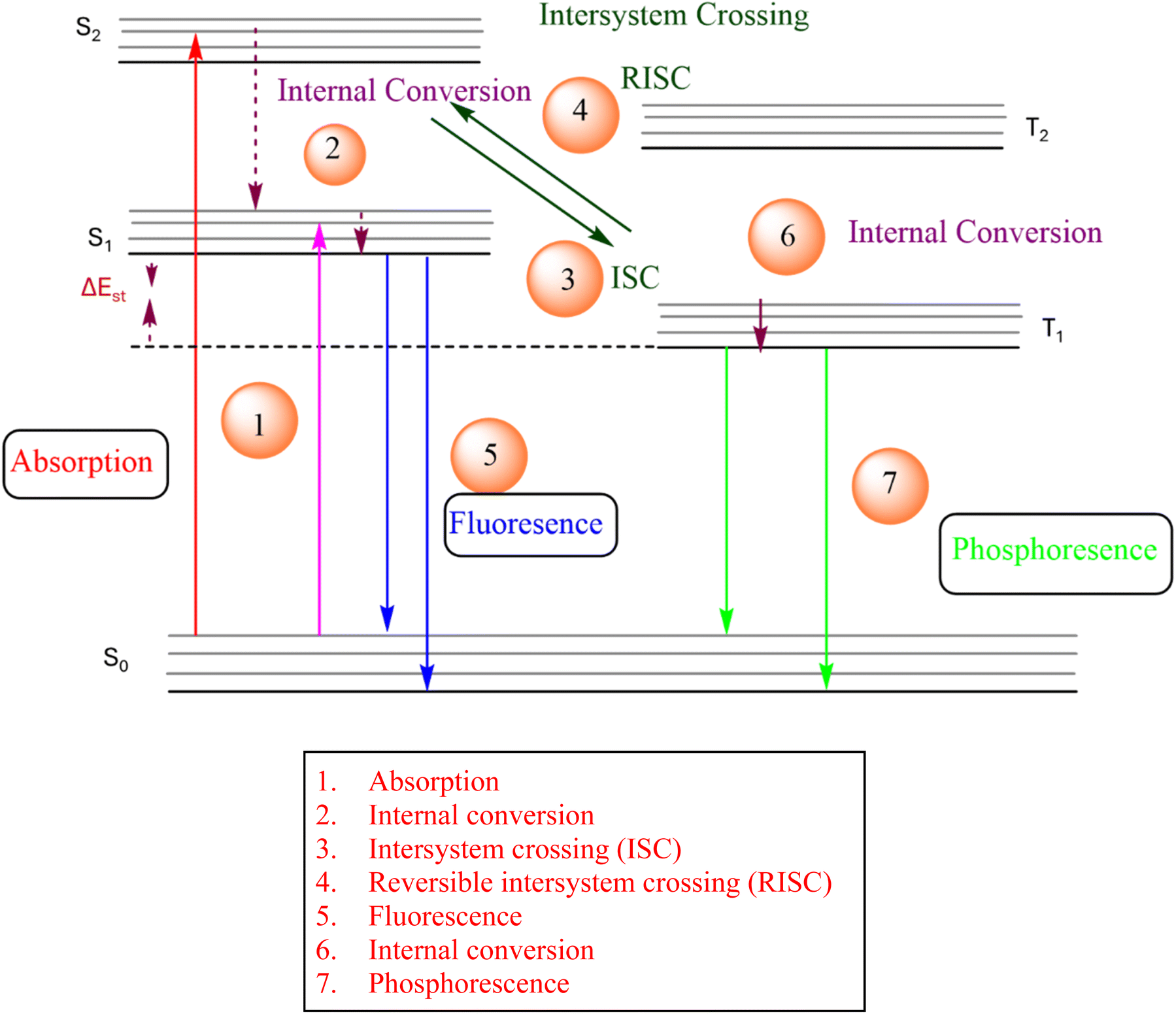 | ||
| Fig. 2 Process of transitions of photo-excited photons for different phenomena. | ||
Organic luminescent molecules with energy gaps above 0.5 eV between the singlet S1 and triplet T1 states (ΔEST) show fluorescence instead of phosphorescence. For example, metal complexes demonstrate large ΔEST values, due to increased ISC and spin–orbit coupling caused by the presence of metal atoms.68 TADF materials have strong radiative transitions of excitons from the S1 state, as evidenced by ISC and a longer delayed emission lifetime compared to phosphorescence. Fig. 2 shows that reverse intersystem crossing (RISC) from the T1 state back to the S1 state causes the release of singlet excitons, which is required for the TADF process. RISC can occur after the addition of thermal energy, resulting in the TADF mechanism being an exothermic process because S1 is a higher-energy state than T1. Consequently, TADF exhibits stronger emissions at elevated temperatures.
2.1. Influence of molecular design on the excited-state lifetime in TADF materials
The microsecond-level excited-state lifetime is a defining characteristic of TADF materials, as it reflects the efficiency of the RISC process. A prolonged excited-state lifetime typically indicates a slow RISC rate, whereas a shorter lifetime suggests efficient upconversion from the triplet to the singlet state. The molecular design of TADF emitters plays a crucial role in determining this parameter.64,69By systematically tailoring molecular design parameters, TADF emitters with controlled excited-state lifetimes can be developed, striking a balance between efficient RISC and high quantum yield. These insights provide a deeper understanding of the structure–property relationships governing TADF behavior, guiding the design of next-generation high-performance OLED materials.
TADF was first described in organic molecules in the 1960s, including organic metal complexes and fullerenes 1.84 In 1961, the pure organic molecule eosin Y was reported to exhibit TADF, initially termed “E-type” delayed fluorescence (DF). This long-lived emission, identified as a secondary band alongside phosphorescence, exhibited a fluorescence band in the red region of the spectra for eosin Y 2. Consequently, Parker and Hatchard attributed this emission to DF from the same excited singlet state.85
McMillin and coworkers suggested an alternative mechanism from early studies of copper(I) complexes involving two excited states in thermal equilibrium.86 During initial studies, the TADF mechanism was only partially impactful due to the relatively large energy gap for eosin 2 (ΔEST = 0.37 eV). Moreover, no evidence was found for a molecular strategy to enhance TADF efficiency, which posed a significant challenge to the fabrication of TADF materials. Consequently, before 2009, TADF materials had few applications and only their photophysical properties were studied.
In 2009, Endo et al. were the first to use TADF to produce singlet and triplet excitons in OLEDs. The excitation was achieved by applying short electrical pulses in an OLED containing a Sn(IV)–porphyrin complex 3, as described in Table 1.88 In 2010, Peters et al. constructed OLEDs with an emissive layer doped with a Cu(I) complex. These OLEDs showed outstanding performance and an external quantum efficiency (EQE) of 16.1%.89 A major milestone for TADF molecules in OLED applications was documented by Adachi et al. in 2011, using purely organic molecules.92 Through a quantum mechanical analysis, they found that a modest ΔEST could be achieved by an efficient TADF mechanism that reduced the overlap between the lowest unoccupied molecular orbital (LUMO) and the highest occupied molecular orbital (HOMO). Based on these results, they suggested a molecular design that entailed creating molecules with a twisted electron donor–acceptor (D–A) structure (D shown in blue and A shown in red) (Table 1). This structure was an effective TADF mechanism with a low ΔEST of 0.11 eV because of the bulky substituents causing steric hindrance. Emitting layers in OLEDs containing were shown to have a high EQE of 5.3%, which is comparable to the theoretical limit of ordinary fluorescent molecules. Adachi and coworkers continued to work on the development of improved TADF-based materials for their applications in OLEDs, focusing on optimizing the radiative decay rate of the final S1 excitons and exceptionally low ΔEST. They documented a set of functionalized TADF emitters utilizing carbazolyl dicyanobenzene (CDCB) as the core component in 2012.17 These emitters exhibited significant photoluminescence (PL) efficiency and could emit a spectrum of colors from orange to sky blue. Applications of the material 4CzIPN 6 in multilayer OLEDs for green OLEDs resulted in a calculated EQE of 19.3%, while orange OLEDs achieved an EQE of 11.2% and sky-blue OLEDs had an EQE of 8.0%. These results marked a significant advancement over traditional fluorescence-based OLEDs, proving that utilizing organic TADF materials to harvest triplet excitons through DF could achieve high efficiency OLEDs. Around the same time, Adachi and his colleagues also describe the application of exciplexes 7 synthesized using a donor and an acceptor (shown in Table 1 in blue and red, respectively), for a molecule to obtain efficient OLED efficiency via efficient DF, with an EQE as high as 10.0%.91,93 Different types of organic-based TADF materials 1–7 described above are illustrated in Table 1.






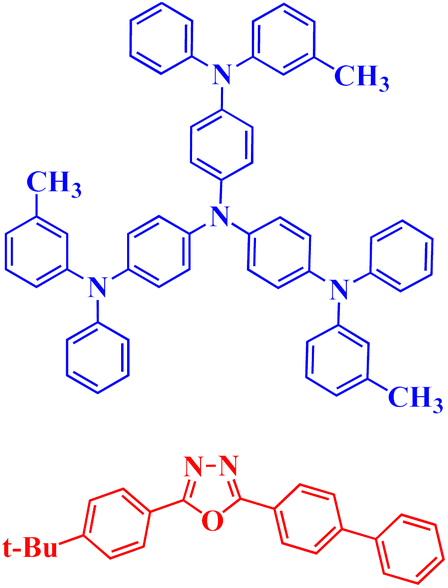
Moreover, unlike OLEDs relying on costly noble metals, pure organic materials can be cost-effective alternatives. However, traditional fluorescence-based OLEDs face a critical limitation in their low EQE, with a theoretical maximum of approximately 5%. TADF has therefore introduced an innovative method for generating triplet excitons in OLEDs, greatly enhancing OLED performance using simple and cost-effective organic molecules. Modern OLEDs employing organic TADF materials in their emitting layers, including organic compounds and exciplexes, can achieve EQE values over 20%, and sometimes even exceed 30%.94–96 TADF materials are now regarded as 3rd-generation emitters, representing a significant advancement in OLED technology.97
3. Principles for designing TADF molecules
TADF properties rely on a small ΔEST, which enables the RISC process (Fig. 3). On the other hand, achieving TADF OLEDs with highly efficient electroluminescence (EL) requires a large radiative decay rate (kr) of S1 excitons, which conflicts with the need for a small ΔEST. | ||
| Fig. 3 Principles for designing D–A system of TADF molecules. | ||
Currently, organic TADF materials reported in the literature can be separated into two classes: organic D–A systems and organic exciplexes consisting of two separate D–A materials.
Huang and colleagues (2015)98 outlined key principles for the fabrication of D–A type TADF molecules, particularly focusing on their applications in high-performance OLEDs. These guidelines include:
(i) In order to achieve a smaller ΔEST, adopting a twisted D–A structure as a result of heavy substituents or a spiro-junction.
(ii) Heavily integrating donor and acceptor groups while enhancing molecular rigidity to improve radiative luminescence efficiency.
(iii) Controlling the length of π-conjugated systems.
(iv) Managing the oxidation-reduction potentials of the donors and acceptors by selectively interrupting conjugation.
In recent years, various innovative methodologies for designing more efficient TADF systems have been introduced. It is desirable to minimize ΔEST in order to increase the radiative decay rate from the S1 to the S0 state. Enhancing the overlap in the density distribution between the electronic wavefunctions of the ground and excited states promotes S1–S0 radiative decay, thereby improving the photoluminescence quantum yield (PLQY).99 To reduce the radiative decay rate of fluorescence (kf) and the ΔEST, it is necessary to achieve significant delocalized molecular orbitals with a clear gap between the HOMO and LUMO.100 The introduction of different molecular systems through physical separation of D–A units,101 utilizing X-shaped structures,102 incorporating dual D or A units,103 or leveraging multiple resonance effects104 can also lead to separating the HOMO and LUMO and therefore minimization of ΔEST. A summary of these principles is shown in Fig. 3 and Table 2 shows D–A systems 8–11 used for TADF molecules.105
| Compound number | Name | Structure |
|---|---|---|
| 6 | 4CzIPN |  |
| 8 | ACRFLCN |  |
| 9 | PxPmmBPX |  |
| 10 | DABNA-2 |  |
3.1. Organic exciplexes with two separate donor and acceptor materials
Organic exciplexes have received tremendous attention from researchers considering their remarkable properties and luminescent applications. These include organic optical waveguides, lasers, various nonlinear optical devices, OLEDs, and even light-emitting transistors. Extensive research has been conducted on exciplexes. Stable exciplexes are generated when one component of the D–A pair is excited directly or locally from its ground state and forms a complex with the other component, allowing the initial component to maintain stability in its excited state.106,1073.2. Generation of exciplexes by photoexcitation
Two distinct classes of excited-state complexes have been produced by progressively depositing p-type materials containing various electron-donating units and n-type materials containing electron-accepting units in a single layer, a process known as co-doping,108–110 as shown in Fig. 4. The combination of bipolar and n-type materials could lead to the formation of excited-state complexes which, in turn, gave rise to a sizable hole/electron barrier between the two materials.111 It was also noted that the radiative electron transitions from the LUMO of the acceptor to the HOMO of the donor resulted in emissions from the excited-state complexes. Similar to exciplexes, electroplexes can arise from the donor carrying a hole or the acceptor carrying an electron. The donor–acceptor gap in an exciplex of planar aromatic compounds is less than that of an electroplex (∼0.4–0.7 nm).112 Consequently, the structural properties of the materials determine whether exciplexes or electroplexes form. Emissions from exciplexes have been reported for EL as well as PL spectra and could be produced using photoexcitation.113–115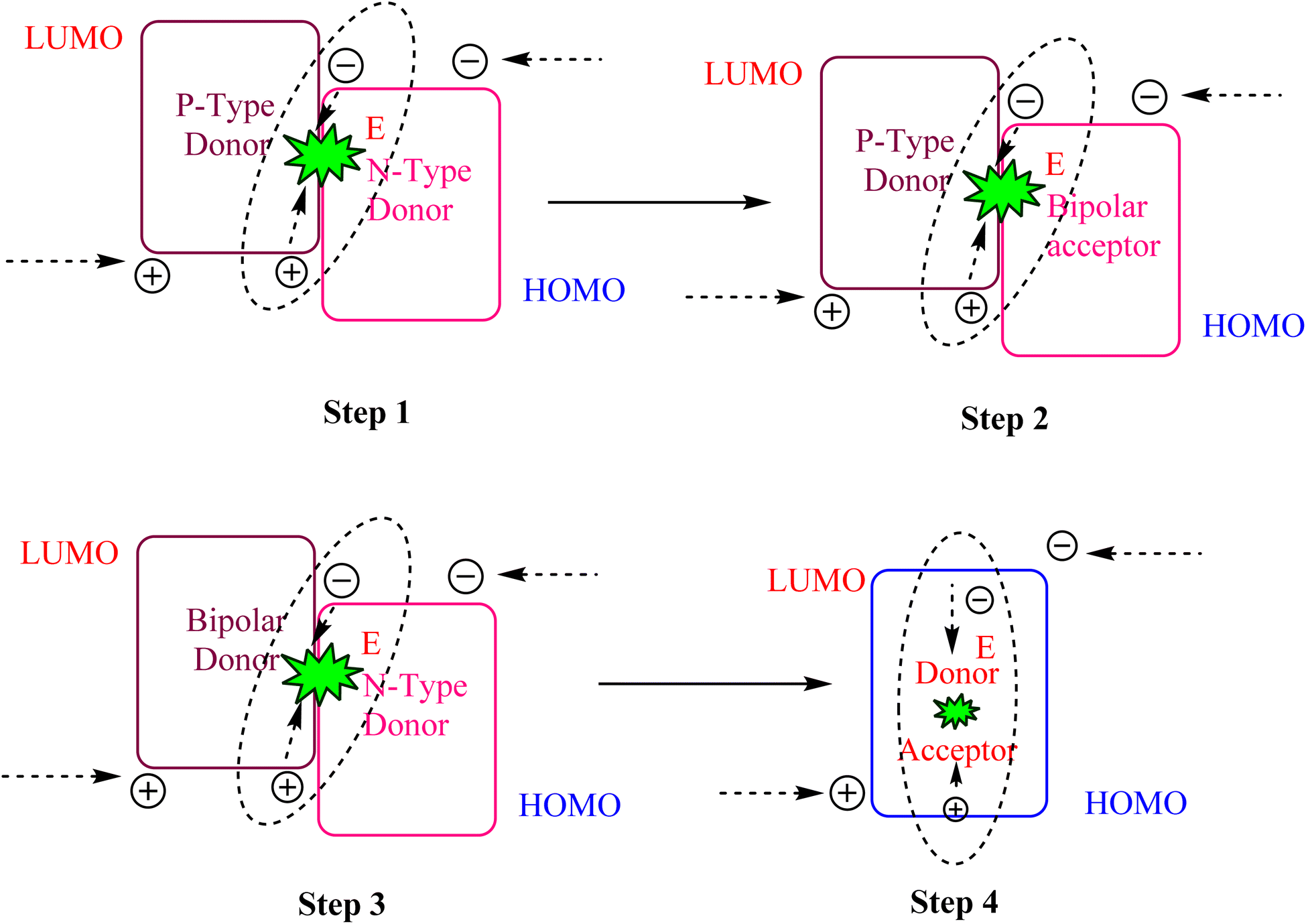 | ||
| Fig. 4 Mechanism for generation of exciplexes by co-doping. | ||
3.3. Electrically excited generation of exciplexes
In type II heterojunctions, the generation of exciplexes is intimately linked to the generation of intermolecular charge transfer (CT) excitons within non-ionic S0 ground state complexes.116 During the process of electrical excitation, electrons are introduced into the LUMO of the acceptor, while holes are introduced into the HOMO of the donor. The substantial energy difference between the LUMOs and HOMOs leads to the accumulation of both electrons and holes at the donor–acceptor interface. This accumulation results in the formation of CT excitons, as depicted in Fig. 5.117 | ||
| Fig. 5 Electrically excited generation of exciplexes. | ||
Once CT excitons are generated, they undergo decay to the ground state either through radiative or non-radiative recombination, which is based on specific mechanisms and time. Exciplex emission phenomena result from a radiative recombination of CT excitons releasing photons. However, exciplexes emit exclusively through the recombination of singlet CT excitons, typically resulting in less efficient PL.118 Exciplexes may exhibit TADF properties using singlet and triplet excitons by rapid and DF decay channels. Further, TADF can exceed the internal quantum efficiency (IQE) of about 25% for singlet excitons in the absence of heavy atoms at atmospheric temperature, theoretically allowing the IQE to reach 100%. When the ΔEST between the lowest triplet and singlet states is minimal, the RISC mechanism is efficiently activated, improving the IQE, as shown in eqn (1) below.
| ΔEst = Es − Et = 2j | (1) |
Furthermore, the fabrication of TADF materials based on exciplexes possessed extremely high quantum yields with the following necessary conditions:
To ensure efficient charge injection, D–A materials must exhibit effective electron and hole transport.
(1) The difference in energy between the HOMO and LUMO of the donor and acceptor must be more than 0.4 eV to that cause accumulation of effective charge on the surface.
(2) A D–A unit with higher triplet energy (T1) levels must be selected to prevent energy transfer back to the exciplex system.119
3.4. Design principles for effective TADF exciplex emitters
Both surface and bulk TADF exciplex emitters' EL properties have been enhanced in the presence of an efficient D–A molecule combination. However, even with these developments, creating effective TADF exciplex emitters remains challenging. As a result, there has been a considerable amount of research done to fully comprehend TADF exciplex systems and several novel ideas have been put out to direct the creation of exciplex emitters with increased efficiency.3.5. Regulation of excited state energy alignment
For an efficient TADF mechanism, the energy levels of both singlet and triplet states should be nearly equal. However, the system also includes local excited states, which can impact the energy transfer during the formation of the exciplex. Particularly, the local triplet states of the donor and acceptor may have different relative energies compared to the exciplex's CT state. Therefore, to ensure efficient TADF in exciplexes, it is essential to analyze and align the excited triplet states of the donor and acceptor with the excited singlet state of the exciplex to minimize loss of energy and prevent energy leakage.79,120,121 Several TADF donor and acceptor-based exciplexes 12–25 separated into their donor and acceptor groups are described in Table 3.119| Name | Donor | Name | Acceptor |
|---|---|---|---|
| mPXPT |  |
POZ-DBPHZ |  |
| mPXTMP |  |
BFPD |  |
| CzSi |  |
OXD-7 |  |
 |
 |
||
| TPD |  |
PIM-TRAZ |  |
| TASBPA |  |
A2 |  |
| TCBPA |  |
A3 |  |
4. Applications of thermally activated delayed fluorescence materials
TADF materials have a wide variety of applications in OLEDs and other industries. They are used in a variety of OLEDs, such as dendrimer-based OLEDs, non-doped TADF-based OLEDs, white OLEDs (WOLEDs), solution-processed OLEDs, and doped TADF-based OLEDs. In addition to OLEDs wide range of cutting-edge applications, TADF materials are also used in electrogenerated chemiluminescence (ECL) cells, fluorescence imaging, organic hybrid microwire radial heterojunctions, organic ultraviolet photodetectors (UVPODs), multicolor luminescent micelles, mechanoluminescence (ML) and mechanoluminochromism (MLC), photocatalysis, photosensitizers, triplet–triplet annihilation (TTA) sensitizers, and biomedical applications.105 A few representative examples of these applications will be discussed below.4.1. Biomedical applications of TADF
Metal-free luminophore-based TADF materials have been used in biomedical applications including time-resolved sensing and imaging, generation of singlet oxygen (1O2) for photodynamic therapy (PDT), and conventional fluorescence imaging in various biological systems (Fig. 6).18,25,122,123 | ||
| Fig. 6 Biomedical application of metal-free TADF luminophores. | ||
In 2015, Adachi et al. explored fluorescent organic nanoparticles, specifically TADF nano-probes based on the anthracene derivative 2,6-bis[4-(diphenylamino)phenyl] anthraquinone (TPAAQ), using self-assembly methods as shown in Fig. 7A and B.55 They discovered that these nanoparticles emitted strong red light, unlike TPAAQ molecules in THF. In particular, the aggregation-induced emission (AIE) properties of NFO-NPs displayed a significant Stokes shift of 177 nm, which makes them ideal for bioimaging in light of their superior signal-to-noise ratio. The study also assessed the long-term cellular tracing abilities of these nanoparticles, which showed sustained red signals after a single application. They analyzed how TPAAQ solution concentration affected NFO-NP precipitation, finding that TPAAQ 25 did not dissolve in THF beyond a certain concentration. At 1 mg mL−1, the NFO-NPs were uniformly sized at 93.5 nm with a polydispersity index (PDI) of 0.148. These particles had low cytotoxicity and demonstrated AIE in aqueous solutions, indicating a potential for targeting A549 cancer cells via endocytosis. Additionally, NFO-NPs exhibited superior resistance to photodegradation and photobleaching compared to dyes like fluorescein sodium, making them promising fluorescent probes for cellular tracing and bioimaging.131
 | ||
| Fig. 7 (A) Illustration of the synthesis of NFO nanoparticles (NFO-NPs) using the reprecipitation technique. (B) SEM image of the NFO-NPs and TEM images (C) Dynamic Light Scattering (DLS) measurements and polydispersity index (PDI) of the NFO-NPs. (D) Zeta potential measurement of NFO-NPs. (E) Photographs showing TPAAQ under UV light (e1) and NFO-NPs dispersed in water (e2 and e3). Reproduced from ref. 131 with permission from the Royal Society of Chemistry. | ||
In 2016, the same research team explored novel fluorescent organic nanoprobes based on electron-based donor–acceptor (D–A) carbazole derivatives for cellular imaging, aiming to address the shortcomings identified in their 2015 study.122 The earlier research had revealed issues such as oxygen sensitivity, low water stability, and challenges associated with TADF emitters, which included potential cell or tissue damage from short-wavelength excitation, background interference from autofluorescence, and limited optical penetration depth. To mitigate these issues, Adachi and his colleagues developed new carbazole-based nanoprobes that emitted red, blue, orange, and green light, suitable for both one- and two-photon cellular imaging techniques, as illustrated in Fig. 8(A and B) 6, 26–27. Despite having identical chemical structures, these TADF materials exhibited diverse structures and optical properties, which were associated with D–A acceptor supramolecular interactions arising from variations in their molecular frameworks.
 | ||
| Fig. 8 (A) Schematic diagram for fabrication of blue/red/green fluorescent nanoprobe for cellular imaging in one- and two-photon imaging systems (B) one- and two-photon imaging showing fluorescence and cellular localization of the three-fluorescent organic nanoprobes (FONs) in A549 cells. Reproduced from ref. 122 with permission from American Chemical Society, Copyright © 2016. | ||
In 2019, the same research group investigated organic nanoparticles exhibiting a high photoluminescence quantum yield (PLQY) of approximately 94%, along with a long-lived delayed emission of 3.1 μs, originating from TADF.132 These nanoparticles developed into glassy state particles in oil or water emulsions under pressures below 20 bar. The resulting organic nanoparticles demonstrated remarkable dispersion capabilities, photostability, and excellent uptake properties in living cells. When incorporated into a glassy host matrix, luminophores such as 4CzIPN achieve an impressive photoluminescence quantum yield (PLQY) of 94%, a substantial enhancement compared to the 13% PLQY observed in pure 4CzIPN O-dots in aqueous solution. This significant improvement highlights their potential as promising agents for both in vivo and in vitro studies. However, a notable drawback of these TADF-emitting glassy organic nanoparticles is their insensitivity to oxygen. The glassy host matrix restricts oxygen diffusion to the emitters after nanoparticle fabrication, and organic nanoparticles produced under air-saturated conditions did not perform effectively. Fig. 9 illustrates various chemical structures (6, 28–30) used in the preparation of these organic nanoparticles.
 | ||
| Fig. 9 Chemical structures of molecules 6, 28, 29 and 30 used in the preparation of TADF-emitting glassy organic nanoparticles. Reproduced from ref. 132 with permission from the Royal Society of Chemistry. | ||
In 2020, Li et al. developed an economical method using galactose, a glucose isomer, to synthesize Gal-PEG-DSPE. This compound was employed to modify TADF liposomes to target HepG2 cells, enhancing the accumulation of TADF materials within cancer cells. This approach aimed to create a targeted fluorescent probe for detecting malignant cells.133 The fluorescence intensity of liposomes modified with Gal-PEG-DSPE showed a significant increase compared to Blank-PEG-DSPE liposomes (P < 0.05). This enhancement is attributed to the Warburg effect, which involves the overexpression of glucose transporter proteins (GLUT) in malignant cells. The presence of galactose ligands on the liposomes therefore facilitated greater uptake by tumor cells compared to conventional liposomes without this modification, as shown in Fig. 10A. Both Gal-PEG-DSPE- and Blank-PEG-DSPE-modified liposomes encapsulating the TADF probe exhibited excitation at 488 nm and emission at 520 nm, as illustrated in Fig. 10B. Fig. 10C shows that the Gal-PEG-DSPE-modified liposomes showed significantly increased fluorescence intensity compared to Blank-PEG-DSPE-modified liposomes. This finding was consistent with the flow cytometry analysis results showing cellular uptake. Confocal imaging revealed that the liposomes were primarily localized in the cytoplasm, with significant accumulation surrounding the nucleus, but no detectable fluorescence signal was detected in the nucleus itself.133
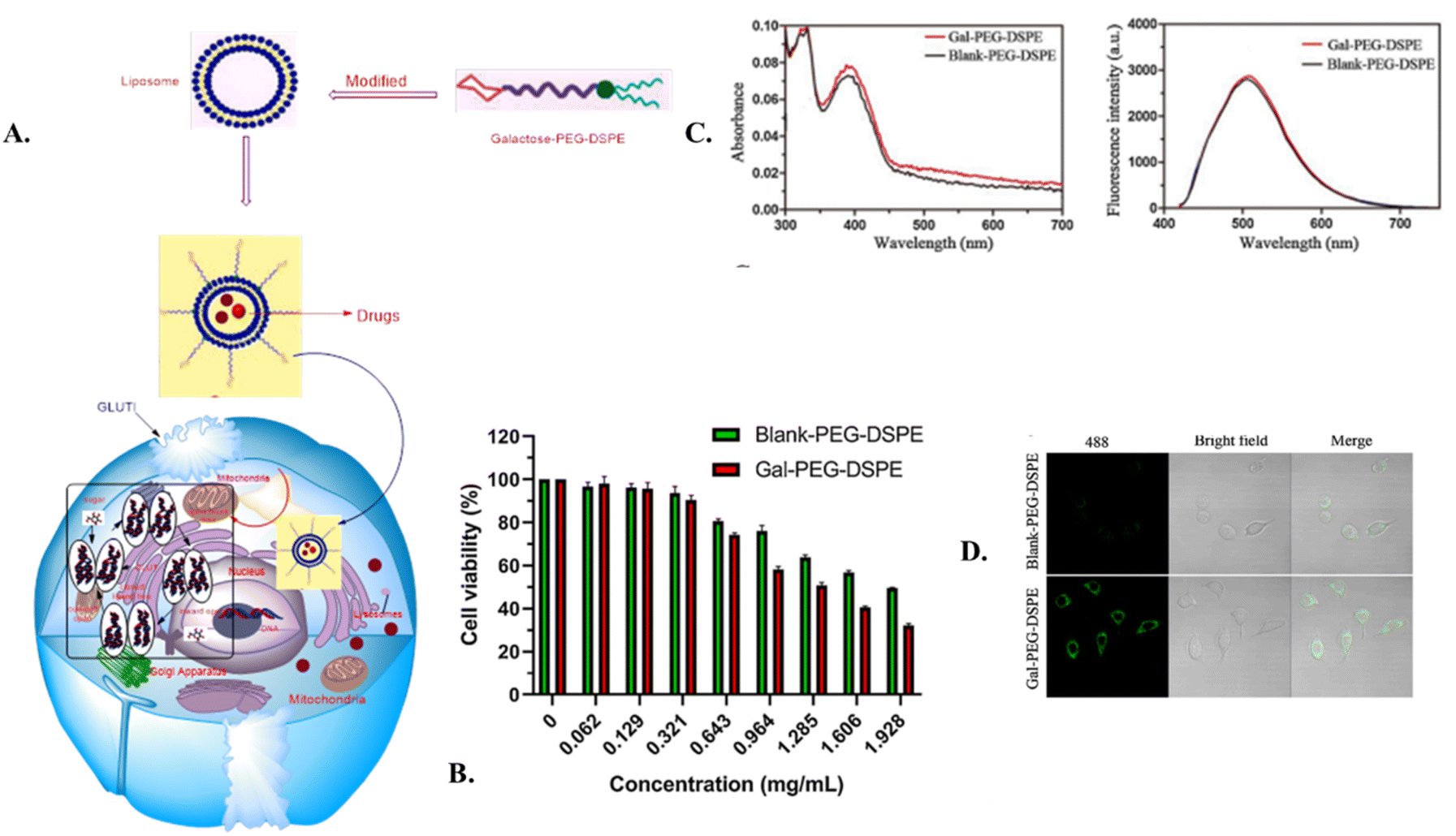 | ||
| Fig. 10 (A) Schematic representation of galactose-modified liposomes transport via GLUT. (B) Cell viability at various concentrations of Gal-PEG-DSPE- and Blank-PEG-DSPE-modified liposomes in HepG2 cells (C) UV-vis absorption spectra for absorbance (left) and fluorescence. (D) Confocal fluorescence images of HepG2 cells after incubation solely with Blank-PEG-DSPE and Gal-PEG-DSPE modified liposomes in PBS for 2 h. Adapted from ref. 133 with permission from Elsevier, Copyright © 2020. | ||
 | ||
| Fig. 11 The fabrication process of TXO NPs through nanoprecipitation. Reproduced from ref. 159 with permission from John Wiley & Sons, Copyright © 2018, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
Research on TRLI has focused on enhancing optical diagnosis and sensing by focusing on luminescence lifetime, thereby increasing specificity and sensitivity. In biological systems, this technique effectively eliminates short-lived background signals, thus enhancing the signal-to-noise ratio. By using a suitable time-gating mechanism among the excitation laser and luminescence detection, TRLI collects only long-lived signals.134,160,161 TADF dye materials are crucial in advancing TRLI.159,162–167
In 2017, Tang and coworkers investigated a set of luminogens based on phenoxazine (PXZ) that displayed both aggregation-induced emission (AIE) and DF.168 These luminogens, detailed in Fig. 12 as compounds 31 and 32, exhibit strong emission in their aggregated state and long-lived fluorescence. Both compounds also formed water-soluble nanoparticles (NPs) suitable for bioimaging when encapsulated by a matrix of bovine serum albumin (BSA). These NPs showed low levels of cytotoxicity and efficiently stained living cells, emitting robust fluorescence in the green or yellow regions. Additionally, the NPs performed well in fluorescence lifetime imaging, clearly displaying intracellular viscosity based on varying fluorescence lifetimes. While the use of luminogens with AIE and DF for fluorescence lifetime bioimaging has been relatively unexplored, these results show that they hold significant promise, particularly in addressing the limitations of luminescent metal complexes, such as the presence of toxic heavy metals.
 | ||
| Fig. 12 Molecular structures of the luminogens and crystal structures of BP-2PXZ (31) and BP-PXZ (32). Reproduced from ref. 168 with permission from the Royal Society of Chemistry. | ||
Huang and coworkers developed and studied two small molecular organic TADF probes with specific targeting for organelles.42 These probes show the longest average fluorescence lifetimes observed in TRFI within cells. Two TADF probes, the mitochondria-targeting AI-Cz-MT (35) and the lysosome-targeting AI-Cz-LT (36) were synthesized and displayed high biocompatibility and organelle-specificity. This was accomplished by attaching a triphenylphosphonium (TPP) group and 2-morpholinoethylamine to AI-Cz-CA (34) to yield AI-Cz-MT (35) and AI-Cz-LT (36), respectively, as shown in Fig. 13A. To assess the influence of organelle-targeting groups on photophysical properties, the team examined and compared the UV-vis absorption and fluorescence emission spectra of 35 and 36 with those of AI-Cz (33). In toluene, all three probes showed similar absorption at 335 nm for the carbazole donor unit and 390 nm due to ICT between the carbazole units and the aromatic imide. The emission spectra displayed identical band shapes, with a maximum emission wavelength at 520 nm, characteristic of TADF compounds with large Stokes shifts, as depicted in Fig. 13B. This strategy sought to mitigate the quenching of TADF fluorophores by oxygen by attaching organelle-targeting groups to TADF probes, thereby preserving their extended fluorescence lifetimes for time-resolved fluorescence imaging (TRFI) in cellular environments. The synthesized 35 and 36 demonstrated high fluorescence quantum yields and precise localization within mitochondria and lysosomes. Their extended fluorescence lifetimes were notable both in solution (6.8 μs for 35 and 6.3 μs for 36) and in TRFI experiments in HepG2 cells (16.7 μs for 35 and 28.0 μs for 36) in Fig. 14A and B. This study represents a notable advancement in organelle-specific TADF probes showing average fluorescence lifetimes in the microsecond range, as measured by TRFI. These developments could allow for the expansion of the potential bioimaging applications of TADF compounds.
 | ||
| Fig. 13 (A) The synthesis of organelle-specific fluorescent probes 35 and 36. (B) The absorption and emission spectra of AI-Cz (33), AI-Cz-MT (35), and AI-Cz-LT (36). Reproduced from ref. 42 with permission from American Chemical Society, Copyright © 2023. | ||
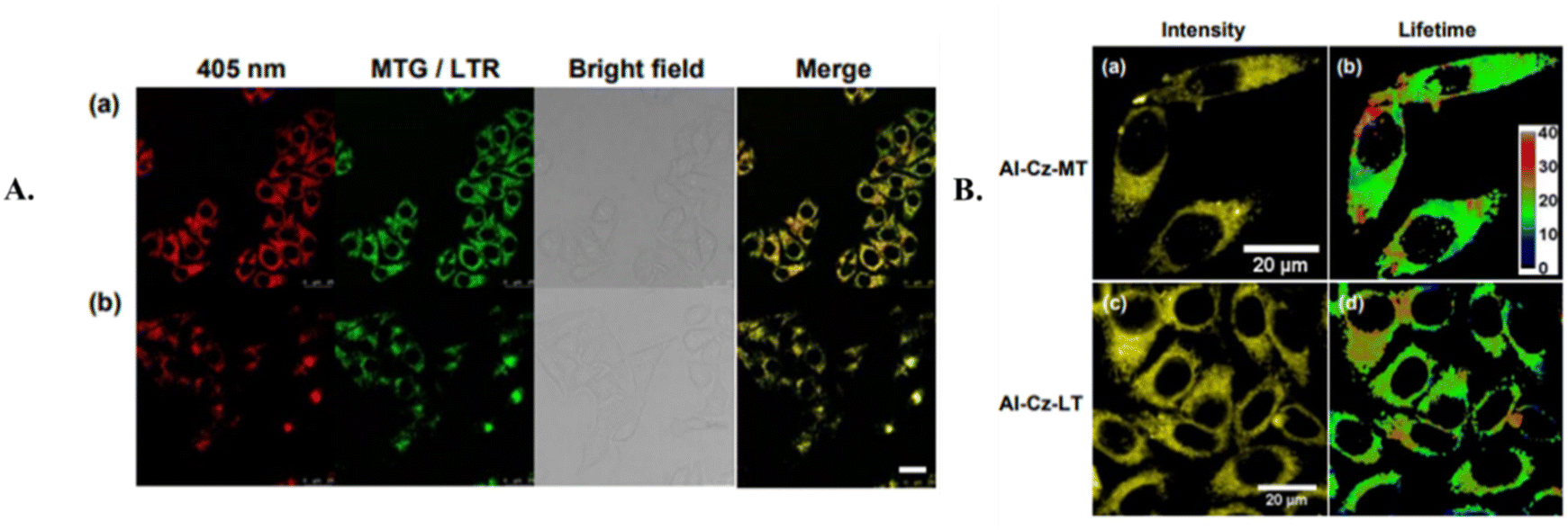 | ||
| Fig. 14 The subcellular localization of AI-Cz-MT (35) (A) and AI-Cz-LT (36) (B) in HepG2 cells investigated using fluorescence microscopy. Reproduced from ref. 42 with permission from American Chemical Society, Copyright © 2023. | ||
In 2020, Ni et al. introduced an innovative approach utilizing in situ aggregation-induced TADF turn-on response for cellular imaging.159 Unlike previous methods that either required external oxygen exclusion or pre-prepared aggregates to limit oxygen exposure, their strategy leverages in situ aggregation to enhance TADF emission. The team synthesized the luminophore PXZT, featuring a D–A structure with PXZ as the donor and terpyridine as the acceptor. PXZT exhibited promising emission characteristics and characteristic TADF behavior. Coordination of PXZT with Zn allowed for the formation of the ZnPXZT1 complex, which significantly enhanced ICT and quenched PXZT's intrinsic fluorescence, reducing background signals. The water-soluble ZnPXZT1 was utilized for time-resolved imaging in HeLa and 3T3 cells by the release of PXZT through dissociation within the cells. This method showed effective long-lived emission from PXZT during TRLI, demonstrating the practicality of ZnPXZT1 as a cellular imaging probe (Fig. 15) 37, 38. The oxygen sensitivity of PXZT was assessed by comparing its emission spectra in toluene under degassed and aerated conditions. The ZnPXZT1 complex proved to be highly effective in detecting ethylenediaminetetraacetic acid (EDTA) with a remarkable 2000-fold increase in fluorescence intensity (Fig. 16). The use of ZnPXZT1 for time-resolved imaging in HeLa and 3T3 cells successfully minimized background signals. This new method, based on dissociation reactions and aggregation processes, offers a practical and efficient platform for time-resolved luminescence imaging compared to previous techniques. This approach, which enhances delayed fluorescence through aggregation, is expected to advance the development of TADF emitters for time-resolved imaging applications.
 | ||
| Fig. 15 Proposed mechanism for regulation of TADF using Zn2+. Reproduced from ref. 159, https://doi.org/10.1039/C8SC01485J, under the terms of the CC BY-NC 3.0 license, https://creativecommons.org/licenses/by-nc/3.0/. | ||
 | ||
| Fig. 16 HeLa cells incubated with 10 μM ZnPXZT1 visualized using (a–c) steady-state imaging, (d) luminescence lifetime imaging, and (e and f) time-gated imaging. Reproduced from ref. 159, https://doi.org/10.1039/C8SC01485J, under the terms of the CC BY-NC 3.0 license, https://creativecommons.org/licenses/by-nc/3.0/. | ||
In 2020, Hu and colleagues developed a novel time-resolved fluorescence nanoprobe, AI-Cz-NP, by self-assembling a single-component amphiphilic monomer, AI-Cz-AM.163 This monomer integrated a hydrophilic chain with a positively charged terminal into the lipophilic TADF emitter, AI-Cz (Fig. 17) 33–39. The resulting AI-Cz-NP, formed in aqueous solutions, demonstrated water solubility and biocompatibility due to its amphiphilic nature. The nanoprobe's positive surface charges enhanced cellular uptake, while the aggregation of TADF units within AI-Cz-NP reduced oxygen diffusion to the core, minimizing triplet state quenching. This design aimed to provide prolonged fluorescence lifetimes even in oxygen-rich environments, making AI-Cz-NP suitable for TRLI.
 | ||
| Fig. 17 Structure of amphiphilic TADF monomer 39 and the synthesis of TADF nanoprobe AI-Cz-NP. Reproduced from ref. 163 with permission from the Royal Society of Chemistry. | ||
Three new D–A type TADF materials, PXZ-NI (40), PTZ-NI (41), and Lyso-PXZ-NI (42), were synthesized by Nguyen and colleagues in 2020.165 These materials have aggregation-induced TADF features that make them appropriate for time-resolved imaging and fluorescence in living cells (Fig. 18A) 40–42. Temperature-dependent time-resolved photoluminescence (TRPL) tests revealed that 40, 41, and 42 had exceptionally low activation energy (Ea) values of 8.3, 13.2, and 7.8 meV, respectively (Fig. 18B). This research gave rise to a viable new path toward the development of TADF materials with delayed fluorescence caused by aggregation.
 | ||
| Fig. 18 (A) Structures of TADF organic materials PXZ-NI (40), PTZ-NI (41), and Lyso-PXZ-NI (42). (B) Temperature-dependent TRPL signals for (a) PXZ-NI (40), (b) PTZ-NI (41), and (c) Lyso-PXZ-NI (42), and corresponding Arrhenius plots for the TRPL signals for (d) PXZ-NI (40), (e) PTZ-NI (41), and (f) Lyso-PXZ-NI (42). Reproduced from ref. 165 with permission from American Chemical Society, Copyright © 2020. | ||
TADF probes are widely recognized in bioimaging, yet most offer only a single emission signal. This reduces the amount of biological information that can be conveyed as well as compromising fluorescence intensity in complex microenvironments. To overcome this challenge, Zhu and colleagues fabricated a dual M–1 TADF probe featuring an asymmetrical D–A–D′ configuration.169 This innovative design provides complementary dual TADF-band characteristics, significantly reducing twists in TRLI (Fig. 19a). Dual-TADF emission is effective in both monomeric solutions and aggregated states. Encapsulation of the M–1 TADF probe in the amphiphilic block copolymer Pluronic F-127 resulted in nanoprobes that mitigate oxygen quenching, enhancing TRLI (Fig. 19b). HeLa cells treated with the nanoprobe showed mean PL lifetimes of 33 μs in the DAPI channel and 36 μs in the FITC channel, respectively (Fig. 19c). This dual-channel capability allows for adjusting signal intensities through TRLI parameters. Dual-channel mean lifetime mapping showed a 30–40% decrease in TRLI distortion compared to single-channel mapping, thereby improving the use of TADF materials in medicine (Fig. 19d).
 | ||
| Fig. 19 (a) The molecular scaffold of M–1 showcases dual TADF emission in the monomeric and aggregated states. (b) Pretreatment involves encapsulating M–1 within NPs (c) TRLI imaging in HeLa cells (d) Integration of TRLI data from dual channels significantly reduces distortion by 30–40%, improving imaging precision. Reproduced from ref. 169 with permission from John Wiley & Sons, Copyright © 2020, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
In 2020, Huang's team incorporated organic afterglow emission properties into the TADF framework, achieving up to 45% afterglow efficiency and enhancing the photophysical properties of TADF compounds for biological imaging, as shown in Fig. 20a and b. They developed a twisted D–A system with DCzB by combining difluoroboron β-diketonate with carbazole. This structure enabled a tri-modal organic afterglow that depends on the thermally activated afterglow (TAA) mechanism, which includes: (1) organic ultralong room temperature phosphorescence (OURTP) from the  state; (2) delayed phosphorescence (DP) through thermally activated exciton release (TAER) due to a narrow exciton trapping depth (ETD); and (3) trapping of S1 excitons by DP via RISC with a small ΔEST. DCzB nanoparticles were created by encasing DCzB in the phospholipid F127 (Fig. 20c and d). Phosphorescence lifetime imaging (PLIM) and TRLI revealed that the DCzB nanoparticles were well-suited for higher specificity for cell imaging, exhibiting a longer lifetime for phosphorescence, approximately 500 μs (Fig. 20e). Additionally, DCzB's temperature-sensitive afterglow color change was explored for visual temperature detection, showing a transition from blue-green to green-yellow in crystals upon cessation of UV light emission, while the DCzB powder exhibited minimal color change from 300 K to 77 K (Fig. 20f).170
state; (2) delayed phosphorescence (DP) through thermally activated exciton release (TAER) due to a narrow exciton trapping depth (ETD); and (3) trapping of S1 excitons by DP via RISC with a small ΔEST. DCzB nanoparticles were created by encasing DCzB in the phospholipid F127 (Fig. 20c and d). Phosphorescence lifetime imaging (PLIM) and TRLI revealed that the DCzB nanoparticles were well-suited for higher specificity for cell imaging, exhibiting a longer lifetime for phosphorescence, approximately 500 μs (Fig. 20e). Additionally, DCzB's temperature-sensitive afterglow color change was explored for visual temperature detection, showing a transition from blue-green to green-yellow in crystals upon cessation of UV light emission, while the DCzB powder exhibited minimal color change from 300 K to 77 K (Fig. 20f).170
 | ||
| Fig. 20 (a) Illustration of TAA emission mechanism involving TAER RISC processes. (b) Structure of DCzB. (c) DCzB NPs prepared by encapsulating DCzB. (d) DLS & TEM images of the DCzB NPs. (e) Treatment of PLIM and TRLI of cells in the presence of DCzB NPs. (f) Images of DCzB in powdered form after UV irradiation (365 nm) at various temperatures. Reproduced from ref. 170, https://doi.org/10.1038/s41467-020-14669-3, under the terms of the CC BY 4.0 license, https://creativecommons.org/licenses/by/4.0/. | ||
Zhou and colleagues have made significant advancements in the field of pure organic two-photon excited room temperature phosphorescent (RTP) materials with extended emission lifetimes and high resolution, which were desirable properties for time-resolved imaging (Fig. 21).171 These aromatic carbonyl-containing materials exhibit both AIE and RTP properties. The incorporation of aromatic carbonyl groups alters the LUMO, resulting in a substantial Stokes shift and enhanced spin–orbit coupling, which facilitates intersystem crossing. For aggregation-induced phosphorescence (AIP), aggregation increases the ΔE between S1 and T1 states, thereby reducing the rate of RISC in the solid state. This aggregation also suppresses nonradiative decay and promotes the emission of phosphorescence. This research affords crucial insights for the synthesis of AIP materials. These compounds show promise as two-photon excitation–aggregation-induced phosphorescence (TPE-AIP) materials for time-resolved two-photon excited luminescence imaging applications.
 | ||
| Fig. 21 (A) Two-photon absorption spectra of TePP, TPM, TPM-CL, 2TPA-DPM, and 2TPA-DPM-CL. (B) (a) One-photon absorption spectra, as well as (b) the one-photon absorption (Abs), fluorescence (FL), and phosphorescence (PH) spectra, with arrows showing the Stokes shift for compounds TPM and TPM-CL in various states. (C) Structures of the investigated compounds. Reproduced from ref. 171 with permission from American Chemical Society, Copyright © 2022. | ||
Yang and colleagues introduced a novel in situ aggregation-induced delayed fluorescence (AIDF) technique for time-resolved luminescence sensing (TRLS).172 They developed a new luminophore, FAc-Py, which exhibits a highly twisted structure and AIDF properties, enabling emission with a long lifetime suitable for time-resolved applications (Fig. 22A) 43–48. A derivative, FAc-Py-Ester, was also synthesized. While non-emissive and liposoluble, it can be used for DF via turn-on sensing of carboxylesterase. This enzyme facilitates the hydrolysis of FAc-Py-Ester, releasing the hydrophobic FAc-Py, which in turn activates the AIDF emission that is insensitive to air. FAc-Py-Ester was used as a luminophore in living cells to assess both the carboxylesterase sensing capability and recovery of delayed fluorescence. HeLa cells treated with various concentrations of FAc-Py-Ester for two hours exhibited significant green luminescence. Given that cellular autofluorescence lifetimes are typically under 8 ns, short-lived fluorescence was excluded using delay times of 20 and 30 ns. Luminescence signals with lifetimes exceeding these thresholds were concluded to be due to the long-lived DF of aggregated FAc-Py, which is likely a result of carboxylesterase-mediated release of FAc-Py of FAc-Py-Ester during cellular imaging (Fig. 22B).
 | ||
| Fig. 22 (A) Preparation of FAc-Py (45) and FAc-Py-Ester (48). (B) Time-gated imaging of carboxylesterase activity in HeLa cells incubated with varying concentrations of FAc-Py-Ester (48). Reproduced from ref. 172 with permission from Elsevier, Copyright © 2022. | ||
Although DF research has grown in importance for biological imaging, it is still difficult to develop small molecule DF probes sensitive to certain analytes. To create analyte-responsive DF probes, Zhang et al. presented a novel technique called excited-state intramolecular proton transfer-delayed fluorescence (ESIPT-DF), as shown in Fig. 23A.42 By using different donor moieties with the ESIPT-based fluorophore 2-(2′-hydroxyphenyl)benzothiazole (HBT) as an acceptor and creating twisted donor–acceptor pairs inside each molecule, their method allowed them to develop ESIPT-DF luminophores. The compounds HBT-PXZ and HBT-PTZ were particularly noteworthy since they showed strong ESIPT and DF properties. Their solid-state lifetimes were 3.65 μs and 5.37 μs, respectively. Furthermore, as seen in Fig. 23B, a caged probe was created by including an ESIPT-DF blocking agent together with a hydrophilic D-galactose group, resulting in the probe HBT-PXZ-Ga, which was used for the specific identification of the enzyme β-galactosidase (β-gal). Using this probe, the impact of metal ions on the activity of surface β-gal in the bacteria Streptococcus pneumoniae was examined. According to the study, Zn2+ ions reduced β-gal activity by 68%. Through modification of the functional group, while retaining the main structure, this TADF “turn-on” method may be modified for the detection of different analytes.
 | ||
Fig. 23 (A) Schematic illustration of the synthesis and β-gal sensing mechanism of HBT-PXZ-Ga. (B) Fluorescence lifetime imaging of HBT-PXZ-Ga-treated S. pneumoniae by (a) fluorescence intensity and (b) lifetime. Excitation wavelength λex = 405 nm (25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Hz) Reproduced from ref. 42 with permission from American Chemical Society, Copyright © 2023. 000 Hz) Reproduced from ref. 42 with permission from American Chemical Society, Copyright © 2023. | ||
Wu and colleagues prepared red TADF nanoparticles designed for real-time TRLI, overcoming challenges related to low efficiency and short lifetimes.173 The study involved doping TPAAQ into a CBP (4,4′-bis(carbazol-9-yl)biphenyl) host matrix, as demonstrated in Fig. 24A. At low TPAAQ concentrations, the nanoparticles displayed significant TADF characteristics, achieving luminescence lifetimes greater than 0.1 ms and a PLQY of up to 35% (Fig. 24B). These nanoparticles substantially improved both luminescence efficiency and lifetime, with TADF lifetimes extending to several hundred microseconds. This enabled rapid LLIM using a time-resolved luminescence microscope. The efficient luminescence facilitated in vivo time-gated luminescence imaging, effectively eliminating interference from biological autofluorescence, as illustrated for an example zebrafish subject (Fig. 24C–E). This technique allowed for both long-lived luminescence and cost-effective imaging, which provides opportunities for real-time detection of biological processes in various organisms, even in the presence of high autofluorescence.
 | ||
| Fig. 24 (A) (a) Representation of TPAAQ/CBP/CPP TADF nanoparticles (b) DLS histograms showing the size of the nanoparticles and inset TEM images showing their morphology. (c) Photographs of the nanoparticle solutions under UV irradiation. (B) Time-gated emission spectra of the nanoparticles at various delay times. (C) Imaging of zebrafish incubated with TPAAQ/CBP/CPP nanoparticles, where the bright field is displayed on the left side of the figure, the steady-state luminescence is shown in the middle, and the time-gated luminescence is displayed on the right. Reproduced from ref. 173 with permission from John Wiley & Sons, Copyright © 2024, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
Zhou and colleagues investigated aggregation-regulated TADF. They proposed new methodologies and strategies for designing two-photon fluorescent TADF materials tailored for time-resolved biological imaging, as depicted in Fig. 25.174 They found that aggregation impacted the internal conversion decay rate and affected both the ΔEST and rate of ICS. By carefully selecting substituent groups, they suggested that materials with conventional fluorescence could be engineered into long-lifetime TADF or phosphorescent materials, offering a method for creating extended-afterglow materials. To improve TADF properties for in vivo time-resolved imaging, they developed a new two-photon excited TADF material, NI-metaCz, incorporating π-extension in its fluorophore structure. NI-metaCz exhibited a significant two-photon absorption cross-section of 112 GM at 775 nm, which is suitable for biological applications, as well as nearly 100% TADF efficiency, a significant Stokes shift (153 nm), and an extended fluorescence lifetime (50.75 μs). These features make NI-metaCz a highly promising candidate for time-resolved two-photon fluorescence imaging for biological detection.
 | ||
| Fig. 25 (A) Design of aggregation-regulated TADF materials (B) diverse architectures of aggregation-regulated TADF materials (C) spectra for (a) one-photon absorption and (b) one-photon fluorescence (c) one-photon absorption and fluorescence of the fluorophores and (d) two-photon absorption. All investigated scaffolds were measured in aqueous solution. Reproduced from ref. 174 with permission from American Chemical Society, Copyright © 2023. | ||
Hudson and coworkers synthesized luminescent glassy organic dots (g-Odots), as potential bioimaging nanoparticles due to their exceptional luminescence intensity and stability afforded by their rigid glassy structure (Fig. 26A).175 The emission wavelength of g-Odots can be easily adjusted by encapsulating different hydrophobic fluorophores within the glassy matrix. This versatility is particularly beneficial in combination with phosphorescent or TADF dyes, enabling time-gated fluorescence imaging while protecting the emitters from oxygen. However, controlling the size of g-Odots, a critical factor for cellular uptake and metabolism, has not been thoroughly explored. In this study, methods for controlling g-Odot size while maintaining their glassy interior were investigated. The approaches included precipitation into “aggregated Odots” (a-Odots), then annealing the glassy host material post-synthesis to maintain the glass transition temperature (Tg) of the particles, and adjusting the host-to-surfactant ratios to achieve tunable particle sizes. These findings enhanced the tunability of g-Odot sizes for bioimaging experiments, where narrow size distributions and small average sizes are crucial (Fig. 26B). Additionally, these probes have potential applications beyond cellular imaging, including imaging vascular structures or in lateral flow assays for point-of-care diagnostics, where robust bright particles are required.
 | ||
| Fig. 26 (A) (Left) Synthetic procedure for g-Odots, showing a suspension of particles under 365 nm excitation in water (5.3 × 109 particles per mL). (Right) SEM image of obtained particles: 2.00 kV accelerating voltage, 40240× magnification, 300 nm scale bar (B) SEM images of (A) g-Odots synthesized by the standard oil-in-water emulsion method, (B) a-Odots prepared by nanoprecipitation, and (C) annealed a-Odots. Reproduced from ref. 175 with permission from American Chemical Society, Copyright © 2024. | ||
Platinum(II) and palladium(II) benzoporphyrin complexes with alkylsulfone groups have been synthesized from inexpensive materials by Borisov et al. (2017).176 Attaching alkylsulfone groups to meso-tetraphenyltetrabenzoporphyrin (TPTBP) significantly improves its solubility in organic solvents, photostability, and electron-deficiency, resulting in a redox potential increase of up to 0.65 V compared to the unmodified porphyrin. These modified dyes show strong absorption in the blue (440–480 nm) and red (620–650 nm) regions and exhibit intense room-temperature phosphorescence in the near-infrared (NIR) spectrum, with quantum yields reaching up to 30% in solution, as illustrated in Fig. 27. Palladium(II) complexes of these dyes display efficient TADF at room temperature, with TADF efficiency rising with temperature to a maximum of 27% at 120 °C for the Pd–O–S and Pd–T–I complexes. The TADF observed is notably stronger than that of advanced porphyrin complexes due to a reduced singlet–triplet energy gap. The ratiometric temperature dependence of the TADF emission allows for dual temperature and oxygen sensing using a single emitter. The photophysical properties of these porphyrins render them suitable for various photonic applications, including organic light-emitting diodes (OLEDs), photovoltaics, and two-photon imaging of oxygen in biological samples. The TADF feature is particularly advantageous for imaging, as it separates the emission spectrum from the NIR excitation spectrum. This prevents overlap between the excitation and emission spectra, which is a limitation of current phosphorescent probes.
 | ||
| Fig. 27 Absorption and emission spectra of (a) Pt(II) and (b) Pd(II) complexes. (c) Pd–O–S emission spectra in anoxic toluene. (d) Schematic illustration of the photoluminescence processes of the tested complexes. Reproduced from ref. 176 with permission from American Chemical Society, Copyright © 2017. | ||
Hudson and coworkers developed a series of TADF-emitting monomers, achieving quantum yields up to 96% and a range of colors from blue to orange (see Fig. 28).177 These monomers contained an oxadiazole core as the acceptor and were polymerized using Cu(0)-mediated reversible deactivation radical polymerization (Cu(0)-RDRP). This technique resulted in copolymers with low dispersity (1.10–1.45) and high molecular weights (Mn > 20 kDa) using a copper wire catalyst at room temperature. The design incorporated structural constraints to reduce donor–acceptor orbital overlap, which facilitated TADF in the materials. Among the synthesized dendrimer materials, dimethylacridine, phenothiazine, phenoxazine, and carbazole exhibited strong delayed fluorescence, while methylphenazine showed weaker emissions. The phenothiazine-based emitter demonstrated dual emission properties suitable for ratiometric oxygen sensing at 516 nm relative to 396 nm for varying oxygen concentrations. The results demonstrate how the emission intensity at 516 nm changes with different levels of oxygen, showcasing the material's sensitivity to oxygen concentration. The researchers also created water-soluble polymer dots by coprecipitation with amphiphilic surfactants. The photophysical properties of these polymer dots in the precipitated state were similar to those in the solid state, displaying ratiometric oxygen sensing and longer fluorescence lifetimes. Overall, these oxadiazole-based polymers present a flexible platform for developing multicolor TADF materials using a simple, scalable approach.
 | ||
| Fig. 28 (Left) TADF polymer dots (PDots) with the green-emitting PTZ-ODA0.15 (in green) and blue-emitting PSMA components (in blue). (Right) The emission intensity of PTZ-ODA0.15 at 516 nm relative to 396 nm was measured under varying oxygen concentrations. Reproduced from ref. 177 with permission from American Chemical Society, Copyright © 2020. | ||
Hudson and collaborators also developed a TADF monomer by integrating a naphthalimide (NAI) acceptor along with a DMAC (dimethylacridine) donor, resulting in TADF emission ranging from orange to deep red.179 The copolymerization of NAI-DMAC monomer (shown in red) with tBuODA (shown in blue) and a matrix of N-isopropylacrylamide (NIPAM) was employed to prepare a series of thermoresponsive polymers (Fig. 29A). Using Cu(0)-RDRP, they produced TADF-emitting polymers with quantum yields up to 31% in the solid state and 58% in the solution. These dual-emission polymers were capable of switching from red TADF to blue fluorescence by changing the temperature from room temperature to 70 °C, with a ratiometric thermal sensitivity of 32 ± 4% K−1 (Fig. 29B). These characteristics make these red TADF polymers suitable for applications in temperature sensing, drug delivery vehicles, biosensing and bioimaging.
 | ||
| Fig. 29 (A) Illustration of the structure of NAI-based TADF polymers P14 and P15 and their colors at temperatures between 10–80 °C. (B) Emission spectra for polymers P14 and P15 at a range of temperatures. (C) The fluorescence intensity ratio I460/I660 versus temperature for both P14 and P15. (D) CIE chromaticity plot for polymer P14. Reproduced from ref. 179 with permission from American Chemical Society, Copyright © 2020. | ||
Hu and colleagues created a water-soluble TADF probe named AI-Cz-Neo by attaching a TADF emitter to neomycin, which specifically targets bacterial 16S ribosomal RNA.180 This probe (49) was effectively used for dual-mode detection of bacterial 16S rRNA, employing both conventional fluorescence imaging and fluorescence lifetime imaging. The probe could be detected in Staphylococcus aureus bacteria via confocal fluorescence microscopy and TRFI in cells and tissues (Fig. 30). This innovation provides a dependable method for diagnosing bacterial infections through dual-mode imaging.
 | ||
| Fig. 30 Schematic diagram depicting synthesis and dual-mode detection of AI-Cz-Neo to image bacterial 16S rRNA in tissue samples. Adapted from ref. 180 with permission from American Chemical Society, Copyright © 2020. | ||
Zhu and colleagues recently developed a 3D ratiometric luminescent sensor by incorporation of a D–A–D′ moiety.181 These TADF molecules are capable of emitting both fluorescence and DF phenomena. This sensor distinguishes environmental polarity by varying the TADF wavelength and lifetime while keeping fluorescence constant. To assess the sensor's practical diagnostic potential, they fabricated simulated membranes with self-assembly into micelle nanostructures by phospholipids (PLs) and cholesterol (Chol) with TADF-3, while varying the Chol content to alter the polarity. The luminescence of TADF-3 in these micelles enabled quantitative detection of polarity changes, as illustrated in a 3D plot diagram (Fig. 31). This TADF-based sensing approach was also validated at the cellular level, demonstrating its potential for detecting cholesterol-related membrane abnormalities.
 | ||
| Fig. 31 (Top) Design of TADF-3-based nanostructured membranes with varying cholesterol contents to self-assemble into micelles containing phospholipids (PLs) and cholesterol (Chol). (Bottom) A 3D plot diagram of sensing using TADF-3-based micelles within simulated membrane environments. Reproduced from ref. 181, https://doi.org/10.1038/s41467-019-08684-2, under the terms of the CC BY 4.0 license, https://creativecommons.org/licenses/by/4.0/. | ||
Lee and colleagues have pioneered organic nanostructures incorporating TADF emitters featuring a small ΔEST, which facilitates efficient ISC and were employed to generate photo-excited singlet oxygen (1O2).195 They prepared donor–acceptor composite NPs by combining two non-TADF donors with an acceptor. While both NPs exhibited high exciplex emission, only one demonstrated TADF, leading to enhanced 1O2 production. This research shows that D:A composite NPs with TADF properties can be derived from non-TADF components, expanding the range of available TADF materials without additional synthesis. In vitro MTT assays confirm the potential of these TADF-based nanostructures for photodynamic therapy (PDT). These findings suggest that water-dispersible TADF nanostructures could significantly advance the development of novel photosensitizers for biomedical applications (Fig. 32a and b).
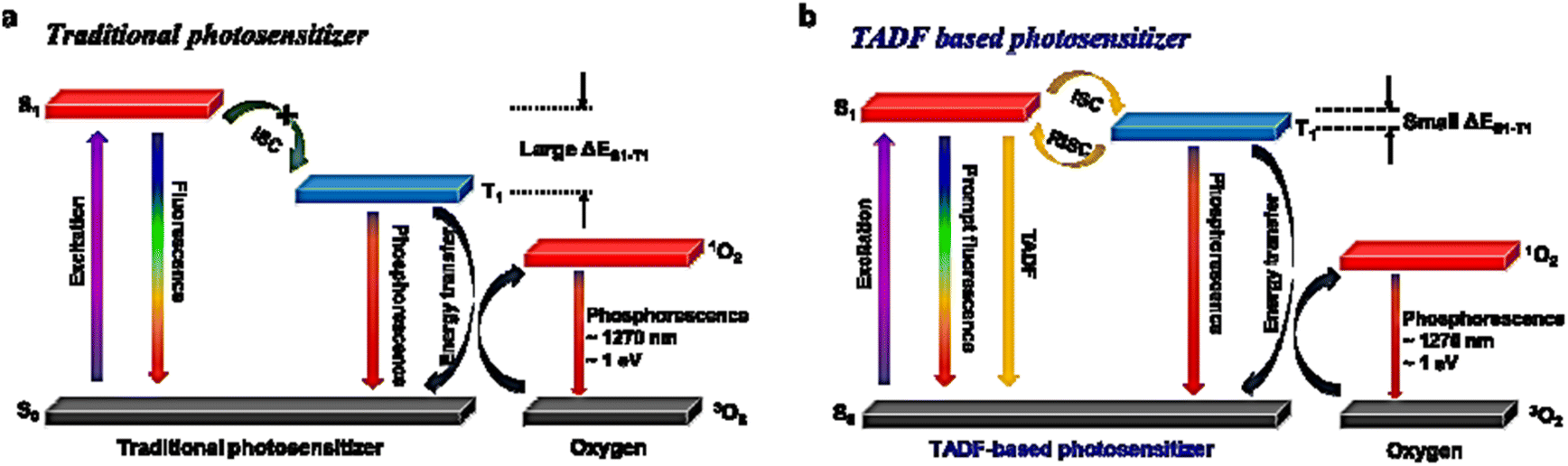 | ||
| Fig. 32 Modified Jablonski diagram illustrating singlet oxygen production by (a) conventional photosensitization and (b) TADF photosensitization. Reproduced from ref. 195 with permission from the Royal Society of Chemistry. | ||
To address challenges such as inadequate singlet oxygen (1O2) quantum yields, limited optical penetration depth, and insufficient organelle targeting in photodynamic therapy (PDT), Lee and colleagues explored TADF emitters with small ΔEST for developing high-performance, metal-free photosensitizers.196 Their research introduced two types of TADF emitter-based nanoparticles (TADF NPs) specifically designed for two-photon PDT and fluorescence imaging, as illustrated in Fig. 33A. These TADF NPs offer several key advantages: (1) a high quantum yield (52%) for 1O2, (2) enhanced penetration of NIR due to two-photon activation and (3) effective targeting of mitochondria without additional modifications. These properties enable significant production of reactive oxygen species (ROS) localized to the mitochondria and effective cancer cell cytotoxicity at low light intensities. The development of multifunctional single molecule-based NPs with TADF emission represents a significant advancement in PDT, offering new avenues for creating potent photosensitizers for biomedical applications. As shown in Fig. 33B, two-photon excitation (TPE) imaging of TADF NPs in A549 lung tumor cells exhibited enhanced NIR light penetration relative to single-photon excitation imaging. Additionally, the ability of the TADF NPs to specifically target mitochondria enhances the effectiveness of PDT in cancer therapy.
 | ||
| Fig. 33 (A) Diagram depicting the design of TADF NPs for TPE fluorescence imaging and PDT (B) single-photon excitation and TPE penetration depth images of A549. Adapted from ref. 196 with permission from American Chemical Society, Copyright © 2019. | ||
Lysosomes have emerged as a significant target for improving the effectiveness of PDT. The ROS produced during PDT can damage lysosomes, causing the release of various acid hydrolases. This disruption triggers programmed cell death known as apoptosis in cancer cells, enhancing the overall therapeutic impact of PDT.197–199 The Peng group synthesized two TADF fluorescein scaffolds, 50 and 51, for targeted fluorescence imaging and PDT as shown in Fig. 34A and B.198 These TADF compounds, which specifically target lysosomes, exhibited markedly higher PDT efficacy compared to traditional porphyrin-based photosensitizers even under mild hypoxic conditions. In the absence of NTR inhibition by dicoumarin, 51 is enzymatically converted by nitroreductase (NTR) into 52, which then activates fluorescence in tumor cells. Additionally, 51 demonstrated remarkable PDT performance and high biosafety in HeLa tumor-bearing mice, as seen in Fig. 34D.
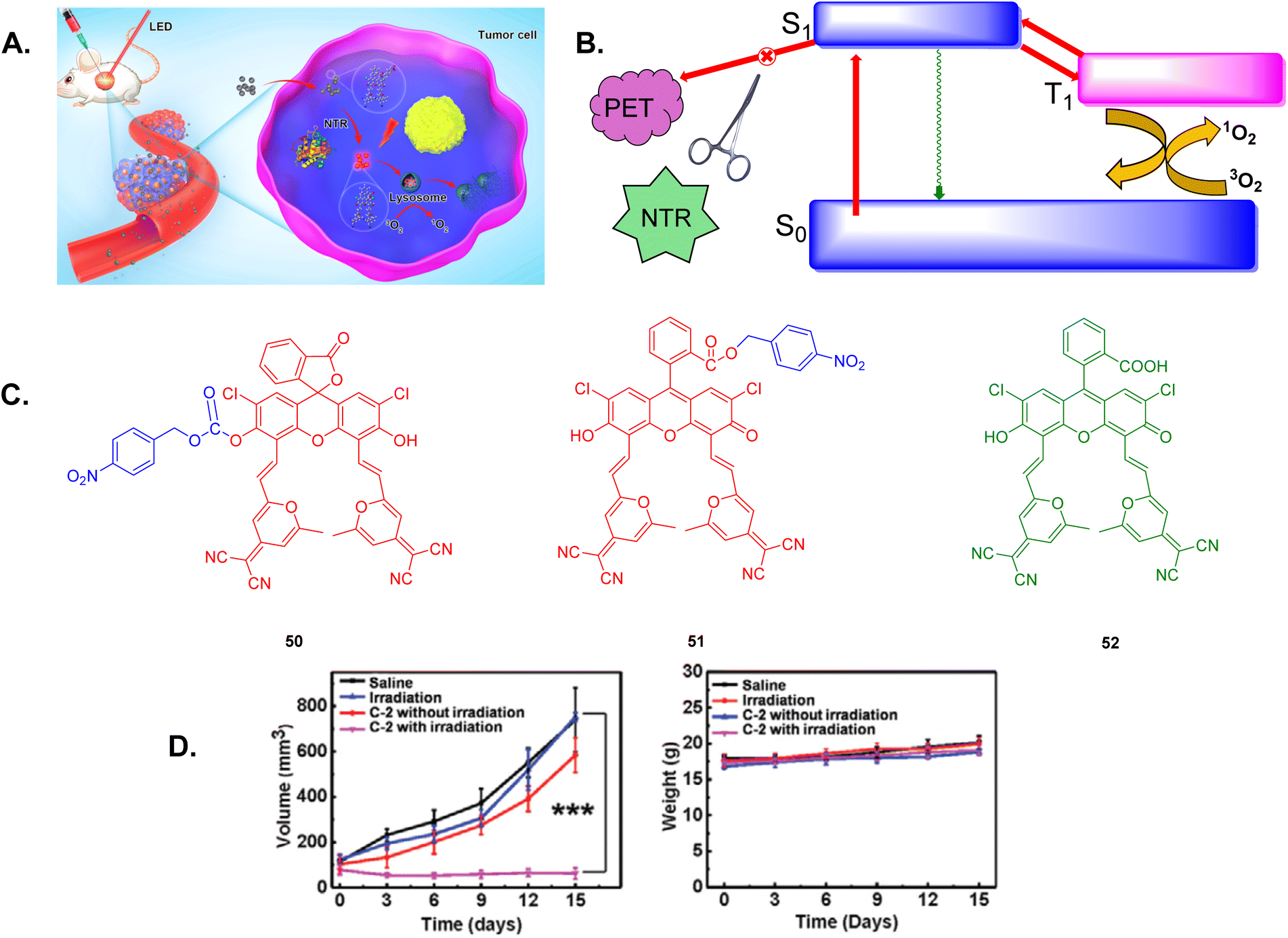 | ||
| Fig. 34 (A) Diagram depicting TADF fluorescein scaffolds for fluorescence imaging and PDT. (B) PDT process of the TADF in theranostics molecule. (C) Structure of the chemical scaffolds of 50, 51, and 52. (D) PDT effectiveness of 51 in mice.198 Reproduced from ref. 198 with permission from American Chemical Society, Copyright © 2019. | ||
By optimization of exciton dynamics for adjustment of energy distribution in TADF NPs with D–A configurations, their photochemical characteristics can be improved to balance fluorescence imaging and PDT. Lee and colleagues developed two types of TADF nanoparticles, PT NPs and AT NPs, each of which possessed different electron donating groups for modification of ΔEST and oscillator strength (f). When incorporated into nanotheranostic agents, PT and AT NPs demonstrated distinct efficiencies in two-photon excitation (1O2) generation and fluorescence emission (Fig. 35A–C). Specifically, PT NPs, with a smaller ΔEST and higher f, exhibited superior PDT effectiveness due to more efficient ISC compared to AT NPs, which displayed stronger fluorescence intensity. These nanoparticles could be used to design TADF nanotheranostics optimized for either high-performance PDT or enhanced fluorescence imaging by adjusting ΔEST and f in TADF materials.200
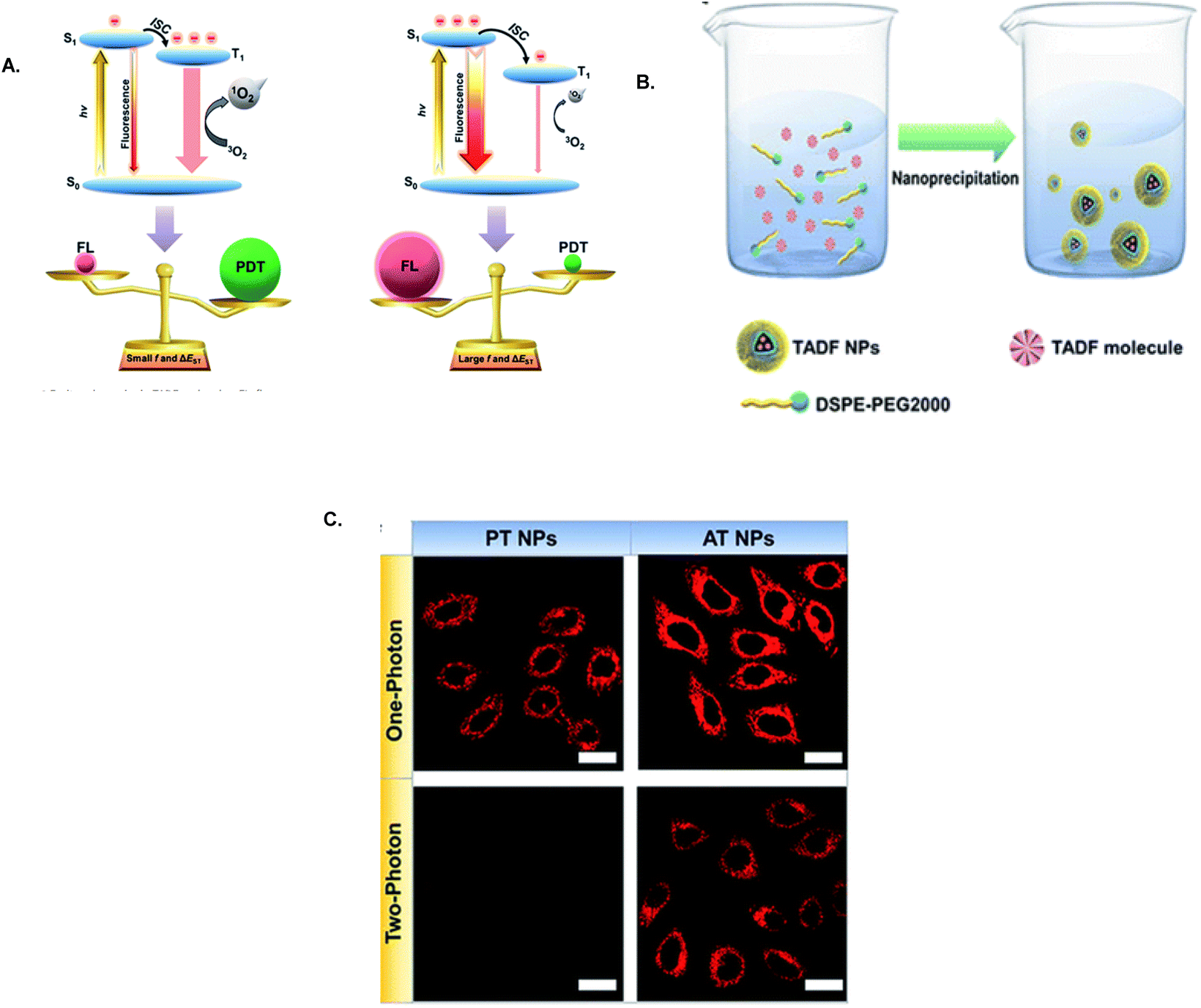 | ||
| Fig. 35 (A) Illustration of exciton dynamics in TADF-NPs. (B) Preparation of TADF nanoparticles (NPs). (C) SPE and TPE images. Reproduced from ref. 200, https://doi.org/10.1039/C9SC05817F, under the terms of the CC BY 4.0 license, https://creativecommons.org/licenses/by/4.0/. | ||
For improvement in the efficacy of TADF materials in PDT, Lee and his team incorporated the effect of heavy atoms in TADF molecule AQCz to create AQCzBr2 photosensitizers, as depicted in Fig. 36.201 Both AQCz 53 and AQCzBr2 54 exhibit a small ΔEST of 0.11 eV. However, AQCzBr2 54, due to the heavy-atom effect, had a spin–orbit coupling constant approximately three times that of AQCz. This increased spin–orbit coupling enhanced the ISC process, resulting in a much higher 1O2 quantum yield for AQCzBr2 54 NPs compared to AQCz NPs (91% vs. 21%). The AQCzBr2 54 NPs achieved a remarkable 91% 1O2 quantum yield and demonstrated safety and effectiveness as both an in vitro and in vivo PDT agent. These findings highlighted that the incorporation of heavy atoms into TADF photosensitizers with inherently small ΔEST gap is a successful strategy for developing highly efficient photosensitizers, offering valuable insights for future advancements in PDT.
 | ||
| Fig. 36 Chemical structures and 1O2 generation process diagrams of AQCz and AQCzBr2. Adapted from ref. 201 with permission from the Royal Society of Chemistry. | ||
Yan and colleagues designed a new TADF material, BCzSFB, by combining the electron acceptor difluoroboron β-diketonate with the electron donor carbazole, linked through a π-bridged styrene unit, as illustrated in Fig. 37.202 This D–π-A–π-D structure, approximately 2.4 nm in length, features an exceptionally small ΔEST of 0.08 eV, a wide absorption spectrum ranging from 350 to 650 nm, and an extended near-infrared (NIR) TADF lifetime when dispersed in aqueous nanoparticles. The singlet oxygen (1O2) quantum yield of BCzSFB was calculated to be 62%, surpassing that of commercial photosensitizers such as eosin blue and methylene blue. Additionally, it effectively targets and destroys Gram-positive bacteria such as S. aureus, with ∼100% of cells within 10 minutes under low-intensity white light (60 mW cm−2).
 | ||
| Fig. 37 (a) Schematic representation of a TADF-based photosensitizer for the generation of 1O2. (b) Structure of the BCzSFB nanomolecule. (c) Computationally derived HOMO and LUMO distributions. Reproduced from ref. 202 with permission from the Royal Society of Chemistry. | ||
Type I photosensitization addresses the challenge of reduced photodynamic therapy (PDT) efficacy in hypoxic tumors by enhancing electron transfer capabilities. Song and colleagues have demonstrated that encapsulating a TADF photosensitizer (PS) with bovine serum albumin (BSA) significantly boosts Type I PDT, leading to increased generation of superoxide anions (O2˙−) as depicted in Fig. 38.203 In this approach, BSA acts as an “electron reservoir” while the PS functions as an “electron pump.” By combining these functions into a single system, they developed the nano photosensitizer PS@BSA. This Type I PDT system effectively targets tumors under hypoxic conditions and shows promising results in tumor-bearing mice. This strategy provides a significant advancement in improving PDT efficiency for hypoxic tumors.
 | ||
| Fig. 38 Illustration of the Type I PDT process. Reproduced from ref. 203 with permission from American Chemical Society, Copyright © 2023. | ||
Tang and colleagues developed a TADF-based zirconium(IV) complex, Zr(MesPDPPh)2, which exhibited unexpected aggregation-induced emission enhancement (AIEE).204 This complex demonstrated long-lived luminescence even in air, likely due to reduced oxygen exposure caused by its aggregation, as described in Fig. 39. This theory was supported by DFT calculations, which indicated that the oscillator strengths remained consistent despite variations in the dihedral angles between the benzene groups and the central plane of the ligands. The emission intensity and lifetime of Zr(MesPDPPh)2 NPs were further improved after deoxygenation. Additionally, DFT calculations showed that the favorable ΔEST and spin–orbit coupling (SOC) rendered Zr(MesPDPPh)2 an effective sensitizer. The study confirmed that Zr(MesPDPPh)2 NPs could function in both Type I and Type II PDT and also act as a photoredox catalyst for NADH oxidation. This catalytic cycle, assisted by the protein cytochrome c, facilitated oxygen-free PDT under hypoxic conditions. In vivo studies demonstrated that Zr(MesPDPPh)2 NPs effectively inhibited tumor growth with good biocompatibility. This work highlights Zr(MesPDPPh)2 as a promising TADF-based photoredox catalyst for overcoming hypoxia in PDT and offers new insights for clinical applications.204
 | ||
| Fig. 39 Schematic representation of the TADF-based photocatalyst Zr(MesPDPPh)2 NPs and its use in PDT. Reproduced from ref. 204 with permission from American Chemical Society, Copyright © 2023. | ||
Photothermal therapy (PTT) with NIR-emitting conjugated polymers has shown significant capability for tumor treatment, but these polymers exhibited inadequate therapeutic efficacy in light of their weak NIR absorbance and low photothermal conversion efficiency (PCE). Zhou and colleagues developed an innovative approach to enhance the performance of NIR polymeric photosensitizers by integrating TPAAQ into the polymer backbone (Fig. 40).205 The resulting polymers, known as PEKBs, demonstrate improved PCE and strong NIR absorbance. Among these, PEKB-244, with an optimal proportion of TPAAQ units, exhibits the highest NIR absorbance and a PCE of 64.5%. Both in vitro and in vivo studies confirm that these NPs can efficiently destroy cancer cells after irradiation by an NIR laser. This new molecular design strategy promises to advance the design of effective NIR photosensitizers for photothermal therapy of cancer.
 | ||
| Fig. 40 Molecular scaffold of NPs, absorption spectra of PEKB-244 and PEKB-064, and an illustration depicting PEKB-244 NP development and phototheranostic uses. Reproduced from ref. 205 with permission from American Chemical Society, Copyright © 2023. | ||
Hudson and colleagues described the first synthesis of multifunctional copolymers containing both a TADF luminophore (DMAc) and an oxygen sensitizer (BDP) for theranostics applications.206 To tune the biological behavior of these materials, “stealthy” polyethylene glycol (PEG) and cell-penetrating guanidinium (BGN) repeat units were incorporated as hydrophilic corona blocks via ring-opening metathesis polymerization (ROMP) as shown in Fig. 41. This allowed for the straightforward combination of these components through sequential addition during polymerization. These amphiphilic copolymers can self-assemble into polymer dots (Pdots) that simultaneously shield the TADF luminophore from the environment while exposing the sensitizer, enabling both delayed emission and singlet oxygen generation. By localizing the BDP sensitizer (1O2 quantum yield = 76%) in the hydrophilic corona of the Pdots, this design achieves an IC50 with only ∼0.05–0.13 μg mL−1 of photosensitizer loading and can lead to over 96% cell death upon irradiation (30–50 min, 23 mW cm−2), while maintaining negligible dark toxicity for both PEG- and BGN-based Pdots. Remarkably, Pdots with PEG versus BGN coronas exhibited completely different cellular uptake results, despite the high efficiency of both types in killing cancer cells. Furthermore, the presence of the oxygen sensitizer did not affect the TADF properties of DMAc in the core of the Pdots, achieving orange-red delayed emission (ΦPL = 8–18%) even when exposed to air. The multifunctional BGN Pdots could be used to image HeLa cells following cellular uptake and showed the potential of these Pdots in time-gated spectroscopy to effectively eliminate the influence of background autofluorescence in bioimaging. The use of ROMP also provided a modular synthetic approach where both the emitter and photosensitizer can be selected to absorb and emit at wavelengths best suited for particular clinical applications. Future avenues for exploration include immunolabeling the PEG Pdots to target specific cells or tissues and altering the chemical composition of the parent copolymers to allow for the imaging probe and the oxygen sensitizer to be excited independently.
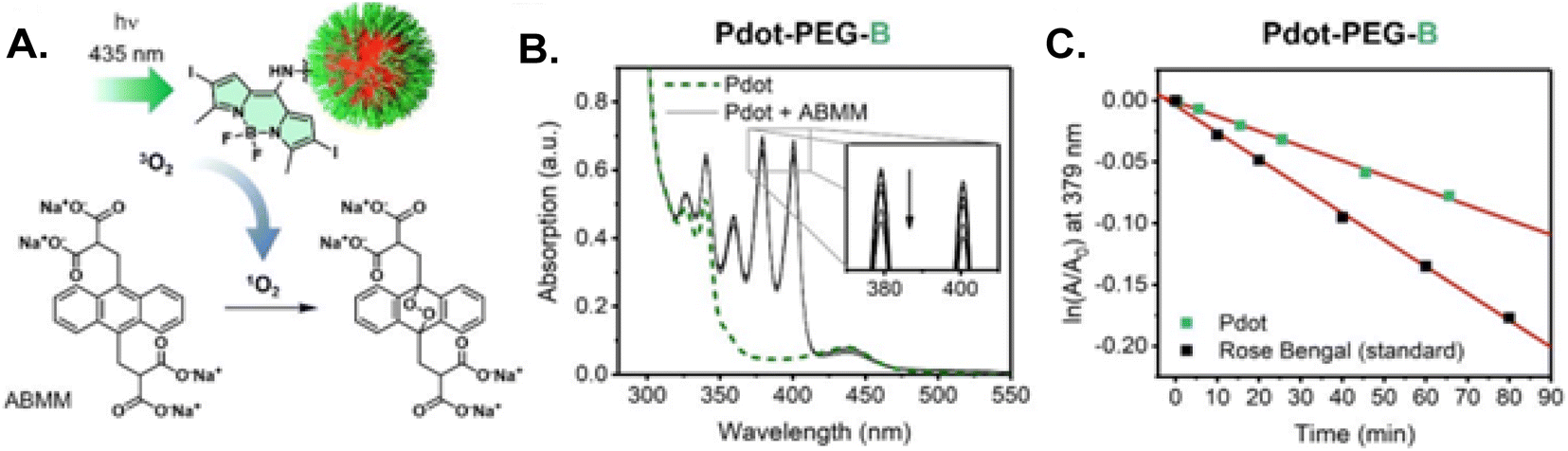 | ||
| Fig. 41 Determination of the 1O2 quantum yield of Pdot-PEG-B: (A) diagram illustrating the photoexcitation of the Pdots and the subsequent degradation of an anthracene-based chemical acceptor (ABMM); (B) UV/vis spectra of a mixture of ABMM and Pdot-PEG-B in water after various irradiation times; (C) comparison of the first-order kinetics of ABMM degradation (λmax = 379 nm) in the presence of either Pdot-PEG-B or Rose Bengal. Reproduced from ref. 206, https://doi.org/10.1002/anie.202400712, under the terms of the CC BY 4.0 license, https://creativecommons.org/licenses/by/4.0/. | ||
Barman and colleagues developed a donor–acceptor system featuring a carbazole donor and a 2H-chromene coumarin derivative as the acceptor, which exhibits both AIE and TADF in the solid state, as illustrated in Fig. 42.207 This system also included a core with a selective affinity for the coronavirus spike protein for antiviral activity against SARS-CoV-2. The team conducted comprehensive photophysical studies to explore the structure–activity relationship structure of this system, focusing on the effect of a cyclic sp3 spacer between the carbazole and the chromene. This spacer-induced TADF phenomenon had not been extensively investigated previously. Additionally, they modified the molecular framework to incorporate an emissive AIEgenic rotor donor, using cyano (–CN) and ester (–CO2Et) groups in the acceptor while preserving the overall structure. Their experimental and theoretical work revealed that TADF occurs through two isoenergetic states: 1CT–3CT and 1CT–3LE–3CT. To explore antiviral potential against SARS-CoV-2, they performed molecular docking simulations and selected the fused 2H-chromene coumarin core due to its strong binding with the virus spike protein. This research offers a novel approach to designing AIE-TADF-based antiviral agents by utilizing advanced concepts of aggregation and triplet-state harvesting in purely organic materials.
 | ||
| Fig. 42 Synthesis of CrCz and ECrCz starting from 9H-carbazole: (i) 4F-Ph-CHO, K2CO3, N,N-dimethylformamide (DMF), 130 °C (ii) CH2(CN)2, EtOH, reflux; (iii) ethyl cyanoacetate, EtOH, reflux; (iv) 4-hydroxycoumarin, Ni–Np, EtOH.207 | ||
4.2. Thermally delayed fluorescence materials as photocatalysts
Detailed applications of TADF material as a photocatalyst have previously been described by Zysman-Colman et al. in a comprehensive review in 2021.208 In this section, we will instead describe new research into applications of TADF materials as photocatalysts from 2021 to 2024.Upon light absorption, the photocatalyst transitions to the excited state PC*, allowing photoinduced electron transfer (PET) and photoinduced energy transfer (PEnT) between the excited photocatalyst and reactant, with the PET and single electron transfer (SET) mechanisms involved as shown in Fig. 43 for photocatalysts 62–66. When PET is active, the process is known as photoredox catalysis. Photocatalysts (PC) undergo selective photoexcitation in the presence of organic reactants, which leads to the formation of products. As many organic substrates are transparent in these regions, it is preferred to use a PC that absorbs in the visible or near-UV regions. Using PCs that require higher energy for photoexcitation increases the risk of direct homolytic bond cleavage in the PC as well as organic substrates, leading to undesired side reactions and reduced process efficiency. Maximizing the yield of excited state formation requires consideration of both the quantum yield and the absorption cross-section of the photocatalyst, which is influenced by its concentration as well as its molar extinction coefficient (ε). For the reactions that do not involve the radical chain mechanism, their quantum yield Φ(λ) ideally approaches 1 (100%) for a photocatalytic process as given in eqn (2).212
 | (2) |
 | ||
| Fig. 43 Photocatalytic cycles of a typical PC with general donor D and acceptor A. Reproduced from ref. 208, https://doi.org/10.1039/D1CS00198A, under the terms of the CC BY-NC 3.0 license, https://creativecommons.org/licenses/by-nc/3.0/. | ||
Additionally, the excited state PC* should have a lifetime on the nanosecond scale or greater to overcome the diffusion processes which lead to the formation of encounter complexes in biomolecular reactions.213–217
Upon photoexcitation, the S1 state is quickly filled. Radiative decay then takes place with the assistance of DF, defined as prompt fluorescence (PF) with a lifetime on the nanosecond scale, similar to typical fluorescent compounds. The competing pathways include ISC or the triplet layer non-radiative decay back to the S0 to populate the T0 to T1 states. Phosphorescence from the triplet state is spin-forbidden and not observed at room temperatures due to a large ΔES0−T1 energy gap (eqn (3)).
 | (3) |
However, RISC from T1 to S1 can occur at atmospheric temperatures as a result of low ΔEST, which can be thermally overcome, resulting in DF that manifests on a timescale similar to phosphorescent complexes, usually in the microseconds range.17 The RISC mechanism is facilitated by spin-vibronic coupling as depicted in Fig. 44. Fig. 45 shows that the rate constant for prompt fluorescence (kPF) is larger compared to those for ISC (kISC) and non-radiative decay from the singlet state (kSnr). DF occurs because kRISC is more rapid than both the radiative (kTr) and non-radiative (kTnr) decay rates from the triplet state.208
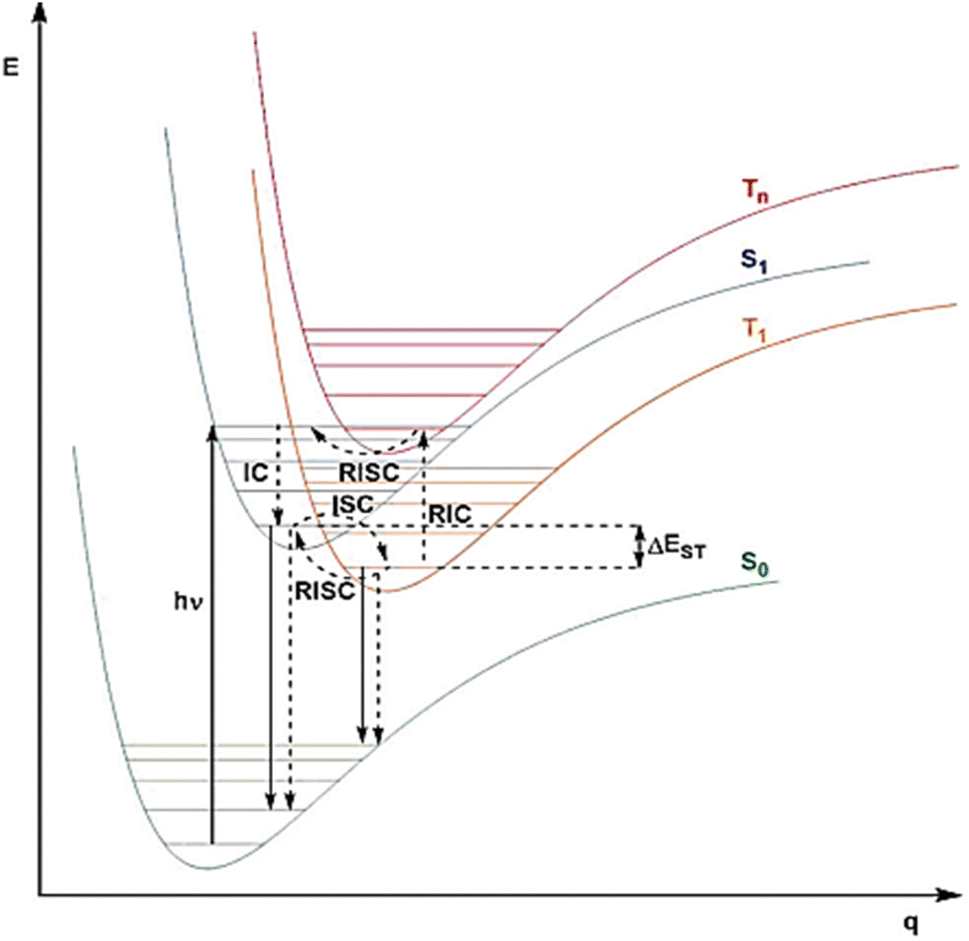 | ||
| Fig. 44 Jablonski-type graph along Morse potentials illustrating the photophysical mechanism in TADF molecules. Reproduced from ref. 208, https://doi.org/10.1039/D1CS00198A, under the terms of the CC BY-NC 3.0 license, https://creativecommons.org/licenses/by-nc/3.0/. | ||
 | ||
| Fig. 45 Jablonski diagram and potential photochemical pathways for excited states of luminescent compounds such as TADF materials. Reproduced from ref. 208, https://doi.org/10.1039/D1CS00198A, under the terms of the CC BY-NC 3.0 license, https://creativecommons.org/licenses/by-nc/3.0/. | ||
For organic TADF compounds, the kISC is generally below 10−7 s−1. However, kISC can vary significantly depending on the specific compound. In these materials, kISC is typically slower than the diffusion rate constant (kdiff), which enhances the likelihood of PET and PEnT from the S1. In TADF compounds with a faster rate of kISC, the triplet state manifold can also contribute to photocatalytic processes. In such scenarios, the quantum yields for PF (ΦPF) and DF (ΦDF) are crucial for assessing the availability of S1 and T1 excited states and thereby determining the probabilities of PET and PEnT from each state.208
Photocatalysis occurs from an excited state that depends on the kISC and kRISC in TADF PCs. The reaction rates that are related to ISC/RISC are defined by the given mathematical eqn (3), where the rate constants depend on both the ΔEST and spin–orbit coupling. Organic TADF compounds are characterized by ΔEST values lower than 0.3 eV,220 which influence the extent of state mixing.221 The ΔEST is directly related to the J (exchange integral) between the excited singlet and triplet states, as expressed in eqn (4).220,221 The dependence of J on the orbital overlap of the FMO involves transitions to S1 and T1, as shown in eqn (5). Here, Ψ and Φ typically represent the HOMO and LUMO wavefunctions and integration into S0–S1 and S0–T1 transitions, respectively, and e represents the charge on the electron.
| ΔEST = ES − ET = 2J | (4) |
From eqn (5), it is evident that a reduction in the J will occur by minimizing the overlap between the HOMO and the LUMO. Consequently, this will result in a smaller ΔEST.220 Decoupled donor–acceptor and spatially separated compounds, characterized by significant torsion between the donor and acceptor units, are typically used to design TADF emitters.222
 | (5) |
A notable TADF compound used as a PC was 2,4,5,6-tetra(carbazol-9-yl)benzene-1,3-dicarbonitrile (4CzIPN), which was first synthesized by Adachi et al. in 2012 (Fig. 46A) 6.17 4CzIPN consisted of four carbazole donors with an isophthalonitrile acceptor. Fig. 46B shows the structure of [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 67, where (dF(CF3)ppy) stands for 2-(2,4-difluorophenyl)-5-trifluoromethylpyridinato and dtbbpy stands for 4,4′-di-tert-butyl-2,2′-bipyridine, which has been utilized. The mixed charge transfer from ligand to ligand or metal to ligand features a radical cation associated with dp(dF(CF3)ppy). Here, the iridium center was oxidized to Ir(IV), and the ligand (dtbbpy) was reduced to a radical cation.
 | ||
| Fig. 46 Chemical scaffold of (A) 4CzIPN and (B) [Ir(dF(CF3)ppy)2(dtbbpy)]PF6. | ||
As illustrated in Fig. 47, all compounds 62–66 showed charge-transfer (CT) excited states. The excited-state redox properties of 4CzIPN are noteworthy, as it is a strong photo-oxidizing and photo-reducing PC compared to other commonly used visible-light PCs, such as [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 67, Ir(ppy)3 68, [Ru(bpy)3](PF6)2 69, Eosin Y 70, [Ir(dF(CF3)ppy)2(bpy)]PF6 71, Rose Bengal 72, [Acr-Mes]ClO4 73, [Ir(ppy)2(dtbbpy)]PF6 74, [Ir(dF(Me)ppy)2(dtbbpy)]PF6 75, [Ir(F(Me)ppy)2(dtbbpy)]PF6 76, and 4CzIPN 6, as shown in Fig. 48 and 49. The main advantage of 4CzIPN is its low-cost synthesis.208
 | ||
| Fig. 47 Photocatalytic cycle comparison of redox and photophysical properties of 4CzIPN. Adapted from ref. 208, https://doi.org/10.1039/D1CS00198A, under the terms of the CC BY-NC 3.0 license, https://creativecommons.org/licenses/by-nc/3.0/. | ||
 | ||
Fig. 48  of common visible light PCs. Reproduced from ref. 208, https://doi.org/10.1039/D1CS00198A, under the terms of the CC BY-NC 3.0 license, https://creativecommons.org/licenses/by-nc/3.0/. of common visible light PCs. Reproduced from ref. 208, https://doi.org/10.1039/D1CS00198A, under the terms of the CC BY-NC 3.0 license, https://creativecommons.org/licenses/by-nc/3.0/. | ||
 | ||
Fig. 49  of common visible light PCs. Reproduced from ref. 208, https://doi.org/10.1039/D1CS00198A, under the terms of the CC BY-NC 3.0 license, https://creativecommons.org/licenses/by-nc/3.0/. of common visible light PCs. Reproduced from ref. 208, https://doi.org/10.1039/D1CS00198A, under the terms of the CC BY-NC 3.0 license, https://creativecommons.org/licenses/by-nc/3.0/. | ||
The effectiveness of a PC is typically evaluated based on the highest product yield achieved.225 However, it is not always confirmed whether the reaction reaches complete conversion, leading to potential variations in yield due to differences in reaction kinetics and reactivity. To address this, a threshold is established for yield comparison, where variations of less than 5% are considered statistically insignificant across different PCs.208 Additionally, isolated yields, often reported, can be more variable due to experimental discrepancies, whereas NMR yields provide a more precise measure of product formation.
A quantitative approach to measuring the effectiveness is the Φ(λ) eqn (2); however, these are separately documented from the product yield.226 Quantum yields are often used to infer the presence of a radical chain mechanism in a reaction. When the quantum yield exceeds one, it suggests that one mole of photons can produce more than one mole of product, indicative of a radical chain mechanism. Conversely, a quantum yield of less than one suggests the radical chain mechanism may not function, although this interpretation is simplistic. Reactions can still involve radical mechanisms even if their quantum yields are below one. In cases where the initiation step of the chain mechanism is highly inefficient, estimating the length of the radical chain may be possible by analyzing the quenching fraction, though such analyses are infrequently reported in photocatalysis studies.227
Throughout the fabrication or formation of a PC, various sources of excitation may be used, ranging from traditional compact fluorescent lightbulbs to high-powered LED lamps.228 Furthermore, photoreactors can catch and reflect all photons from the source of excitation towards the reaction vessel to enhance reaction efficacy.
It is crucial to recognize that the proposed mechanisms for the reactions discussed in this review are largely speculative and frequently lack robust supporting evidence. Mechanistic proposals frequently rely on comparing the thermodynamic properties of PCs and reactants. While this provides an overall assessment of PC effectiveness, it often neglects the importance of reaction kinetics. For example, Wolf and colleagues studied how photoexcited state lifetime influences the yield of product in the trifluoromethylation of quinoline.229 They discovered that longer lifetimes for the excited state of PCs led to faster SET, but the second SET was also discovered to be the rate-limiting step rather than the first SET. Similarly, Scaiano and coworkers230 noted that, although suitable redox potentials were required for the PCs to start the target SET, the success of photocatalysis was determined by reaction kinetics, which are less frequently studied. Lamentably, kinetics studies like these are infrequently conducted.
4CzIPN is a highly investigated TADF-based PC, predominantly involved in photoredox catalysis reactions through PET. However, there are a few instances, which will be discussed below, where the photocatalysis occurs through PEnT.208
 | ||
| Fig. 50 The catalytic cycle for photoinduced oxidative decarboxylation via a radical addition mechanism.232 | ||
Examples of radical addition include the synthesis of arylhydroxylamines (87) through photoredox catalysis. This process involves the reaction between carboxylic acids (85) and nitrosoarenes (86), as illustrated in Fig. 51.233 Another example is the addition of vinyl boronic esters (88) to radicals generated from α-amino carboxylic acids (89) to produce γ-amino boronic esters (100), which is outlined in Fig. 52.234 1,2-Amidoalkynylation of alkenes can be performed using the addition of alkenes (91) to amidyl radicals derived from 2,2,2-trichloroethoxycarbonyl-protected α-aminoxy acids (92) and ethynylbenziodoxolone (93) to yield the alkynylated products (94) as shown in Fig. 53.235 The addition of alkyl radicals generated from amino acids or peptides (95) to a methyleneoxazolidinone analogue of dehydroalanine (96) has been employed to produce peptide derivatives (97) as illustrated in Fig. 54.236 Finally, a hydrosilylation reaction can be achieved by the addition of silyl radicals formed from silacarboxylic acids (98) to alkenes (99) to yield hydrosilylated alkanes (100) as seen in Fig. 55.237
 | ||
| Fig. 51 The synthesis of arylhydroxylamines via photoredox catalysis. Reproduced from ref. 233 with permission from Georg Thieme Verlag KG, Copyright © 2018. | ||
 | ||
| Fig. 52 Synthesis of γ-amino boronic esters.234 | ||
 | ||
| Fig. 53 1,2-Amidoalkynylation of alkenes. Adapted from ref. 235 with permission from John Wiley & Sons, Copyright © 2018, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
 | ||
| Fig. 54 Derivatization of amino acids.236 | ||
 | ||
| Fig. 55 Hydrosilylation of alkene.237 | ||
In other reactions utilizing organic PCs, no products were formed when eosin Y and 9-mesityl-10-methylacridinium perchlorate ([Mes-Acr]ClO4) were used.238 The 1,2-aminoalkynation of alkenes (Fig. 53), with 2-methylpent-1-ene as the alkene, produced a 77% yield using 4CzIPN as a PC. Other organic PCs produced yields of 7% to 33%, such as eosin Y which is effective and produces a yield of about 33%.235 The difference in yield was correlated with the ground-state reduction potential of the PCs 4CzIPN, eosin Y, and [Mes-Acr]ClO4.238 This suggests that the reduction potential of the PC significantly influences its success.
In alkene hydrosilylation (Fig. 55), 4CzIPN outperformed ruthenium and iridium PCs, achieving an 82% yield.237 This superior performance was attributed to the exceptional photo-oxidizing competence of 4CzIPN, which had an  of 1.35 V compared to [Ir(dF(CF3)ppy)2(dtbbpy)]PF6, which had an
of 1.35 V compared to [Ir(dF(CF3)ppy)2(dtbbpy)]PF6, which had an  = 1.21 V. This is crucial for the oxidation of
= 1.21 V. This is crucial for the oxidation of  . This high efficiency was notably observed in the derivatization of amino acids (Fig. 54).236 4CzIPN and [Ir(dF(Me)ppy)2(dtbbpy)]PF6 provided almost equal yields (such as 89% and 90%, respectively), prompting the selection of 4CzIPN as the PC for further investigation.
. This high efficiency was notably observed in the derivatization of amino acids (Fig. 54).236 4CzIPN and [Ir(dF(Me)ppy)2(dtbbpy)]PF6 provided almost equal yields (such as 89% and 90%, respectively), prompting the selection of 4CzIPN as the PC for further investigation.
Unfortunately, 4CzIPN and [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 is an ineffective PC for synthesizing the hydroxylamine product (Fig. 51), but [Mes-Acr]ClO4 and [Mes-Acr]BF4 proved to be an effective PC for this reaction, due to their exceptional oxidizing ability in the excited state with a reduction potential of approximately 2.06 V.
Fig. 52 shows the preparation of γ-amino boronic esters using four PCs: [Ir(ppy)2(dtbbpy)]PF6, [Ir(dF(CF3)ppy)2(dtbbpy)]PF6, [Ru(phen)3]Cl2, and 4CzIPN. Among these, [Ir(ppy)2(dtbbpy)]PF6 was low-cost and produced a yield of about 88% of the final product in the presence of Boc-Pro-OH and the addition of Cs2CO3. In contrast, the yield of 4CzIPN is 66% without Cs2CO3, and its yield with the additive was not reported, making direct comparison challenging. The superior performance of [Ir(ppy)2(dtbbpy)]PF6 over 4CzIPN was likely due to their relative ground-state reduction potentials. The lower Ered of the iridium PCs led to the formation of the intermediate α-boryl radical. A mechanistic approach-related example secluded the functionalization of amides and amines (101) with the help of electrophilic nitrogen radicals (102) to form the functionalized amides/amines (103) (Fig. 56).239 This reaction generates an electrophilic amidyl radical through the decarboxylation of carboxylic acid scaffolds. The resulting radical then undergoes a 1,5-hydrogen atom transfer (HAT), leading to the formation of a distal alkyl radical as shown in Fig. 57.239 This radical subsequently undergoes radical cleavage or group transfer in the presence of polarized SOMOphile (SOMO = singly occupied molecular orbital), resulting in the formation of remotely substituted amines or amides. 4CzIPN demonstrated higher yields in various reactions, including γ-chlorination, thioetherification, cyanation, and alkynylation, although there was no comparative data for an iridium photocatalyst in the final reaction.
 | ||
| Fig. 56 Reaction illustration for the functionalization of amines and amides.239 | ||
 | ||
| Fig. 57 Generalized cycle for group transfer in C–H functionalization and decarboxylation.239 | ||
Carboxylic acids can serve as radical precursors for acetylation of alkynyl bromides (106) (Fig. 58).240 The reaction proceeds via decarboxylation, generating an acetal radical (109) that subsequently forms a bromoalkenyl radical (111) by addition to the alkynyl bromide (Fig. 59).240 The removal of a bromyl radical, which is removed by the PC (66), leads to the formation of the product.
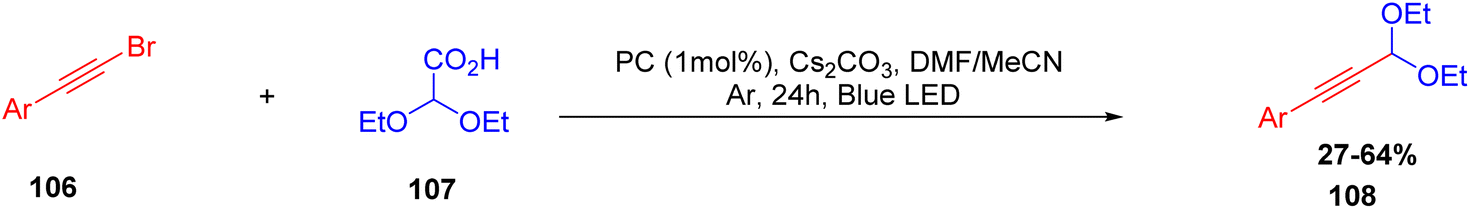 | ||
| Fig. 58 Synthetic approach for acetalation of alkynyl bromides.240 | ||
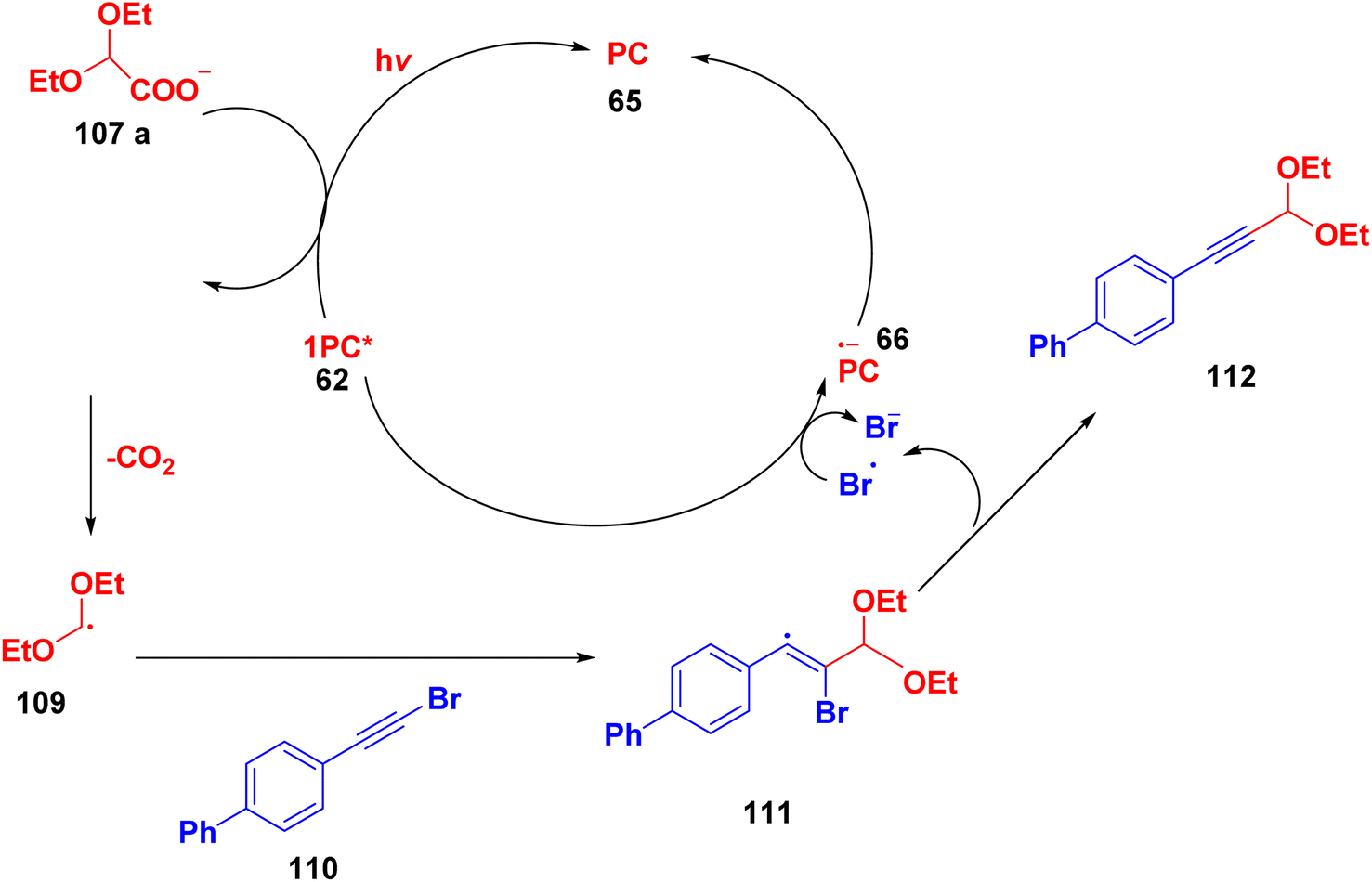 | ||
| Fig. 59 Synthetic approach for the photocatalytic acetalation of alkynyl bromides.240 | ||
Radical capturing experiments revealed the inclusion of the acetal radical, and further radical suppression studies supported the radical-based nature of the process. Among the PCs tested, 4CzIPN and [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 yielded only 5% and 36%, respectively, while [Ru(bpy)3](PF6)2 and eosin Y did not produce any product. Wang et al. attributed this to the photooxidizing abilities of the PCs. The cesium salt of 2,2-diethoxyacetic acid, with Eox of 0.95V, was studied241 and the results indicate that for a reaction to be thermodynamically feasible, the PCs should have a more positive reduction potential than that of substrate.241 Both [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 and 4CzIPN exhibited sufficiently high photooxidizing abilities, which likely accounted for the efficient performance of 4CzIPN, but [Ru(bpy)3](PF6)2 and eosin Y were not effective for this single-electron transfer process. Additionally, oxidative decarboxylation has been employed in cyclization reactions, such as the synthesis of functionalized cyclopropanes (115 and 116) from carboxylic acids (95) and a carboxylate ester (113) or boronic ester (114) (Fig. 60A),242 intramolecular arene alkylation from N-(acyloxy) phthalimides (117) to alkylated arene (118) (Fig. 60B),243 and the fabrication of 2-substituted piperazines (121) by addition of a glycine-based diamine (119) to aldehydes (120) (Fig. 60C).244

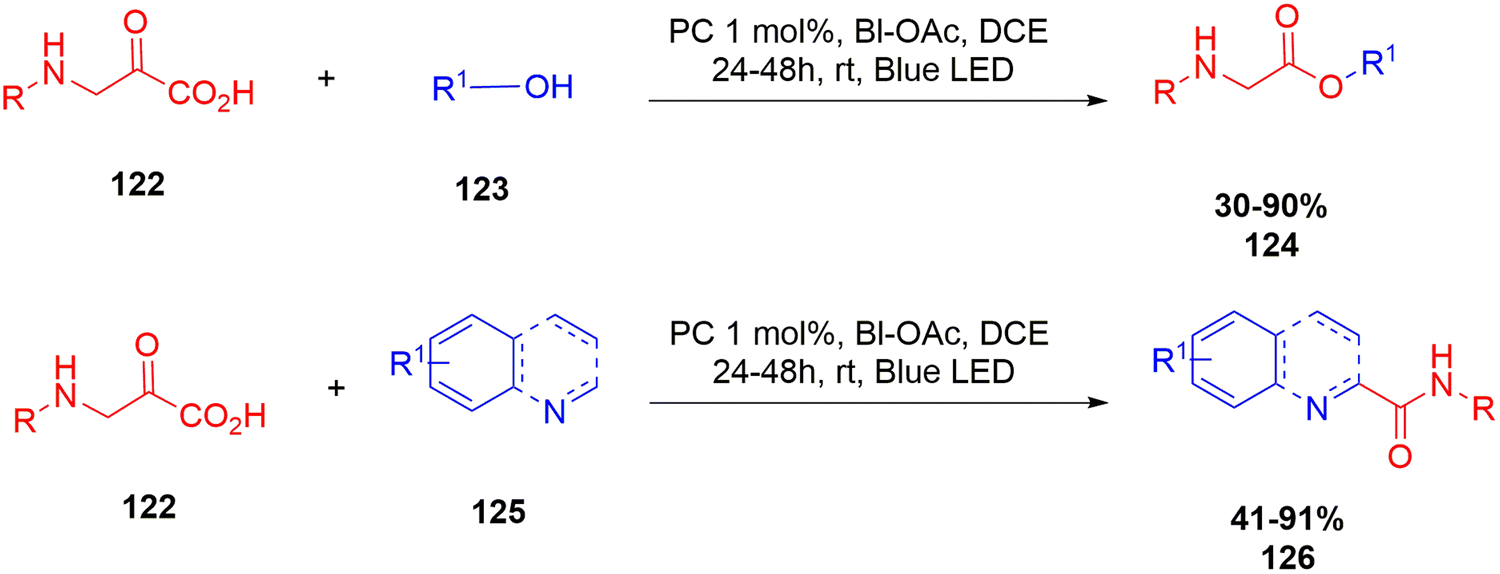 | ||
| Fig. 61 Synthetic approach by oxidative decarboxylation of oxamic acids.245 | ||
 | ||
| Fig. 62 Oxidative decarboxylation of oxamic acids using photocatalysis.245 | ||
The excited PC can also be used for the Minisci reaction.247 This reaction begins with oxidative quenching in the presence of N-(acyloxy)phthalimides (NAPs) (131), which leads to the generation of an alkyl radical. The addition of the radical to a heteroarene (132) leads to the functionalized compound (133), as described in Fig. 63.247 Ir PCs and 4CzIPN produced similar yields in the reaction but 4CzIPN was chosen as the PC to prevent potential toxicity associated with the use of Ir-based PCs by utilizing an organocatalyzed process.
 | ||
| Fig. 63 Minisci reaction in the use of carboxylic acids in the presence of NAPs.247 | ||
An analogous synthetic approach includes the formation of carbon-fluorine bonds with N-hydroxyphthalimide esters (134) to form alkyl fluorides (135) (Fig. 64) 134–135.248
 | ||
| Fig. 64 Nucleophilic fluorination of redox-active esters.248 | ||
The mechanism of this reaction used a PC* oxidative quenching of redox-active ester 134, which results in an alkyl radical 136 (Fig. 65).248 In this reaction, the alkyl radical is oxidized in the presence of PC*. This results in a carbocation (137) instead of the addition of a double bond or coupling to another radical. This carbocation is then captured by a fluoride ion, leading to the formation of an alkyl fluoride (135). The quantum yield of the reaction was 37%, revealing that this process does not have any involvement in the radical chain mechanism. Stern–Volmer quenching studies revealed effective quenching of the excited photocatalyst by the phthalimide ester. The presence of the alkyl radical was validated by the inhibition of product formation by adding TEMPO (2,2,6,6-tetramethylpiperidin-1-oxyl), leading to the TEMPO adduct. Additional radical capturing experiments with different precursors such as styrene and methyl acrylate supported the presence of alkyl radical. The formation of the carbocation was further validated by capture experiments in the presence of different nucleophiles such as alcohols and 1,3,5-trimethoxybenzene. Among the tested photocatalysts, 4CzIPN achieved a 91% yield, which is comparable to that of Ir PCs.
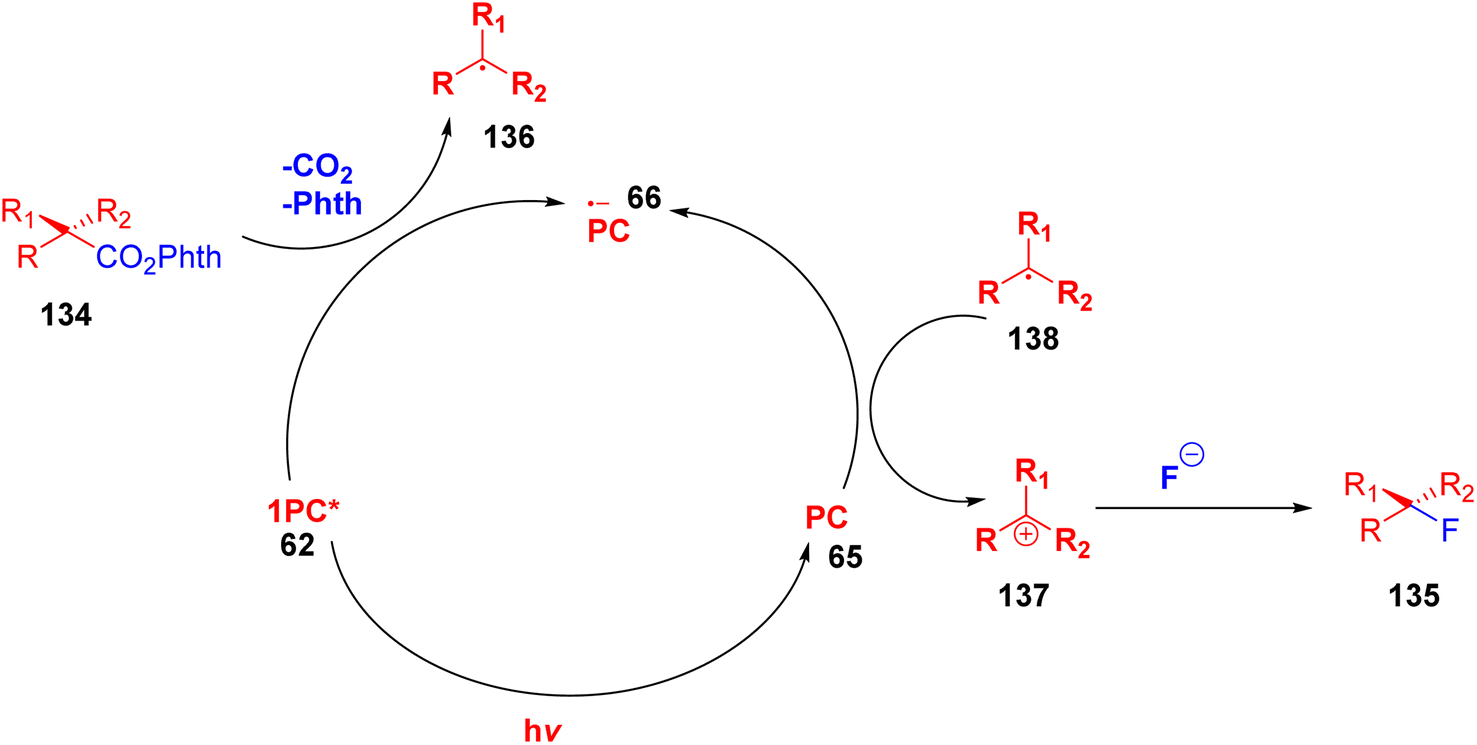 | ||
| Fig. 65 Detailed mechanism of nucleophilic fluorination of redox-active esters using a photocatalyst.248 | ||
Excitation of the PC is typically performed by oxidative quenching with carboxylic acid derivatives, resulting in alkyl radicals. Alternatively, the generation of alkyl radicals can be attributed to the oxidized photocatalyst itself. This mechanism is evident in different types of photocatalytic reactions such as synthesis of aldehydes and ketones (140) from arylacetic acids (139) (Fig. 66A),249 a δ-C–H mono- and difluorination reaction of an alkoxy radical precursor (141) to form the difluorinated products 142 (Fig. 66B) 141–142,250 intermolecular hydroalkylative dearomatization of electron-deficient indole scaffolds (143) via reaction with phenylglycine (143) to yield the dearomatized product (145) (Fig. 66C),251 1,2-hydroalkylation for the intermolecular dearomatization of naphthalene scaffolds (146) via reaction with phenylglycine (147) to yield the dearomatized products (148 and 149) (Fig. 66D),252 and the synthesis of 1,4-amino alcohols (152) by decarboxylative radical addition using a reaction between aliphatic aldehydes (120), carboxylic acids (150) and aromatic aldehydes (151) (Fig. 66E) 120, 150–152.253 In the given examples 4CzIPN is employed as a PC because it is commercially available TADF material, wide functional group captability and reactions under mild conditions. In these examples, a 99% yield is obtained as compared to other transition metal-based PCs such as iridium PCs which is why 4CzIPN is used as the best photocatalyst in these reactions.
 | ||
| Fig. 66 Examples of different photocatalytic reactions: (A) synthesis of aldehydes and ketones,249 (B) δ-fluorination,250 (C) dearomatization of indoles,251 (D) dearomatization of naphthalene,252 and (E) synthesis of 1,4-amino alcohols.253 | ||
The fluorination reaction of the radical 163 was achieved using Selectfluor (164), leading to the formation of 142 and reduction of the Selectfluor radical cation (165) by the excited PC* (62). The fluorination process was completed in just 10 minutes in the presence of PC with a 1% molar concentration compared to the chlorination reaction that involved 18 hours with a molar concentration of PC of 3 mol%. Quantum yield experiments indicated values of 7.0% for fluorination and 1.6% for chlorination, suggesting the unavailability of a radical chain mechanism. The greater efficiency of the fluorination reaction, even with lower PC loading, was corroborated by radical clock experiments, which showed that the quenching of alkoxy radical was 53 times faster in fluorination as compared to chlorination. Among the tested PCs, the yields were 67% for 4CzIPN and 71% for [Ir(dF(CF3)ppy)2(dtbbpy)]PF6, respectively. Despite the comparable results, 4CzIPN was favored for its superior efficiency.
In the dearomatization of the indole scaffolds reaction mechanism (Fig. 66C), the PC* was essential for oxidative quenching, which formed a new radical.251 The α-amino radical is then formed through oxidative decarboxylation of the scaffold (144) catalyzed in the presence of the oxidized PC, which then couples with a protonated intermediate to form the final product. Stern–Volmer quenching experiments showed that indole quenched the PC* with more efficacy than glycine, with constants of 1460 and 173, respectively. Among the photocatalysts tested, 4CzIPN achieved a yield of nearly 99%, surpassing transition metal-based photocatalysts such as fac-Ir(ppy)3, [Ru(bpy)3]Cl2 and [Ir(ppy)2(dtbbpy)]PF6 due to their oxidation potential in the excited state.
For the 1,2-hydroalkylation reaction used for intermolecular dearomatization of naphthalene derivatives (Fig. 66D), 4CzIPN was the most effective among the tested PCs, achieving a 90% yield with a 2:1 ratio of uncyclized to cyclized products.252 In contrast, [Ir(ppy)2(dtbbpy)]PF6 achieved only a 60% yield with a 3.7:1 ratio. The discrepancy in yield remains unexplained, as both catalysts have similar Ered = −1.45 V for the naphthalene derivative.
Finally, for producing 1,4-amino alcohols by the decarboxylative radical addition bifunctionalization process, as shown in Fig. 66E.253 In this process formation of ketyl radical occurred through oxidative quenching of aldehyde in the presence of PC*. The oxidized PC produces the alkyl radical from the carboxylate in a similar fashion to the previous examples. The reaction of an alkyl radical with olefins generated a new radical that subsequently coupled with a ketyl radical. Radical protonation leads to the formation of the final product (152). The proposed photocatalytic cycle was confirmed by experiments using the radical scavenger TEMPO and deuterium labeling. Among the eight PCs tested, the yield was 90% for 4CzIPN and was 42% for [Ir(dF(CF3)ppy)2(dtbbpy)]PF6. This is attributed to their strong photoreducing abilities compared to [Ru(bpy)3]Cl2, which did not produce any product. The authors also suggest an alternative reductive quenching mechanism. However, the absence of a clear mechanistic understanding makes it difficult to explain why 4CzIPN demonstrated superior performance compared to the other photocatalysts.
The preparation of aldehydes and ketones from arylacetic acids was achieved via the coupling of the alkyl radical with a superoxide radical causing the generation of SET by the excited PC*.249 This coupling led to dehydration and protonation, resulting in the synthesis of the final product (Fig. 67). Stern–Volmer quenching studies demonstrated higher luminescence quenching of the PC by oxygen, highlighting its function. Electron paramagnetic resonance (EPR) spectroscopy confirmed the superoxide radicals' presence during the reaction. The introduction of TEMPO interrupted the reaction, suggesting the involvement of radical intermediates. Among the organic PCs tested, the yield of 4CzIPN was 91% but eosin Y and Rose Bengal yielded significantly less product. This disparity was attributed to their oxidation potentials. For example, Eosin Y and Rose Bengal, with oxidation potentials of 0.78 V and 0.84 V, respectively, are insufficient for oxidizing the carboxylate. In contrast, 4CzIPN, with a higher oxidation potential of 1.52 V, effectively facilitates the reaction. Additionally, by reducing the concentration of PC slightly results increased the yield of product from 91% to 98%.
 | ||
| Fig. 67 Photocatalytic formation of ketones and aldehydes in the presence of carboxylic acids.249 | ||
For δ-C–H mono- and dehalogenation reactions (Fig. 66B), the oxidation of PC facilitates the decarboxylation of carboxylic acid substrates, resulting in the loss of CO2 and MeCN. This process involves alkoxy radical formation by a 1,5 hydrogen atom transfer (HAT) mechanism (Fig. 68).250
 | ||
| Fig. 68 Fluorination of carboxylic acid scaffolds in a photocatalytic reaction.250 | ||
 | ||
| Fig. 69 Non-dearomative and dearomative intramolecular [2 + 2] photocycloaddition reactions.254 | ||
The photodegradation of TADF-active halo-isophthalonitriles is a crucial aspect to address for their application as photocatalysts and for designing appropriate photoredox-promoted chemical transformations. Cozzi et al. (2022) showed that the photoconversion of the halo-isophthalonitrile 3DPAFIPN produces an isolable carbazole-1,3-dicarbonitrile derivative with significant photophysical properties, such as TADF, as depicted in Fig. 70A.255 These properties include a prolonged delayed fluorescence lifetime and modulated redox potentials in both ground and excited states, which are advantageous for photocatalysis using CoBr2. Dual metal and photoredox catalysis were utilized for the effective allylation of aldehydes, yielding homoallylic alcohols with good efficiency.
 | ||
| Fig. 70 (A) TADF PC for cobalt-mediated allylations and (B) its proposed mechanism.255 | ||
Zysman-Colman et al. (2022) have discovered a donor–acceptor TADF compound, pDTCz-DPmS, that could rival the commonly used 4CzIPN in various photocatalytic reactions, as illustrated in Fig. 71.256 Their results consistently highlight the photocatalytic potential of pDTCz-DPmS in comparison to 4CzIPN. Due to its significantly stronger photoreducing ability, pDTCz-DPmS is expected to be a more effective photocatalyst in oxidative quenching mechanisms, especially when reductive quenching pathways are not feasible. Furthermore, due to its higher triplet energy (ET) of approximately 0.4 eV greater than that of 4CzIPN, pDTCz-DPmS is expected to perform well in photoinduced energy transfer reactions involving substrates with high ETs. This is comparable to benzophenone (ET = 3.00 eV), a well-established efficient energy transfer PC. Furthermore, under the studied reaction conditions, pDTCz-DPmS showed much better photostability than 4CzIPN. Thus, our work shows that TADF donor–acceptor compounds that are not limited to 4CzIPN or the carbazolyl dicyanobenzene (CDCB) family could be used and could even outperform this widely used organic photocatalyst in terms of efficiency.
 | ||
| Fig. 71 Investigated photocatalysis reactions including energy transfer, oxidative quenching, dual metallaphotocatalysis and reductive quenching via Ni(II) cocatalyst.256 | ||
As seen in Fig. 72, Song et al. (2023) prepared two PCs based on an organic TADF molecule for the oxidation of sulfides into sulfoxides in aqueous media.257 Cross-linking between the two PCs was thought to be responsible for their catalytic activity. By forming hydrophobic domains by cross-linking, the photocatalyst FL-DNS was able to increase its catalytic activity and prolong the duration of its triplet state. In addition, FL-DNS is both recyclable and reusable, making it suitable for aquatic settings or other applications for green chemistry. Mechanistic investigations demonstrated that superoxide  and singlet oxygen (1O2) were both involved in the catalytic process. The study highlighted the value of delayed fluorescence and the advantages of photocatalysts based on TADF for catalytic applications.
and singlet oxygen (1O2) were both involved in the catalytic process. The study highlighted the value of delayed fluorescence and the advantages of photocatalysts based on TADF for catalytic applications.
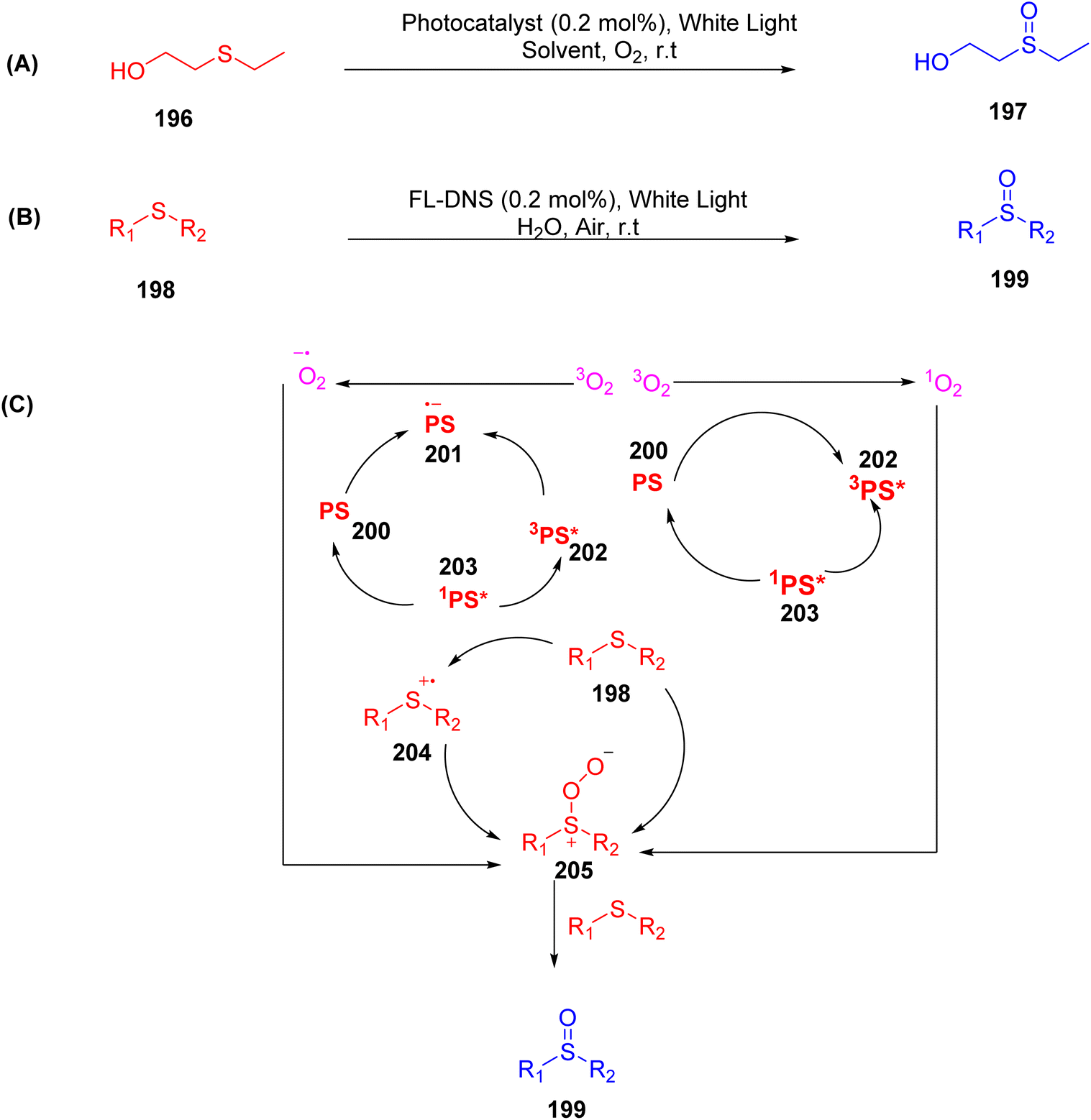 | ||
| Fig. 72 (A) Oxidation of sulfide to sulfoxide via photocatalysis using white light. (B) Oxidation of sulfides via FL-DNS. (C) Photocatalytic mechanism of oxidation of sulfide into sulfoxide via FL-DNS.257 | ||
Hudson et al. (2023) synthesized a set of TADF compounds by incorporating a new imidazo-phenothiazine (IPTZ) acceptor to enhance spin–orbit coupling and excited-state lifetimes by structural constraints and the heavy atom effect.258 These materials were employed as PCs in various visible light-driven energy transfer reactions. These included [2 + 2] cycloaddition, etherification, disulfide–ene reactions, Ni-mediated esterification, and amination. They generally displayed superior activity to ACR-IMAC and 2CzPN, as illustrated in Fig. 73 and 74. The high concentration of locally excited triplet states in these materials was key to effective photocatalysis in MeCN, as it minimized solvatochromic effects responsible for lower triplet energy. SACR-IPTZ emerged as an especially effective triplet sensitizer, noted for its high energy transfer efficiency, significant locally excited triplet state character, long-lived excited states in polar solvents, broad visible-range absorption, and excellent photostability. This study highlighted the potential of the effect of heavy atoms in creating TADF materials with exceptional photocatalytic capabilities. Future research should focus on further optimizing TADF PCs with enhanced visible-range absorption, minimized solvatochromic shifts, and improved photostability and solubility in polar solvents. Additionally, IPTZ-based materials could be used in SET-mediated reactions to expand their applications, leveraging the success of phenothiazine-based PCs in sulfur-centered radical photoredox reactions.
 | ||
| Fig. 73 (A) Reaction [2 + 2] cycloaddition. (B) Reaction scheme for disulfide–ene and (C) amination in the presence of IPTZ PCs. (D) Esterification, (E) etherification, and (F) generalized mechanism for energy transfer occurring in the Ni-catalyzed cross-coupling reaction.258 | ||
 | ||
| Fig. 74 (A–C) Optimizing Ni cross-coupling reactions (D) Ni-catalyzed amination through SACR-IPTZ with reduced loading of catalyst.258 | ||
Wang et al. (2024) developed a technique for producing α-fluoracrylates (226) from gem-difluorostyrenes (224) utilizing formate salts (225) as a carbon source, as shown in Fig. 75.259 This method produces the (Z)-type α-fluoracrylate framework without using transition metals and with high regioselectivity. Mechanistic investigations found that the carbon sources for the defluorinative carboxylation include sodium formate and potassium carbonate, with successive photoexcitation presumably performing a part in the SET defluorination of gem-difluoroalkene. Furthermore, 4DPAIPN and its chloro- or bromo-substituted variants were efficient photocatalysts in this process. Future studies will focus on researching photocatalytic reactions using halogen-containing dyes.259
 | ||
| Fig. 75 Production of α-fluoracrylates (226) via a redox photocatalytic process.259 | ||
Strehmel et al. (2024) worked on the confinement of carbon dots (CDs), which resulted in light-sensitive materials capable of forming triplet states, evidenced by the prolonged afterglow of TADF and RTP.260 This enabled metal-free photo-ATRP (afterglow room temperature phosphorescence) based on a reductive mechanism as shown in Fig. 76. Triplet states play a crucial role in the mechanistic setup, as their spin orientation delays electron back-transfer reactions. Photo-CIDNP (photochemically induced dynamic nuclear polarization) studies could enhance understanding in this area. Notably, this approach worked well with confined carbon dots derived from cellulose, while materials without confinement, such as CMCCDs, did not exhibit either TADF or RTP. No heavy atoms were involved in triplet formation. Future work could focus on altering the confinement matrix to increase TADF and RTP efficiency. Materials with ordered structures, whether from synthetic or biomaterials, might be promising candidates. Photopolymerization experiments also demonstrated the distinct behavior of these heterogeneous systems compared to those in homogeneous environments. More exploratory studies are needed to understand the surface morphology of particles used for polymer synthesis. Unwanted chain growth could be mitigated by adding small amounts of CuBr2/L, which improved selectivity during polymerization. However, this compromised the advantage of a metal-free photo-ATRP system, which could be beneficial for some electronic applications, even though small amounts of CuBr2/L drastically alter the system. Future research should clarify how this metal cocatalyst interacts with the confined material at the surface, explaining the reactivity differences between a metal-free system and one containing CMCCDs@SiO2/CuBr2/L. Additionally, strategies should be developed to keep the photocatalyst surface active throughout the process. Recycling is another intriguing feature offered by these confined materials. CDs do not offer this feature due to their much smaller size, making this aspect worthy of more attention in future work to design greener systems.
 | ||
| Fig. 76 Participation of confined CMCCDs@SiO2 (ConCDs) in a metal-free photopolymerization following (a) an oxidative mechanism or (b) a reductive mechanism in an ATRP setup. (c) The addition of Cu2+ to ConCDs.260 | ||
4.3. Applications of TADF materials in OLEDs
The TADF emitter component of OLEDs is generally distributed at a low concentration in an appropriate substrate matrix. This technique is aimed at reducing aggregation-caused quenching (ACQ) and TTA, resulting in improved EL performance. As a result, it is vital to construct host materials with specific properties:(i) A high triplet energy (ET) in the host material is crucial to inhibit energy transfer from the TADF emitter back towards the host, not dependent on emission color.
(ii) Proper alignment of HOMO and LUMO is essential to confirm efficient charge transfer from adjacent layers.
(iii) Maximizing direct charge capture on the doped emitter, by increasing the energy gap between the HOMO and LUMO.
(iv) Both positive and negative charge transport properties are critical for achieving an equitable balance of electrons and holes in the emitting layer.
(v) Exhibiting high structural stability and excellent film properties.104
Numerous materials, notably bipolar host materials, have attracted substantial interest for their potential in efficient TADF-based OLEDs and have become a key aspect of OLED research.261
TADF materials have gained significant attention due to their ability to harness triplet excitons for high-efficiency OLEDs. A progressive approach to their application necessitates a structured classification based on emission color, quantum yield enhancement strategies, and mechanisms facilitating efficient RISC.
➢ Rigidity enhancement through π-extended frameworks to reduce non-radiative decay.
➢ Host–guest interactions in OLED devices to suppress aggregation-induced quenching.
➢ Excited-state engineering via intramolecular charge-transfer tuning to balance radiative and non-radiative pathways.265,266
➢ Minimizing ΔEST through optimal D–A spatial separation while maintaining strong orbital overlap.
➢ Utilizing high-spin–orbit coupling (SOC) elements or second-order vibronic coupling to accelerate the intersystem crossing dynamics.
➢ Designing multi-resonance TADF molecules to achieve fast and efficient RISC via intermediate charge-transfer states.267
By integrating these progressive aspects into TADF material design, researchers continue to refine the structure–property relationships governing OLED efficiency, paving the way for next-generation display and lighting technologies.
In 2017, Chi et al. reviewed the applications of TADF-emitting molecules in OLEDs.105 In this section, the applications of TADF molecules in OLEDs from 2019 to 2024 are examined.
Wang and his coworkers (2024) studied multiple resonance TADF materials which represent remarkable applications such as high color purity and high-resolution OLEDs display. They worked on two new MR-TADF emitters Me-PABCZ 227 and Me-PABDPA 228 with PLQYs of 94.6% and 91.8% respectively, that are synthesized by using one-shot electrophilic C–H borylation reaction. The ideal device had narrow emission spectra with an FWHM of 36 nm, CIE coordinates of 0.14, and 0.57, and a maximum EQE of 27.9% as shown in Fig. 77. This study extends the versatile use of MR-TADF luminogens in electroluminescent devices and offers a novel concept for their molecular structure construction.268
 | ||
| Fig. 77 (A) Chemical structures of PAB, Me-PABCZ, and Me-PABDPA (B) EL characteristics of Me-PABCZ based OLEDs using different doping concentrations: current J–V–L characteristics; EQE–luminance curves; current efficiency–luminance–power efficiency curves; normalized EL spectra at 6 V. | ||
In this study, Kwon and colleagues (2024) employed a molecular design strategy focused on rigidifying the boron-embedded polycyclic heteraborin core and regulating molecular orbitals to achieve ultra-pure green emission. Two MR emitters, BpIC-DPA and BpIC-Cz, were produced by combining fused curvilinear indolocarbazoles with peripheral tert-butyl substituted carbazole/diphenylamine donors. The produced symmetrical heteraborins had incredibly narrow FWHMs of 25 and 22 nm, respectively, along with exceedingly rigid molecular structures and pure green emission at 527 and 534 nm. With matching CIE coordinates of (0.21, 0.73), BpIC-DPA demonstrated the purest green emission of the two materials, meeting the BT2020 green color criteria. A high PLQY of more than 84% is displayed by both emitters. The designed OLEDs based on these emitters have optimum efficiencies of over 22.0% and ultra-roll-off characteristics. The BpIC-Cz-based device had an operational lifetime (LT90) of 291 hours at an initial brightness of 1000 cd m−2. This is the longest lifetime achieved for pure green narrowband OLEDs. In the future these design tactics will continue to steer the investigation of extremely efficient and reliable MR-TADF heteraborins with full-color narrowband emissions, resulting in highly efficient OLEDs with great color purity (Fig. 78).269
 | ||
| Fig. 78 The molecular structures of green emissive B/N type MR emitters and designed polycyclo-heteraborin MR-TADF materials BpIC-DPA and BpIC-Cz. | ||
Cheng et al. (2018) worked on two rod-like D–A–D-type DBA derivatives with carbazole groups, CzDBA and tBuCzDBA, which were produced through a facile two-step method for DBA compounds (Fig. 79). Excellent PLQYs and TADF properties, such as very tiny ΔEST, short, delayed fluorescence lifespan τd, and high horizontal emission dipole orientations, are displayed by the two DBA derivatives. Green electroluminescent device A (CIE 1931 coordinates (0.31, 0.61)) has achieved a record-low efficiency roll-off (only 0.3% roll-off of the EQE at 1000 cd m−2) and a record-high EQE of 37.8 ± 0.6% as shown in Fig. 79. For TADF materials, the DBA core is an exceptional electron-accepting group that can provide excellent PLQY. It offers a foundation for the next molecular designs of extremely effective TADF-based OLEDs.270
 | ||
| Fig. 79 Synthetic route of CzDBA and tBuCzDBA and along with external quantum efficiency versus luminance electroluminescent spectra.270 | ||
Lee et al. (2020) demonstrated that ortho-donor–acceptor–donor (D–A–D) compounds, incorporating a 9,10-diboraanthracene (DBA) acceptor functionalized with ortho-donor groups (Cz, BuCz, and DMAC), can exhibit strong TADF with emission spanning from green to red. Computational studies corroborated the crystal structure study, which showed twisted conformations between donors and DBA moieties as well as the presence of weak intramolecular N⋯B nonbonding interactions. While DMAC-substituted compound DMACz2oDBA was somewhat emissive (PLQY = 44%) in a stiff state, compounds Cz2oDBA and BuCz2oDBA containing Cz displayed significant PLQYs that approached 100%. Highly effective green-to-red TADF-OLEDs were achieved by using ortho-D–A–D compounds as emitters. Above 100 lm W−1, the device with a green emitter Cz2oDBA demonstrated exceptional power efficiency and a very high ECE of 26.6%. With an EL peak at 615 nm, a red OLED device based on emitter DMACz2oDBA attained a maximum EQE of 10.1%, which was like that of red OLEDs doped with boron that has been reported. The creation of extremely effective green-to-red TADF-OLEDs that utilize emitters containing boron (Fig. 80).271
 | ||
| Fig. 80 Different diboron-based D–A TADF systems.271 | ||
For the first time, a new class of Cu(I) exciplexes was created by combining Cu(I) CMA complexes with DMIC-TRZ studied by Gong et al. (2024). Because of the intermolecular through-space charge transfer process, these Cu(I) exciplexes displayed unique TADF characteristics. The emission colors and exciton durations of the Cu(I) exciplexes can be controlled by adjusting the donor ligands of Cu(I) CMA complexes. The copper atom is essential to the production of these exciplexes, as demonstrated by experimental research and theoretical computations. This is because the core copper atom subtly participates in border molecular orbitals. Additionally, it has been shown that certain named conformations of CMA complexes promote the creation of exciplexes. Both emitters and hosts for OLEDs can be found in the class of Cu(I) exciplexes. The OLED that used Cu(I) exciplex as the emitter among them produced a high EQE of 10.9%. With low driving voltages of less than 3 V, the OLEDs that used MR-TADF molecules as guest emitters and Cu(I) exciplexes as host materials produced remarkable EVEs of up to 24.9% as shown in Fig. 81. In the future, this work opens up a wide range of possible uses for coinage-metal-based exciplexes in addition to presenting a novel design approach for the creation of Cu(I)-based emitters using intermolecular charge transfer.272
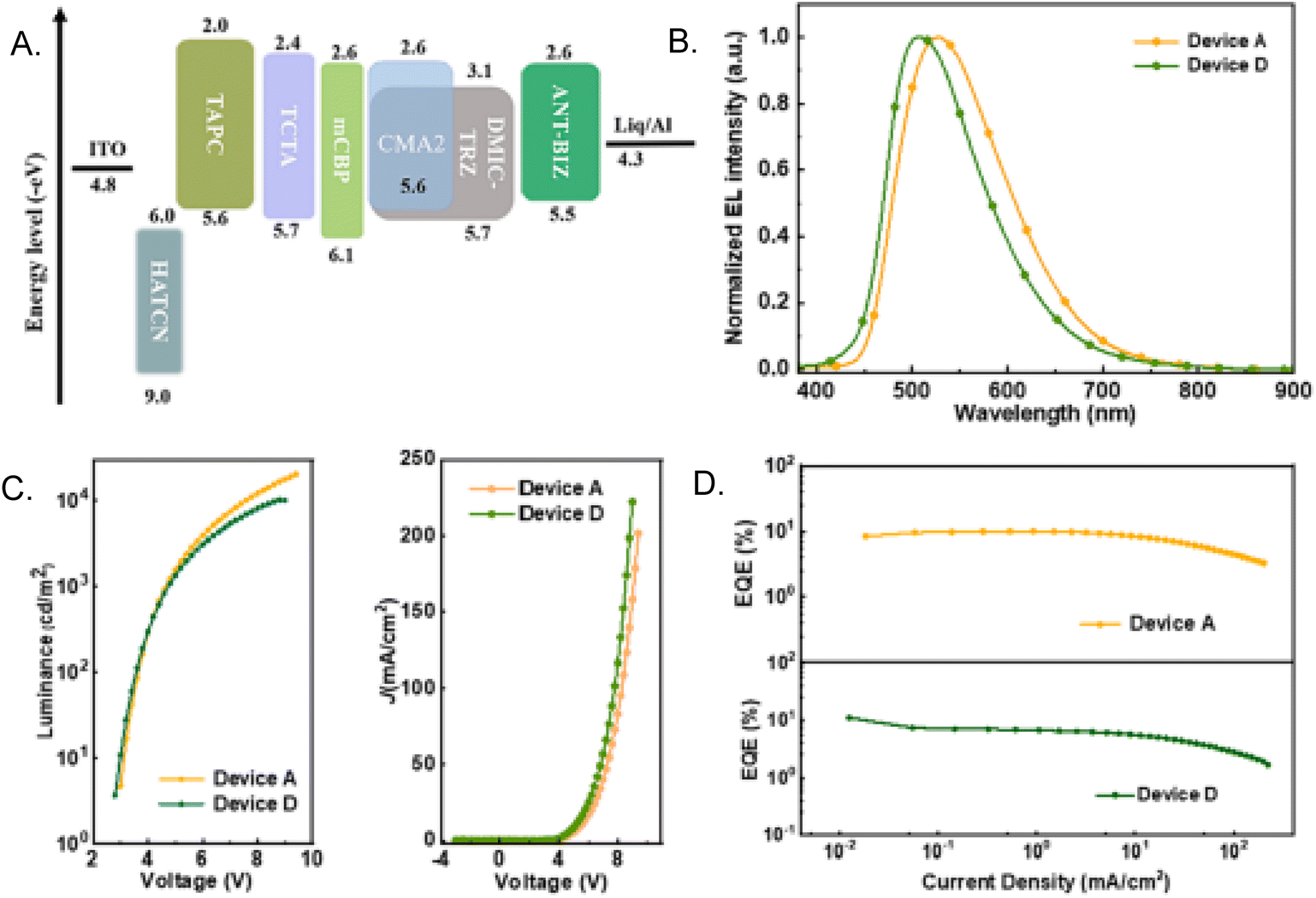 | ||
| Fig. 81 (A) Device structure and energy level diagram. (B) EL spectra of devices A and D. (C) Luminance–voltage, current density–voltage, and (D) EQE–current density curves of devices A and D. | ||
The ink optimization for printing tBuCzDBA as an emitting layer in OLEDs has been documented in this study by Mariano et al. (2024). To get the ideal printed thin film, a suitable blend of three solvents (toluene, chloroform, and o-dichlorobenzene) with varying surface tensions was used. This created Marangoni flows in the wet film and facilitated a consistent and quick drying process. Since chlorinated solvents and other widely used printing solvents are hazardous to both humans and the environment, they can undoubtedly hinder industrial scalability. However, they are still a necessary component of industrial processes and the creation of organic materials for optoelectronic devices. With the optimized solvent formulation, the printed tBuCzDBA emissive layer demonstrated excellent morphology and minimal roughness. This film exhibits TADF features, according to the time-resolved photophysical analysis. The thin film produced by inkjet printing was used as an emissive layer in an OLED structure, adjusting the tBuCzD concentration in the ink to achieve the ideal film thickness and device functionality. The device with 18 nm of tBuCzDBA demonstrated an EQE of 10%, a maximum luminance of 32000 cd m−2 at 13 V, and a max-CE of 27.5 cd A−1 as shown in Fig. 82. The presented findings show that the ideal ink formulation can be regarded as a potent instrument for effortless inkjet printing small molecule active layers in OLEDs, attaining superior optical, electro-optical, and morphological characteristics. The design of an OLED can be made simpler by the potential use of a self-hosted material in the emissive layer. Furthermore, OLED technology can now be used in a wider range of industries at much lower costs because of solution-based, inexpensive, and industrially scalable manufacturing procedures.273
 | ||
| Fig. 82 Characterization of OLED device with different thicknesses of inkjet printed tBuCzDBA film as the active layer (A) device architecture and energy level diagram of the prepared OLEDs (B) current density versus voltage, (C) luminance versus voltage, and (D) efficiency versus luminance characteristics. | ||
By integrating a blue MR-TADF unit into the main polycarbazole chain, a general and efficient method for creating blue narrowband TADF conjugated polymers was studied by Yang et al. (2024). In the toluene solution, the conjugated polymers PCzDBNx 229 effectively produced narrowband blue emissions with emission peaks at 474 nm and FWHMs of 23 nm. With an EQEmax of 17.9% and an FWHM of only 28 nm, the devices based on these polymers demonstrated outstanding EL characteristics, leveraging the narrowband emission and good TADF features as shown in Fig. 83. This is the first instance of a narrowband TADF conjugated polymer in the blue area that they are aware and think that intriguing outcomes and the molecular design idea will direct the development of narrowband emissive TADF polymers in the future, leading to high-performance solution-processable OLEDs.274
 | ||
| Fig. 83 Chemical structure of (A) device architecture and energy level diagram of the prepared OLEDs (B) current density versus voltage, (C) luminance versus voltage, and (D) current efficiency–luminance–power efficiency characteristics (CE–L–PE) (E) external quantum efficiency luminance plots (F) summary of the EQE versus EL peak wavelength and FWHM plots for the reported narrowband emissive TADF polymer OLEDs. | ||
Yang et al. (2024) showed that creating green MR-TADF 230 conjugated polymers is possible as shown in Fig. 84. This method entailed embedding the acceptor moiety into the conjugated backbone and concurrently attaching a bluish-green MR emitting moiety as a pendant to an acceptor triphenyltriazine. Gaining from the improved ICT interaction with the addition of triphenyltriazine to the B atom's para carbon in the MR unit, as well as prolonged. All produced polymers showed red-shifted PL spectra in comparison to the pendant MR emitting moiety, indicating a conjugation effect along the polymeric backbone. These polymers' narrowband green emissions were indicated by their PL emission peaks, which ranged from 505 to 517 nm, and corresponding FWHMs, which were in the 46–48 nm range. With a record-high EQEmax of 22.2% among the MR-TADF polymer-based narrowband OLEDs, the EL devices based on these polymers demonstrated exceptional EL performance. It is anticipated that this work will encourage further researchers to investigate the MR-TADF polymers, which are incredibly effective.275
 | ||
| Fig. 84 Green MR-TADF and its attachment with MR emitting moiety acts as a pendant to an acceptor triphenyltriazine and simultaneously embeds acceptor moiety into polycarbazole backbone. | ||
The effective implementation of a molecular design strategy for high-performance blue MR-TADF materials involved affixing a planar electron-acceptor group to the MR framework's HOMO-distributed carbon atom designed by Yang et al. (2024) as shown in Fig. 85. The produced emitter 3QCzBN was provided with narrowband blue emission, high PLQY, and good TADF qualities inherited from the parent MR molecule due to the special capabilities of the decorating DPTRZ group. Additionally, it achieved increased molecule planarity, which resulted in an almost perfect Θ// of 98%. The outcome was a blue emission with a peak at 476 nm, a high EQEmax value of 25.2%, and an efficient suppression of efficiency roll-off in the manufactured non-sensitized binary OLED using 3QCzBN as an emitter. Notably, 3QCzBN-based HF OLED achieved a notable improvement in EQE with the help of a TADF sensitizer, reaching 34.8% at its maximum and sustaining 26.5% at a brightness of 1000 cd m−2. The 3QCzBN-based devices' two EQEmax values significantly outperformed their parent MR molecule equivalent. This work presents a straightforward yet effective method for creating high-performance MRTADF materials and emphasizes the critical role that molecule orientation plays in determining the EQE of OLEDs.276
 | ||
| Fig. 85 Chemical structure and molecular design strategy of 3QCzBN. | ||
Bi et al. (2024) suggested using the FMOE strategy to control several pyrimidine derivatives to create orange-red MR-TADF emitters with decreased aggregation-caused quenching simply and practically. Consequently, the newly designed emitters successfully prevented spectrum and aggregation quenching. Because of their twisted molecular topologies caused by the addition of large terminal substituents on the MR framework, they exhibit broadening characteristics in the solid state. Furthermore, by altering the electronic structure to varied degrees with the electron-withdrawing ability of peripheral acceptor substitutes, the emitter's PL spectra could be realized with precise orange-red emission while maintaining the narrowband emission and small ΔESTS characteristics. Solution-processed OLEDs with sensitizers showed good EL performance, with maximum ECEs of 19.0%, 20.4%, and 20.4% for BN-R1 235, BN-R2 236, and BN-R3 237 respectively, as well as high color purity orange-red light with CIE coordinates of 0.663, 0.337, 0.606, 0.393, and 0.553, 0.44 as shown in Fig. 86. To satisfy the requirements of ultra-high-definition displays, our work offers an effective route for creating orange-red MR-TADF emitters and is anticipated to stimulate more research efforts toward the production of a wider spectrum of narrowband emitters.277
 | ||
| Fig. 86 Molecular design strategy of BN-Cz-based MR-TADF emitters and chemical structures of BN-R1, BN-R2 and BN-R3. | ||
Zhu et al. (2024) resolved the question of why certain emitters appear to exhibit a negative singlet–triplet energy gap. Three carefully tailored organic emitters—DPS-m-bAc, DPS-p-bAc, and DPS-OAc—were created and thoroughly characterized for this purpose. The TADF and AIEE characteristics of all three compounds were evident. While DPS-m-bAc and DPS-p-bAc have two ICT states—the ICThigh and ICTlow states—DPS-OAc only has the ICThigh state, making it a reference for the 1ICThigh and 3ICThigh energies, according to an examination of the absorption spectra in solution. The through-bond nature of this ICT state results in a relatively large gap between the 1ICThigh and 3ICThigh states, whereas time-resolved photoluminescence studies (conducted at both 5 and 80 K) show that 3ICTlow and 1ICTlow are nearly isoenergetic due to some through-space contribution to this ICT state. Crucially, internal conversion between the higher and lower 3ICT states is slower than phosphorescence from 3ICThigh, and the 3ICThigh state has a larger energy total than both 1ICTlow and 3ICTlow. Therefore, in addition to the fluorescence from 1ICTlow, 77 K measurements also show phosphorescence from 3ICThigh. This could be mistakenly interpreted as a negative singlet–triplet gap, even though all of the compounds have very small but positive singlet–triplet gaps, as demonstrated by thorough time-resolved photoluminescence measurements conducted at 5 K. Because of its large TSCT impact and relatively modest TBCT effect, solid-state photophysical investigations reveal that DPS-m-bAc exhibits the greatest kRISC, with a value on the order of 107 s−1. Accordingly, with a slight efficiency roll-off, the solution-processed OLEDs based on DPS-m-bAc had the greatest EQE of 21.7% as shown in Fig. 87.278
 | ||
| Fig. 87 The energy levels, molecular structures in the device, and device performance. (a) energy levels of the device; (b) molecular structures; (c) EL spectra of the devices; (d) EQE–current density curves. (Inset) The images of the devices (left is DPS-m-bAc, and right is DPS-p-bAc). | ||
Park et al. (2024) studies Encasing the planar MR core (CzBN) with large phenyl-fluorene (Fl) units effectively produced a solution-processable, ACQ-free MR emitter with strong PLQY and narrowband emission.
Even in highly doped films, it was discovered that the MR core of the target chemical, 4FlCzBN 239, effectively inhibited interchromophore interactions. The spectrum broadening of 4FlCzBN in doped films, the red shift of emission spectra, and the ACQ were all greatly decreased because of this sterically shielded design. At different doping concentrations (2–16 weight percent), 4FlCzBN-based OLEDs produced by the solution technique demonstrated narrowband emission (28–30 nm) and EQEmax values of 10.1–10.9%. Additionally, it was discovered that 4FlCzBN-based devices with TADF sensitizer greatly reduced the efficiency roll-off and raised the EEQEmax by up to 12.2% as shown in Fig. 88. In the future present approach involves creating solution processable and ACQ-free MR emitters offers important insights into the creation of several novel compounds with related functions.279
 | ||
| Fig. 88 Chemical representation and advantages of 4FICzBN and EQE electroluminescence characteristics of the material. | ||
For the first time Cheng et al. (2024) worked on donor–acceptor type tetradentate Pd(II) emitters that exhibit strong TADF emission in solution and thin film, with quantum yields as high as 99% and kr as high as 7.3 × 105 s−1 at ambient temperature as shown in Fig. 89. Such Pd(II)-TADF emitters are very efficient and adaptable, producing sky-blue TADF OLEDs and blue TADF-sensitized OLEDs with maximum EQEs of 24.8% and 23.1%, respectively. They also have low-efficiency roll-off at 1000 cd m−2. Tetradentate Pd(II)-TADF complexes are a viable option for producing stable blue OLED emitters with great performance.280
 | ||
| Fig. 89 Chemical representation of Pd–B-2 and tetradentate Pd(II) emitters. | ||
Yan et al. (2024) have developed a novel deep blue hybridized local and charge-transfer (HLCT) fluorophore, PICZ2F 244, characterized by high oscillator strength, a fast radiative decay rate (kr), and bipolar charge transport properties. The doped device had an EQE of 7.10% with the desired CIE coordinate (0.155, 0.068), while the non-doped device had an EQE of 6.16% with reduced roll-off at a high brightness level of 15000 cd m−2 (EQE 15000: 5.80%). A high-performance single-cell all-fluorescent CT-WOLED with a single-doped single-emissive-layer structure was accomplished by using PICZ2F as both a blue emitter and a universal host for the yellow TADF emitter. Along with a huge CCT span of 3749–18279 K, this device demonstrated remarkable efficiencies of 89.5 lm W−1, 79.8 cd A−1, and 23.73%, respectively as shown in Fig. 90. This work not only shows the significant potential of HLCT-type deep-blue materials as all-fluorescent WOLED emitters and hosts, but it also offers a workable design approach for achieving straightforward and effective CT-WOLEDs, which could open the door for the use of all-fluorescent WOLEDs with color tuning in real-world applications.281
 | ||
| Fig. 90 Molecular representation and design concept of PICZ2F along with EL characteristics. | ||
Yang et al. (2024) produced two deep blue-emitting di-nuclear TADF-Au(I) complexes 246–247 using an MR-type B/N ligand. On the one hand, the participation of Au Orbitals in the S1 state. Significantly improves the SOC effect between S1 and T1 states. The complexes had 8-fold lower delayed fluorescence lifetimes than the parent molecule. However, adding two Au atoms to the B/N MR skeleton's periphery positions does not affect color purity. The OLED with iPrAuBN emitter produces pure blue light with a maximum at 442 nm and FWHM of 19 nm, combining the CIE coordinates (0.154, 0.036) and EQEmax of 14.8% as shown in Fig. 91. This research suggests that combining MR skeletons with heavy metal atoms can create non-sensitized, high-performance ultrapure blue emitters.282
 | ||
| Fig. 91 Chemical representation of MeBN and di-nuclear Au(II) complexes (A) electroluminescence curve (B) CIEy coordinate vs. emission wavelength which indicates deep blue phosphorescent and MR-TADF OLEDs. | ||
Yan et al. (2024) designed MR-TADF emitters with diverse electron-donating components to study the impact of LRCT and heavy-atom effects on excited states as shown in Fig. 92. They developed MR-TADF emitters by combining electron-donating units with the MR skeleton to study the impact of LRCT and heavy-atom effects on excited-state properties and energy levels. Photophysical properties, including quantum states and energy levels. Photophysical properties and quantum chemistry computations show that radiative decay and exciton-up conversion can be modulated. MR-TADF emitters have covalently bonded MR segments that donate electrons. The electron-donating ability of moieties allows for precise control over their excited-state properties. Enhanced LRCT in tBuCzBN-PXZ allows for spin-flipping of triplet excitons, quickening the RISC process while retaining narrow emission of the S1 state. Research shows that LRCT is more effective than heavy-atom effects in influencing exciton dynamics in MR-TADF emitters with strong electron-donating units.
 | ||
| Fig. 92 Chemical representation of different TADF emitters. | ||
Adding a phenyl bridge between the MR segment and donor moiety weakens LRCT strength. The photophysical properties and quantum chemical simulations show that increasing the number of atoms leads to a significant increase in kRISC values, indicating that the heavy-atom effect can accelerate the spin-flip of triplet excitons in MR-TADF emitters without modulatory LRCT states. Both tBuCzBN-PXZ 248 and tBuCzBN-Ph-PSeZ 253 have significantly higher kRISC values (>2.0 × 105 s−1) and near-unity photoluminescence quantum yields. The EQEmax values of tBuCzBN-PXZ and tBuCzBN-Ph-PSeZ-based solution-processable OLEDs reach 21.3% and 19.4%, respectively, placing them in the top tier of reported solution-processable MR TADF binary OLEDs without additional sensitizers. This study suggests a method for controlling the photoelectric characteristics of MR-TADF emitters by carefully managing LRCT and heavy-atom effects.283
4.4. Miscellaneous applications of TADF materials
Because TADF-based materials can effectively convert triplet excitons into singlet excitons, they are mainly utilized in OLEDs to improve performance. In addition to OLEDs, these materials find use in a few cutting-edge applications, including organic hybrid microwire radial heterojunctions, TTA sensitizers, ECL cells, UVOPDs, LEECs, multicolor luminous micelles, ML, and MLC. This adaptability highlights the wide range of applications for TADF-based materials in cutting-edge technological domains, which will be outlined below.Due to their beneficial properties, many TTA-based materials have been investigated as OLED emitters.286 Beyond their role in OLED technology, TTA materials also show promise in biomedical applications and sensing.287 Their tunable excitation and emission wavelengths, strong visible light absorption, and high upconversion quantum yields make them particularly useful in energy-related applications such as photocatalysis and solar cells.288
There is existing literature discussing the role of TADF molecules in TTA mechanisms.105,289–296 This section provides an overview of recent studies on the role of TADF molecules in TTA mechanisms from 2023–2024.
Chen et al. (2023) introduced a novel TTA upconversion system using the newly developed TADF molecule DMACPDO 254 as a triplet photosensitizer, paired with diphenylanthracene (DPA) 255 as an annihilator (Fig. 93).297 Efficient upconversion of fluorescence was reported in toluene, benzene, and chlorobenzene, with upconversion quantum yields (ΦUC) of 22.3%, 21.9%, and 8.1%, respectively. Further, the effect of solvent properties on ISC and triplet–triplet energy transfer (TTET) processes was explored. The critical factor for TTA upconversion was determined to be the solvent polarity, as it could alter the energy of the T2 triplet state.
 | ||
| Fig. 93 Diagram depicting the TTA upconversion process between the TADF photosensitizer DMACPDO (254) and the annihilator DPA (255). Adapted from ref. 297 with permission from the Royal Society of Chemistry. | ||
Li et al. (2023) created the asymmetrically structured diphenylsulfone derivative 3,4′-SOAD (256), as shown in Fig. 94, to efficiently use triplet excitons through the RISC process. A combination of low ΔEST and increased singlet–triplet spin-orbital coupling was caused by an asymmetrical packing of 3,4′-SOAD that resulted in alternating strengths of intermolecular interactions. As a result, the triplet excitons could be promoted from TADF to TTA upconversion. A sky blue device based on 3,4′-SOAD was produced and showed high electroluminescence efficiencies and low-efficiency roll-off, with a maximum power efficiency (PEmax) of 33.5 lm W−1, a maximum EQEmax of 14.7%, and a maximum current efficiency (CEmax) of 32.0 cd A−1.298
 | ||
| Fig. 94 Molecular representation of 3,4′-SOAD (256).298 | ||
Tang et al. (2024) employed magneto-electroluminescence to study a “hot exciton” channel in OLEDs doped with 1% rubrene and a TADF sensitizer, CzDBA, at room temperature, as depicted in Fig. 95.299 This high-level triplet–triplet annihilation (HL-TTA) process involves transitions from T2 to S1. The presence of CzDBA enhances Dexter energy transfer pathways between two sets of triplets: the host-sensitizer and sensitizer-guest triplets, thereby increasing excitons in the T2 state to promote HL-TTA. Similar to the conventional TTA process, the HL-TTA process is enhanced by a higher bias current, but unlike the conventional TTA process, the HL-TTA process is diminished by decreased temperature. These findings provide valuable insights into the mechanisms of HL-TTA processes, also referred to as “hot exciton” channels A deeper understanding of these processes could contribute to the development of high-performance OLEDs.
 | ||
| Fig. 95 Diagram showing room-temperature energy transfer mechanisms in device B1 without a CzDBA sensitizer. Schematic illustration of low-temperature energy transfer mechanisms in device B1 without a CzDBA sensitizer. Adapted from ref. 299, with the permission of AIP Publishing. | ||
Using Stille coupling reactions between bromoanthracene and corresponding stannanes, Bezvikonnyi et al. (2024) synthesized three carbazolyl phenyl anthracenyl benzonitriles with distinct substituents (tert-butyl, methoxy, and methoxyethoxy) at the C-3 and C-6 positions of the carbazole moiety, as shown in Fig. 96A.300 The planar anthracene with π-conjugation enhanced molecular packing in the solid state and the resulting π-stacking interactions favored TTA. The compounds emitted DF on photoexcitation, which improved their thermal stability and PLQY values, and the t-butyl substituent was the most efficient. The t-butyl compound was therefore used to fabricate host-free and guest–host emitting layers in OLEDs. With the use of TTA upconversion and a bicomponent host–guest system, a hyperfluorescent OLED could also be produced without the requirement for a sensitizer. As can be observed in Fig. 96B, the non-doped TTA OLEDs achieved a 5.9% maximum EQE, while the hyperfluorescent OLEDs using TTA upconversion achieved a 7.1% maximum EQE, and a long-lived emission characteristic of TTA was detected for the device using the t-butyl-substituted emitter.
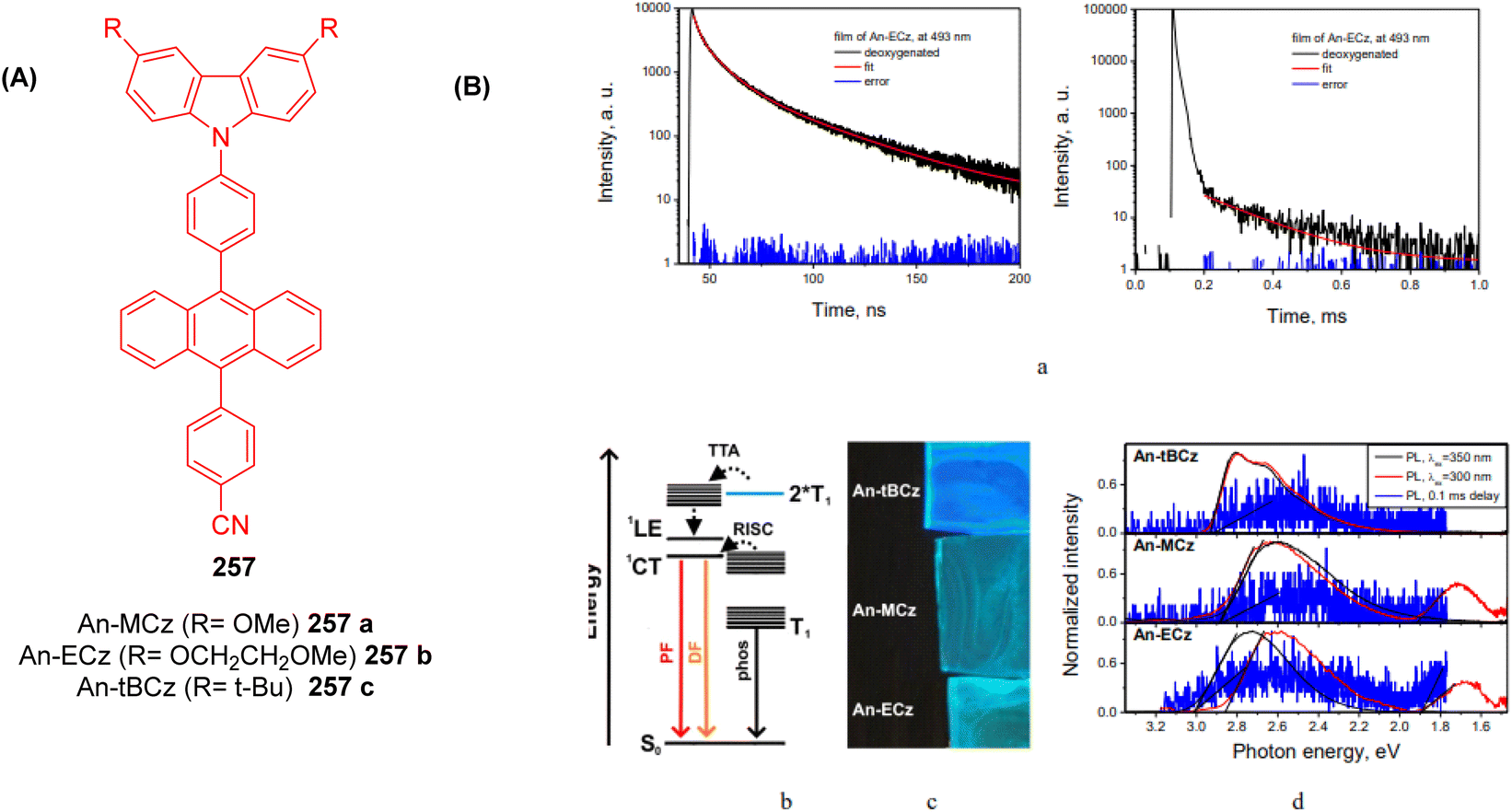 | ||
| Fig. 96 (A) Molecular structure of An-MCz (257a), An-MCz (257b), and An-tBCz (257c) (B) (a) PL decay curves of the films (b) diagram illustrating the electronic states and transitions of the molecules (c) a photograph of the films of the compounds produced by deposition on drop casting on FTO substrates (d) PL spectra with varying delays. Reproduced from ref. 273, https://doi.org/10.1021/acsaelm.4c00533, under the terms of the CC BY 4.0 license, https://creativecommons.org/licenses/by/4.0/. | ||
Zhao et al. (2024) examined the TADF molecule o-BTH-DMF (seen in Fig. 97a), using both time-resolved spectroscopy and computational modeling employing DFT calculations, to study the dynamics of both the singlet and triplet excited states.301 Transient absorption (TA) spectroscopy was done using two different timescales: femtoseconds (fs-TA) and nanoseconds (ns-TA). The fs-TA spectra indicated that the S1 state has a lifetime similar to that of transient fluorescence as well as a long-lived triplet state. On the other hand, the ns-TA spectra showed that the larger energy gap between the singlet excited state S1 and excited triplet states above T1 (Tn), in addition to the local emission properties between triplet states, meant that the internal conversion from Tn to T1 was favored over RISC. The accumulation of excitons in the T1 state was then confirmed using sensitization experiments confirmed that the observed triplet state in ns-TA is the T1 state as shown in Fig. 97d and e. The RISC processes in the o-BTH-DMF molecules were validated using transient fluorescence emission spectroscopy. These findings were supported by the DFT calculations for HOMO and LUMO shown in Fig. 97b. This research provides greater insight into the o-BTH-DMF luminescence mechanism, which could be useful in applications of this TADF molecule for OLEDs.
 | ||
| Fig. 97 (a) Molecular structure of o-BTH-DMF. (b) Theoretical calculations showing the HOMO and LUMO distributions of o-BTH-DMF. (c) UV-vis absorption and PL spectra along with phosphorescent spectra of o-BTH-DMF/PMMA doped thin film. (d and e) Delayed fluorescence lifetime measurements of o-BTH-DMF (d) nitrogen conditions and (e) oxygen conditions. (f) Simplified photophysical diagram illustrating the photophysical processes in o-BTH-DMF, including PF, DF, and phosphorescence. Reproduced from ref. 301 with permission from American Chemical Society, Copyright © 2024. | ||
Ahmad et al. (2024) integrated strontium titanate (STO) into the triplet sensitizer-emitter (TSE) system composed of a 4CzIPN/p-terphenyl thin film to form the TSE/STO photovoltaic cell. The STO significantly enhanced the EQE under visible light, as depicted in Fig. 98, which was further enhanced by increased irradiance intensity.302 The TSE upconversion was dependent on the thickness of the film, resulting in higher efficiency without saturation in EQE/EQE0, potentially allowing these cells to maximize absorption of the high photon flux. The efficiency of the cells was further optimized by preparation and characterization in an inert gas environment with N2 bubbling to deoxygenate the TSE solution. Overall, the TSE system demonstrated promise as a thin film for photovoltaic cells for effectively using photon absorption from longer wavelengths while suppressing “parasitic absorption” processes from the electron transport layer of the cell.
 | ||
| Fig. 98 Visible light-assisted charge extraction in high bandgap SrTiO3 through the integration of triplet sensitizer-emitter thin film. Reproduced from ref. 302 with permission from American Chemical Society, Copyright © 2024. | ||
 | ||
| Fig. 99 (a) Absorption spectra of PVK:2CzPN, PVK:4CzPN, and PVK:4CzIPN blend films. (b) Molecular scaffolds of 2CzPN, 4CzPN, and 4CzIPN and device architecture. Reproduced from ref. 303 © IOP Publishing. Reproduced with permission. All rights reserved. | ||
To develop materials that could be used in devices with activity as both OLEDs and UVOPDs, Wang et al. (2023) synthesized two blue-fluorescent emitters, TPA-AN and 2mTPA-AN. These emitter molecules were composed of an acceptor unit, benzonitrile, and a donor unit, triphenylamine (TPA) or 4-methyl-N-phenyl-N-(p-tolyl)-aniline (MeTPA), with anthracene (AN) as a bridge as shown in Fig. 100.304
 | ||
| Fig. 100 Molecular structures of the employed materials.304 | ||
Zeng and coworkers (2024) demonstrated that TADF triplet state excitons generated by 4CzIPN can significantly enhance the performance of organic photodetectors (OPDs). These OPDs were prepared by a combination of 4CzIPN with a high-mobility PBTTT-C14 polymer via a bulk type-II heterojunction, as illustrated in Fig. 101a.305 The long-lived spin-triplet excitons of 4CzIPN, which are nearly isoenergetic with the spin-singlets, improve free carrier extraction and photogating effect, leading to a fast response and high gain, as illustrated in the energy level diagram in Fig. 101b. The PBTTT-C14/4CzIPN UVOPD device was able to operate at a low dark current (∼pA) with a detectivity of ∼1011 Jones while demonstrating a quick reaction time of ∼79 ms, a high EQE of ∼1.0 × 104, and a high photogain (∼103). In addition, the gate photoresponse range was broad (250–600 nm) and tunable.
 | ||
| Fig. 101 (a) Diagram of the device structure and (b) energy level diagram in PBTTT-C14 polymer and 4CzIPN bulk heterojunction phototransistor. Reproduced from ref. 305 with permission from John Wiley & Sons, Copyright © 2024, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
Adachi and colleagues 2014 researched TADF molecules, such as 2CzPN, 4CzPN, 4CzIPN, and 4CzTPN, that emit ECL with high efficiency, producing light ranging from green to red.307 Notably, 4CzIPN in MeCN achieved an ECL efficiency of approximately 15%, which is about three times higher than the efficiency observed for the [Ru(bpy)3]2+ system and significantly exceeds its PLQY. This result indicates the significant advantage of spin upconversion through thermal activation for ECL as shown in Fig. 102. Therefore, TADF molecules show great promise for use in ECL applications such as immunoassays and liquid light-emitting devices due to their high ECL efficiency.
 | ||
| Fig. 102 Molecular structures of four TADF molecules with ECL emission.307 | ||
A study by Mizuno and coworkers in 2014 described a four-color microfluidic ECL system using straightforwardly designed ECL cells, capable of both single and multi-color emission as shown in Fig. 103.308 In this configuration, the TADF compound 4CzIPN was utilized for green ECL. For the fabrication of yellow, blue, and red ECLs, conventional fluorescent materials were employed including rubrene, DPA, and DBP-doped rubrene, respectively. This study underscores the considerable potential of this multicolor microfluidic device for applications in liquid-based light emission.
 | ||
| Fig. 103 (a) Photographs for emission of light and emissions of light from DPA, 4CzIPN, rubrene and DBP-doped rubrene in simple ECL cells (b) ECL spectra from the simple-structured ECL cells. Reproduced from ref. 308 with permission from Elsevier, Copyright © 2014. | ||
Kasahara et al. designed a yellow phosphorescent ECL cell prepared using a mediator in solution, namely the carbazolyl dicyanobenzene derivative 2CzPN 29, as shown in Fig. 104.309 2CzPN 29 has deeper HOMO and LUMO levels compared to Ir(BT)2(acac) 262. This configuration achieved the highest maximum luminance (Lmax) of 113 cd m−2 and efficiency CEmax of 2.84 cd A−1 reported for iridium complex-based cells. The study suggested that the developed ECL system could be suitable for both fluorescent and phosphorescent materials. The authors suggested potential future improvements for this system, including using combinations of iridium complexes with these mediators to form multicolor phosphorescent cells, encapsulating the ECL cells to inhibit phosphorescence quenching caused by oxygen molecules with a triplet ground state, and coating the electrodes with conductive films to enhance the ECL performance.
 | ||
| Fig. 104 Molecular representation of 2CzPN (29) and Ir(BT)2(acac) (262) along with the microfluidic ECL cell with a schematic cross-section. Reproduced from ref. 309, https://doi.org/10.5796/electrochemistry.23-00147, under the terms of the CC BY 4.0 license, https://creativecommons.org/licenses/by/4.0/. | ||
 | ||
| Fig. 105 (top) Diagram representing the process for the synthesis of ternary radial heterojunction microwires and (below) photoconductivity process in TCTA/PCBM/4CzIPN. Reproduced from ref. 310 with permission from the Royal Society of Chemistry. | ||
 | ||
| Fig. 106 Synthetic route of micellar ensemble P-M, with the encasement of the block copolymer P by diphenyl-diacetylene monomer M to form oligodiacetylene trimers (P-T) and dimers (P-DT and P-DT2). Reproduced from ref. 311, https://doi.org/10.1039/C5SC04253D, under the terms of the CC BY 4.0 license, https://creativecommons.org/licenses/by/4.0/. | ||
In 2015, Chi and Xu's group fabricated an asymmetric D–A–D′ type TADF material by modification of the acceptor unit with benzophenone, resulting in the synthesis of carbazolyl- and phenothiazinyl-substituted benzophenone (OPC).316 OPC possessed characteristics such as dual emission, aggregation-induced emission (AIE) TADF and mechanofluorochromic properties. In its crystalline form, OPC emitted white light with dual photoluminescence peaks in the blue and yellow regions of the spectrum, but after grinding, only the yellow peak remained, as shown in Fig. 107. This study demonstrated that these asymmetric molecules present a promising approach for creating novel organic materials that emit white light with dual emission properties and mechanofluorochromic behavior, which makes them suitable for various applications.
 | ||
| Fig. 107 PL spectrum of OPC in the solid state before and after grinding. Reproduced from ref. 316 with permission from John Wiley & Sons, Copyright © 2015, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
Cheng and coworkers demonstrated a TADF material in 2016 that possessed two meta-carbazolyl groups on the phenyl ring of benzoyl-4-pyridine mDCBP (263). The MLC properties and chemical structure of mDCBP (263) are shown in Fig. 108.317 Upon grinding, the material's luminance color was changed from blue to green. The original blue color could be returned by exposing the material to dichloromethane vapor, demonstrating the reversible nature of mDCBP's emission properties.
 | ||
| Fig. 108 Chemical structure and photographs of the material in different states. Adapted from ref. 317 with permission from the Royal Society of Chemistry. | ||
 | ||
| Fig. 109 Schematic diagram of a general LEEC device. | ||
There has been a great deal of research into describing the role TADF materials can play in LEECs.320–329
Wong et al. (2015) introduced charged organic TADF-based OLEDs and LEEC devices. This TADF material could function both as a charge transporter and emitter, as depicted in Fig. 110.330 The cyano (–CN) group, commonly used as an electron acceptor, plays a crucial role in lowering the Lowest Unoccupied Molecular Orbital (LUMO) energy level, enhancing the charge-transfer characteristics. By incorporating strong acceptor groups like cyano, the energy gap between the singlet and triplet excited states (ΔEST) is minimized. This facilitates reverse intersystem crossing (rISC), where triplet excitons are converted back to the singlet state, enabling delayed fluorescence. The donor groups, on the other hand, such as carbazole, triphenylamine, or other electron-rich moieties, raise the Highest Occupied Molecular Orbital (HOMO) energy level. This donor–acceptor configuration ensures efficient charge separation in the excited state, promoting TADF. The maximum EQE of the LEECs was improved by lowering the current density, leading to a value of 0.39% for this emitter. The OLEDs produced from this emitter were color-stable and able to be processed in solution. In addition, OLEDs using pristine films of the emitter demonstrated superior performance compared to OLEDs using a doped EML, which is consistent with the findings for the LEECs, demonstrating the potential of these emitters for non-doped TADF devices.330
 | ||
| Fig. 110 The chemical structure of the TADF compound superimposed on a photograph of its LEEC. Reproduced from ref. 330, https://doi.org/10.1021/acs.chemmater.5b03245, under the terms of the CC BY 4.0 license, https://creativecommons.org/licenses/by/4.0/. | ||
Pei and coworkers (2017) presented a single-layer LEEC based on a TADF host, illustrated in Fig. 111.331 The single active layer of the LEEC is comprised of a solution-processed mixture of a TADF host material (PIC-TRZ, 5), three dopants 264, 265, and 266 that were used to emit green, yellow and red phosphorescence, respectively, polyvinylcarbazole (PVK, 267) to improve the morphology of the layer, and an electrolyte (268). By varying the ratios of the dopants, different color emissions could be achieved, including green, yellow, red, and warm white light, as well as blue-green in the non-doped device. However, the non-doped device only emitted less than 300 cd m−2 with a maximum efficiency of 0.5 cd A−1, while the green, yellow, red, and white LEECs reached 6000, 4200, 2300, and 1200 cd m−2 with a peak efficiency of 7.1 cd A−1. The green LEEC even achieved an exceptional luminance of 35000 cd m−2 for a short period, although the effect was short-lived. These LEECs were able to leverage the optical and transport properties of TADF materials, which allowed them to surpass the efficiency and brightness of conventional LEECs using phosphorescent hosts. Further, the simple single-layer solution-processed structure would be more cost-effective for the production of lighting sources with high luminance.
 | ||
| Fig. 111 Chemical structures of the TADF host (5), PD molecules (264–266), morphology modifier (267), and electrolyte (268) along with emission mechanisms in TADF-LEECs. Reproduced from ref. 331, https://doi.org/10.1038/s41598-017-01812-2, under the terms of the CC BY 4.0 license, https://creativecommons.org/licenses/by/4.0/. | ||
Moore et al. (2018) synthesized and investigated three first-generation carbazole (Cz)-based ionic host materials for usage in LEEC devices.332 The carbazole (Cz)-based ionic host materials were shown to have broad band gaps characterized by reversible oxidations and irreversible reductions. PL experiments showed that the luminescence of films containing the carbazole (Cz)-based host materials and an ionic transition metal complex (iTMC) guest was enhanced compared to pristine films of the host materials alone. The LEECs were therefore made with thin films of the pristine hosts, pristine guests, and 90%/10% host/guest blends. Of these, the system containing PBI-CzH (269) host and its guest Ir(ppy)2(dtb-bpy) (270) (as shown in Fig. 112), resulted in the best device performance, with an EQE of 3.80% and a luminance maximum of 624 cd m−2.
 | ||
| Fig. 112 Chemical structure of first-generation carbazole (Cz)-based ionic host material (270).332 | ||
Edman et al. (2019) prepared a metal-free LEEC device from a single layer of active material between air-stable electrodes that efficiently produced vivid TADF emission.333 The active material was composed mostly of a polymeric blend host and less than 1% of the TADF-active guest emitter. Three TADF guest emitters were studied: 4CzIPN, 2-[4-(diphenylamino)phenyl]-10,10-dioxide-9H-thioxanthen-9-one (TXO-TPA), and the orange-emitting 7,10-bis(4-(diphenylamino)phenyl)-2,3-dicyanopyrazinophenanthrene (TPA-DCPP), as shown in Fig. 113g. The host allowed for uniform film morphology, effective electronic transport and a balance of electrochemical doping, while the low guest concentration ensured optimal energy transfer from host to guest and TADF emission while reducing guest–guest aggregation. To optimize the electrochemical doping, the electrolyte tetrahexylammonium tetrafluoroborate (THABF4) was included as part of the polymeric blend. The most efficient of the resulting TADF LEECs produced using this system was a yellow-emitting LEEC with a maximum EQE was 7.0% and peak current efficiency (CEmax), as well as a luminance of 120 cd m−2, as shown in Fig. 113.
 | ||
| Fig. 113 Voltammograms were recorded on thin films of the host compounds (a and b), the blend host (c), and the guest compounds (d–f), as identified in the insets. (g) Structure of three TADF guest emitters. (h) Energy levels of host and guest compounds. (i) Normalized absorbance of the guest compound (left y-axis) and normalized PL of the host (right y-axis) for various films. Reproduced from ref. 333, https://doi.org/10.1038/s41598-017-01812-2 https://doi.org/10.1038/s41467-019-13289-w, under the terms of the CC BY 4.0 license, https://creativecommons.org/licenses/by/4.0/. | ||
Shanmugasundaram et al. (2019) synthesized a carbazole-phenothiazine compound (CPC, 271) as depicted in Fig. 114.334 CPC (271) was shown to have high thermal stability with blue emission both in thin films and in solution. This blue emission was also observed for a LEEC with CPC as the active material, with CIE coordinates (0.16, 0.33). The incorporation of an ionic liquid improved the electroluminescent performance of the LEEC, resulting in blue emission centered at 485 nm, with a brightness of 454 cd m−2, as well as a current efficiency of 1.33 cd A−1 and an EQE of 1.76%.
 | ||
| Fig. 114 Molecular structure of CPC (271).334 | ||
Su et al. and coworkers designed TADF LEECs using a phosphorescent complex 272 as the host and a red TADF emitter, APDC-DTPA 273, which emits deep-red TADF, as the guest (Fig. 115).335 Excitons from both the S1 and T1 states of 301 were involved in the Förster energy transfer between the host and guest, which was confirmed by PL and photophysical experiments. Guest triplet excitons were also recycled by Dexter energy transfer from the host to guest followed by RISC. This process was supported by the observed DF decay. The LEEC exhibited a maximum EQE of 5.11%, which the authors noted was one of the highest for deep-red emitting LEECs at the time. Thanks to the efficient energy transfer and RISC processes, the resulting LEEC was able to use over 73% of the excitons produced in the guest molecules for light emission. On the other hand, if the device current or concentration of guest doping increased, the efficiency was still limited by TTA in the guest molecules.
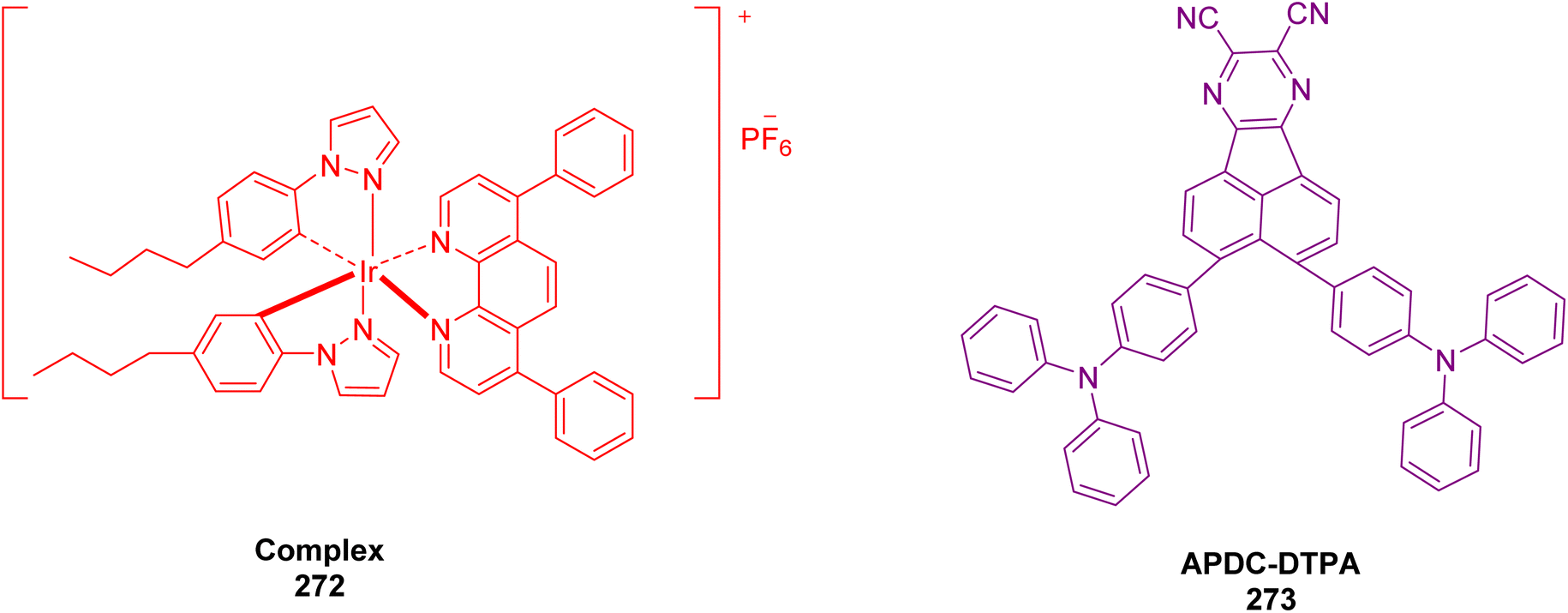 | ||
| Fig. 115 Chemical structure of phosphorescent complex 272 and APDC-DTPA (273).335 | ||
He et al. (2022) prepared two intrinsically-ionic molecules (274) using a donor, 3,6-di-tert-butylcarazole (tBuCAZ), and an acceptor, 2,4-diphenylpyrimidine (dPhPM),336 in which the tBuCAZ and dPhPM groups were at para-positions for H1 and ortho- for H2, as described in Fig. 116. H1 displayed no TADF, as a consequence of its low triplet energy and correspondingly large ΔEST. Conversely, H2 exhibited high TADF due to higher triplet energy and correspondingly small ΔEST. Therefore, H2 was used as a host to construct blue-emitting LEECs with [Ir(buoppy)2(dmapzpy)]PF6 as guest material. The H2-based LEECs were color-stable and showed a peak brightness of 1078 cd m−2, a peak EQE of 8.6%, and a half-lifetime (t1/2) of 66 minutes under a constant-current driver. The addition of LiPF6 as an electrolyte to the active layer improved the performance to a brightness of 918 cd m−2 with a t1/2 of 2 hours.
 | ||
| Fig. 116 (top) Chemical structure of tBuCAZ and (bottom) (a) peak EL intensity, (b) time-dependent brightness and (c) time-dependent voltage. Adapted from ref. 336 with permission from Elsevier, Copyright © 2022. | ||
In 2022, Su et al. synthesized two pyridinium-based orange-red TADF emitters, Pym-CZ 275a and Pym-tBuCZ 276b, for high-efficiency LECs, as depicted in Fig. 117.337 The pyridine moiety of the emitter was both the ionic component of the LEEC as well as the electron acceptor for TADF. The pyridine was connected to a donor, either CZ or t-BuCZ in a meta-position. TADF emission was detected by delayed fluorescence lifetimes of 2887 ns for Pym-CZ 275a and 1746 ns for Pym-tBuCZ 276b. The LEEC containing Pym-CZ 276a exhibited a maximum EQE of 1.2%. As the ionic component of the LEECs is pyridine, these are the first purely organic ionic TADF materials described for orange-red LEECs, which may make them useful as a new low-cost approach for the fabrication of these devices.
 | ||
| Fig. 117 Chemical structure of Pym-CZ (275a) and Pym-tBuCZ (276b).337 | ||
Zysman-Colman and coworkers (2024) synthesized TPPO-tBu-DiKTa 277 and TPA-tBu-DiKTa 278, the two novel MR-TADF emitters seen in Fig. 118.338 According to photophysical investigations and DFT calculations, the excited states of both TPPO-tBu-DiKTa and TPA-tBu-DiKTa exhibited short-range charge transfer (SRCT). The intermolecular charge transfer in TPA-tBu-DiKTa sped up the RISC processes, resulting in a kRISC of 1.8 × 104 s−1 in a 2 wt% doped mCP film, compared to 4.0 × 103 s−1 for TPPO-tBu-DiKTa in the same film. OLEDs were produced with different concentrations of doped TPPO-tBu-DiKTa or TPA-tBu-DiKTa in a mCP:PPT cohost. The devices containing 2% TPPO-tBu-DiKTa or 5% TPA-tBu-DiKTa OLED containing 2% TPPO-tBu-DiKTa showed a blue emission at 480 nm with an EQEmax of 24.4%, while the OLED containing 5% TPA-tBu-DiKTa showed a green emission at 508 nm with an EQEmax of 31.0%. The ortho-substituted geometry in both emitters also prevents aggregation of DiKTa moieties leading to ACQ, even with increases in the dopant concentration. For example, a device with 25% TPA-tBu-DiKTa had a broader emission while retaining a high EQEmax, and reduced efficiency roll-off. The emitters were also observed to produce narrowband emission via the facilitation of electrochemical doping. For example, a LEEC with optimized TPA-tBu-DiKTa concentration in a 26DCzPPy:TCTA host matrix was able to produce steady narrowband blue emission with a luminance of 425 cd m−2 with an EQEmax of 2.6%.
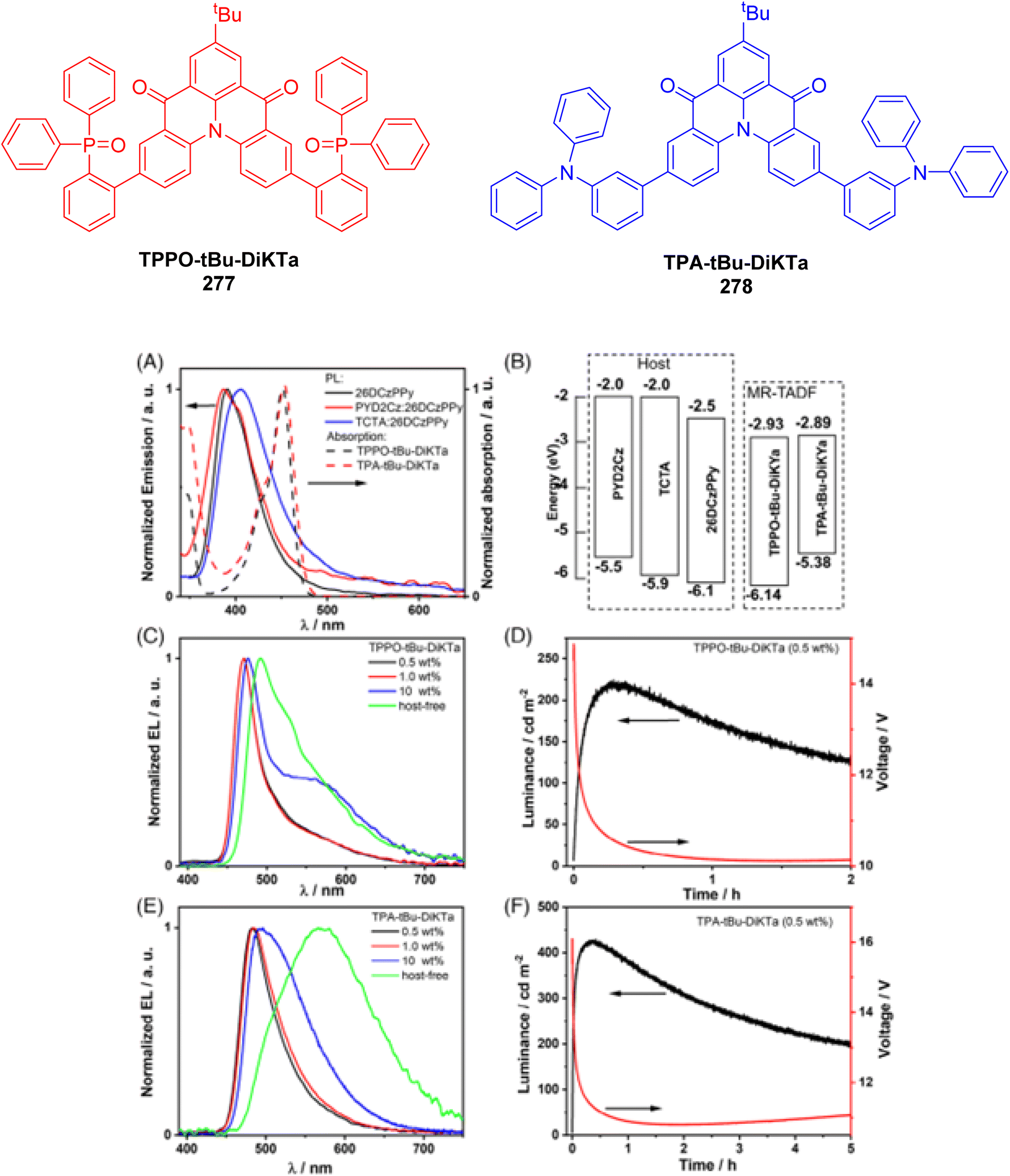 | ||
| Fig. 118 (A) Normalized PL spectra of the three host systems and the normalized absorption spectra of the two MR-TADF emitters (B) electron energy diagram of the three host compounds and the two MR-TADF emitters (C) normalized EL spectra and (D) temporal evolution of luminance and the voltage of the host-free triphenylphosphine oxide (TPPO)-tBu-DiKTa LECs (0.5 wt%). (E) Normalized EL spectra and (F) temporal evolution of the luminance and the voltage of the host-free triphenylamine (TPA)-tBu-DiKTa LECs (0.5 wt%). Reproduced from ref. 338, https://doi.org/10.1002/agt2.571https://doi.org/10.1038/s41598-017-01812-2, under the terms of the CC BY 4.0 license, https://creativecommons.org/licenses/by/4.0/. | ||
5. Conclusions and future outlook
Research into TADF has attracted significant global interest as an efficient method for the production of triplet excitons in pure organic molecules for EL applications. TADF materials as a host material in OLEDs the next generation of OLED technology, and have achieved high device functioning as comparable to phosphorescent OLEDs fabricated using noble metal complexes. Improvement in the functioning of TADF OLEDs, including solution-processed devices, is essential for real-world display technologies. Beyond electroluminescence, TADF materials have shown potential for photocatalysis, facilitating organic transformations under milder conditions than traditional catalytic methods. New organic photocatalysts like 4CzIPN offer advantages such as lower toxicity, cost-effectiveness, and structural diversity. While the development of TADF photocatalysts has lagged behind TADF materials for OLEDs, the field is rapidly evolving, providing valuable insights into structural design and mechanisms. TADF materials are also being explored for various applications, including TTA sensitizers, organic UVPODs, fluorescence probes for biomedical applications, and mechanoluminochromism, leveraging their small ΔEST and efficient RISC processes. Development of TADF materials is still in a nascent stage and further advances in molecular design and theoretical frameworks are needed to enhance their photophysical characteristics and move beyond D–A type systems. Future research should focus on developing new models and innovative approaches to control excitons through the TADF process, potentially expanding these materials into other research fields and applications. In conclusion, TADF materials represent a continuously advancing field of study with significant potential across various applications. The continued exploration and development of TADF materials promise to drive advancements across a spectrum of technologies, from healthcare to environmental sustainability, unlocking new potentials and innovative solutions in various scientific and industrial fields.Data availability
All the data is provided in the manuscript file.Conflicts of interest
The authors have no conflict of interest to declare.Acknowledgements
The authors extend their appreciation to Umm Al-Qura University, Saudi Arabia for funding this research work through grant number: 25UQU4320545GSSR01. This research work was funded by Umm Al-Qura University, Saudi Arabia under grant number: 25UQU4320545GSSR01.References
- R. Gao, M. S. Kodaimati and D. Yan, Chem. Soc. Rev., 2021, 50, 5564–5589 RSC.
- N. Armaroli and H. J. Bolink, Top. Curr. Chem., 2016, 374, 1–3 CrossRef CAS PubMed.
- B. Santra, S. Pal and A. Kanjilal, ACS Appl. Opt. Mater., 2024, 2, 687–696 CrossRef CAS.
- J. Hölsä, Electrochem. Soc. Interface, 2009, 18, 42 CrossRef.
- A. Mesaros, Appl. Sci., 2023, 13, 11221 CrossRef CAS.
- M. G. Brik and A. M. Srivastava, Luminescent Materials: Fundamentals and Applications, Walter de Gruyter GmbH & Co KG, 2023 Search PubMed.
- E. Kim, Y. Lee, S. Lee and S. B. Park, Acc. Chem. Res., 2015, 48, 538–547 CrossRef CAS PubMed.
- J. Zhou, Q. Liu, W. Feng, Y. Sun and F. Li, Chem. Rev., 2015, 115, 395–465 CrossRef CAS PubMed.
- M. D. Allendorf, C. A. Bauer, R. Bhakta and R. Houk, Chem. Soc. Rev., 2009, 38, 1330–1352 RSC.
- D. T. Thielemann, A. T. Wagner, E. Rösch, D. K. Kölmel, J. G. Heck, B. Rudat, M. Neumaier, C. Feldmann, U. Schepers and S. Bräse, J. Am. Chem. Soc., 2013, 135, 7454–7457 CrossRef CAS PubMed.
- S. Silvi and A. Credi, Chem. Soc. Rev., 2015, 44, 4275–4289 RSC.
- D. Wang and B. Z. Tang, Acc. Chem. Res., 2019, 52, 2559–2570 CrossRef CAS PubMed.
- Kenry, K. C. Chong and B. Liu, Acc. Chem. Res., 2019, 52, 3051–3063 CrossRef CAS PubMed.
- S. Datta and J. Xu, ACS Appl. Bio Mater., 2023, 6, 4572–4585 CrossRef CAS PubMed.
- Y. Wang, J. Nie, W. Fang, L. Yang, Q. Hu, Z. Wang, J. Z. Sun and B. Z. Tang, Chem. Rev., 2020, 120, 4534–4577 CrossRef CAS PubMed.
- D. Barman, K. Narang, R. Parui, N. Zehra, M. N. Khatun, L. R. Adil and P. K. Iyer, Aggregate, 2022, 3, e172 CrossRef CAS.
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234–238 CrossRef CAS PubMed.
- X. Xiong, F. Song, J. Wang, Y. Zhang, Y. Xue, L. Sun, N. Jiang, P. Gao, L. Tian and X. Peng, J. Am. Chem. Soc., 2014, 136, 9590–9597 CrossRef CAS PubMed.
- J. Mei, N. L. Leung, R. T. Kwok, J. W. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718–11940 CrossRef CAS PubMed.
- X. Chen, L. Sun, A. A. Sukhanov, S. Doria, L. Bussotti, J. Zhao, H. Xu, B. Dick, V. K. Voronkova and M. Di Donato, Chem. Sci., 2024, 15, 10867–10881 RSC.
- J. Perrin, Lumière et Réactions Chimiques, Gauthier-Villars, 1926 Search PubMed.
- R. Delorme and F. Perrin, J. Phys. Radium, 1929, 10, 177–186 CrossRef CAS.
- D. Huang and J. M. Cole, Sci. Data, 2024, 11, 80 CrossRef CAS PubMed.
- Y. Xiong, J. Gong, J. Liu, D. Wang, H. Wu, Z. Zhao, M. Fang, Z. Li, D. Wang and B. Z. Tang, J. Mater. Chem. C, 2022, 10, 10009–10016 RSC.
- F. Fang, L. Zhu, M. Li, Y. Song, M. Sun, D. Zhao and J. Zhang, Adv. Sci., 2021, 8, 2102970 CrossRef CAS PubMed.
- A. F. Suleymanova, M. Z. Shafikov, A. C. Whitwood, R. Czerwieniec and D. W. Bruce, J. Mater. Chem. C, 2021, 9, 6528–6535 RSC.
- A. F. Suleymanova, M. Z. Shafikov, X. Chen, Y. Wang, R. Czerwieniec and D. W. Bruce, Phys. Chem. Chem. Phys., 2022, 24, 22115–22121 RSC.
- D. Chen, F. Tenopala-Carmona, J. A. Knöller, A. Mischok, D. Hall, S. Madayanad Suresh, T. Matulaitis, Y. Olivier, P. Nacke and F. Gießelmann, Angew. Chem., Int. Ed., 2023, 62, e202218911 CrossRef CAS PubMed.
- A. Farokhi, S. Lipinski, L. M. Cavinato, H. Shahroosvand, B. Pashaei, S. Karimi, S. Bellani, F. Bonaccorso and R. D. Costa, Chem. Soc. Rev., 2025,(54), 266–340 RSC.
- Y. Zhang, K. Aslan, M. J. Previte, S. N. Malyn and C. D. Geddes, J. Phys. Chem. B, 2006, 110, 25108–25114 CrossRef CAS PubMed.
- A. Abdurahman, T. J. Hele, Q. Gu, J. Zhang, Q. Peng, M. Zhang, R. H. Friend, F. Li and E. W. Evans, Nat. Mater., 2020, 19, 1224–1229 CrossRef CAS PubMed.
- A. P. M. Reponen, G. Londi, C. S. Matthews, Y. Olivier, A. S. Romanov, N. C. Greenham and A. J. Gillett, Angew. Chem., Int. Ed., 2024, 63, e202402052 CrossRef CAS PubMed.
- Y. Feng, X. Zhuang, Y. Xu, J. Xue, C. Qu, Q. Wang, Y. Liu and Y. Wang, Chem. Eng. J., 2023, 478, 147123 CrossRef CAS.
- Y. Hu, P. Wu, H. Jiang, H. Wu, W. Li, D. Gu and T. Zhang, Opt. Mater., 2022, 134, 113071 CrossRef CAS.
- M. Mońka, P. Pander, D. Grzywacz, A. Sikorski, R. Rogowski, P. Bojarski, A. P. Monkman and I. E. Serdiuk, ACS Appl. Mater. Interfaces, 2025, 17(6), 9635–9645 CrossRef PubMed.
- W. Xiong, C. Zhang, Y. Fang, M. Peng and W. Sun, Polymers, 2022, 15, 98 CrossRef PubMed.
- V. Ferraro, C. Bizzarri and S. Bräse, Adv. Sci., 2024, 11, 2404866 CrossRef CAS PubMed.
- K. M. Rao, R. Rajavaram, D. Kolli, S. P. Vattikuti and M. Godumala, J. Mater. Chem. C, 2025,(13), 3091–3122 Search PubMed.
- X. Chen, X. Zhang, X. Xiao, Z. Wang and J. Zhao, Angew. Chem., Int. Ed., 2023, 62, e202216010 CrossRef CAS PubMed.
- Q. Xue and G. Xie, Adv. Opt. Mater., 2021, 9, 2002204 CrossRef CAS.
- X.-K. Chen, D. Kim and J.-L. Brédas, Acc. Chem. Res., 2018, 51, 2215–2224 CrossRef CAS PubMed.
- T. Zhang, Y. Xiao, H. Wang, S. Kong, R. Huang, V. K. -M. Au, T. Yu and W. Huang, Angew. Chem., Int. Ed., 2023, 62, e202301896 CrossRef CAS PubMed.
- Y. Bao, Langmuir, 2024, 40, 3275–3282 CrossRef CAS PubMed.
- B. Cui, J. Zhou, C. Duan and H. Xu, Adv. Opt. Mater., 2025, 2403295 CrossRef.
- X.-F. Luo, X. Xiao and Y.-X. Zheng, Chem. Commun., 2024, 60(9), 1089–1099 RSC.
- X. Xiaoyang, L. Meiyan, L. Chenglong, W. Xiaoming and L. Xuguang, Chin. J. Org. Chem., 2023, 43, 3826 CrossRef.
- R. Wang, C.-S. Lee and Z. Lu, J. Organomet. Chem., 2023, 984, 122564 CrossRef CAS.
- J. Shi, Z. Ran, F. Peng, M. Chen, L. Li, L. Ji and W. Huang, J. Mater. Chem. C, 2022, 10, 9165–9191 RSC.
- P. Keerthika and R. K. Konidena, Adv. Opt. Mater., 2023, 11, 2301732 CrossRef CAS.
- K. P. CP, K. R. Naveen and J. Hur, Mater. Chem. Front., 2024, 8, 769–784 RSC.
- C. Lv, X. Wang, Q. Zhang and Y. Zhang, Mater. Chem. Front., 2023, 7, 2809–2827 RSC.
- K. Chu, Z. Ding and E. Zysman-Colman, Chem.–Eur. J., 2023, 29, e202301504 CrossRef CAS PubMed.
- P. Ledwon, Org. Electron., 2019, 75, 105422 CrossRef CAS.
- B. Wex and B. R. Kaafarani, J. Mater. Chem. C, 2017, 5, 8622–8653 RSC.
- J. Zhang, R. Chen, Z. Zhu, C. Adachi, X. Zhang and C. S. Lee, Highly Stable Near-Infrared Fluorescent Organic Nanoparticles with a Large Stokes Shift for Noninvasive Long-Term Cellular Imaging, ACS Appl. Mater. Interfaces, 2015, 7(47), 26266–26274 CrossRef CAS PubMed.
- N. Jiang, C.-Y. Zhu, K.-X. Li, Y.-H. Xu and M. R. Bryce, Macromolecules, 2024, 57, 5561–5577 CrossRef CAS PubMed.
- Y.-X. Che, X.-N. Qi, Q. Lin, H. Yao, W.-J. Qu, B. Shi, Y.-M. Zhang and T.-B. Wei, J. Mater. Chem. C, 2022, 10, 11119–11174 RSC.
- J.-T. Ye, L. Wang, H.-Q. Wang, H.-M. Xie and Y.-Q. Qiu, Org. Electron., 2019, 70, 193–204 CrossRef CAS.
- D.-Y. Qin, M. Zhang, Y.-N. Hu, Y.-X. Miao, J. Ye, C.-J. Zheng, J. Zhang, W. Xu, J.-C. Li and K. Wang, Chem. Eng. J., 2022, 450, 138174 CrossRef CAS.
- J.-T. Ye, H.-Q. Wang, Y. Zhang and Y.-Q. Qiu, J. Phys. Chem. C, 2019, 124, 921–931 CrossRef.
- S. Sharma and S. Sengupta, Org. Chem. Front., 2023, 10, 6087–6095 RSC.
- S. Kumar, M. Kumar, H. Bhambri, S. K. Mandal and V. Bhalla, ACS Appl. Mater. Interfaces, 2024, 16, 67683–67696 CrossRef CAS PubMed.
- Y.-H. Mao, M.-K. Hung, S.-T. Chung, S. Sharma, K.-W. Tsai and S.-A. Chen, ACS Appl. Mater. Interfaces, 2024, 16, 60715–60731 CrossRef CAS PubMed.
- S. Garain and F. Würthner, Chem. Commun., 2025,(61), 3081–3092 RSC.
- N. J. Turro, V. Ramamurthy and J. C. Scaiano, Modern Molecular Photochemistry of Organic Molecules, University Science Books Sausalito, CA, 2010 Search PubMed.
- A. Köhler and H. Bässler, Electronic Processes in Organic Semiconductors: an Introduction, John Wiley & Sons, 2015 Search PubMed.
- P. L. dos Santos, M. K. Etherington and A. P. Monkman, J. Mater. Chem. C, 2018, 6, 4842–4853 RSC.
- G. Jiang, F. Li, J. Fan, Y. Song, C.-K. Wang and L. Lin, J. Mater. Chem. C, 2020, 8, 98–108 RSC.
- S. J. Yoon, J. H. Kim, W. J. Chung and J. Y. Lee, Chem.–Eur. J., 2021, 27, 3065–3073 CrossRef CAS PubMed.
- A. Danos, D. Gudeika, N. A. Kukhta, R. Lygaitis, M. Colella, H. F. Higginbotham, A. N. Bismillah, P. R. McGonigal, J. V. Grazulevicius and A. P. Monkman, J. Mater. Chem. C, 2022, 10, 4737–4747 RSC.
- Y. Olivier, M. Moral, L. Muccioli and J.-C. Sancho-García, J. Mater. Chem. C, 2017, 5, 5718–5729 RSC.
- X.-T. Liu, W. Hua, H.-X. Nie, M. Chen, Z. Chang and X.-H. Bu, Natl. Sci. Rev., 2022, 9, nwab222 CrossRef CAS PubMed.
- J. M. Kaminski, T. Böhmer and C. M. Marian, J. Phys. Chem. C, 2024, 128, 13711–13721 CrossRef CAS.
- H. Miranda-Salinas, Y.-T. Hung, Y.-S. Chen, D. Luo, H.-C. Kao, C.-H. Chang, K.-T. Wong and A. Monkman, J. Mater. Chem. C, 2021, 9, 8819–8833 RSC.
- D. Zhong, Y. Yu, L. Yue, X. Yang, L. Ma, G. Zhou and Z. Wu, Chem. Eng. J., 2021, 413, 127445 CrossRef CAS.
- H.-Y. Zhang, H.-Y. Yang, M. Zhang, H. Lin, S.-L. Tao, C.-J. Zheng and X.-H. Zhang, Mater. Horiz., 2022, 9, 2425–2432 RSC.
- T. J. Penfold and J. Gibson, Highly Efficient OLEDs: Materials Based on Thermally Activated Delayed Fluorescence, 2018, pp. 297–330 Search PubMed.
- M. Hempe, N. A. Kukhta, A. Danos, M. A. Fox, A. S. Batsanov, A. P. Monkman and M. R. Bryce, Chem. Mater., 2021, 33, 3066–3080 CrossRef CAS PubMed.
- M. K. Etherington, J. Gibson, H. F. Higginbotham, T. J. Penfold and A. P. Monkman, Nat. Commun., 2016, 7, 13680 CrossRef CAS PubMed.
- H. L. Lee, S. O. Jeon, I. Kim, S. C. Kim, J. Lim, J. Kim, S. Park, J. Chwae, W.-J. Son, H. Choi and J. Y. Lee, Adv. Mater., 2022, 34, 2202464 CrossRef CAS PubMed.
- T. J. Penfold, E. Gindensperger, C. Daniel and C. M. Marian, Chem. Rev., 2018, 118, 6975–7025 CrossRef CAS PubMed.
- N. Li, F. Ni, X. Lv, Z. Huang, X. Cao and C. Yang, Adv. Opt. Mater., 2022, 10, 2101343 CrossRef CAS.
- S. Jhulki, M. W. Cooper, S. Barlow and S. R. Marder, Mater. Chem. Front., 2019, 3, 1699–1721 RSC.
- K. Shizu and H. Kaji, Commun. Chem., 2022, 5, 53 CrossRef CAS PubMed.
- C. Parker and C. Hatchard, Trans. Faraday Soc., 1961, 57, 1894–1904 RSC.
- J. R. Kirchhoff, R. E. Gamache Jr, M. W. Blaskie, A. A. Del Paggio, R. K. Lengel and D. R. McMillin, Inorg. Chem., 1983, 22, 2380–2384 CrossRef CAS.
- M. N. Berberan-Santos and J. M. Garcia, J. Am. Chem. Soc., 1996, 118, 9391–9394 CrossRef CAS.
- A. Endo, M. Ogasawara, A. Takahashi, D. Yokoyama, Y. Kato and C. Adachi, Adv. Mater., 2009, 21, 4802–4806 CrossRef CAS PubMed.
- J. C. Deaton, S. C. Switalski, D. Y. Kondakov, R. H. Young, T. D. Pawlik, D. J. Giesen, S. B. Harkins, A. J. Miller, S. F. Mickenberg and J. C. Peters, J. Am. Chem. Soc., 2010, 132, 9499–9508 CrossRef CAS PubMed.
- Y. Liu, X.-L. Chen, X.-Y. Li, S.-S. Zhu, S.-J. Li, Y. Song, L.-B. Qu and B. Yu, J. Am. Chem. Soc., 2020, 143, 964–972 CrossRef PubMed.
- K. Goushi, K. Yoshida, K. Sato and C. Adachi, Nat. Photonics, 2012, 6, 253–258 CrossRef CAS.
- A. Endo, K. Sato, K. Yoshimura, T. Kai, A. Kawada, H. Miyazaki and C. Adachi, Appl. Phys. Lett., 2011, 98, 083302 CrossRef.
- J.-H. Lee, PhD thesis, Seoul National University Graduate School, 2014 Search PubMed.
- S. Y. Kim, W. I. Jeong, C. Mayr, Y. S. Park, K. H. Kim, J. H. Lee, C. K. Moon, W. Brütting and J. J. Kim, Adv. Funct. Mater., 2013, 23, 3896–3900 CrossRef CAS.
- J. W. Sun, J. H. Lee, C. K. Moon, K. H. Kim, H. Shin and J. J. Kim, Adv. Mater., 2014, 26, 5684–5688 CrossRef CAS PubMed.
- D. R. Lee, B. S. Kim, C. W. Lee, Y. Im, K. S. Yook, S.-H. Hwang and J. Y. Lee, ACS Appl. Mater. Interfaces, 2015, 7, 9625–9629 CrossRef CAS PubMed.
- C. Adachi, Jpn. J. Appl. Phys., 2014, 53, 060101 CrossRef.
- Y. Tao, K. Yuan, T. Chen, P. Xu, H. Li, R. Chen, C. Zheng, L. Zhang and W. Huang, Adv. Mater., 2014, 26, 7931–7958 CrossRef CAS PubMed.
- K. Shizu, H. Noda, H. Tanaka, M. Taneda, M. Uejima, T. Sato, K. Tanaka, H. Kaji and C. Adachi, J. Phys. Chem. C, 2015, 119, 26283–26289 CrossRef CAS.
- S. Hirata, Y. Sakai, K. Masui, H. Tanaka, S. Y. Lee, H. Nomura, N. Nakamura, M. Yasumatsu, H. Nakanotani and Q. Zhang, Nat. Mater., 2015, 14, 330–336 CrossRef CAS PubMed.
- K. Kawasumi, T. Wu, T. Zhu, H. S. Chae, T. Van Voorhis, M. A. Baldo and T. M. Swager, J. Am. Chem. Soc., 2015, 137, 11908–11911 CrossRef CAS PubMed.
- S. Y. Lee, T. Yasuda, I. S. Park and C. Adachi, Dalton Trans., 2015, 44, 8356–8359 RSC.
- Y. J. Cho, S. K. Jeon, B. D. Chin, E. Yu and J. Y. Lee, Angew. Chem., Int. Ed., 2015, 54, 5201–5204 CrossRef CAS PubMed.
- T. Hatakeyama, K. Shiren, K. Nakajima, S. Nomura, S. Nakatsuka, K. Kinoshita, J. Ni, Y. Ono and T. Ikuta, Adv. Mater., 2016, 28, 2777–2781 CrossRef CAS PubMed.
- Z. Yang, Z. Mao, Z. Xie, Y. Zhang, S. Liu, J. Zhao, J. Xu, Z. Chi and M. P. Aldred, Chem. Soc. Rev., 2017, 46, 915–1016 RSC.
- T. Mori and Y. Inoue, Chem. Soc. Rev., 2013, 42, 8122–8133 RSC.
- S. A. Jenekhe and J. A. Osaheni, Science, 1994, 265, 765–768 CrossRef CAS PubMed.
- M. Cocchi, D. Virgili, G. Giro, V. Fattori, P. Di Marco, J. Kalinowski and Y. Shirota, Appl. Phys. Lett., 2002, 80, 2401–2403 CrossRef CAS.
- S. Lee, K. H. Kim, D. Limbach, Y. S. Park and J. J. Kim, Adv. Funct. Mater., 2013, 23, 4105–4110 CrossRef CAS.
- D. Chen, G. Xie, X. Cai, M. Liu, Y. Cao and S.-J. Su, Adv. Mater., 2015, 28, 239–244 CrossRef PubMed.
- D. Chen, Z. Wang, D. Wang, Y.-C. Wu, C.-C. Lo, A. Lien, Y. Cao and S.-J. Su, Org. Electron., 2015, 25, 79–84 CrossRef CAS.
- X. Jiang, R. A. Register, K. A. Killeen, M. E. Thompson, F. Pschenitzka, T. R. Hebner and J. C. Sturm, J. Appl. Phys., 2002, 91, 6717–6724 CrossRef CAS.
- M. Zhang, Z. Chen, L. Xiao, B. Qu and Q. Gong, Appl. Phys. Express, 2011, 4, 082105 CrossRef.
- P. Pikna, V. Skoromets, C. Becker, A. Fejfar and P. Kužel, Appl. Phys. Lett., 2015, 107, 233901 CrossRef.
- H. W. Lee, H. J. Kim, Y. S. Kim, J. Kim, S. E. Lee, H. W. Lee, Y. K. Kim and S. S. Yoon, J. Lumin., 2015, 165, 99–104 CrossRef CAS.
- H.-B. Kim and J.-J. Kim, J. Soc. Inf. Disp., 2019, 20, 105–121 CrossRef CAS.
- D. Y. Zhang, Y. Zheng, H. Zhang, J. H. Sun, C. P. Tan, L. He, W. Zhang, L. N. Ji and Z. W. Mao, Adv. Sci., 2018, 5, 1800581 CrossRef PubMed.
- J. F. Wang, Y. Kawabe, S. E. Shaheen, M. M. Morrell, G. E. Jabbour, P. A. Lee, J. Anderson, N. R. Armstrong, B. Kippelen and E. A. Mash, Adv. Mater., 1998, 10, 230–233 CrossRef CAS.
- Q. Wang, Q.-S. Tian, Y.-L. Zhang, X. Tang and L.-S. Liao, J. Mater. Chem. C, 2019, 7, 11329–11360 RSC.
- J. Gibson, A. P. Monkman and T. J. Penfold, ChemPhysChem, 2016, 17, 2956–2961 CrossRef CAS PubMed.
- T. Hosokai, H. Matsuzaki, H. Nakanotani, K. Tokumaru, T. Tsutsui, A. Furube, K. Nasu, H. Nomura, M. Yahiro and C. Adachi, Sci. Adv., 2017, 3, e1603282 CrossRef PubMed.
- J. Zhang, W. Chen, S. Kalytchuk, K. F. Li, R. Chen, C. Adachi, Z. Chen, A. L. Rogach, G. Zhu and P. K. Yu, ACS Appl. Mater. Interfaces, 2016, 8, 11355–11365 CrossRef CAS PubMed.
- V.-N. Nguyen, A. Kumar, M. H. Lee and J. Yoon, Coord. Chem. Rev., 2020, 425, 213545 CrossRef CAS.
- J. Huang, J. Li, Y. Lyu, Q. Miao and K. Pu, Nat. Mater., 2019, 18, 1133–1143 CrossRef CAS PubMed.
- Q. Miao, D. C. Yeo, C. Wiraja, J. Zhang, X. Ning, C. Xu and K. Pu, Angew. Chem., 2018, 130, 1270–1274 CrossRef.
- J. Zhang, S. Li, F.-F. An, J. Liu, S. Jin, J.-C. Zhang, P. C. Wang, X. Zhang, C.-S. Lee and X.-J. Liang, Nanoscale, 2015, 7, 13503–13510 RSC.
- J.-T. Hou, K.-K. Yu, K. Sunwoo, W. Y. Kim, S. Koo, J. Wang, W. X. Ren, S. Wang, X.-Q. Yu and J. S. Kim, Chem, 2020, 6, 832–866 CAS.
- F. Fang, M. Li, J. Zhang and C.-S. Lee, ACS Mater. Lett., 2020, 2, 531–549 CrossRef CAS.
- M. Lan, J. Zhang, X. Zhu, P. Wang, X. Chen, C.-S. Lee and W. Zhang, Nano Res., 2015, 8, 2380–2389 CrossRef CAS.
- T. Zhang, C. Ma, T. Sun and Z. Xie, Coord. Chem. Rev., 2019, 390, 76–85 CrossRef CAS.
- J. Zhang, R. Chen, Z. Zhu, C. Adachi, X. Zhang and C.-S. Lee, ACS Appl. Mater. Interfaces, 2015, 7, 26266–26274 CrossRef CAS PubMed.
- Y. Tsuchiya, K. Ikesue, H. Nakanotani and C. Adachi, Chem. Commun., 2019, 55, 5215–5218 RSC.
- C. Wang, Z. Chen, X. Tang, X. Liu, W. Na, W. Li and T. Liu, Photodiagnosis Photodyn. Ther., 2020, 32, 102014 CrossRef CAS PubMed.
- K. Y. Zhang, Q. Yu, H. Wei, S. Liu, Q. Zhao and W. Huang, Chem. Rev., 2018, 118, 1770–1839 CrossRef CAS PubMed.
- S. Huo, S. Jin, X. Ma, X. Xue, K. Yang, A. Kumar, P. C. Wang, J. Zhang, Z. Hu and X.-J. Liang, ACS Nano, 2014, 8, 5852–5862 CrossRef CAS PubMed.
- X. H. Wang, H. S. Peng, L. Yang, F. T. You, F. Teng, L. L. Hou and O. S. Wolfbeis, Angew. Chem., 2014, 126, 12679–12683 CrossRef.
- J. G. Huang, T. Leshuk and F. X. Gu, Nano Today, 2011, 6, 478–492 CrossRef CAS.
- X. Ma, J. Jia, R. Cao, X. Wang and H. Fei, J. Am. Chem. Soc., 2014, 136, 17734–17737 CrossRef CAS PubMed.
- F. Fang, D. Zhao, Y. Zhang, M. Li, J. Ye and J. Zhang, ACS Appl. Bio Mater., 2020, 3, 5103–5110 CrossRef CAS PubMed.
- P. Dong, J. Stellmacher, L. M. Bouchet, M. Nieke, A. Kumar, E. R. Osorio-Blanco, G. Nagel, S. B. Lohan, C. Teutloff and A. Patzelt, Angew. Chem., Int. Ed., 2021, 60, 14938–14944 CrossRef CAS PubMed.
- G. Hong, A. L. Antaris and H. Dai, Nat. Biomed. Eng., 2017, 1, 0010 CrossRef CAS.
- J. Joo, X. Liu, V. R. Kotamraju, E. Ruoslahti, Y. Nam and M. J. Sailor, ACS Nano, 2015, 9, 6233–6241 CrossRef CAS PubMed.
- R. Liu, J. Tang, Y. Xu and Z. Dai, ACS Nano, 2019, 13, 5124–5132 CrossRef CAS PubMed.
- T. S. Blacker, Z. F. Mann, J. E. Gale, M. Ziegler, A. J. Bain, G. Szabadkai and M. R. Duchen, Nat. Commun., 2014, 5, 3936 CrossRef CAS PubMed.
- I. Georgakoudi, B. C. Jacobson, M. G. Muller, E. E. Sheets, K. Badizadegan, D. L. Carr-Locke, C. P. Crum, C. W. Boone, R. R. Dasari and J. Van Dam, Cancer Res., 2002, 62, 682–687 CAS.
- H. Shi, H. Sun, H. Yang, S. Liu, G. Jenkins, W. Feng, F. Li, Q. Zhao, B. Liu and W. Huang, Adv. Funct. Mater., 2013, 23, 3268–3276 CrossRef CAS.
- F. Ni, N. Li, L. Zhan and C. Yang, Adv. Opt. Mater., 2020, 8, 1902187 CrossRef CAS.
- Y. Jiang, J. Huang, X. Zhen, Z. Zeng, J. Li, C. Xie, Q. Miao, J. Chen, P. Chen and K. Pu, Nat. Commun., 2019, 10, 2064 CrossRef PubMed.
- Y. Tanigawa, J. Li, J. M. Justesen, H. Horn, M. Aguirre, C. DeBoever, C. Chang, B. Narasimhan, K. Lage and T. Hastie, Nat. Commun., 2019, 10, 4064 CrossRef PubMed.
- Q. Miao, C. Xie, X. Zhen, Y. Lyu, H. Duan, X. Liu, J. V. Jokerst and K. Pu, Nat. Biotechnol., 2017, 35, 1102–1110 CrossRef CAS PubMed.
- Y. Lyu, D. Cui, J. Huang, W. Fan, Y. Miao and K. Pu, Angew. Chem., Int. Ed., 2019, 58, 4983–4987 CrossRef CAS PubMed.
- X. Zhen, Y. Tao, Z. An, P. Chen, C. Xu, R. Chen, W. Huang and K. Pu, Adv. Mater., 2017, 29, 1606665 CrossRef PubMed.
- Y. Fan, P. Wang, Y. Lu, R. Wang, L. Zhou, X. Zheng, X. Li, J. A. Piper and F. Zhang, Nat. Nanotechnol., 2018, 13, 941–946 CrossRef CAS PubMed.
- Z. Zhu, B. Song, J. Yuan and C. Yang, Adv. Sci., 2016, 3, 1600146 CrossRef PubMed.
- M. Delbianco, V. Sadovnikova, E. Bourrier, G. Mathis, L. Lamarque, J. M. Zwier and D. Parker, Angew. Chem., 2014, 126, 10894–10898 CrossRef.
- S. M. King, S. Claire, R. I. Teixeira, A. N. Dosumu, A. J. Carrod, H. Dehghani, M. J. Hannon, A. D. Ward, R. Bicknell and S. W. Botchway, J. Am. Chem. Soc., 2018, 140, 10242–10249 CrossRef CAS PubMed.
- T. He, C. Ren, Z. Li, S. Xiao, J. Li, X. Lin, C. Ye, J. Zhang, L. Guo and W. Hu, Appl. Phys. Lett., 2018, 112, 211102 CrossRef.
- Y. Liu, C. Li, Z. Ren, S. Yan and M. R. Bryce, Nat. Rev. Mater., 2018, 3, 1–20 CrossRef.
- W. Hu, L. Guo, L. Bai, X. Miao, Y. Ni, Q. Wang, H. Zhao, M. Xie, L. Li and X. Lu, Adv. Healthcare Mater., 2018, 7, 1870062 CrossRef.
- N. R. Paisley, C. M. Tonge and Z. M. Hudson, Front. Chem., 2020, 8, 229 CrossRef CAS PubMed.
- F. Ni, N. Li, L. Zhan and C. Yang, Adv. Opt. Mater., 2020, 8, 1902187 CrossRef CAS.
- Q. Zhang, S. Xu, M. Li, Y. Wang, N. Zhang, Y. Guan, M. Chen, C.-F. Chen and H.-Y. Hu, Chem. Commun., 2019, 55, 5639–5642 RSC.
- R. Wei, L. Zhang, S. Xu, Q. Zhang, Y. Qi and H.-Y. Hu, Chem. Commun., 2020, 56, 2550–2553 RSC.
- S. Gan, J. Zhou, T. A. Smith, H. Su, W. Luo, Y. Hong, Z. Zhao and B. Z. Tang, Mater. Chem. Front., 2017, 1, 2554–2558 RSC.
- S. Qi, S. Kim, V.-N. Nguyen, Y. Kim, G. Niu, G. Kim, S.-J. Kim, S. Park and J. Yoon, ACS Appl. Mater. Interfaces, 2020, 12, 51293–51301 CrossRef CAS PubMed.
- Y. Wu, L. Jiao, F. Song, M. Chen, D. Liu, W. Yang, Y. Sun, G. Hong, L. Liu and X. Peng, Chem. Commun., 2019, 55, 14522–14525 RSC.
- Y. Wu, F. Song, W. Luo, Z. Liu, B. Song and X. Peng, ChemPhotoChem, 2017, 1, 79–83 CrossRef CAS.
- S. Gan, J. Zhou, T. A. Smith, H. Su, W. Luo, Y. Hong, Z. Zhao and B. Tang, Mater. Chem. Front., 2017, 2554 RSC.
- M. Luo, X. Li, L. Ding, G. Baryshnikov, S. Shen, M. Zhu, L. Zhou, M. Zhang, J. Lu, H. Ågren, X.-d. Wang and L. Zhu, Angew. Chem., Int. Ed., 2020, 59, 17018–17025 CrossRef CAS PubMed.
- J. Jin, H. Jiang, Q. Yang, L. Tang, Y. Tao, Y. Li, R. Chen, C. Zheng, Q. Fan, K. Y. Zhang, Q. Zhao and W. Huang, Nat. Commun., 2020, 11, 842 CrossRef CAS PubMed.
- X.-L. Hao, A.-M. Ren and L. Zhou, J. Phys. Chem. Lett., 2022, 13, 11745–11752 CrossRef CAS PubMed.
- F. Ni, M. Xie, T. Liu, X. Zhou, Z. Chen, K. Zheng, Y. Wu, Q. Zhao and C. Yang, Chem. Eng. J., 2022, 437, 135396 CrossRef CAS.
- Z. Zhu, Z. Luo, Y.-Q. Xie, Y. Sun, L. Xu and Q. Wu, Adv. Funct. Mater., 2024, 34, 2313701 CrossRef CAS.
- X.-L. Hao, A.-M. Ren and L. Zhou, J. Phys. Chem. Lett., 2023, 14, 10309–10317 CrossRef CAS PubMed.
- W. L. Primrose, P. Hu and Z. M. Hudson, ACS Appl. Nano Mater., 2024, 12673–12681 CrossRef CAS.
- P. W. Zach, S. A. Freunberger, I. Klimant and S. M. Borisov, ACS Appl. Mater. Interfaces, 2017, 9, 38008–38023 CrossRef CAS PubMed.
- C. M. Tonge, N. R. Paisley, A. M. Polgar, K. Lix, W. R. Algar and Z. M. Hudson, ACS Appl. Mater. Interfaces, 2020, 12, 6525–6535 CrossRef CAS PubMed.
- C. A. DeRosa, J. Samonina-Kosicka, Z. Fan, H. C. Hendargo, D. H. Weitzel, G. M. Palmer and C. L. Fraser, Macromolecules, 2015, 48, 2967–2977 CrossRef CAS PubMed.
- C. J. Christopherson, D. M. Mayder, J. Poisson, N. R. Paisley, C. M. Tonge and Z. M. Hudson, ACS Appl. Mater. Interfaces, 2020, 12, 20000–20011 CrossRef CAS PubMed.
- S. Xu, Q. Zhang, X. Han, Y. Wang, X. Wang, M. Nazare, J.-D. Jiang and H.-Y. Hu, ACS Sens., 2020, 5, 1650–1656 CrossRef CAS PubMed.
- X. Li, G. Baryshnikov, C. Deng, X. Bao, B. Wu, Y. Zhou, H. Ågren and L. Zhu, Nat. Commun., 2019, 10, 731 CrossRef CAS PubMed.
- T. J. Dougherty, C. J. Gomer, B. W. Henderson, G. Jori, D. Kessel, M. Korbelik, J. Moan and Q. Peng, J. Natl. Cancer Inst., 1998, 90, 889–905 CrossRef CAS PubMed.
- Y. Wan, G. Lu, W.-C. Wei, Y.-H. Huang, S. Li, J.-X. Chen, X. Cui, Y.-F. Xiao, X. Li and Y. Liu, ACS Nano, 2020, 14, 9917–9928 CrossRef CAS PubMed.
- J. Li and K. Pu, Chem. Soc. Rev., 2019, 48, 38–71 RSC.
- J. Zhang, Y.-C. Liang, X. Lin, X. Zhu, L. Yan, S. Li, X. Yang, G. Zhu, A. L. Rogach and P. K. Yu, ACS Nano, 2015, 9, 9741–9756 CrossRef CAS PubMed.
- X. Cui, J. Zhang, Y. Wan, F. Fang, R. Chen, D. Shen, Z. Huang, S. Tian, Y. Xiao and X. Li, ACS Appl. Bio Mater., 2019, 2, 3854–3860 CrossRef CAS PubMed.
- A. P. Castano, P. Mroz and M. R. Hamblin, Nat. Rev. Cancer, 2006, 6, 535–545 CrossRef CAS PubMed.
- Y. Wan, G. Lu, J. Zhang, Z. Wang, X. Li, R. Chen, X. Cui, Z. Huang, Y. Xiao and J. Chelora, Adv. Funct. Mater., 2019, 29, 1903436 CrossRef.
- S. M. Mousavi, S. A. Hashemi, M. Y. Kalashgrani, N. Omidifar, S. Bahrani, N. Vijayakameswara Rao, A. Babapoor, A. Gholami and W.-H. Chiang, Polymers, 2022, 14, 617 CrossRef CAS PubMed.
- X. Li, F. Fang, B. Sun, C. Yin, J. Tan, Y. Wan, J. Zhang, P. Sun, Q. Fan and P. Wang, Nanoscale Horiz., 2021, 6, 177–185 RSC.
- D. E. Dolmans, D. Fukumura and R. K. Jain, Nat. Rev. Cancer, 2003, 3, 380–387 CrossRef CAS PubMed.
- S. S. Lucky, K. C. Soo and Y. Zhang, Chem. Rev., 2015, 115, 1990–2042 CrossRef CAS PubMed.
- J. Zhao, W. Wu, J. Sun and S. Guo, Chem. Soc. Rev., 2013, 42, 5323–5351 RSC.
- X. Cui, G. Lu, S. Dong, S. Li, Y. Xiao, J. Zhang, Y. Liu, X. Meng, F. Li and C.-S. Lee, Mater. Horiz., 2021, 8, 571–576 RSC.
- J. Zhang, W. Chen, R. Chen, X.-K. Liu, Y. Xiong, S. V. Kershaw, A. L. Rogach, C. Adachi, X. Zhang and C.-S. Lee, Chem. Commun., 2016, 52, 11744–11747 RSC.
- J. Zhang, F. Fang, B. Liu, J.-H. Tan, W.-C. Chen, Z. Zhu, Y. Yuan, Y. Wan, X. Cui, S. Li, Q.-X. Tong, J. Zhao, X.-M. Meng and C.-S. Lee, ACS Appl. Mater. Interfaces, 2019, 11, 41051–41061 CrossRef CAS PubMed.
- Z. Liu, W. Shi, G. Hong, W. Chen, B. Song, X. Peng, X. Xiong and F. Song, J. Contr. Release, 2019, 310, 1–10 CrossRef CAS PubMed.
- Z. Liu, F. Song, W. Shi, G. Gurzadyan, H. Yin, B. Song, R. Liang and X. Peng, ACS Appl. Mater. Interfaces, 2019, 11, 15426–15435 CrossRef CAS PubMed.
- M. Li, J. Xia, R. Tian, J. Wang, J. Fan, J. Du, S. Long, X. Song, J. W. Foley and X. Peng, J. Am. Chem. Soc., 2018, 140, 14851–14859 CrossRef CAS PubMed.
- Y.-F. Xiao, J.-X. Chen, S. Li, W.-W. Tao, S. Tian, K. Wang, X. Cui, Z. Huang, X.-H. Zhang and C.-S. Lee, Chem. Sci., 2020, 11, 888–895 RSC.
- Y.-F. Xiao, J.-X. Chen, W.-C. Chen, X. Zheng, C. Cao, J. Tan, X. Cui, Z. Yuan, S. Ji, G. Lu, W. Liu, P. Wang, S. Li and C.-S. Lee, Chem. Commun., 2021, 57, 4902–4905 RSC.
- Z. Li, X.-G. Yang, H. Zhang, J.-R. Zhang, X.-K. Tian, J.-H. Qin, L.-F. Ma and D. Yan, Inorg. Chem. Front., 2022, 9, 4281–4287 RSC.
- W. Chen, Z. Wang, M. Tian, G. Hong, Y. Wu, M. Sui, M. Chen, J. An, F. Song and X. Peng, J. Am. Chem. Soc., 2023, 145, 8130–8140 CrossRef CAS PubMed.
- J. Zhang, J. Ma, S. Zhang, X. Lou, Y. Ding, Y. Li, M. Xu, X. Xie, X. Jiao and X. Dou, ACS Nano, 2023, 17, 23430–23441 CrossRef CAS PubMed.
- Y. Xu, B. Chen, D. Su, J. Li, Q. Qi, Y. Hu, Q. Wang, F. Xia, X. Lou, Z. Zhao, J. Dai, X. Dong and J. Zhou, ACS Appl. Mater. Interfaces, 2023, 15, 56314–56327 CrossRef CAS PubMed.
- B. T. Luppi, W. L. Primrose and Z. M. Hudson, Angew. Chem., Int. Ed., 2024, 63, e202400712 CrossRef CAS PubMed.
- G. S. Nongthombam, D. Barman and P. K. Iyer, ACS Appl. Bio Mater., 2024, 7(3), 1899–1909 CrossRef CAS PubMed.
- M. A. Bryden and E. Zysman-Colman, Chem. Soc. Rev., 2021, 50, 7587–7680 RSC.
- D. A. Nicewicz and D. W. MacMillan, Science, 2008, 322, 77–80 CrossRef CAS PubMed.
- M. A. Ischay, M. E. Anzovino, J. Du and T. P. Yoon, J. Am. Chem. Soc., 2008, 130, 12886–12887 CrossRef CAS PubMed.
- J. M. Narayanam, J. W. Tucker and C. R. Stephenson, J. Am. Chem. Soc., 2009, 131, 8756–8757 CrossRef CAS PubMed.
- D. M. Arias-Rotondo and J. K. McCusker, Chem. Soc. Rev., 2016, 45, 5803–5820 RSC.
- N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS PubMed.
- R. Marcus, J. Phys. Chem., 1989, 93, 3078–3086 CrossRef CAS.
- R. A. Marcus, Discuss. Faraday Soc., 1960, 29, 21–31 RSC.
- D. Rehm, A. Weller and B. Bunsenges, Phys. Chem., 1969, 73, 834–839 CAS.
- R. A. Marcus, J. Chem. Phys., 1956, 24, 966–978 CrossRef CAS.
- M. Bouzrati-Zerelli, N. Guillaume, F. Goubard, T.-T. Bui, S. Villotte, C. Dietlin, F. Morlet-Savary, D. Gigmes, J. P. Fouassier and F. Dumur, New J. Chem., 2018, 42, 8261–8270 RSC.
- Y. Zhang, T. S. Lee, J. M. Favale, D. C. Leary, J. L. Petersen, G. D. Scholes, F. N. Castellano and C. Milsmann, Nat. Chem., 2020, 12, 345–352 CrossRef CAS PubMed.
- F. B. Dias, T. J. Penfold and A. P. Monkman, Methods Appl. Fluoresc., 2017, 5, 012001 CrossRef PubMed.
- M. Y. Wong and E. Zysman-Colman, Adv. Mater., 2017, 29, 1605444 CrossRef PubMed.
- Y. Im, M. Kim, Y. J. Cho, J.-A. Seo, K. S. Yook and J. Y. Lee, Chem. Mater., 2017, 29, 1946–1963 CrossRef CAS.
- V. Srivastava and P. P. Singh, RSC Adv., 2017, 7, 31377–31392 RSC.
- A. Penzkofer, A. Beidoun and M. Daiber, J. Lumin., 1992, 51, 297–314 CrossRef CAS.
- S. Grotjahn and B. König, Chem. Commun., 2024, 60, 12951–12963 RSC.
- L. Buzzetti, G. E. Crisenza and P. Melchiorre, Angew. Chem., Int. Ed., 2019, 58, 3730–3747 CrossRef CAS PubMed.
- M. Cismesia and T. Yoon, Chem. sci., 2015, 6(10), 5426–5434 RSC.
- R. C. McAtee, E. J. McClain and C. R. Stephenson, Trends Chem., 2019, 1, 111–125 CrossRef CAS PubMed.
- J. R. Ochola and M. Wolf, Org. Biomol. Chem., 2016, 14, 9088–9092 RSC.
- S. P. Pitre, C. D. McTiernan and J. C. Scaiano, Acc. Chem. Res., 2016, 49, 1320–1330 CrossRef CAS PubMed.
- J. Xuan, Z. G. Zhang and W. J. Xiao, Angew. Chem., Int. Ed., 2015, 54, 15632–15641 CrossRef CAS PubMed.
- L. M. Reid, T. Li, Y. Cao and C. P. Berlinguette, Sustain. Energy Fuels, 2018, 2, 1905–1927 RSC.
- J. Davies, L. Angelini, M. A. Alkhalifah, L. M. Sanz, N. S. Sheikh and D. Leonori, Synthesis, 2018, 50, 821–830 CrossRef CAS.
- A. Noble, R. S. Mega, D. Pflästerer, E. L. Myers and V. K. Aggarwal, Angew. Chem., Int. Ed., 2018, 57, 2155–2159 CrossRef CAS PubMed.
- H. Jiang and A. Studer, Chem.–Eur. J., 2019, 25, 516–520 CrossRef CAS PubMed.
- O. Zhang and J. W. Schubert, J. Org. Chem., 2020, 85, 6225–6232 CrossRef CAS PubMed.
- N. X. Xu, B. X. Li, C. Wang and M. Uchiyama, Angew. Chem., 2020, 132, 10726–10731 CrossRef.
- H. Huang, C. Yu, Y. Zhang, Y. Zhang, P. S. Mariano and W. Wang, J. Am. Chem. Soc., 2017, 139, 9799–9802 CrossRef CAS PubMed.
- S. P. Morcillo, E. M. Dauncey, J. H. Kim, J. J. Douglas, N. S. Sheikh and D. Leonori, Angew. Chem., Int. Ed., 2018, 57, 12945–12949 CrossRef CAS PubMed.
- X.-L. Lyu, S.-S. Huang, H.-J. Song, Y.-X. Liu and Q.-M. Wang, RSC Adv., 2019, 9, 36213–36216 RSC.
- H. Huang, X. Li, C. Yu, Y. Zhang, P. S. Mariano and W. Wang, Angew. Chem., Int. Ed., 2017, 56, 1500–1505 CrossRef CAS PubMed.
- C. Shu, R. S. Mega, B. J. Andreassen, A. Noble and V. K. Aggarwal, Angew. Chem., 2018, 130, 15656–15660 CrossRef.
- T. C. Sherwood, H.-Y. Xiao, R. G. Bhaskar, E. M. Simmons, S. Zaretsky, M. P. Rauch, R. R. Knowles and T. M. Dhar, J. Org. Chem., 2019, 84, 8360–8379 CrossRef CAS PubMed.
- R. Gueret, L. Pelinski, T. Bousquet, M. Sauthier, V. Ferey and A. Bigot, Org. Lett., 2020, 22, 5157–5162 CrossRef CAS PubMed.
- G. G. Pawar, F. Robert, E. Grau, H. Cramail and Y. Landais, Chem. Commun., 2018, 54, 9337–9340 RSC.
- S. Sharma and A. Sharma, Org. Biomol. Chem., 2019, 17, 4384–4405 RSC.
- T. C. Sherwood, N. Li, A. N. Yazdani and T. M. Dhar, J. Org. Chem., 2018, 83, 3000–3012 CrossRef CAS PubMed.
- E. W. Webb, J. B. Park, E. L. Cole, D. J. Donnelly, S. J. Bonacorsi, W. R. Ewing and A. G. Doyle, J. Am. Chem. Soc., 2020, 142, 9493–9500 CrossRef CAS PubMed.
- S. He, X. Chen, F. Zeng, P. Lu, Y. Peng, L. Qu and B. Yu, Chin. Chem. Lett., 2020, 31, 1863–1867 CrossRef CAS.
- A. N. Herron, D. Liu, G. Xia and J.-Q. Yu, J. Am. Chem. Soc., 2020, 142, 2766–2770 CrossRef CAS PubMed.
- X.-L. Huang, Y.-Z. Cheng, X. Zhang and S.-L. You, Org. Lett., 2020, 22, 9699–9705 CrossRef CAS PubMed.
- Y. Z. Cheng, X. L. Huang, W. H. Zhuang, Q. R. Zhao, X. Zhang, T. S. Mei and S. L. You, Angew. Chem., 2020, 132, 18218–18223 CrossRef.
- Q.-F. Bao, M. Li, Y. Xia, Y.-Z. Wang, Z.-Z. Zhou and Y.-M. Liang, Org. Lett., 2021, 23, 1107–1112 CrossRef CAS PubMed.
- E. R. Sauvé, D. M. Mayder, S. Kamal, M. S. Oderinde and Z. M. Hudson, Chem. Sci., 2022, 13, 2296–2302 RSC.
- E. Pinosa, E. Bassan, S. Cetin, M. Villa, S. Potenti, F. Calogero, A. Gualandi, A. Fermi, P. Ceroni and P. G. Cozzi, J. Org. Chem., 2022, 88, 6390–6400 CrossRef PubMed.
- M. A. Bryden, F. Millward, T. Matulaitis, D. Chen, M. Villa, A. Fermi, S. Cetin, P. Ceroni and E. Zysman-Colman, J. Org. Chem., 2022, 88, 6364–6373 CrossRef PubMed.
- G. Hong, Y. Wu, J. An, W. Chen, F. Song and X. Peng, Sustain. Energy Fuels, 2023, 7, 3447–3453 RSC.
- R. Hojo, K. Bergmann, S. A. Elgadi, D. M. Mayder, M. A. Emmanuel, M. S. Oderinde and Z. M. Hudson, J. Am. Chem. Soc., 2023, 145, 18366–18381 CrossRef CAS PubMed.
- C. Sun, Q. Zhou, C.-Y. Li, Z.-W. Hou and L. Wang, Org. Lett., 2024, 26, 883–888 CrossRef CAS PubMed.
- P. Wang, M. Ge, X. Luo, Y. Zhai, N. Meckbach, V. Strehmel, S. Li, Z. Chen and B. Strehmel, Angew. Chem., Int. Ed., 2024, e202402915 CAS.
- K. S. Yook and J. Y. Lee, Chem. Rec., 2016, 16, 159–172 CrossRef CAS PubMed.
- S. Yadav, P. Mittal and S. Negi, Bull. Mater. Sci., 2022, 45, 109 CrossRef CAS.
- S. K. Behera, R. Kainda, S. Basu and Y. S. Chaudhary, Appl. Mater. Today, 2022, 27, 101407 CrossRef.
- X. Fan, X. Hao, F. Huang, J. Yu, K. Wang and X. Zhang, Adv. Sci., 2023, 10, 2303504 CrossRef CAS PubMed.
- Q. Wei, N. Fei, A. Islam, T. Lei, L. Hong, R. Peng, X. Fan, L. Chen, P. Gao and Z. Ge, Adv. Opt. Mater., 2018, 6, 1800512 CrossRef.
- K.-H. Kim and J.-J. Kim, Adv. Mater., 2018, 30, 1705600 CrossRef PubMed.
- X.-F. Luo, X. Xiao and Y.-X. Zheng, Chem. Commun., 2024, 60, 1089–1099 RSC.
- L. Sun, K. Di, R. Guo, Y. Wang, H. Su, Y. Tian, S. Huang and L. Wang, Dyes Pigm., 2024, 222, 111858 CrossRef CAS.
- P. Palanisamy, O. P. Kumar, H. U. Kim, K. R. Naveen, J.-Y. Kim, J.-H. Baek, M. Y. Chae and J. H. Kwon, Chem. Eng. J., 2024, 481, 148781 CrossRef CAS.
- T.-L. Wu, M.-J. Huang, C.-C. Lin, P.-Y. Huang, T.-Y. Chou, R.-W. Chen-Cheng, H.-W. Lin, R.-S. Liu and C.-H. Cheng, Nat. Photonics, 2018, 12, 235–240 CrossRef CAS.
- A. Kumar, H. Y. Shin, T. Lee, J. Jung, B. J. Jung and M. H. Lee, Chem.–Eur. J., 2020, 26, 16793–16801 CrossRef CAS PubMed.
- Y. Tan, A. Ying, Y. Liu, X. Cai, L. Zhan, Z. Bin, J. You, C. Li and S. Gong, Chem. Eng. J., 2024, 487, 150618 CrossRef CAS.
- M. Cinquino, C. T. Prontera, A. Maggiore, A. Zizzari, M. Pugliese, F. Mariano, V. Valenzano, I. E. Palamà, R. Manfredi and G. Gigli, Adv. Electron. Mater., 2024, 10, 2300358 CrossRef CAS.
- W. Luo, T. Wang, Z. Huang, H. Huang, N. Li and C. Yang, Adv. Funct. Mater., 2024, 34, 2310042 CrossRef CAS.
- T. Wang, Z. Huang, H. Zhang, J. Miao and C. Yang, Adv. Funct. Mater., 2024, 2408119 CrossRef CAS.
- Z. Xue, G. Chen, Z. Chen, W. Luo, N. Li, Z. Huang and C. Yang, Adv. Funct. Mater., 2024, 2409244 CrossRef CAS.
- X. Cai, Y. Pan, X. Song, C. Li, Y. Pu, X. Zhuang, H. Bi and Y. Wang, Adv. Opt. Mater., 2024, 12, 2302811 CrossRef CAS.
- X. Chen, S. Bagnich, R. Pollice, B. Li, Y. Zhu, R. Saxena, Y. Yin, W. Zhu, A. Aspuru-Guzik and E. Zysman-Colman, Adv. Opt. Mater., 2024, 12, 2301784 CrossRef CAS.
- N. Peethani, N. Y. Kwon, C. W. Koh, S. H. Park, J. M. Ha, M. J. Cho, H. Y. Woo, S. Park and D. H. Choi, Adv. Opt. Mater., 2024, 12, 2301217 CrossRef CAS.
- M. K. Sit, G. S. M. Tong, T. L. Lam, G. Cheng, F. F. Hung, K. M. So, L. Du, K. O. Choy, K. H. Low and C. M. Che, Adv. Opt. Mater., 2024, 12, 2302308 CrossRef CAS.
- S. Wang, L. Peng, F. He, Y. Ming, H. Qi, Y. Liu, D. Ma, S. Ying and S. Yan, Adv. Opt. Mater., 2024, 12, 2400503 CrossRef CAS.
- X.-F. Song, S. Luo, N. Li, X. Wan, J. Miao, Y. Zou, K. Li and C. Yang, Angew. Chem., 2024, e202413536 Search PubMed.
- S. Li, Z. Yang, Y. Xie, L. Hua, S. Ying, Y. Liu, Z. Ren and S. Yan, Chem. Sci., 2024, 15(44), 18335–18346 RSC.
- T. Komino, H. Nomura, T. Koyanagi and C. Adachi, Chem. Mater., 2013, 25, 3038–3047 CrossRef CAS.
- C. A. Parker and E. J. Bowen, Proc. R. Soc. London, A, 1963, 276, 125–135 CAS.
- L. G. Franca, D. G. Bossanyi, J. Clark and P. L. dos Santos, ACS Appl. Opt. Mater., 2024, 2476–2500 CrossRef CAS PubMed.
- Z. Chen, W. Sun, H. J. Butt and S. Wu, Chem.–Eur. J., 2015, 21, 9165–9170 CrossRef CAS PubMed.
- Y. Y. Cheng, B. Fückel, R. W. MacQueen, T. Khoury, R. G. Clady, T. F. Schulze, N. Ekins-Daukes, M. J. Crossley, B. Stannowski and K. Lips, Energy Environ. Sci., 2012, 5, 6953–6959 RSC.
- M. Hussain, S. S. Razi, T. Tao and F. Hartl, J. Photochem. Photobiol., C, 2023, 100618 CrossRef CAS.
- C. Gao, W. W. Wong, Z. Qin, S. C. Lo, E. B. Namdas, H. Dong and W. Hu, Adv. Mater., 2021, 33, 2100704 CrossRef CAS PubMed.
- L. Zeng, L. Huang, J. Han and G. Han, Acc. Chem. Res., 2022, 55, 2604–2615 CrossRef CAS PubMed.
- P. Bharmoria, H. Bildirir and K. Moth-Poulsen, Chem. Soc. Rev., 2020, 49, 6529–6554 RSC.
- T. Schloemer, P. Narayanan, Q. Zhou, E. Belliveau, M. Seitz and D. N. Congreve, ACS Nano, 2023, 17, 3259–3288 CrossRef CAS PubMed.
- W. Ahmad, J. Wang, H. Li, Q. Ouyang, W. Wu and Q. Chen, Coord. Chem. Rev., 2021, 439, 213944 CrossRef CAS.
- X. Qiao and D. Ma, Mater. Sci. Eng. R Rep., 2020, 139, 100519 CrossRef.
- K. V. Y. Jie, M. F. Ahmad, M. M. Ramli, S. Shaari, A. M. Ismail and Y. Sulaiman, Int. J. Nanoelectron. Mater., 2022, 15, 331–339 Search PubMed.
- M. Zheng, Y. Li, Y. Wei, L. Chen, S. Liu and X. Zhou, J. Phys. Chem. C, 2023, 127, 2846–2854 CrossRef CAS.
- J. Li, Y. Xia, G. Li, M. Chen, J. Zhou, W. Yan, B. Zhao, K. Guo and H. Wang, Chem. Eng. J., 2023, 470, 143966 CrossRef CAS.
- X. Tang, R. Pan, X. Guan, Y. He, S. Jiang, Y. Wang, X. Zhou and Z. Xiong, Appl. Phys. Lett., 2024, 124, 123506 CrossRef CAS.
- O. Bezvikonnyi, A. Bucinskas, P. Arsenyan, A. Petrenko, Z.-Y. Wei, J.-H. Lee, D. Volyniuk, E. U. Rashid, T.-L. Chiu and J. V. Grazulevicius, ACS Appl. Electron. Mater., 2024, 6, 4489–4503 CrossRef CAS.
- F. Zhao, J. Kong, W. Zhang, Z. Kuang and M. Zhou, J. Phys. Chem. Lett., 2024, 15, 2885–2892 CrossRef CAS PubMed.
- K. V. Y. Jie, M. F. Ahmad, A. R. Mohmad, A. M. Ismail, M. N. Norizan, M. M. Ramli, S. Shaari, Y. Sulaiman, S. A. A. Rais and S. Johari, ACS Appl. Energy Mater., 2024, 7, 799–809 CrossRef CAS.
- X. Wang, D. Zhou, H. Wang, H. Jin and J. Yu, J. Phys. D: Appl. Phys., 2018, 52, 025106 CrossRef.
- W. Dong, Y. Bai, Y. Wang, S. Zhao, H. Xu, Y. Miao, Q. Luo, H. Wang and J. Yu, Sci. China Mater., 2024, 67, 197–204 CrossRef CAS.
- M. A. Iqbal, X. Weng, C. Kang, N. Arif, K. Wu, W. Tang, S. Dai, X. Fang, H. Cai and Y. J. Zeng, Laser Photon. Rev., 2024, 18, 2300612 CrossRef CAS.
- M. Hesari, J. Turnbull, C. B. Khadka, F. Weigend, J. F. Corrigan and Z. Ding, Electrochim. Acta, 2016, 210, 79–86 CrossRef CAS.
- R. Ishimatsu, S. Matsunami, T. Kasahara, J. Mizuno, T. Edura, C. Adachi, K. Nakano and T. Imato, Angew. Chem., Int. Ed., 2014, 53, 6993–6996 CrossRef CAS PubMed.
- T. Kasahara, S. Matsunami, T. Edura, R. Ishimatsu, J. Oshima, M. Tsuwaki, T. Imato, S. Shoji, C. Adachi and J. Mizuno, Sens. Actuators, A, 2014, 214, 225–229 CrossRef CAS.
- N. Ichinohe, R. Otsuka, R. Ishimatsu, M. Kobayashi, J. Mizuno, N. Akino and T. Kasahara, Electrochemistry, 2024, 92, 027004 CrossRef CAS.
- J. Zhang, X. Xu, C. Yao, J. Peng, M. Jia and L. Li, J. Mater. Chem. C, 2016, 4, 4505–4511 RSC.
- L. Zhu, M. T. Trinh, L. Yin and Z. Zhang, Chem. Sci., 2016, 7, 2058–2065 RSC.
- Y. Sagara and T. Kato, Nat. Chem., 2009, 1, 605–610 CrossRef CAS PubMed.
- Y. Sagara, S. Yamane, M. Mitani, C. Weder and T. Kato, Adv. Mater., 2016, 28, 1073–1095 CrossRef CAS PubMed.
- Z. Chi, X. Zhang, B. Xu, X. Zhou, C. Ma, Y. Zhang, S. Liu and J. Xu, Chem. Soc. Rev., 2012, 41, 3878–3896 RSC.
- J. Xu and Z. Chi, Mechanochromic Fluorescent Materials: Phenomena, Materials and Applications, Royal Society of Chemistry, 2014 Search PubMed.
- Z. Xie, C. Chen, S. Xu, J. Li, Y. Zhang, S. Liu, J. Xu and Z. Chi, Angew. Chem., Int. Ed., 2015, 54, 7181–7184 CrossRef CAS PubMed.
- P. Rajamalli, N. Senthilkumar, P. Gandeepan, C.-Z. Ren-Wu, H.-W. Lin and C.-H. Cheng, J. Mater. Chem. C, 2016, 4, 900–904 RSC.
- Q. Pei, Science, 1995, 269, 1086 CrossRef CAS PubMed.
- Q. Pei, Y. Yang, G. Yu, C. Zhang and A. J. Heeger, J. Am. Chem. Soc., 1996, 118, 3922–3929 CrossRef CAS PubMed.
- E. Fresta and R. D. Costa, J. Mater. Chem. C, 2017, 5, 5643–5675 RSC.
- S. Tang and L. Edman, Photoluminescent Materials and Electroluminescent Devices, 2017, pp. 375–395 Search PubMed.
- K. Matsuki, J. Pu and T. Takenobu, Adv. Funct. Mater., 2020, 30, 1908641 CrossRef CAS.
- S. Kanagaraj, A. Puthanveedu and Y. Choe, Adv. Funct. Mater., 2020, 30, 1907126 CrossRef CAS.
- B. Pashaei, S. Karimi, H. Shahroosvand, P. Abbasi, M. Pilkington, A. Bartolotta, E. Fresta, J. Fernandez-Cestau, R. D. Costa and F. Bonaccorso, Chem. Soc. Rev., 2019, 48, 5033–5139 RSC.
- B. Pashaei, S. Karimi, H. Shahroosvand and M. Pilkington, Adv. Funct. Mater., 2020, 30, 1908103 CrossRef CAS.
- A. Szlapa-Kula and S. Kula, Energies, 2023, 16, 5194 CrossRef CAS.
- M. Y. Wong and E. Zysman-Colman, in Light-Emitting Electrochemical Cells: Concepts, Advances and Challenges, ed. R. D. Costa, Springer International Publishing, Cham, 2017, pp. 237–266, DOI:10.1007/978-3-319-58613-7_9.
- T.-T. Bui, F. Goubard, M. Ibrahim-Ouali, D. Gigmes and F. Dumur, Appl. Sci., 2018, 8, 494 CrossRef.
- J.-H. Lee, C.-H. Chen, P.-H. Lee, H.-Y. Lin, M.-k. Leung, T.-L. Chiu and C.-F. Lin, J. Mater. Chem. C, 2019, 7, 5874–5888 RSC.
- M. Y. Wong, G. J. Hedley, G. Xie, L. S. Kölln, I. D. Samuel, A. Pertegás, H. J. Bolink and E. Zysman-Colman, Chem. Mater., 2015, 27, 6535–6542 CrossRef CAS.
- J. Liu, J. Oliva, K. Tong, F. Zhao, D. Chen and Q. Pei, Sci. Rep., 2017, 7, 1524 CrossRef PubMed.
- M. D. Moore, M. H. Bowler, J. E. Reynolds III, V. M. Lynch, Y. Shen, J. D. Slinker and J. L. Sessler, ACS Appl. Mater. Interfaces, 2018, 10, 24699–24707 CrossRef CAS PubMed.
- P. Lundberg, Y. Tsuchiya, E. M. Lindh, S. Tang, C. Adachi and L. Edman, Nat. Commun., 2019, 10, 5307 CrossRef CAS PubMed.
- K. Shanmugasundaram, J. C. John, S. Chitumalla, J. Jang and Y. Choe, Org. Electron., 2019, 67, 141–145 CrossRef CAS.
- Y. Chen, Y.-X. Wang, C.-W. Lu and H.-C. Su, J. Mater. Chem. C, 2022, 10, 11211–11219 RSC.
- X. Pang, K. Zhang, Y. Song, Y. Xiu, R. Yu and L. He, Chem. Eng. J., 2022, 450, 137987 CrossRef CAS.
- H.-L. Shen, P.-W. Hsiao, R.-H. Yi, Y.-H. Su, Y. Chen, C.-W. Lu and H.-C. Su, Dyes Pigm., 2022, 203, 110346 CrossRef CAS.
- J. Wang, H. Hafeez, S. Tang, T. Matulaitis, L. Edman, I. D. Samuel and E. Zysman-Colman, Aggregate, 2024, e571 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2025 |