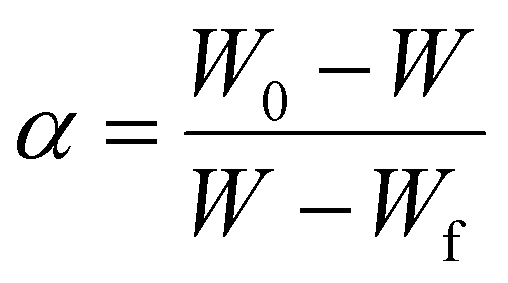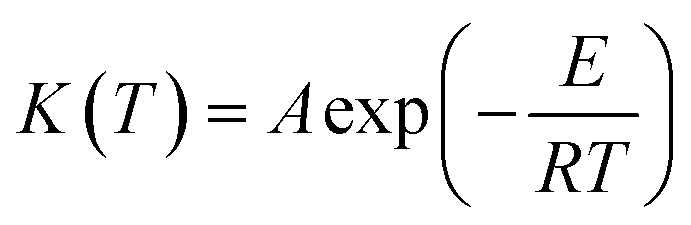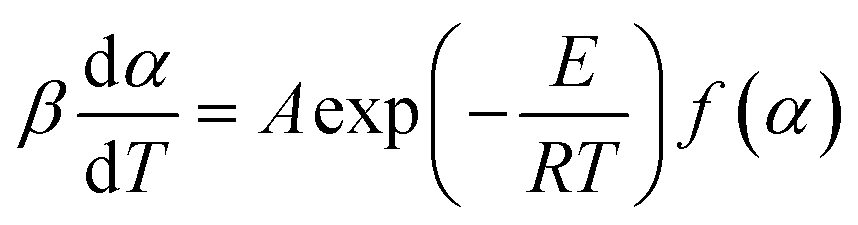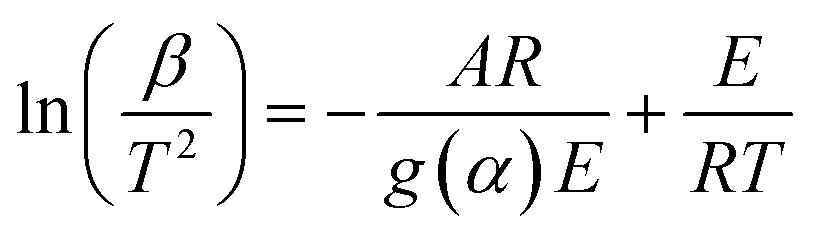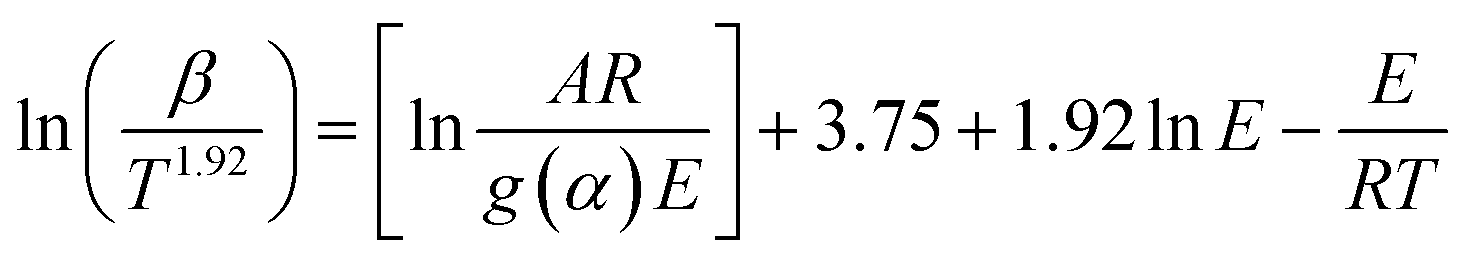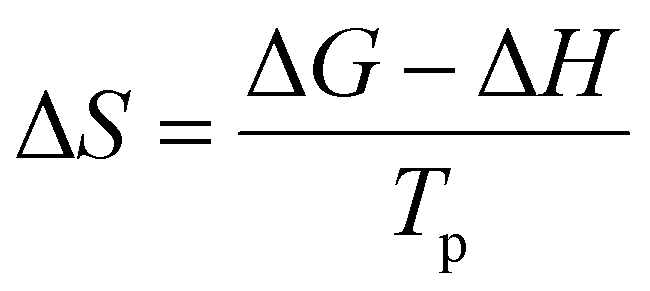DOI:
10.1039/D4RA08978B
(Paper)
RSC Adv., 2025,
15, 4861-4873
Synthesis of acidic–basic Y zeolite from kaolin: catalytic and mechanism study in the cracking of polyethylene†
Received
23rd December 2024
, Accepted 5th February 2025
First published on 13th February 2025
Abstract
Cracking of plastic waste by the acidic FAU-type zeolites faces challenges of low production of light olefins, a high amount of coke formation, and expensive process. Basic zeolites might be a better alternative; however, their preparation is difficult and the related cracking mechanism is unknown. We addressed these difficulties by direct synthesis of a hierarchical acidic–basic FAU-type zeolite (HY–BS) using silica extracted from a low-grade kaolin containing alkaline (earth) metals and investigated the impact of basicity in the linear low density polyethylene (LLDPE) cracking mechanism and products distribution. Compared to an acidic HY zeolite, the HY–BS presented comparable density of Brønsted acidity but a drastically higher density of the basicity. The HY–BS had comparable activity to the reference HY zeolite, but it produced twice light olefins (30.5 vs. 16.2%), triple lighter aromatics (41.0 vs. 13.8%), and less coke in LLDPE cracking. The thermodynamic investigation of thermal and zeolitic cracking of LLDPE was conducted based on the TGA results, along with kinetic analysis using four model-free techniques and a differential method. According to kinetic and thermodynamic parameters and product distribution, the HY–BS exhibited a dual cracking pathway that first followed a carbocation mechanism before switching to a radical mechanism as the reaction progressed, accounting for the reason behind improved light olefins selectivity. Consequently, direct promotion of the basicity of the zeolites synthesized from impure kaolin was effective to overcome difficulties facing plastic waste cracking.
Introduction
Approximately 80% of the plastics produced annually is turned into waste, most ended up in the environment or incinerated to recovery its energy content, leading to severe ecological and environmental crises.1,2 Among all kinds of plastics, polyolefins including low density polyethylene (LDPE), high density polyethylene (HDPE), and polypropylene (PP) accounts for 57% of total municipal solid plastic waste.3 An alternate way for converting plastic waste into chemicals is thermal cracking, but this process uses a lot of energy, generates CO2, and produces products of poor quality.4,5 Catalytic pyrolysis of plastic waste is a more selective process toward desired products like light olefins, and it occurs under relatively milder reaction conditions. This can turn challenges of plastic waste into economic and environmental opportunities by reducing fossil fuel consumption and CO2 emissions.6
Zeolites, among acidic solid catalyst, have shown a better potential for selective cracking of plastic waste into desired products due to their well-defined microporous channels, large surface area and pore volume, and adjustable acidity.2,7,8 However, they still suffer from high cost of preparation, limited selectivity toward desired products of light olefins and light aromatics, and rapid deactivation by coking.9 Synthesis of zeolites from natural aluminosilicates resources has been sought by many research groups to reduce the cost of preparation and avoid various wastes generation,10–12 and they have been used for catalytic cracking of heavy hydrocarbons,13 and plastic waste.14 However, the synthesis of zeolites from natural resources is hindered by phase and elemental impurities that prevent pure synthesis, low yield, and high crystal size of the resulting products,11 which limits their effective applications.
Zeolitic cracking occurs by protonating hydrocarbons to create carbocations, which is followed by bimolecular hydrogen transfer reactions producing alkanes and carbenium ions or by β-scission monomolecular cracking reactions producing smaller olefin intermediates.15 Depending on the physicochemical characteristics of the zeolites, they may undergo further secondary reactions of oligomerization, isomerization, cyclization, deep dehydrogenation, and aromatization, producing liquid oil and coke.2 Previous studies have shown that product selectivity can be tailored to a certain extent by boosting/inhibiting monomolecular or bimolecular hydrogen transfer mechanism in comparison to one another.16 The production of olefins has increased and the formation of coke by zeolites has decreased as a result of stimulating the monomolecular cracking mechanism by decreasing the interaction of olefin intermediates with the acid sites,17 increasing dehydrogenation reactions,18 and by faster diffusion out the intermediate species from the cavities.19–22 Hierarchical and recently developed intrinsic mesoporous zeolites promote cracking intermediates to diffuse out from the particles, which reduces the creation of coke, whereas longer residence of olefin intermediates inside the cavity stimulates secondary reactions leading to coke formation.23–25 In addition to increasing the generation of light olefins (C2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) –C4), altering surface acidity by creating bifunctional acidic–basic zeolites also prevented the creation of coke during crude oil cracking.26
–C4), altering surface acidity by creating bifunctional acidic–basic zeolites also prevented the creation of coke during crude oil cracking.26
Cracking of hydrocarbons by basic catalysts is an obvious option to produce light olefins at extremely low coke formation by reduced bimolecular hydrogen transfer reaction, although these catalysts prefer the high operating temperatures of 600–800 °C.27 Since basic catalysts are unable to protonate hydrocarbons, it is suggested that the pyrolysis of plastics by these catalysts occurs via the free radical mechanism.27 This is similar to the thermal cracking of hydrocarbons that occurs through radical mechanism including initiation, propagation, and termination.2 Wu et al., proposed that the presence of active oxygen in basic catalysts activates C–C and C–H bonds cleavage producing the initial free radicals and subsequent C–C cleavage, producing olefins and smaller alkyl radicals.28 The generated initial free radicals might also produce olefins through β-scissions.29 However, there is no consensus on the exact cracking mechanism over basic catalysts among the researchers. On the other hand, kinetic studies employing thermogravimetric analysis data is helpful to forecast reaction mechanism and product selectivity when the actual cracking mechanism is unknown30 but, these haven't been frequently conducted. Additionally, study of the kinetics and thermodynamics parameters is essential for assessing energy efficiency, constructing reactors for industrial use, and conducting technical and financial feasibility.28,31,32
The primary hypothesis of this study was that bifunctional acidic–basic zeolites might be a superior option for pyrolysis of plastic waste because the acidic sites drive the cracking reactions while basic sites promote olefins production. In this study, a silica solution derived from a low-grade kaolin was used to synthesize a bifunctional acidic–basic hierarchical FAU-type zeolite (HY–BS), and its use in the cracking of linear low-density polyethylene (LLDPE) was examined. Degrading activity, light olefins production, and coking quantity were used to assess the catalytic performance of the bifunctional HY–BS zeolite and a reference acidic HY zeolite in LLDPE cracking. The kinetic and thermodynamic parameters of LLDPE cracking by the HY–BS were first determined using four model-free methods and a differential method. These were then compared with those obtained by thermal cracking and by the reference acidic HY zeolite. Ultimately, the correlation between the anticipated mechanism, the distribution of products, and the acidic–basic characteristics of every zeolite in the LLDPE cracking process was assessed.
Methods
Theoretical background of kinetic study
Plastics can undergo complicated multi-step reactions, including parallel, competitive, and sequential reactions, through thermal and catalytic pyrolysis. The eqn (1) explains the general kinetic equation of pyrolysis.33| |
 | (1) |
The primary premise for plastic deterioration is that the conversion rate is proportional to the reactant concentration. The conversion rate can be expressed by eqn (2):
| |
 | (2) |
where the conversion rate
α is defined by:
| |
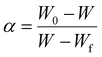 | (3) |
where
w is the weight of the sample at time
t, so that
W0 and
Wf are the initial and final weight, respectively.
K(
T) and
f(
α) are functions of temperature and the conversion, respectively.
K(T) the temperature dependence of the weight loss rate which is expressed by the Arrhenius equation:
| |
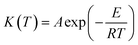 | (4) |
where
E is the activation energy,
A is the pre-exponential factor and
R is the gas constant.
By a combination of eqn (2) and (4), the reaction rate can be written as follows:
| |
 | (5) |
The E was then estimated by using different model-free methods (Table 1),34 of Flynn–Wall–Ozawa (FWO),35 Kissinger–Akahira–Sunose (KAS),36 Starink,37 and Tang,38 and a differential model of Friedman.39 For a given degree of conversion (α) and different heating rates (β), a linear relationship was obtained by plotting ln![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) β, ln(β/T2), ln(β/T1.92), ln(β/T1.89) and ln(dα/dt) versus 1/T, and E was obtained from the slope of the straight line of different models.
β, ln(β/T2), ln(β/T1.92), ln(β/T1.89) and ln(dα/dt) versus 1/T, and E was obtained from the slope of the straight line of different models.
Table 1 Different model-free equations applied for kinetic analysis34
| Model name |
Model equation |
Plot |
Formula number |
| FWO |
 |
 |
(6) |
| KAS |
 |
 |
(7) |
| Starink |
 |
 |
(8) |
| Tang |
 |
 |
(9) |
| Friedman |
 |
 |
(10) |
Theoretical background of thermodynamic study
Next, thermodynamic parameters were calculated based on E value obtained from the kinetic analysis. These parameters, including Gibbs free energy changes (ΔG), enthalpy changes (ΔH) and entropy changes (ΔS) are obtained from the following equations.40,41| |
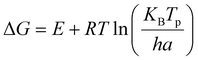 | (12) |
| |
 | (13) |
| |
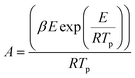 | (14) |
Results and discussion
Synthesis and characterization of physical properties of the zeolites
The synthesis of FAU-type zeolite from kaolin involved adding sodium aluminate and seed to the basic solution of the extracted silica at 4 °C (Fig. S1 and Table S1†), which was then aged for 24 h at room temperature and followed by an additional 24 h at 50 °C (details of the synthesis in ESI† and Fig. 1A). Sodium aluminate was added to the extracted silica solution at a low temperature in order to facilitate the nucleation and crystallization process by promoting the creation of 4- and 6-membered rings building units and inhibiting the formation of complicated aluminosilicates.42 The obtained material, named NaY–BS was characterized by relevant techniques and compared with those of a commercially available reference NaY zeolite. The powder XRD of NaY–BS presented diffraction peaks of the FAU-type structure at the 2θ of 15.7°, 18.7°, 20.4°, 23.7°, 27.1°, 30.8°, 31.5°, and 34.2° (Fig. 1B). The N2 sorption isotherms of the HY–BS, proton-exchanged of the NaY–BS, presented a significant amount of adsorption at the relative pressure (P/P0) < 0.1, indicating large microporosity of 0.15 cm3 g−1, total pore volume of 0.25 cm3 g−1, and total surface area of 326.82 m2 g−1 (Table S2† and Fig. 1C). The HY–BS also presented a hysteresis loop at 0.42 < P/P0 < 1 with capillary evaporation at P/P0 of 0.42 indicating ink-bottle cavity with the entrance size <5 nm,43 and a large external surface area of 87.85 m2 g−1 and a mesopore volume of 0.11 cm3 g−1. The pore size distribution (PSD) obtained from the N2 adsorption isotherm showed mean pore width of 4.40 nm and 15.92 nm, which may represent the size of the entrance and cavity, respectively (Fig. 1D). The FTIR spectra of the NaY–BS showed absorption band at 459, 574, 790, and 1017 cm−1 bands representing tetrahedral TO4 units (primary building unit), double-six-membered rings (D6R), symmetric stretching vibrations of Si–O–Al in the outer framework, and the asymmetric stretching vibrations of tetrahedral atoms, respectively, similarly to those by the reference NaY zeolite (Fig. 1E).44,45 Accordingly, these findings demonstrated the effective synthesis of a hierarchical FAU-type zeolite employing silica extracted from an impure kaolin.
 |
| | Fig. 1 (A) Schematic of the procedure of NaY–BS synthesis, (B) XRD patterns, (C) N2 sorption isotherms of the reference HY zeolite and HY–BS, (D) pore size distribution, (E) FTIR spectra, (F and G) FE-SEM images of the reference NaY zeolite and NaY–BS, (H–J) HRTEM study of microtomed cross-section of HY–BS shown in different magnification, and (K–O) TEM-EDS mapping of the HY–BS. The double-headed arrows indicate mesopores. | |
The FE-SEM, HRTEM, and HRTEM-EDS mapping techniques were used to examine the morphology, chemical composition, and intra-particle porosity of the (Na)HY–BS (Fig. 1F–O). The FE-SEM images of the reference NaY zeolite exhibited octahedral particles with smooth surfaces and sharp edges, but the ball-milled seed and NaY–BS showed spherical particles with rough surfaces (Fig. S2† and 1G). The size distribution histogram of the reference NaY zeolite, seed, and NaY–BS sub-particles presented average sizes of 750, 510, and 435 nm, respectively (inserted figures; Fig. 1F, G and S2F†). The HRTEM analysis of the HY–BS sample, which had previously been microtomed, revealed well-defined intra-crystalline mesopores with an average size of 10.3 nm (Fig. 1H–J), being smaller than that obtained by the N2 isotherm due to the fact that it is not a bulk technique. Furthermore, TEM-EDS mapping of the HY–BS was conducted showing the presence of Ca in large extent on the surface sample, originating from kaolin used for silica extraction (Fig. 1K, L and O). The XRF analysis, in accordance with the TEM-EDS study, confirmed the presence of a high amount of Ca but a much lower amount of K, Fe, and Ti in the NaY–BS sample. The silicon-to-aluminum (SAR) ratio of the reference NaY zeolite and NaY–BS obtained by the XRF were 10.72 and 4.33, respectively (Table S1†).
Acidic–basic properties of the zeolites
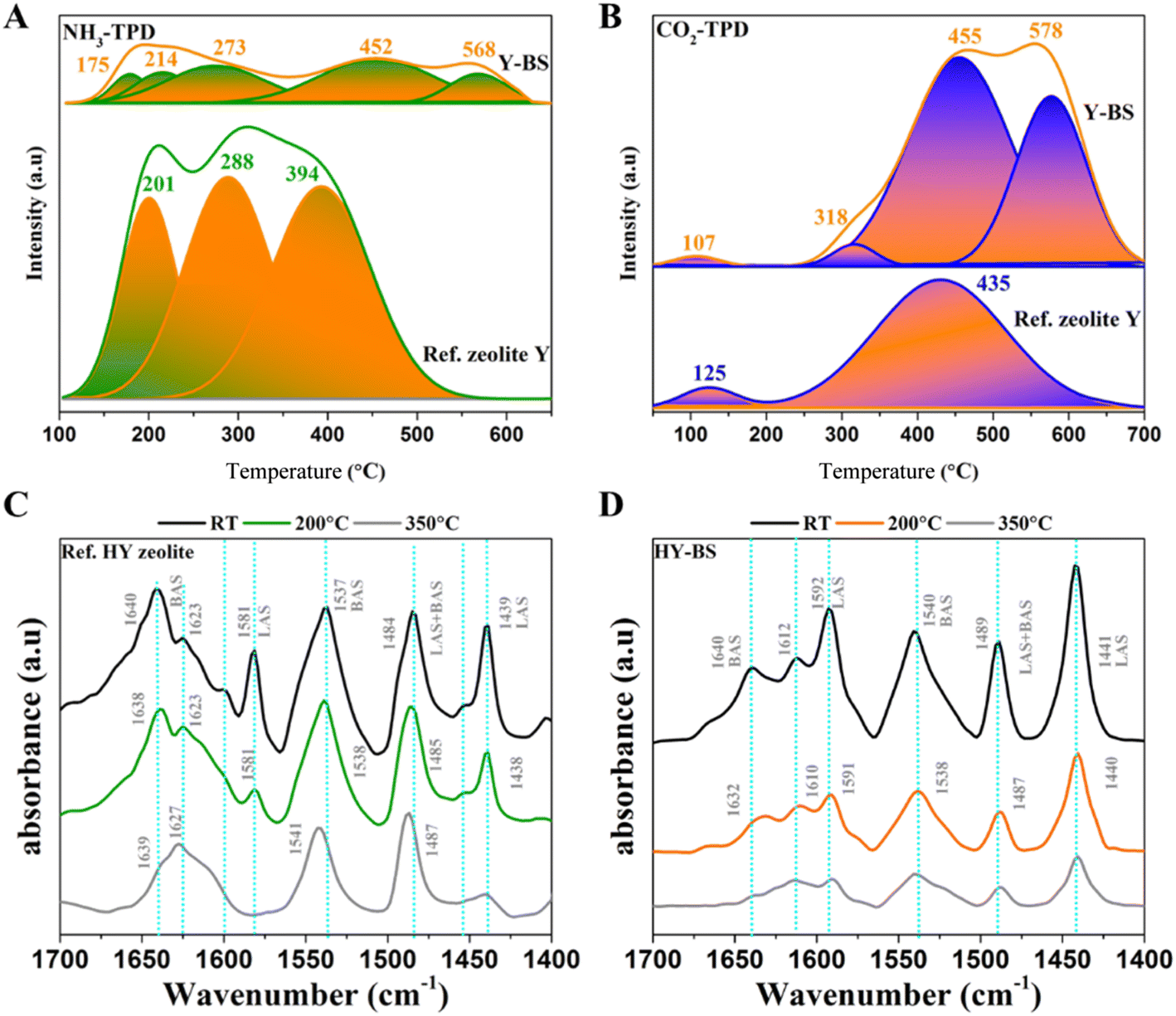
Acidity and basicity were assessed using NH3-TPD and CO2-TPD, respectively, and then Brønsted acidity (BAS) and Lewis acidity (LAS) were distinguished using pyridine-IR spectroscopy (LAS; Fig. 2). The NH3-TPD of reference HY zeolite displayed desorption peaks at 201, 288, and 394 °C indicating weak, medium, and strong Brønsted acidity, respectively, while that of the HY–BS displayed desorption peaks at 175, 214, and 273 °C for weak-to-medium strength acidity and at 452 and 568 °C for strong acidity. The BAS density, total BAS content/surface area, of the reference HY zeolite and HY–BS was 3.50 and 3.36 μmole-NH3 per m2, respectively, while the corresponding total BAS content was 2931 and 1100 μmole-NH3 per g-zeolite (Table S3†). The CO2-TPD by the reference HY zeolite displayed desorption peaks at 125 and 435 °C, demonstrating a lower basic strength than the HY–BS, which displayed more desorption peaks at higher temperatures of 107, 318, 455, and 578 °C. The density of basicity of the HY–BS and reference HY zeolite was 2.92 and 0.88 μmole-CO2 per m2, respectively, and their total basicity was 957.2 and 735 μmole-CO2 per g-catalyst (Fig. 2B). The fact that CaO was evenly deposited on the surface (Fig. 1L) has contributed to the much higher level of stronger basicity by HY–BS.
 |
| | Fig. 2 (A) NH3-TPD and (B) CO2-TPD spectra of the reference HY zeolite and the HY–BS, (C and D) Py-IR of the reference HY zeolite and the HY–BS. | |
The Py-IR was conducted to determine content and strength of BAS and LAS sites of the zeolites by desorbing pyridine at room temperature, 200, and 350 °C (Fig. 2C, D and Table S4†). The Py-IR spectra of the reference HY zeolite showed five main distinct bands at 1439, 1484, 1553, 1581, and 1640 cm−1 and a shoulder band at 1623 cm−1 and that of the HY–BS showed bands at 1441, 1489, 1540, 1592, 1612, and 1640 cm−1 related to different BAS and LAS sites. The bands centered at about 1440 and 1596 cm−1 and those at 1541 and 1636 cm−1 indicate the LAS and BAS sites, respectively.46 The band at 1492 cm−1 is structure insensitive. The bands at 1445 and 1545 cm−1 were used to quantify the LAS and BAS, respectively. The most noticeable difference between both catalysts was the sharp decline in the peak at 1440 cm−1 associated with the LAS site of reference HY zeolite, whereas the peak of HY–BS persisted more prominently, indicating that the latter had stronger LAS sites. We then calculated the density of acidic sites by the reference HY and HY–BS zeolites. The density of the BAS sites by the HY zeolite at room temperature, 200, and 350 °C were 0.93, 0.73, and 0.43 μmole-BAS per m2-zeolite while those by the HY–BS zeolite were 1.31, 0.75, and 0.42 μmole-BAS per m2-zeolite, respectively. These results demonstrated comparable, robust BAS sites that were consistent with the NH3-TPD. The HY–BS had a density of 1.1, 0.59, and 0.33 μmole-LAS per m2-zeolite, while the reference HY zeolite had a density of 0.20, 0.11, and 0.04 μmole-LAS per m2-zeolite at ambient temperature, 200 °C, and 350 °C, respectively. The well-distributed calcium on the surface of HY–BS may have contributed to its increased Lewis acidity, as previously observed,47 by the hydrolysis of the Si–OH–Al BAS units, creating extra-framework Al with the LAS feature.26 The density of the total acidic sites by the reference HY zeolite were 1.12, 0.84, and 0.48 μmole-acidity per m2-zeolite at ambient temperature, 200 °C, and 350 °C, respectively, while those by the HY–BS were 2.40, 1.34, and 0.73 μmole-acidity per m2-zeolite. The reference HY zeolite thus displayed a strong acidic character, whereas the HY–BS displayed a significantly larger density of stronger LAS sites and similar BAS sites.
Catalytic study of the zeolites in LLDPE cracking
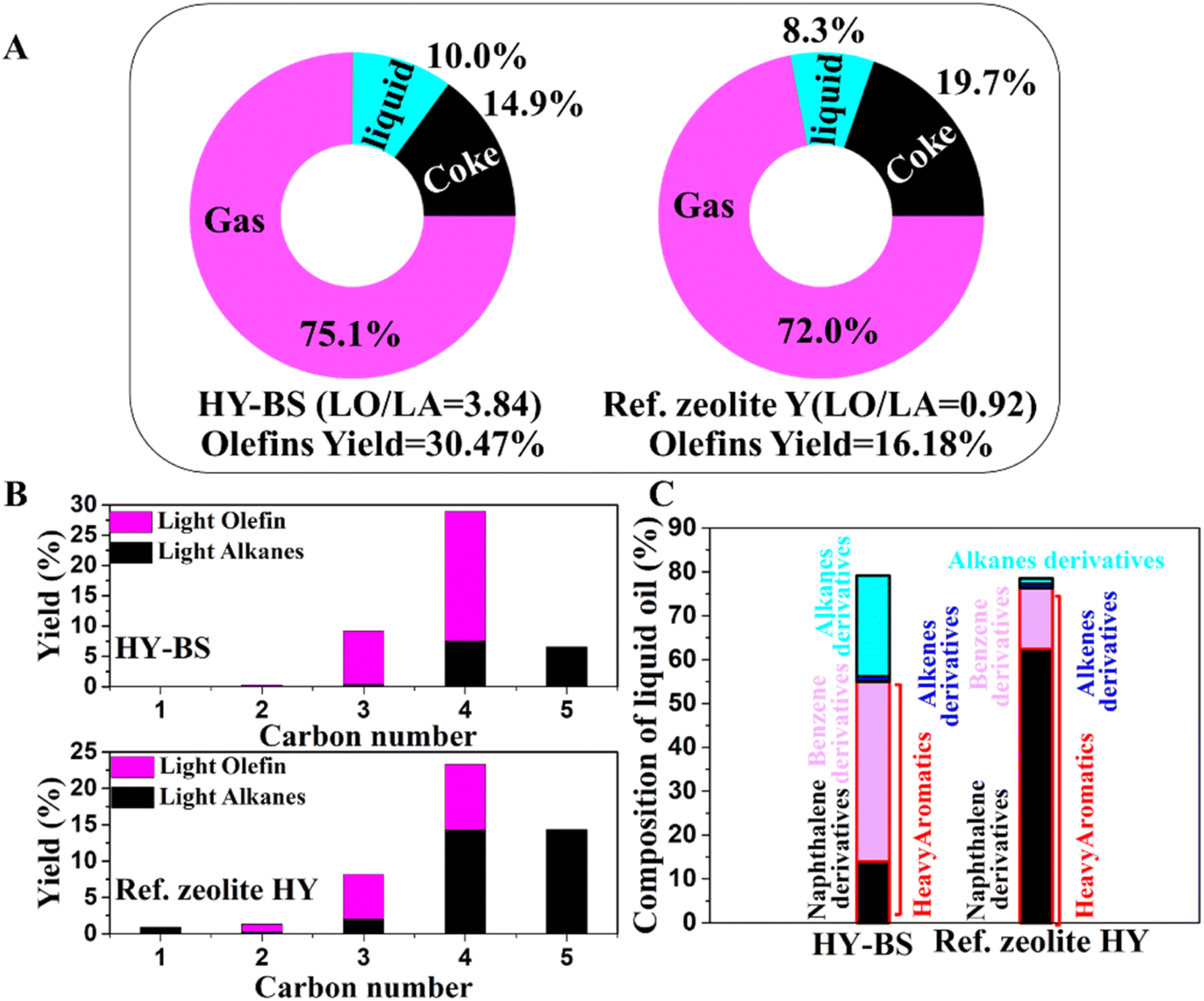
The pyrolysis of LLDPE in the presence and absence of the zeolites was conducted, and the gas, liquid, and coke that were produced were examined using a variety of methods (Fig. S3†). The yield of gas, liquid, and coke by the HY–BS were 75.1, 10.0, 14.9%, respectively and those by the HY zeolite were 72, 8.3, 19.7%, respectively (Fig. 3A). Although these two catalysts produced a comparatively similar amount of various products, their compositions differed greatly from one another. The light olefins-to-light alkanes (C2–C4/C1–C4; LO/LA) and light olefins yield by the HY zeolite were 0.92 and 16.18%, respectively while those by HY–BS were 3.84 and 30.47% (Fig. 3A and Table S5†). To forecast how a potential cracking mechanism could affect product selectivity, the hydrogen transfer coefficient (HTC = (n-C4 + i-C4)/C4) was computed. The HY zeolite and HY–BS had HTC of 1.59 and 0.35, respectively (Table S5†), indicating that monomolecular cracking mechanism was dominant by the latter while in case of the former the bimolecular hydrogen transfer mechanism was dominant. This is because the basic sites of the HY–BS weakened the C–H bond and promoted dehydrogenation,28 and inhibited bimolecular hydrogen transfer reaction.48 Additionally, alkaline (earth) metals enhanced the exterior surface area and produced mesoporosity,30 which promoted diffusion of the olefins intermediates away from the particles that dominated the monomolecular process. As a result, both basicity and mesoporosity contributed in enhanced light olefins production by HY–BS. Conversely, the strong BAS sites of HY zeolite promoted bimolecular hydrogen transfer reaction and strong acid-catalyzed secondary reactions leading to less light olefins production and higher coke formation.2 The impact of alkaline (earth) metals on zeolites properties depend on its preparation approach, loading, and dispersion.49 Therefore, compared with the previous study, the better improvement of product selectivity toward light olefins by HY–BS might be related to well-distribution of CaO on the surface.26 We furthermore conducted thermal cracking of LLDPE in the absence of any catalyst at 550 °C, producing a high amount of 70% waxes which is in accordance with those reported previously.50
 |
| | Fig. 3 Yield (%) of (A) total gas, liquid, and coke, (B) light olefins and light alkanes components of the gas fractions, and (C) and liquid oil components obtained from LLDPE cracking by the HY zeolite and HY–BS. | |
The liquid oils collected from LLDPE cracking were analyzed by the GC-mass spectrometry. The liquid oil obtained from LLDPE cracking by the HY zeolite contained 13.8% benzene derivatives, 62.4% naphthalene derivatives, 1.0% long-chain alkenes, and 1.2% long-chain alkanes but that by the HY–BS contained 41.0% benzene derivatives, 14.0% naphthalene derivatives, 1.3% long chains alkene, and 14.4% long chain alkane. According to the findings, the aromatics became lighter and there was less coke after the acidic–basic zeolite by the HY–BS was established (Fig. 3C, S4 and Table S6†). These might come from that the high density of basicity and LAS sites slowed down secondary reactions by the HY–BS and the hydrogen radicals generated by the active basic sites inhibit the condensation reaction resulting in lighter aromatics.28,51
Study of thermal and catalytic degradation of LLDPE by the TGA
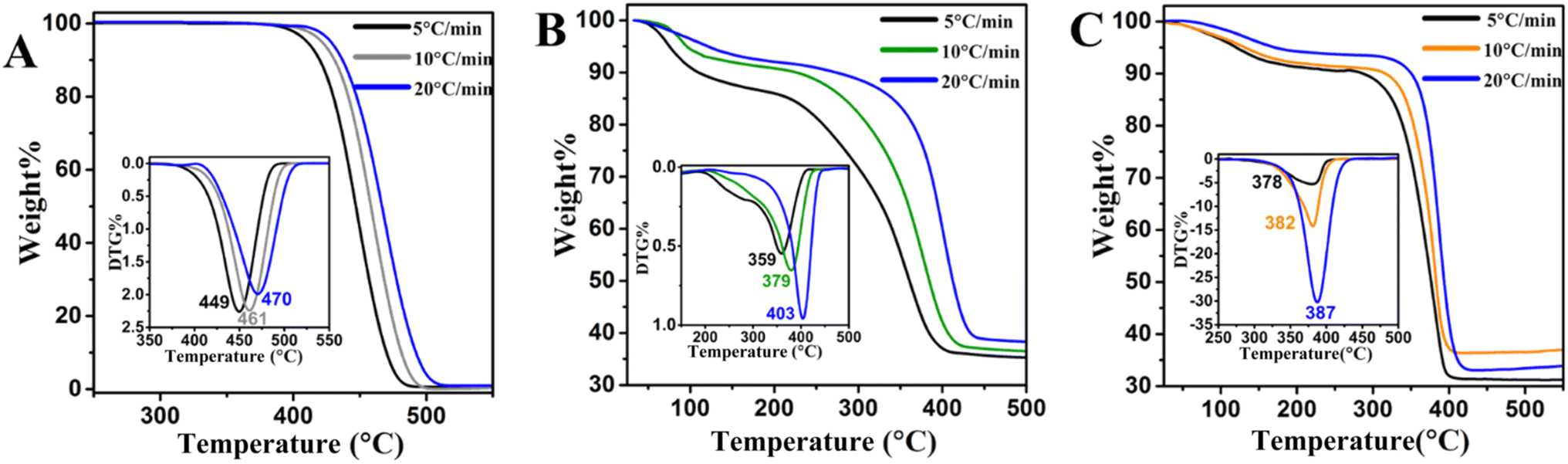
Thermogravimetric analysis (TGA) is a powerful approach to ascertain the kinetic and thermodynamic parameters of plastic waste cracking.52 Consequently, TGA was carried out on the LLDPE alone and with zeolites at varying temperature rates of 5, 10, and 20 °C min−1 while N2 flowed at a rate of 10 ml min−1. This is because conducting a kinetic analysis utilizing experimental α–T (temperature) data gathered at a minimum 3–5 heating rate is reliable, as recommended by the ICTAC Kinetics Committee.34 The TGA curves of LLDPE, LLDPE + HY zeolite, and LLDPE + HY–BS, at various heating rates of 5, 15, and 20 °C min−1, demonstrated a comparable weight loss trend; however, the starting, maximum, and completion temperatures varied. As the rate of heating rose, the Tmax shifted to the higher temperature at which 50% of the LLDPE degradation occurs. For example, when the temperature rate increased from 5 °C min−1 to 20 °C min−1, the Tmax for the sole LLDPE, LLDPE + HY zeolite, and LLDPE + HY–BS changed from 449 to 470 °C, 359 to 403 °C, and 378 to 387 °C, respectively (Table S7† and Fig. 4). This is due to the fact that when the heating rate was increased from 5 to 20 °C min−1, the sample took less time to reach the appropriate temperature, which causes LLDPE degradation at the higher temperature.53 Temperature gradients are also impacted by the quantity of heat transmission from the furnace to the sample, which varies with heating rates.54
 |
| | Fig. 4 TGA and dTGA of LLDPE decomposition of (A) sole LLDPE, (B) LLDPE + HY zeolite, and (C) LLDPE + HY–BS at heating rate of 5, 10, and 20 °C min−1. The weight ratio of LLDPE and zeolite was 60:40. | |
Kinetic analysis for activation energy determination
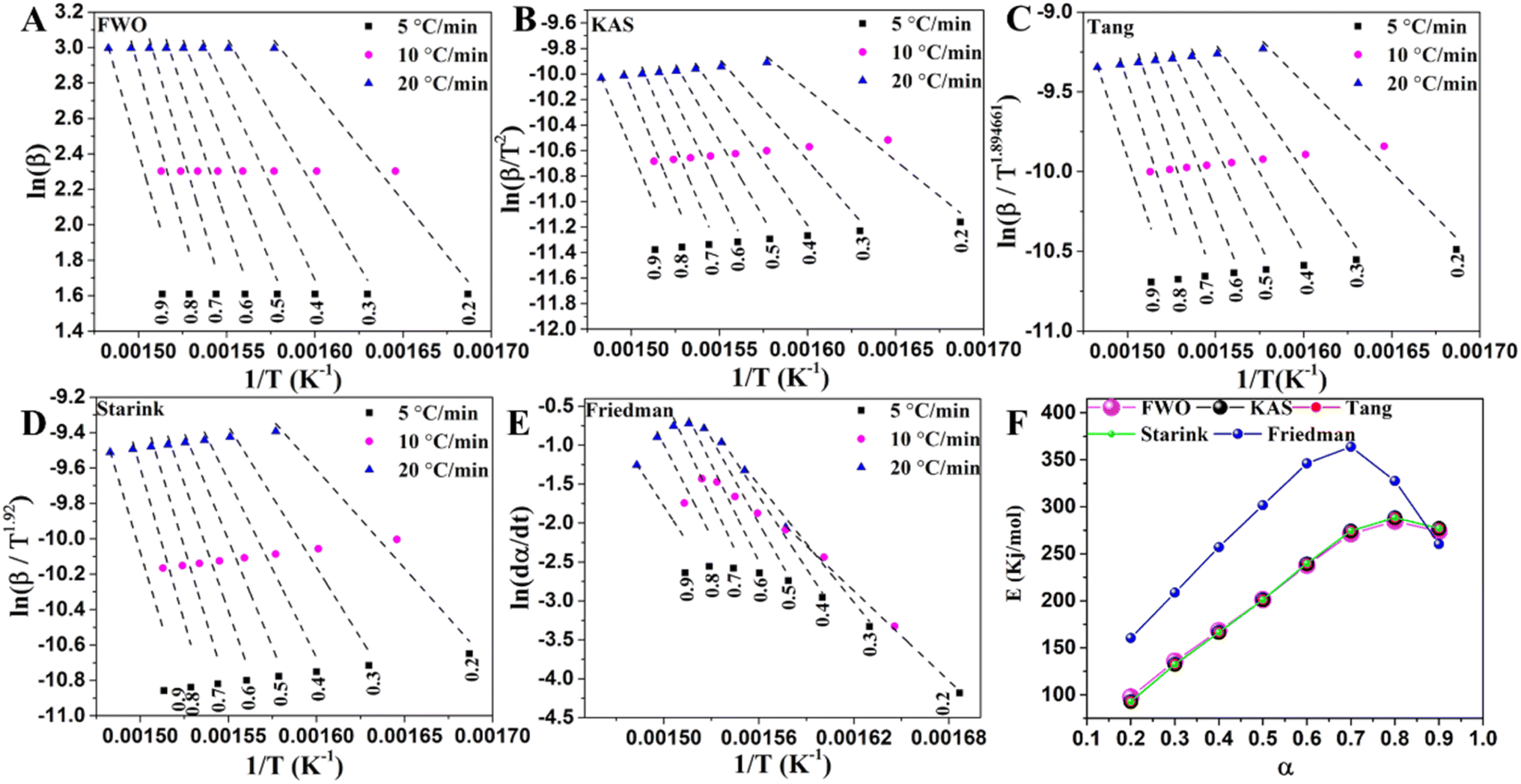
The activation energy of LLDPE cracking by the TGA system was determined using a differential isoconversional (Friedman) approach and four integral isoconversional equations (FWO, KAS, Starink, and Tang). According to previous studies, Friedman's differential technique is more accurate than the integral techniques since it does not rely on an approximation, and the accuracy of the integral approximation equations is in the order of Starink < Tang < KAS < FWO.34 The use of the differential method to analyze TGA data, however, requires numerical derivative which causes inaccuracy. The activation energy of LLDPE, LLDPE + HY zeolite, and LLDPE + HY–BS was determined using the indicated models, and the results are shown in Tables S8–S10† and Fig. 5–7. The kinetic models demonstrated a high degree of accuracy in fitting the experimental data, as evidenced by the average correlation coefficient (R2) values for all samples by various methods exceeding 0.92, with the exception of HY–BS, which had an R2 = 0.89 of the Friedman method (Fig. 5–7). The activation energy value of each cracking system obtained by the FWO, KAS, Tang, and Starink models was similar, but they were different from those by the differential Friedman method. The R2 values which shows the accuracy of fitting of experimental with theoretical models were used to evaluate the best model among the others. The most accurate model among all of them was the FWO model with the highest value of R2, which was used for further study. The activation energy range based on α ranging from 0.2 to 0.9 (and average) for cracking sole LLDPE, LLDPE + HY–BS, and LLDPE + HY zeolite was 284.2 to 308.0 kJ mol−1 (average: 295.3 kJ mol−1), 97.7 to 273.8 kJ mol−1 (average: 208.8 kJ mol−1), and 18.2 to 117.9 kJ mol−1 (average: 67.6 kJ mol−1), respectively. This average of activation energy followed the order of LLDPE > LLDPE + HY–BS > LLDPE + HY zeolite. While the activation energy of sole LLDPE cracking decreased with conversion level (Fig. 5 and Table S8†), the activation energy of LLDPE cracking by the HY zeolite increased with α from 0.2 to 0.9 (Fig. 6, 7 & Tables S8–S10†). As was previously documented, catalyst deactivation by coke formation over the acidic active sites may be the cause of an increase in the activation energy of LLDPE cracking upon conversion by the HY zeolite.55 The decrease in the activation energy of thermal cracking of LLDPE upon conversion was due to that the initial free radical formation is the slowest step, requiring more energy to activate the cleavage of C–C and C–H bonds, whereas the subsequent reactions of propagation and termination require less energy.56 Therefore, once the reaction has been started, less energy is required to proceed. The trend and average activation energy changes of LLDPE cracking by HY–BS differed from those obtained by thermal and HY zeolite, suggesting that HY–BS with acidic–basic feature followed a different cracking mechanism, which is discussed in more detail below.
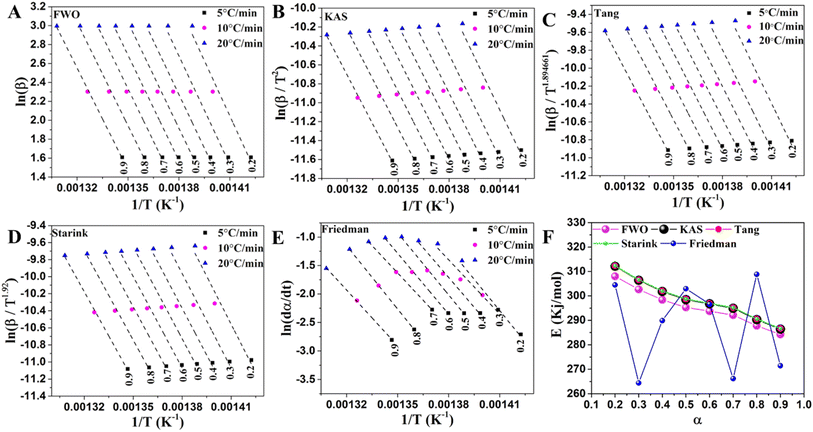 |
| | Fig. 5 The linear fitted curves were obtained from sole LLDPE cracking by (A) FWO, (B) KAS, (C) Tang, (D) Starink, and (E) Friedman methods and (F) variations of activation energy versus conversion level using these methods for cracking of sole LLDPE. | |
 |
| | Fig. 6 The linear fitted curves obtained from LLDPE cracking by the acidic HY zeolite by (A) FWO, (B) KAS, (C) Tang, (D) Starink, and (E) Friedman methods and (F) various of activation energy versus conversion level using these methods for catalytic cracking of LLDPE in the presence of HY acidic zeolite. | |
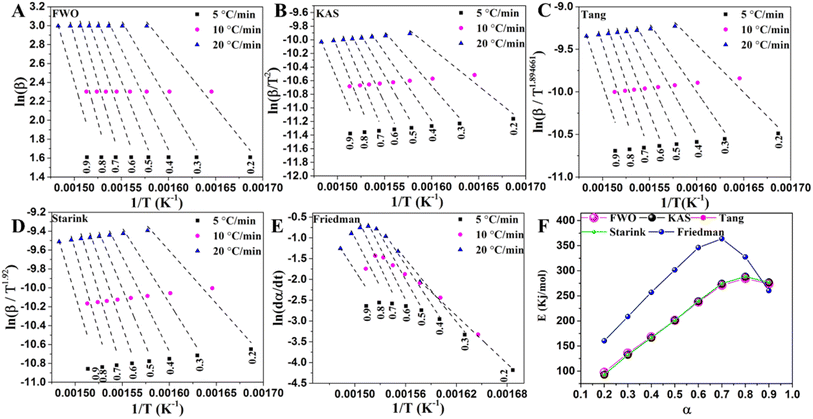 |
| | Fig. 7 The linear fitted curves were obtained from (A) FWO, (B) KAS, (C) Tang, (D) Starink, and (E) Friedman methods and (F) variation of activation energy versus conversion level using these methods for catalytic cracking of LLDPE in the presence of HY–BS. | |
The average activation energy value of LLDPE + HY–BS fell between that of LLDPE + HY zeolite cracking and sole LLDPE; however, the trend of changes followed a combination of LLDPE and that in the presence of HY zeolite. The activation energy of LLDPE cracking by HY–BS was increased similarly to those by the HY zeolite in α < 0.7. This indicated that when the conversion level was less than 0.7, the LLDPE cracking by HY–BS zeolite followed the carbocation mechanism that is recognized for strong acidic zeolites.57 The rate of increase in the average activation energy of LLDPE cracking by HY–BS from α of 0.7 to 0.9 was dropped, presenting similar values and trend to those of thermal cracking of LLDPE. On the other hand, the thermal cracking of hydrocarbons occurs via the radical mechanism, which needs a high degree of activation energy.2 These implied that at greater conversion levels of α > 0.7, the HY–BS cracking of LLDPE followed a catalytic radical mechanism by its basic sites. This shift in the trend of activation energy might come from the fact that once the acidic sites of the HY–BS blocked by the coke, the active oxygen present in basic catalysts activates C–C and C–H bonds cleavage, producing long-chain alkyl radicals, which then undergo further β-scission or hydrogen abstraction reactions, generating olefins.26–28,48 In accordance with the previous studies, the product distribution already demonstrated outstanding olefins production by the HY–BS, as a characteristic of basic catalysts. These findings, understanding the average and change in trend of activation energy upon conversion by kinetic studies, which was supported by the excellent yield of olefins, led us to hypothesize that depending on the conversion level, LLDPE cracking by the HY–BS may follow either carbocation or radical mechanism.
Thermodynamics study of thermal and catalytic cracking of LLDPE
Pre-exponential coefficient (A). The pre-exponential factor (A) and thermodynamic parameters like ΔH, ΔG, and ΔS were calculated using the activation energy obtained from the FWO kinetic model (Tables S8–S10†). The pre-exponential coefficient (A) is the collision frequency of the reactant molecules to be converted into products, the increase of which shows a higher amount of the activation energy required to proceed with the reaction.58,59 As conversion progresses from α of 0.2 to 0.9, the coefficient A for thermal cracking of LLDPE drops from 9.52 × 1019 to 1.78 × 1018 (Tables S11–S13)†. This is because a higher activation energy is required to initiate and split the main chain of the LLDPE into initial radicals at the beginning of the reaction, which should be provided by a high rate of molecular collision (coefficient A).56 However, once radicals are formed, their propagation and termination require less energy because they are highly reactive, and thus less molecular collision is required.Upon conversion from α of 0.2 to 0.9, the catalytic pyrolysis coefficient A for LLDPE + HY zeolite increased from 2.49 × 10−2 to 1.57 × 107 (Tables S11–S13)†. The catalyst deactivation caused by the high quantity of coke deposition may be the cause of the increase in coefficient A upon an increase in the conversion level of LLDPE by the acidic zeolite, as shown in Fig. 3A. This is because, as seen in Fig. 6, coke production reduces the acidic sites, necessitating a larger molecular collision rate to offset the elevated activation energy.60 Notably, lower activation energy by acidic sites resulted in lower average values of coefficient A of LLDPE cracking by acidic HY zeolite than thermal pyrolysis.
The coefficient A for catalytic pyrolysis of polymers with HY–BS ranges from 1.70 × 107 to 5.30 × 1021 (Table S11†). The coefficient A in the conversion level of 0.2–0.7 was significantly lower than that of thermal pyrolysis, but it was closer to that of the HY zeolite. This might indicate that the acidic sites of HY–BS activated C–C bond cleavage of the LLDPE through a carbocation mechanism at the lower conversion levels. The pre-exponential coefficient A at the conversion level of 0.7 to 0.9 was high, but it increased slower, which might be because coke deposition already had deactivated the acidic sites by conversion of 0.7 (Fig. 3A). As a result, the basic sites might use a catalytic radical mechanism to continue conversion at a higher level. Thus, the examination of the coefficient A values indicated that the acidic–basic HY–BS zeolite cracked LLDPE by radical and carbocation mechanisms upon the conversion level.
Enthalpy change (ΔH). The heat needed to transfer a molecule from its ground state to a higher energy state is known as enthalpy change (ΔH), which is used to determine whether a reaction is endothermic or exothermic.61–63 For cracking of LLDPE, LLDPE + HY–BS, and LLDPE + HY zeolite, the corresponding ΔH values were 301.9–287.1, 92.3–268.4, and 12.8–112.5 kJ mol−1 (Fig. 8a–c and Tables S11–S13†) in the α range of 0.2 to 0.9. The endothermic nature of thermal and catalytic cracking was demonstrated by positive enthalpy values; however, thermal pyrolysis requires more energy from an external source to drive the cracking reaction via a free radical mechanism. Faster protonation of LLDPE by strong acidic sites of the HY zeolite was the reason for the lower enthalpy values compared to those by HY–BS and thermal cracking, which were in accordance with the low activation energy by the former. For thermal pyrolysis of LLDPE, the enthalpy changes were almost negligible over the entire conversion levels (Fig. 8a), which was in accordance with the small changes in the activation energy as already shown in Fig. 5F. The catalytic pyrolysis enthalpy of the HY zeolite and HY–BS increased significantly as the conversion level rises, which might originate from coke formation over the acidic sites (Fig. 6, 7 and Tables S11–S13†). The reason behind the decrease in enthalpy change from conversion of 0.8 to 0.9 by the HY–BS might be related to the change in cracking mechanism from carbocation to radical.
 |
| | Fig. 8 ΔH, ΔG, and ΔS of pyrolysis of (A and D) LLDPE, (B and E) LLDPE + HY zeolite, (C and F) LLDPE + HY–BS. | |
Gibbs free energy change (ΔG). The spontaneous and non-spontaneous nature of the reaction during pyrolysis is indicated by the Gibbs free energy change (ΔG), which remains unchanged when a catalyst is present and the product distributions are identical.64,65 The average ΔG values for cracking system of LLDPE, LLDPE + HY zeolite, and LLDPE + HY–BS were 212.8, 195.9, and 190.2 kJ mol−1, respectively (Fig. 8a–c and Tables S11–S13†). The positive ΔG for both thermal and catalytic LLDPE cracking suggests that they are not spontaneous. The notable distinction between the ΔG of thermal and catalytic LLDPE cracking may be due to the noticeable differences in the distribution of their products, as the thermal cracking produced waxes while the latter resulted in liquid, gas, and coke formation.
Entropy change (ΔS). The degree of disorder in the product can be determined by changes in ΔS; lower values of ΔS suggest a more stable system.62 LLDPE, LLDPE + HY–BS, and LLDPE + HY zeolite cracking yielded ΔS ranging from 121.7 to 88.7 J mol−1 K−1, −155.4 to 122.1 J mol−1 K−1, and −290.44 to −122.00 J mol−1 K−1, respectively (Fig. 8d–f and Tables S11–S13†). For thermal pyrolysis of the LLDPE, the ΔS values were positive at all conversion levels, indicating a significant amount of system disordering as previously described.55 The ΔS of catalytic pyrolysis of LLDPE by the HY zeolite showed negative values in all conversion levels, explaining the low disordering or closeness of the system to equilibrium.55 The ΔS was negative at low conversion (α < 0.5) in the case of HY–BS catalytic pyrolysis of LLDPE, which resembles acidic catalytic cracking, and positive at greater conversion (α > 0.5), which resembles thermal cracking of LLDPE. These indicated that HY–BS cracked LLDPE via a carbocation mechanism when α < 0.5 and a radical mechanism when α > 0.5.In summary, the change of the kinetic and thermodynamic parameters of LLDPE cracking by the HY–BS showed a different behavior than those by the HY zeolite and thermal cracking. Change in activation energy trend was similar to that by the HY zeolite at low conversion levels and different than that at the higher conversion levels. Additionally, the trend of pre-exponential factor (A), enthalpy change (ΔH), and entropy change (ΔS) of LLDPE cracking by the HY–BS was similar to those by the HY zeolite at low conversion levels, and they showed similarity to those by the thermal cracking at the high conversion levels. The product distribution from LLDPE cracking by the HY–BS also showed similarity to those by acidic and basic zeolites. Therefore, we concluded that cracking of the LLDPE by the HY–BS occurs through a carbocation mechanism by acidic sites at low conversion levels and a catalytic radical mechanism at the higher conversion levels.
Conclusion
Acidic zeolites are the key catalysts for cracking of plastic wastes into fuels and chemicals; however, they face challenges of re-adsorption and consumption of olefins intermediates into low value products such as coke. A bifunctional acidic–basic FAU-type zeolite (HY–BS) using silica extracted from a highly impure kaolin containing high amount of alkaline (earth) metals was synthesized. The HY–BS presented high density of acidic sites as well as a high density of strong basic sites originating from homogeneously deposited CaO. Due to its basic feature, the HY–BS presented much higher yield of light olefins and much lighter aromatics-rich liquid oils in LLDPE cracking compared with the reference HY zeolite. Kinetic and thermodynamic study showed that while the thermal cracking of LLDPE underwent via radical mechanism and the acidic HY zeolite underwent the carbocation mechanism; the bifunctional acidic–basic HY–BS followed carbocation mechanism for the first half of the conversion level and then changed to radical mechanism upon progress of conversion. The HY–BS synthesized from a highly impure kaolin enhanced olefins production and decreased coke formation. For the future work, deposition of alkaline (earth) metals on intrinsic mesoporous zeolites might allow better control of loading and dispersion for further enhancement of the light olefins production in the cracking of plastics waste.
Data availability
The data supporting this article have been included as part of the ESI.†
Author contributions
Samira Motamadnejad – conceptualization, data curation, formal analysis, investigation, methodology, project administration, resources, software, validation, visualization, writing – original draft, writing – review & editing. Li Gao: formal analysis, investigation, resources, writing – review & editing. Kourosh Tabar Heydar: data curation, investigation, resources, writing – review & editing. Reza Panahi: resources, visualization, writing – review & editing. Bingsen Zhang: writing – review & editing, resources, visualization. Mozaffar Shakeri: conceptualization, funding acquisition, project administration, resources, supervision, validation, visualization, writing – original draft, writing – review & editing.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors gratefully acknowledge the financial support provided by the Iran National Science Foundation (No. 4001399) and the National Natural Science Foundation of China (No. 52161145403).
References
- A. J. Martín, et al., Catalytic processing of plastic waste on the rise, Chem, 2021, 7(6), 1487–1533 Search PubMed.
- Z. Dong, et al., Understanding the structure–activity relationships in catalytic conversion of polyolefin plastics by zeolite-based catalysts: a critical review, ACS Catal., 2022, 12(24), 14882–14901 CrossRef CAS.
- R. Geyer, J. R. Jambeck and K. L. Law, Production, use, and fate of all plastics ever made, Sci. Adv., 2017, 3(7), e1700782 CrossRef PubMed.
- T. Shan, et al., Study on pyrolysis behavior and kinetics of waste plastics with spent FCC catalyst, Int. J. Chem. Kinet., 2023, 55(2), 87–101 CrossRef CAS.
- A. Angyal, N. Miskolczi and L. Bartha, Petrochemical feedstock by thermal cracking of plastic waste, J. Anal. Appl. Pyrolysis, 2007, 79(1–2), 409–414 CrossRef CAS.
- M. Jahirul, et al., Transport fuel from waste plastics pyrolysis–A review on technologies, challenges and opportunities, Energy Convers. Manage., 2022, 258, 115451 CrossRef CAS.
- H. Yuan, et al., Recent developments on the zeolites catalyzed polyolefin plastics pyrolysis, Fuel Process. Technol., 2022, 238, 107531 CrossRef CAS.
- V. Blay, et al., Engineering zeolites for catalytic cracking to light olefins, ACS Catal., 2017, 7(10), 6542–6566 CrossRef CAS.
- Z. Chen, et al., Deactivation of a Y-zeolite based catalyst with coke evolution during the catalytic pyrolysis of polyethylene for fuel oil, Appl. Catal., A, 2021, 609, 117873 CrossRef CAS.
- T. Pan, Z. Wu and A. C. Yip, Advances in the green synthesis of microporous and hierarchical zeolites: a short review, Catalysts, 2019, 9(3), 274 CrossRef CAS.
- Hartati, et al., A review on synthesis of kaolin-based zeolite and the effect of impurities, J. Chin. Chem. Soc., 2020, 67(6), 911–936 CrossRef CAS.
- Y. He, et al., Research progress on green synthesis of various high-purity zeolites from natural material-kaolin, J. Clean. Prod., 2021, 306, 127248 CrossRef CAS.
- Y. Ghrib, et al., Synthesis of cocrystallized USY/ZSM-5 zeolites from kaolin and its use as fluid catalytic cracking catalysts, Catal. Sci. Technol., 2018, 8(3), 716–725 RSC.
- A. A. Ajibola, J. A. Omoleye and V. E. Efeovbokhan, Catalytic cracking of polyethylene plastic waste using synthesised zeolite Y from Nigerian kaolin deposit, Appl. Petrochem. Res., 2018, 8, 211–217 CrossRef CAS.
- D. Liu, et al., Catalytic conversion of light alkanes to aromatics by metal-containing HZSM-5 zeolite catalysts—A review, Fuel Process. Technol., 2021, 216, 106770 CrossRef CAS.
- X. Sun, et al., On reaction pathways in the conversion of methanol to hydrocarbons on HZSM-5, J. Catal., 2014, 317, 185–197 CrossRef CAS.
- M. Guisnet and P. Magnoux, Organic chemistry of coke formation, Appl. Catal., A, 2001, 212(1–2), 83–96 CrossRef CAS.
- K. Wakui, et al., Cracking of n-butane over alkaline earth-containing HZSM-5 catalysts, Catal. Lett., 2002, 84, 259–264 CrossRef CAS.
- J. Zhao, et al., Synthesis and characterization of mesoporous zeolite Y by using block copolymers as templates, Chem. Eng. J., 2016, 284, 405–411 CrossRef CAS.
- J. Jin, et al., Facile synthesis of mesoporous zeolite Y with improved catalytic performance for heavy oil fluid catalytic cracking, Ind. Eng. Chem. Res., 2014, 53(8), 3406–3411 CrossRef CAS.
- J. García-Martínez, et al., Mesostructured zeolite Y—high hydrothermal stability and superior FCC catalytic performance, Catal. Sci. Technol., 2012, 2(5), 987–994 RSC.
- Z. Qin, et al., A defect-based strategy for the preparation of mesoporous zeolite Y for high-performance catalytic cracking, J. Catal., 2013, 298, 102–111 CrossRef CAS.
- M. Guisnet and P. Magnoux, Coking and deactivation of zeolites: influence of the pore structure, Appl. Catal., 1989, 54(1), 1–27 CrossRef CAS.
- P. Lu, et al., A stable zeolite with atomically ordered and interconnected mesopore channel, Nature, 2024, 636(8042), 368–373 CrossRef CAS PubMed.
- Z. R. Gao, et al., Interchain-expanded extra-large-pore zeolites, Nature, 2024, 628(8006), 99–103 CrossRef CAS PubMed.
- D. Zhang, et al., Catalytic dehydrogenation cracking of crude oil to light olefins by structure and basicity/acidity
adjustment of bifunctional metal/acid catalysts, Fuel, 2023, 334, 126808 CrossRef CAS.
- Q. Wu, Acidic and basic catalytic cracking technologies and its development prospects for crude oil to chemicals, Fuel, 2023, 332, 126132 CrossRef CAS.
- S. Wu, et al., Catalytic dehydrogenation cracking characteristics of vacuum residue over solid basic catalyst, Fuel, 2024, 368, 131578 CrossRef CAS.
- J. H. Song, et al., Catalytic cracking of n-hexane over MoO2, J. Mol. Catal. A: Chem., 2002, 184(1–2), 197–202 CrossRef CAS.
- B. Saha and A. Ghoshal, Model-free kinetics analysis of decomposition of polypropylene over Al-MCM-41, Thermochim. Acta, 2007, 460(1–2), 77–84 CrossRef CAS.
- D. Sangaré, et al., Comparative pyrolysis studies of lignocellulosic biomasses: Online gas quantification, kinetics triplets, and thermodynamic parameters of the process, Bioresour. Technol., 2022, 346, 126598 CrossRef PubMed.
- L. Liu, et al., Pyrolysis of Phragmites hirsuta study on pyrolysis characteristics, kinetic and thermodynamic analyses, Int. J. Energy Res., 2021, 45(10), 15200–15216 Search PubMed.
- Y. Zhang, et al., Kinetics, product evolution, and mechanism for the pyrolysis of typical plastic waste, ACS Sustain. Chem. Eng., 2021, 10(1), 91–103 CrossRef.
- S. Vyazovkin, et al., ICTAC Kinetics Committee recommendations for performing kinetic computations on thermal analysis data, Thermochim. Acta, 2011, 520(1–2), 1–19 Search PubMed.
- J. H. Flynn and L. A. Wall, A quick, direct method for the determination of activation energy from thermogravimetric data, J. Polym. Sci. B Polym. Lett., 1966, 4(5), 323–328 CrossRef CAS.
- H. E. Kissinger, Variation of peak temperature with heating rate in differential thermal analysis, J. Res. Natl. Bur. Stand., 1956, 57(4), 217–221 CrossRef CAS.
- M. Starink, A new method for the derivation of activation energies from experiments performed at constant heating rate, Thermochim. Acta, 1996, 288(1–2), 97–104 CrossRef CAS.
- W. Tang, et al., New approximate formula for Arrhenius temperature integral, Thermochim. Acta, 2003, 408(1–2), 39–43 CrossRef CAS.
- H. L. Friedman, Kinetics of thermal degradation of char-forming plastics from thermogravimetry. Application to a phenolic plastic, in Journal of Polymer Science Part C: Polymer Symposia, Wiley Online Library, 1964 Search PubMed.
- I. Mohan, et al., Screening of modified kaolin catalysts in the catalytic pyrolysis of waste high-density polyethylene: Kinetics and thermodynamic analysis, Artificial Neural Network prediction, batch pyrolysis study and characterization of liquid fuel, J. Environ. Chem. Eng., 2023, 11(5), 111039 Search PubMed.
- G. D. Mumbach, et al., Thermal investigation of plastic solid waste pyrolysis via the deconvolution technique using the asymmetric double sigmoidal function: Determination of the kinetic triplet, thermodynamic parameters, thermal lifetime and pyrolytic oil composition for clean energy recovery, Energy Convers. Manag., 2019, 200, 112031 CrossRef CAS.
- C.-T. Chen, et al., Reaction kinetics regulated formation of short-range order in an amorphous matrix during zeolite crystallization, J. Am. Chem. Soc., 2021, 143(29), 10986–10997 Search PubMed.
- M. Shakeri, Z. Khatami Shal and P. Van Der Voort, An Overview of the challenges and progress of synthesis, characterization and applications of plugged sba-15 materials for heterogeneous catalysis, Materials, 2021, 14(17), 5082 Search PubMed.
- E. M. Flanigen, H. Khatami, and H. A. Szymanski, Infrared Structural Studies of Zeolite Frameworks, ACS Publications, 1971 Search PubMed.
- Y. Huang, et al., Synthesis of hierarchical porous zeolite NaY particles with controllable particle sizes, Microporous Mesoporous Mater., 2010, 127(3), 167–175 CrossRef CAS.
- S. R. Batool, V. L. Sushkevich and J. A. van Bokhoven, Factors affecting the generation and catalytic activity of extra-framework aluminum Lewis acid sites in aluminum-exchanged zeolites, ACS Catal., 2024, 14(2), 678–690 CrossRef CAS.
- S. Kokuryo, et al., LDPE cracking over mono-and divalent metal-doped beta zeolites, Catal. Sci. Technol., 2022, 12(13), 4138–4144 RSC.
- M. Gao, et al., Research progress of basic catalyst used in catalytic cracking for olefin production and heavy oil utilization, Ind. Eng. Chem. Res., 2023, 62(3), 1215–1226 CrossRef CAS.
- G. Yang and J. Yu, Advancements in basic zeolites for biodiesel production via transesterification, Chemistry, 2023, 5(1), 438–451 CrossRef CAS.
- E. Rodríguez, et al., Fuel production by cracking of polyolefins pyrolysis waxes
under fluid catalytic cracking (FCC) operating conditions, Waste Manag., 2019, 93, 162–172 CrossRef PubMed.
- Y. Sun, et al., Coke formation over zeolite catalysts in light alkanes aromatization and anti-carbon-deposition strategies and perspectives: a review, Energy Fuels, 2023, 37(3), 1657–1677 CrossRef CAS.
- P. Negi, et al., Thermo-Kinetics of Chemical Recycling of Real-World Waste Plastics, ACS Sustain. Chem. Eng., 2023, 11(44), 15951–15963 CrossRef CAS.
- H. Liu, et al., Pyrolysis kinetics and thermodynamics of typical plastic waste, Energy Fuels, 2020, 34(2), 2385–2390 CrossRef CAS.
- K. Patidar, et al., Investigation of kinetic and thermodynamic parameters approaches to non-isothermal pyrolysis of mustard stalk using model-free and master plots methods, Mater. Sci. Energy Technol., 2022, 5, 6–14 CAS.
- S. Gupta, P. Patel and P. Mondal, Catalytic pyrolysis of pine needles using metal functionalized spent adsorbent derived catalysts: Kinetics, thermodynamics and prediction modelling using artificial neural network (ANN) approach, Ind. Crops Prod., 2024, 214, 118481 CrossRef CAS.
- B. Basu and D. Kunzru, Catalytic pyrolysis of naphtha, Ind. Eng. Chem. Res., 1992, 31(1), 146–155 CrossRef CAS.
- C. Chizallet, et al., Molecular views on mechanisms of Brønsted acid-catalyzed reactions in zeolites, Chem. Rev., 2023, 123(9), 6107–6196 CrossRef CAS PubMed.
- N. Sbirrazzuoli, Determination of pre-exponential factors and of the mathematical functions f (α) or G (α) that describe the reaction mechanism in a model-free way, Thermochim. Acta, 2013, 564, 59–69 CrossRef CAS.
- S. Gupta and P. Mondal, Catalytic pyrolysis of pine needles with nickel doped gamma-alumina: reaction kinetics, mechanism, thermodynamics and products analysis, J. Clean. Prod., 2021, 286, 124930 CrossRef CAS.
- S. Hooda, R. Lanjewar and P. Mondal, In-depth study of kinetics, thermodynamics, and reaction mechanism of catalytic pyrolysis of disposable face mask using spent adsorbent based catalysts, J. Energy Inst., 2023, 108, 101247 CrossRef CAS.
- D. Rammohan, N. Kishore and R. V. Uppaluri, Pyro–catalytic co–pyrolysis of Delonix regia and butyl rubber tube: Kinetic modelling and thermodynamic insights, Renewable Energy, 2022, 201, 194–203 CrossRef CAS.
- K. Wang, et al., Study on pyrolysis characteristics, kinetics and thermodynamics of waste tires catalytic pyrolysis with low-cost catalysts, Fuel, 2024, 356, 129644 CrossRef CAS.
- J. M. R. Silva, et al., Evaluation of the kinetic and thermodynamic parameters in catalytic pyrolysis process of sunflower oil using Al-MCM-41 and zeolite H-ZSM-5, Fuel, 2023, 333, 126225 CrossRef.
- Z. Yao, et al., Pyrolysis-gasification conversion of waste pharmaceutical blisters: Thermo-kinetic and thermodynamic study, fuel gas analysis and machine learning modeling, Chem. Eng. Sci., 2024, 120583 CrossRef CAS.
- I. Mohan, et al., Co-pyrolysis of Azadirachta indica non-edible seed and waste LDPE: analysis of kinetic models using thermogravimetric analyser and prediction modeling with Artificial Neural Network (ANN), Fuel, 2023, 350, 128765 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: The authors have procedure of synthesis and characterization of the materials, TGA and dTGA curves, details of gas and liquid oils analysis, and Py-IR procedure and the results. See DOI: https://doi.org/10.1039/d4ra08978b |
| ‡ Samira Motamadnejad and Li Gao contributed equally in this manuscript. |
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Reza Panahi
a,
Reza Panahi