
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFirst-principles investigation of thermoelectric performance in KMnZ (Z = Sn, Pb) half-Heusler alloys
Bharti Gurunani and
Dinesh C. Gupta*
and
Dinesh C. Gupta*
Condensed Matter Theory Group, School of Studies in Physics, Jiwaji University, Gwalior 474011, India. E-mail: bgurunani001@gmail.com; sosfizix@gmail.com
First published on 13th February 2025
Abstract
In this study, we performed an in-depth analysis of the structure stability, elasto-mechanical properties, thermophysical characteristics, and thermoelectric behavior of the KMnZ half-Heusler alloy using density functional theory (DFT) implemented through the WIEN2k simulation package. We examined the structural stability across phase types I, II, and III by optimizing their energy configurations. Our results demonstrate that the compound achieves optimal stability in the spin-polarized state of phase type II. Subsequently, we employ density functional perturbation theory (DFPT) to forecast the dynamic behavior of these structured systems. The electronic band structure analysis, conducted using the local spin density approximation (LDA), Perdew–Burke Generalized Gradient Approximation (PBE-GGA), and Tran–Blaha modified Becke–Johnson (TB-mBJ) schemes, reveals that the Heusler alloy exhibits half-metallic properties. Additionally, the calculated second-order elastic parameters confirm the material's ductile nature. To evaluate the thermodynamic and thermoelectric stability under various temperature and pressure conditions, we utilized the Quasi-Harmonic Debye model. The computed magnetic moment is consistent with the Slater-Pauling rule. These findings indicate that the KMnZ half-Heusler alloy is a promising candidate for applications in spintronics and thermoelectrics.
1. Introduction
In recent years, spintronic materials have garnered significant global attention due to their broad applicability across various advanced devices.1–4 As a rapidly evolving field, spintronics canters on the manipulation of electron spin properties to facilitate high-speed information processing and enhanced data storage capabilities.5 The integration of spintronic technology is increasingly becoming vital to the development of modern solid-state devices, which exploit both the spin and charge carrier mobility.6–9 Among the forefront of these technologies is the integration of spin injection and single-electron spin sources, which has emerged as a critical area of research. Spin-polarized charge carriers are fundamental to the advancement of contemporary spintronic devices.10–13 Heusler compounds, distinguished by their superior spin-polarized characteristics,14–17 have established themselves as leading candidates in this domain, enabling the creation of novel devices. The spin-polarized nature of these materials is essential for the efficient processing of large data sets and reliable information storage, underscoring their importance in the ongoing evolution of spintronic technologies.18–21 Heusler compounds are distinguished by their spin-dependent electronic properties, most notably their half-metallic behavior. This unique characteristic is defined by the material's metallic conduction in one spin channel (either spin-up or spin-down) while acting as a semiconductor in the opposite spin channel, resulting in 100% spin polarization.22,23 The extent of this spin polarization is typically determined by analyzing the density of states at the Fermi level (EF) for both spin channels.24Thermoelectricity (TE) represents a cutting-edge technology that harnesses the thermoelectric effect to convert heat into electric current and vice versa, providing an environmentally friendly energy solution. TE technology is particularly effective in capturing and repurposing waste heat from sources such as automobiles, refineries, and various industrial processes, thereby playing a crucial role in efforts to achieve zero carbon emissions. Heusler compounds have emerged as promising candidates for thermoelectric applications due to their cost-efficiency, ease of synthesis, environmental compatibility, and stable crystal structures. These characteristics position them at the forefront of thermoelectric materials research. Heusler compounds are generally categorized into three primary types based on their structural stoichiometry: half-Heusler (HH), full-Heusler (FH), and quaternary Heusler (QH) compounds.
Over the years, a substantial body of research, encompassing both theoretical and experimental approaches, has focused on unravelling the spintronic and thermoelectric properties of Heusler compounds.25–30 These materials have garnered significant attention due to their versatile electronic and magnetic characteristics, which are highly tunable through compositional and structural modifications. The pioneering work by Groot et al.31 marked a critical milestone in this field by experimentally confirming the half-metallic behavior of the NiMnSb half-Heusler compound. This discovery laid the foundation for subsequent investigations into similar materials. Shortly after, Fe2TiSb and Fe2TiAs were reported as half-metallic full Heusler compounds, further expanding the scope of research in this domain.32 The exploration of half-metallicity continued with Luo and colleagues, who conducted extensive theoretical studies on a series of full Heusler compounds, including Mn2FeGa, Mn2FeAl, Mn2FeGe, and Mn2FeSe.33 These studies provided deeper insights into the electronic structures and magnetic properties of these materials, highlighting their potential for spintronic applications. Among the broad spectrum of Heusler compounds, half-Heusler alloys have emerged as a particularly promising class of materials, distinguished by their unique structural attributes and exceptional performance in various applications.34 Transitioning from full Heusler (FH) to half-Heusler (HH) compounds introduces a shift in structural composition and chemical flexibility. Half-Heusler compounds typically adopt the formula XYZ, where X and Y are metallic atoms, often selected from transition metals or the alkaline earth metal family, while Z is typically an element from the sp-block. This structural configuration results in a C1b crystal structure, characterized by three interpenetrating face-centered cubic (FCC) lattices. The key distinction between half-Heusler and full Heusler compounds lies in the presence of a vacant site within the lattice of half-Heuslers, which significantly influences their electronic and magnetic properties. This structural feature not only differentiates them from their full Heusler counterparts but also contributes to their distinct physical properties, making them highly suitable for spintronic and thermoelectric applications.
With the multifaceted growth in spintronic applications, half-Heusler (HH) alloys have emerged as a versatile class of materials, offering significant potential for both spintronic and thermoelectric applications. Numerous HH alloys have been reported in the literature, including XCrSb (X = Fe, Ni),35 KCrZ (Z = Si, Ge),36 KCrZ (Z = S, Se, Te),37 NaZrZ (Z = P, Sb, As),38 LiMnZ (Z = N, Si, P),39 RhCrZ (Z = Ge, Si),40 PtXBi (X = Co, Fe, Mn, Ni),41 IrCrZ (Z = Ge, Sn, As, Sb),42 and VCoSb.43 These compounds are distinguished by their ability to achieve 100% spin polarization, classifying them as half-metallic ferromagnets (HMFs).44 HMFs are particularly valued for their wide-ranging applications in spintronic technologies, including spin injection,45 magnetic tunnel junctions (MTJs),46 spin valve devices,47 tunnelling magnetoresistance (TMR),48 magnetic random-access memory (MRAM),49 and permanent magnets.50 The appeal of HH alloys lies in their unique electronic structures, which enable them to exhibit half-metallic behavior, where one spin channel is metallic while the other is insulating. This characteristic makes them ideal candidates for spintronic devices that rely on high spin polarization. Despite the considerable progress made in identifying and synthesizing HH alloys with desirable properties, the ongoing quest for new half-metallic materials remains critical to advancing the efficiency and performance of spintronic and thermoelectric devices.
In particular, potassium (K)-based half-Heusler compounds have attracted attention due to their structural robustness and potential to exhibit half-metallic ferromagnetism. These materials offer a promising avenue for the fabrication of next-generation spintronic and thermoelectric devices. Their ability to maintain structural integrity while delivering high spin polarization and favorable thermoelectric properties positions them as strong candidates for further research and development. The exploration of these K-based HH compounds could lead to significant breakthroughs in the design and optimization of materials for advanced technological applications, reinforcing their role as a cornerstone in the evolution of spintronic and thermoelectric device technologies.
This research aims to provide a detailed analysis of the physical properties of KMnZ (Z = Sn, Pb) half-Heusler compounds, employing density functional theory (DFT) models integrated with the WIEN2k simulation package. The study emphasizes a multifaceted exploration of these compounds, including structural phase stability, half-metallic ferromagnetism (HMF), mechanical stability, magnetic properties, and thermodynamic behavior. The research objectives include determining the most stable magnetic configuration, evaluating half-metallic ferromagnetism, calculating second-order elastic constants, magnetic moments, and Curie temperatures, as well as investigating electronic properties using the PBE-GGA approximation within the DFT framework. Furthermore, the thermodynamic performance of the KMnZ compounds under varying temperature and pressure conditions will be rigorously analyzed using the BoltzTraP code and quasi-harmonic Debye approximation. This work seeks to deliver a comprehensive computational assessment of the fundamental properties of these half-Heusler compounds, with a particular focus on their potential applications in sustainable spintronics, electronics, thermoelectrics, and other emerging technologies.
2. Computational parameters and convergence criteria
In this study, density functional theory (DFT) was employed to conduct a comprehensive investigation into the structural stability, half-metallic behavior, magnetic properties, and thermodynamic response of the alkaline earth metal-based half-Heusler compound KMnZ. DFT, recognized for its precision and accuracy, was implemented using the full-potential linearized augmented plane wave (FP-LAPW) basis set within the WIEN2k simulation package.51–55 To analyze phonon dispersions and evaluate dynamical stability, we utilized density functional perturbation theory (DFPT) within the Quantum Espresso simulation framework.34 Electron–electron interactions were managed through the generalized gradient approximation with the Perdew–Burke–Ernzerhof (PBE) exchange-correlation potential.56 For a more detailed analysis of the electronic structure, the modified Becke–Johnson (mBJ) potential57 was utilized. Structural stability and magnetic properties of KMnZ were rigorously examined. The simulations involved partitioning the KMnZ unit cell into two distinct regions: the muffin-tin sphere and the interstitial space. The muffin-tin approximation, a well-established method for estimating electron energy states within the crystal lattice, was applied with muffin-tin radii (RMT) of 2.28, 2.31, 2.34, and 2.25 for K, Mn, Sn, and Pb atoms, respectively. Outside the muffin-tin spheres, electron wave functions were represented by plane waves with a cutoff defined as RMT × Kmax = 7, where Kmax denotes the reciprocal lattice vector. A fixed energy of −7.0 Ry was used to differentiate between core and valence electrons, and self-consistent field (SCF) convergence was achieved with a precision of 10−5 Ry. The Brillouin zone was sampled using the Monkhorst–Pack technique58 with a (14 × 14 × 14) k-point mesh. The elastic parameters of KMnZ were analyzed using the IRElast module within the WIEN2k package, and thermodynamic properties were investigated using the BoltzTrap2 code59 with the classical Boltzmann approximation.3. Result and discussions
In this section, we present a detailed analysis of the distinctive properties exhibited by KMnZ half-Heusler compounds, (Z = Sn, Pb).3.1 Crystal structure and optimised lattice parameter
To accurately predict the properties of any material, a thorough understanding of its crystal structure is essential, as the arrangement of atoms within a crystal lattice fundamentally influences its physical, electronic, and mechanical properties. Half-Heusler compounds, such as KMnZ, are particularly notable for their C1b-type crystal structure, which can stabilize in three distinct atomic configurations. These configurations are crucial because they dictate the material's behavior under various conditions, including temperature, pressure, and magnetic fields. The C1b-type structure of these half-Heusler compounds consists of three interpenetrating face-centered cubic (FCC) sublattices, where each sublattice is occupied by different atomic species. The three possible configurations, each defined by specific atomic coordinates and Wyckoff positions, represent different ways in which the constituent atoms (K, Mn, Sn and Pb) can be arranged within the crystal lattice. These variations in atomic positioning not only impact the stability of the crystal structure but also influence the electronic band structure, magnetic properties, and elastic characteristics of the material. Understanding these configurations allows researchers to predict and tailor the material's properties for specific applications, such as in spintronics, thermoelectrics, and other advanced technologies.The specific Wyckoff positions for these three configurations of the KMnZ half-Heusler compound are detailed in Table 1, providing a precise map of atomic locations within the lattice represented in Fig. 1, which is critical for further computational and experimental analysis.
| Alloys | Phase | K | Mn | Z = Sn, Pb |
|---|---|---|---|---|
| KMnZ | Type 1 | (0.25,0.25,0.25) | (0.50,0.50,0.50) | (0,0,0) |
| Type 2 | (0,0,0) | (0.25,0.25,0.25) | (0.50,0.50,0.50) | |
| Type 3 | (0.50,0.50,0.50) | (0,0,0) | (0.25,0.25,0.25) |
 | ||
| Fig. 1 Crystallographic structures of the KMnZ (Z = Sn, Pb) half-Heusler alloys, illustrating three distinct phase configurations: type I, type II, and type III respectively. | ||
The structure stability of the proposed half-Heusler (HH) compound is contingent upon its atomic arrangement within the crystal lattice. Consequently, identifying the most energetically favorable phase among the existing configurations is imperative. Our computational analysis and Fig. 2 reveals that the type I structure of KMnZ HH exhibits the lowest energy relative to types II and III. Subsequently, we determined that KMnZ with the F![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) 3m (#216) space group is more stable in the spin-polarized (SP) phase than in the non-magnetic (NM) phase as reported in Fig. 3. The antiferromagnetic (AFM) configuration of the KMnZ Heusler alloy was thoroughly analyzed to better understand its magnetic and structural properties. As part of this investigation, the relationship between the unit cell volume and the total energy for the AFM state was calculated and graphically represented, as shown in Fig. 4. This plot provides crucial insights into the stability of the material in its antiferromagnetic phase, indicating how energy varies with changes in lattice volume. To ascertain the equilibrium lattice parameters and minimum energy for cubic KMnZ with the stable type-I crystal structure, we employed the Murnaghan equation of state,60 as expressed in equation for volume optimization in the SP phase:
3m (#216) space group is more stable in the spin-polarized (SP) phase than in the non-magnetic (NM) phase as reported in Fig. 3. The antiferromagnetic (AFM) configuration of the KMnZ Heusler alloy was thoroughly analyzed to better understand its magnetic and structural properties. As part of this investigation, the relationship between the unit cell volume and the total energy for the AFM state was calculated and graphically represented, as shown in Fig. 4. This plot provides crucial insights into the stability of the material in its antiferromagnetic phase, indicating how energy varies with changes in lattice volume. To ascertain the equilibrium lattice parameters and minimum energy for cubic KMnZ with the stable type-I crystal structure, we employed the Murnaghan equation of state,60 as expressed in equation for volume optimization in the SP phase:

 | ||
| Fig. 2 Optimized energy–volume curve for the spin-polarized cubic KMnZ (Z = Sn, Pb) half-Heusler alloys in the type I, type II, and type III configuration. | ||
 | ||
| Fig. 3 Optimized energy–volume curve for the spin-polarized cubic KMnZ (Z = Sn, Pb) half-Heusler alloys in the spin-polarised (SP) and non-magnetic (NM) configuration. | ||
 | ||
| Fig. 4 Optimized energy–volume curve for the spin-polarized cubic KMnZ (Z = Sn, Pb) half-Heusler alloys in the anti-spin-polarised (antiferromagnetic AFM) configuration. | ||
| Alloys | Phase | a | V | B |
|
E0 |
|---|---|---|---|---|---|---|
| KMnSn | Type I | 7.49 | 542.43 | 27.80 | 4.37 | −15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 879.57 879.57 |
| Type II | 7.14 | 614.07 | 24.45 | 4.99 | −15879.69 |
|
| Type III | 7.35 | 554.62 | 26.73 | 4.74 | −15879.66 |
|
| NM | 6.69 | 507.18 | 38.99 | 4.57 | −15879.54 |
|
| AFM | 7.23 | 635.47 | 40.58 | 4.69 | −15879.67 |
|
| KMnPb | Type I | 7.58 | 739.65 | 23.98 | 4.72 | −45378.63 |
| Type II | 7.28 | 651.94 | 25.97 | 5.17 | −45378.73 |
|
| Type III | 7.48 | 707.06 | 29.57 | 5.03 | −45378.71 |
|
| NM | 7.12 | 711.56 | 29.12 | 5.31 | −45378.56 |
|
| AFM | 7.35 | 706.78 | 28.92 | 5.12 | −45378.64 |

| Ecoh = EKMnZtotal − (EKatom + EMnatom + EZatom) |
Cohesive energy represents the amount of energy necessary to separate the KMnZ (Z = Sn, Pb) crystal into its individual atoms, providing an indication of the material's bonding strength. From the data in Table 3, it is evident that all cohesive energy values are positive. This suggests that the bonds in both alloys are sufficiently robust to maintain the integrity of their atomic structures, emphasizing their structural stability. In summary, the positive cohesive energy values confirm the strong molecular structure and the cohesive forces that hold the atoms together in the KMnZ compounds.
| Alloys | ECoh | EFor | ΔH |
|---|---|---|---|
| KMnSn | 2.38 | −0.64 | −4.59 |
| KMnPb | 2.76 | −0.43 | −3.87 |
| EFor = EKMnZtotal − (EKbulk + EMnbulk + EZbulk) |
The formation energy, represented as ΔEFor, quantifies the difference between the total energy of a crystal EKMnZbulk and the summed energies of its elemental components EKbulk, EMnbulk, and EZbulk under standard conditions of 300 K temperature and 0 GPa pressure. A negative formation energy value indicates that the compound is thermodynamically stable. As shown in Table 3, both compounds exhibit negative formation energy values, demonstrating their potential for experimental synthesis. This highlights their favorable energetic properties and supports the possibility of synthesizing these materials under standard laboratory conditions.
| ΔH = ETotal − aEA − bEB − dEX |
 | ||
| Fig. 5 Phonon dispersion curves were calculated for KMnSn and KMnPb along the high-symmetry direction of their respective Brillouin zones. | ||
3.2 Mechanical and elastic properties
Elastic and mechanical properties of a material are characterized by its elastic parameters, which quantify the relationship between stress and strain. These parameters determine the material's deformation under external forces and its subsequent return to its original state. For assessing mechanical stability, elastic constants are crucial, particularly at optimized conditions. The mechanical attributes of solids, which underlie their practical applications, are directly linked to these elastic constants.49 Given that the KMnZ half-Heusler is stable in the cubic type II symmetry, we calculated the values of three independent elastic parameters: C11, C12, and C44. C11 measures longitudinal expansion, while C12 measures transverse deformation. To ensure mechanical stability, these independent elastic constants must satisfy the Born stability criteria for cubic symmetry, which are as follows:| C11 + 2C12 > 0; C11 − C12 > 0; C12 > 0; C11 > 0 and C44 > 0. |
As shown in Table 4, the KMnZ half-Heusler compound unequivocally satisfies the Born stability criteria, thereby confirming its robust mechanical stability. Furthermore, the table presents other pertinent parameters, including the shear modulus (G) and bulk modulus (B), which respectively characterize compressibility and stiffness. The resistance to plastic deformation, a measure of the opposing force to fracture, can be quantified by the G and B parameters. These values are calculated using the following equations:


| Parameters | KMnSn | KMnPb |
|---|---|---|
| C11 | 32.39 | 41.94 |
| C12 | 21.44 | 17.65 |
| C44 | 2.02 | 9.17 |
| B | 25.09 | 25.74 |
| GV | 3.40 | 10.36 |
| GR | 2.70 | 10.16 |
| G | 3.05 | 10.26 |
| E | 8.79 | 27.17 |
| CP | 19.42 | 8.48 |
| B/G | 8.22 | 2.50 |
| σ | 0.44 | 0.32 |
| A | 0.36 | 0.75 |
| Vl (m s−1) | 3.16 | 10.07 |
| Vt (m s−1) | 1.02 | 5.14 |
| Vm (m s−1) | 1.16 | 5.76 |
| θD | 121.18 | 232 |
The subscripts R and V in above equation denote the Reuss and Voigt bounds, respectively. The values of GR and BV can be determined using the equations below:

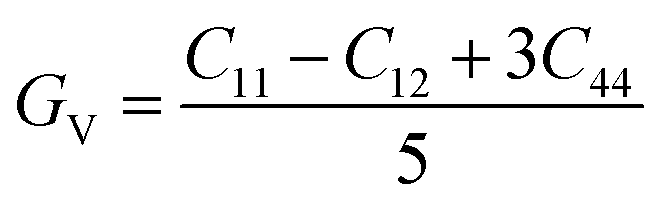
Table 4 summarizes the calculated values of these elastic parameters. The calculated results not only confirm the mechanical stability of the KMnZ compound but also provide valuable insights into its resistance to deformation and its potential applications in various structural and engineering contexts. The bulk modulus (B) value derived from above bulk modulus eqn, as presented in Table 4, is consistent with the values obtained through our calculations. Young's modulus (E), which defines the material's stiffness and is the ratio of stress to strain, was determined using the following relationship:

This relation connects the bulk modulus (B) and shear modulus (G) to provide insight into the material's elastic properties. To evaluate the material's brittleness or ductility, the Cauchy pressure (CP) is a crucial factor. The Cauchy pressure is calculated using the following relation:
| CP = C11 − C12 |
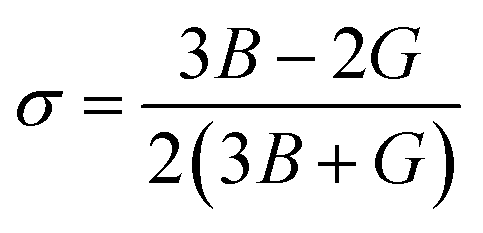
A higher Poisson's ratio typically indicates a material that is more compressible and has more ductile behavior. However, for the KMnZ alloy, the Poisson's ratio suggests that the material is relatively incompressible, supporting the conclusion that it is mechanically stable and ductile. From the exploration of these elastic properties, it can be concluded that the KMnZ HH alloy exhibits brittleness and incompressibility, characteristics that affirm its mechanical stability. These properties are essential for the practical use of the alloy, particularly in applications requiring high structural integrity under stress. Given the growing interest in half-Heusler (HH) alloys for potential spintronic applications, a thorough understanding of their mechanical stability is crucial during the device fabrication process.
The Debye temperature (θD) is a fundamental property of solid materials, representing the temperature at which the vibrational energy of atoms within the material equals their thermal energy. It is calculated using the average sound velocity (vm)42


Utilizing Navier's equation41 and the calculated bulk and shear moduli, we determined the longitudinal velocity (vl) and shear velocity (vs). The resulting values are tabulated in Table 4.
The Debye temperatures for KMnSn and KMnPb alloys were found to be 121.18 K and 232 K, respectively. When a material's Debye temperature exceeds room temperature, it signifies that the vibrational energy of its constituent atoms surpasses its thermal energy.
Another significant physical property is the anisotropic factor (A), which characterizes the nature of bonding in different crystallographic directions and is defined by:

A value of A = 1 indicates an isotropic crystal, while values less than one suggest elastic anisotropy. The computed data in Table 4 reveals that both half-Heusler compounds exhibit anisotropic behavior.
3.3 Electronic properties
To investigate the electronic properties of half-Heusler alloys KMnZ (Z = Sn, Pb), we calculated spin-polarized electronic band structures at the equilibrium lattice constant in the ferromagnetic (FM) phase. These calculations, performed along the high-symmetry directions of the first Brillouin zone (BZ), used LDA, GGA-PBE, and TB-mBJ, approximations at 0 K and 0 GPa. The spin-resolved band structures (BS) and density of states (DOS), shown in Fig. 6–10, provide essential insights into the electronic behavior. These results reveal how spin-up and spin-down states contribute to the electronic structure, highlighting the material's half-metallic and semiconducting characteristics. Such findings are crucial for spintronic applications, where controlling spin-polarized currents is key. | ||
| Fig. 6 Spin polarised band structure for KMnSn and KMnPb in LDA approximation. | ||
 | ||
| Fig. 7 Spin polarised band structure for KMnSn and KMnPb in GGA approximation. | ||
 | ||
| Fig. 8 Spin polarised band structure for KMnSn and KMnPb in mBJ approximation. | ||
 | ||
| Fig. 9 Spin-polarized TDOS of KMnZ (Z = Sn, Pb) HH alloys by LDA, GGA, and mBJ approximation. | ||
 | ||
| Fig. 10 Spin-polarized PDOS of KMnZ (Z = Sn, Pb) HH alloys by mBJ approximation. | ||
3.4 Magnetic properties
According to the Galanakis model42 the spin magnetic moment of half-Heusler compounds is defined as the difference between the occupied energy bands corresponding to the up and down spin states. This relationship is particularly pertinent for ternary 1:1:1 half-Heusler compounds characterized by a C1b crystal structure, such as the prototype Cu2MnAl. Kubler was notably the first to establish the Slater-Pauling 18-electron rule, which articulates that the total magnetic moment (MTot) is given by the equation:| MTot = 8 − ZTot |
:1:1 half-Heusler alloys, we have determined a total magnetic moment (MTot) of approximately 4 μB. Our findings are summarized in Table 5, which outlines the total magnetic moment alongside contributions from atomic resolved and interstitial magnetic moments specific to the compounds KMnZ (Z = Sn, Pb). Significantly, our analysis reveals that the predominant contributions to the total magnetic moment arise from the manganese (Mn) atom in these half-Heusler compounds. This observation underscores the critical influence of the Mn atom in shaping the magnetic properties of the material, aligning well with the predictions made by the Slater-Pauling rule.
| Alloys | Methods | K | Mn | Sn/Pb | Int. | Total | Band gap |
|---|---|---|---|---|---|---|---|
| KMnSn | LDA | 0.05 | 3.79 | −0.10 | 0.17 | 3.91 | 0.55 |
| GGA | 0.05 | 3.91 | −0.13 | 0.15 | 3.98 | 0.72 | |
| mBJ | 0.04 | 4.06 | −0.19 | 0.08 | 4.00 | 1.09 | |
| KMnPb | LDA | 0.01 | 4.11 | −0.14 | 0.01 | 4.00 | 0.56 |
| GGA | 0.01 | 4.21 | −0.17 | −0.05 | 4.00 | 0.73 | |
| mBJ | 0.01 | 4.33 | −0.20 | −0.13 | 3.99 | 1.25 |
3.5 Curie temperature
The Curie temperature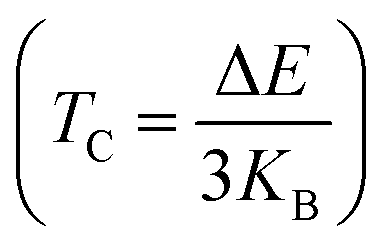 for the alloys KCrSi and KCrGe was determined using a well-established equation derived from the Heisenberg model,.43 In this context, ΔE represents the energy difference between ENM (the total energy of the nonmagnetic state) and EFM (the total energy of the ferromagnetic state). This parameter captures the energy gap associated with the stability of magnetic ordering in the material. The calculation also incorporates the Boltzmann constant (KB), which connects the energy to the thermal properties of the system. The parameter ΔE is a fundamental quantity that reflects the minimum energy barrier required for a system to transition between states, such as magnetic phases or other physical transformations. This energy difference provides significant insights into the thermodynamic stability and kinetic feasibility of these transitions. Specifically, it quantifies the degree of stability of the ferromagnetic state relative to the nonmagnetic state. At the Curie temperature (TC), the thermal energy in the material becomes comparable to ΔE, enabling the disruption of the ferromagnetic spin alignment. This marks a critical point where the material undergoes a phase transition from a ferromagnetic to a paramagnetic state. Consequently, ΔE plays a central role in determining the Curie temperature and, by extension, the thermal stability of the magnetic ordering. It defines the threshold energy at which magnetic interactions are overcome by thermal agitation, providing an essential parameter for evaluating the magnetic behavior of the material under varying thermal conditions. The Curie temperature not only indicates the temperature at which a material loses its magnetic ordering but also serves as a benchmark for the thermal stability of ferromagnetic phases. For the alloys studied, the predicted values of TC are 814.89 K for KMnSn and 821.42 K for KMnPb. These results highlight the robustness of the ferromagnetic state in these materials and emphasize the influence of ΔE on their magnetic and thermal properties. Furthermore, this analysis underscores the significance of understanding the interplay between the energy states of a material and its magnetic behavior. The ability to estimate TC from first principles, using the Heisenberg model and energy state calculations, offers a reliable framework for predicting and analyzing the performance of magnetic materials in applications requiring thermal stability, such as spintronics and thermoelectric devices. By correlating ΔE with TC, this approach provides valuable insights into the material's ability to maintain ferromagnetic order under varying temperature conditions.
for the alloys KCrSi and KCrGe was determined using a well-established equation derived from the Heisenberg model,.43 In this context, ΔE represents the energy difference between ENM (the total energy of the nonmagnetic state) and EFM (the total energy of the ferromagnetic state). This parameter captures the energy gap associated with the stability of magnetic ordering in the material. The calculation also incorporates the Boltzmann constant (KB), which connects the energy to the thermal properties of the system. The parameter ΔE is a fundamental quantity that reflects the minimum energy barrier required for a system to transition between states, such as magnetic phases or other physical transformations. This energy difference provides significant insights into the thermodynamic stability and kinetic feasibility of these transitions. Specifically, it quantifies the degree of stability of the ferromagnetic state relative to the nonmagnetic state. At the Curie temperature (TC), the thermal energy in the material becomes comparable to ΔE, enabling the disruption of the ferromagnetic spin alignment. This marks a critical point where the material undergoes a phase transition from a ferromagnetic to a paramagnetic state. Consequently, ΔE plays a central role in determining the Curie temperature and, by extension, the thermal stability of the magnetic ordering. It defines the threshold energy at which magnetic interactions are overcome by thermal agitation, providing an essential parameter for evaluating the magnetic behavior of the material under varying thermal conditions. The Curie temperature not only indicates the temperature at which a material loses its magnetic ordering but also serves as a benchmark for the thermal stability of ferromagnetic phases. For the alloys studied, the predicted values of TC are 814.89 K for KMnSn and 821.42 K for KMnPb. These results highlight the robustness of the ferromagnetic state in these materials and emphasize the influence of ΔE on their magnetic and thermal properties. Furthermore, this analysis underscores the significance of understanding the interplay between the energy states of a material and its magnetic behavior. The ability to estimate TC from first principles, using the Heisenberg model and energy state calculations, offers a reliable framework for predicting and analyzing the performance of magnetic materials in applications requiring thermal stability, such as spintronics and thermoelectric devices. By correlating ΔE with TC, this approach provides valuable insights into the material's ability to maintain ferromagnetic order under varying temperature conditions.
3.6 Thermodynamics properties
The calculation of thermodynamic properties is essential for understanding the behavior of materials under conditions of pressure and temperature that differ from their standard state. This enables the prediction of thermal, mechanical, and chemical stability across a range of environments. Theoretical modeling of these properties provides critical insights, serving as key indicators for assessing the potential industrial scalability of such compounds. In this study, the quasi-harmonic Debye model, as implemented in the Gibbs2 program,25 is employed to predict the thermodynamic behavior of Heusler KMnZ (Z: Sn and Pb) alloys as a function of temperature. The properties are evaluated over a temperature range of 0–900 K and at pressures of 0, 5, 10, and 15 GPa, within the framework of the GGA approximation.The heat capacity at constant volume (Cv) is a fundamental thermodynamic property that measures a material's ability to absorb heat without experiencing changes in volume. This property is deeply linked to the vibrational behavior of a material's atomic structure, influencing its thermal, mechanical, and electronic characteristics. A thorough understanding of these vibrational properties is essential for optimizing materials used in a wide range of applications, including thermoelectric devices, superconductors, and high-temperature technologies. Fig. 11(a) illustrates the evolution of CV with temperature for KMnSn and KMnPb alloys, highlighting key trends in their thermal responses. At temperatures below 300 K, CV exhibits a strong dependence on both temperature and pressure, following a T3 relationship in accordance with the Debye T3 law.37 This behavior, typical of low-temperature crystalline solids, suggests that the heat capacity is primarily governed by acoustic phonon vibrations. As temperature rises above 300 K, CV increases more slowly, approaching the Dulong–Petit limit,38 which represents the maximum possible contribution of atomic vibrations to heat capacity at high temperatures. Furthermore, an inverse relationship between CV and pressure is observed at constant temperatures, indicating that increasing pressure reduces the material's ability to absorb thermal energy. This suggests that pressure restricts atomic vibrations, diminishing the overall heat capacity. The results also indicate that temperature has a more significant influence on CV than pressure, highlighting the dominant role of thermal excitations in determining a material's thermal behavior. Understanding these interactions is crucial for predicting the performance of KMnSn and KMnPb alloys under various operational conditions in industrial applications.
 | ||
| Fig. 11 (a) Specific heat as a function of temperature for different pressure values for KMnZ (Z = Sn, Pb). (b) Bulk modulus as a function of temperature for different pressure values for KMnZ (Z = Sn, Pb). (c) Thermal expansion coefficient as a function of temperature for different pressure values for KMnZ (Z = Sn, Pb). (d) Thermal expansion coefficient as a function of temperature for different pressure values for KMnZ (Z = Sin, Pb). | ||
The variation of the bulk modulus (B) with temperature exhibits a nearly identical trend for both KMnSn and KMnPb alloys, as depicted in Fig. 11(b). The data consistently show that B decreases linearly with increasing temperature, a pattern observed across all applied pressure conditions. This linear reduction indicates that the material becomes more compressible as temperature rises, a behavior typically associated with thermal expansion and the weakening of atomic bonds at elevated temperatures. Moreover, the results demonstrate that at any specific temperature, the bulk modulus increases proportionally with pressure, signifying reduced compressibility as pressure rises. This suggests that higher pressures enhance the material's resistance to deformation by reinforcing atomic cohesion, while increasing temperature has the opposite effect, making the material more compressible. The contrasting influence of temperature and pressure on the bulk modulus underscores their opposing roles in shaping the material's mechanical properties—pressure strengthens atomic interactions, whereas temperature diminishes them. The values of the bulk modulus at 0 K and 0 GPa closely match those obtained from the Birch–Murnaghan equation of state and are consistent with those derived from the elastic constants. This alignment confirms the accuracy of the theoretical modeling employed in the study, reinforcing confidence in the calculated mechanical properties. The findings highlight the significance of comprehending the combined effects of temperature and pressure when assessing the mechanical stability and compressibility of these alloys, which is crucial for their potential industrial and technological applications.
The thermal expansion coefficient (α) characterizes how a material's dimensions respond to temperature fluctuations, providing critical insight into its structural stability under thermal stress. Fig. 11(c) illustrates the variation of α as a function of temperature and pressure for the half-Heusler (HH) alloys KMnZ (Z = Sn, Pb) within a temperature range of 0 to 1000 K and pressure levels of 0, 5, 10, and 15 GPa. The data show that at lower temperatures, particularly below 300 K, α increases sharply as temperature rises, indicating rapid dimensional expansion driven by intensified atomic vibrations as thermal energy accumulates. As the temperature exceeds 300 K, the rate of increase in α becomes more gradual, suggesting that the material approaches a thermal equilibrium where expansion is less pronounced. This behavior is characteristic of many solids, as the thermal expansion rate tends to stabilize at higher temperatures once atomic vibrations reach a maximum amplitude. The shift from rapid to moderate growth in α reflects the material's nuanced response to temperature changes across different thermal regimes, with the lower range displaying more dynamic expansion behavior. Additionally, at constant temperatures, α exhibits a linear decrease as pressure increases, highlighting an inverse relationship between thermal expansion and pressure. This suggests that increasing pressure suppresses atomic vibrations, thereby limiting the material's capacity to expand under thermal stress. The reduction in α with rising pressure underscores the stabilizing effect of compressive forces on material dimensions. Understanding the combined impact of temperature and pressure on α is crucial for predicting the behavior of KMnSn and KMnPb alloys in applications requiring high-dimensional stability across varied operational environments.
The Grüneisen parameter (γ) is a key thermodynamic quantity that provides critical insight into the behavior of phonon frequencies in response to variations in crystal volume. It is instrumental in assessing the anharmonic properties of a material, as it directly correlates with the degree of anharmonicity inherent in the system. Specifically, γ serves to quantify how phonons—quanta of lattice vibrations—are influenced by changes in both temperature and pressure. In this work, the dependence of the Grüneisen parameter on temperature and pressure is illustrated in Fig. 11(d). At lower temperatures, the alloys exhibit an exponentially decreasing trend in γ, indicating a significant sensitivity of phonon frequencies to anharmonic effects as temperature increases from low values. This decline in γ suggests that as temperature rises, anharmonic interactions grow stronger, leading to a reduction in phonon stability. However, at higher temperatures, the Grüneisen parameter plateaus, reflecting a stabilization of anharmonicity where further increases in temperature have little to no impact on phonon behavior. Conversely, the influence of pressure on the Grüneisen parameter is markedly subdued. As depicted in Fig. 11(d), γ shows minimal variation under increasing pressure, suggesting that pressure has a negligible effect on the phonon dynamics of these alloys when compared to temperature. This contrast highlights the dominance of thermal effects over pressure in governing the anharmonic behavior of the material. Thus, the temperature effect far outweighs the pressure effect, indicating that thermal expansion and vibrational properties are more strongly governed by temperature variations.
3.7 Thermoelectric properties
Utilizing the WIEN2k software and the BoltzTrap code, we conducted a comprehensive analysis of the thermoelectric properties of KMnZ (Z = Sn, Pb) compounds. BoltzTrap enabled the accurate calculation of the Seebeck coefficient (S), electrical conductivity (σ), power factor (PF), thermal conductivity (κ), and figure of merit (ZT), for both spin-up and spin-down channels over a temperature range of 50 K to 900 K. To ensure computational accuracy, a dense k-mesh grid of 50 × 50 × 50 was employed in conjunction with the mBJ approximation. This rigorous approach provides a solid foundation for understanding the thermoelectric performance of these alloys and their potential applications.
This property is intimately linked to the material's electronic structure near the Fermi level. As depicted in Fig. 12(a), KMnZ (Z = Sn, Pb) alloys exhibit a pronounced positive Seebeck coefficient in the spin-up state at room temperature, characteristic of p-type semiconductors. Specifically, KMnSn and KMnPb display Seebeck coefficients of 163.20 μV K−1 and 159.09 μV K−1, respectively. Conversely, the spin-down channel of these alloys demonstrates a decreasing Seebeck coefficient with increasing temperature, indicative of semiconductor behavior. At room temperature, KMnSn and KMnPb in the spin-down state exhibit Seebeck coefficients of 396.17 μV K−1 and 452.76 μV K−1, respectively. These findings underscore the distinct thermoelectric properties of these alloys and their potential for applications in energy conversion and management.
 | ||
| Fig. 12 (a) Variation of the Seebeck coefficient with temperature for KMnZ (Z = Sn, Pb) half-Heusler alloys in both spin channels. (b) Variation of the electrical conductivity with temperature for KMnZ (Z = Sn, Pb) half-Heusler alloys in both spin channels. (c) Variation of the thermal conductivity (κ) with temperature for KMnZ (Z = Sn, Pb) half-Heusler alloys. (d) Variation of the Power factor (PF) and figure of merit (ZT) with temperature for KMnZ (Z = Sn, Pb) half-Heusler alloys. | ||
To achieve reduced thermal conductivity, we employ Slack's model, which emphasizes the critical role of acoustic phonon modes in heat transport in semiconductors.52 Slack's model is mathematically represented as:60

4. Conclusion
In this work, we employ the FP-LAPW method within the density functional theory (DFT) framework to conduct an in-depth analysis of the half-Heusler (HH) alloys KMnZ (Z = Sn, Pb). Our results identify the type II configuration as the most energetically favorable, with these alloys exhibiting half-metallic ferromagnetic (HM-FM) characteristics and band gaps of 1.09 eV for KMnSn and 1.25 eV for KMnPb. The half-metallic nature and complete spin polarization, as evidenced by DOS and band structure analyses, underscore their suitability for spintronic applications. The calculated total magnetic moments (MM) of the KMnZ alloys align closely with established theoretical values, validating the accuracy of our approach. Mechanical property assessments confirm the alloys' stability and ductility, making them viable for engineering and industrial purposes. Furthermore, transport property evaluations indicate a ZT value of 1, highlighting their potential for thermoelectric applications, specifically in converting waste heat to electricity. Thermal stability analyses also confirm their robustness under extreme conditions, supporting their applicability in practical scenarios. Collectively, these findings position HH KMnZ alloys as promising materials for advanced technological applications, particularly in spintronic heterostructures, energy harvesting, and other next-generation devices, given their comprehensive electronic, mechanical, and thermal attributes.Data availability
Data will be made are available from the corresponding author upon reasonable request.Author contributions
Bharti Gurunani: writing – review & editing, writing – original draft, methodology. Dinesh C. Gupta: supervision, software, writing – review & editing.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
Any agency does not fund the research work.References
- T. Ghellab, H. Baaziz, Z. Charifi, H. Latelli, M. J. Ahmad, M. Telfah, A. Alsaad, A. Telfah, R. Hergenröder and R. Sabirianov, First-principles calculations of the high-pressure behavior, electronic, magnetic, and elastic properties of praseodymium pnictides: PrX (X= P, As and Bi), J. Magn. Magn. Mater., 2022, 546, 168919 CrossRef.
- X. P. Wei, Y. D. Chu, X. W. Sun, J. B. Deng and Y. Z. Xing, Stability, electronic, magnetic and pressure effect of half-Heusler alloys CNaCa and SiNaCa: a first-principles study, Superlattices Microstruct., 2014, 74, 70–77 CrossRef CAS.
- J. F. Gregg, R. P. Borges, E. Jouguelet, C. L. Dennis, I. Petej, S. M. Thompson and K. Ounadjela, Spin injection efficiency in spin electronic devices, J. Magn. Magn. Mater., 2003, 265(3), 274–289 CrossRef CAS.
- A. Kumar, S. Singh, S. A. Sofi, T. Chandel and N. Thakur, Robustness in half-metallicity, thermophysical and structural properties of Co2YAl (Y= Pd, Ag) Heuslers: a first-principles perspective, Mol. Phys., 2022, 120(18), e2120839 CrossRef.
- Z. Bai, L. E. Shen, G. Han and Y. P. Feng, Data storage: review of Heusler compounds, Spin, 2012, 2, 1230006 CrossRef.
- V. Mishra, V. Barwal, L. Pandey, N. K. Gupta, S. Hait, A. Kumar, N. Sharma, N. Kumar and S. Chaudhary, Investigation of spin gapless semiconducting behaviour in quaternary CoFeMnSi Heusler alloy thin films on Si (1 0 0), J. Magn. Magn. Mater., 2022, 547, 168837 CrossRef CAS.
- S. Dubey, K. Dubey, V. Sahu, A. Modi, R. A. Parry, G. Pagare and N. K. Gaur, Structural, electronic and elastic properties of FeCrAs Half-metallic ferromagnetic Half-Heusler Alloy: A First-Principles study, Mater. Today: Proc., 2022, 67, 12–19 CAS.
- I. Galanakis and P. Mavropoulos, Spin-polarization and electronic properties of half-metallic Heusler alloys calculated from first principles, J. Phys.: Condens. Matter, 2007, 19(31), 315213 CrossRef CAS PubMed.
- N. A. Ganie, S. A. Mir and D. C. Gupta, Tantalum half-Heusler alloys RbTaSi and RbTaGe: potential candidates for desirable thermoelectric and spintronic applications, RSC Adv., 2023, 13(11), 7087–7101 RSC.
- E. Gürbüz, M. Tas, E. Şaşıoğlu, I. Mertig, B. Sanyal and I. Galanakis, First-principles prediction of energy bandgaps in 18-valence electron semiconducting half-Heusler compounds: Exploring the role of exchange and correlation, J. Appl. Phys., 2023, 134(20), 1–10 CrossRef.
- A. O. Moghaddam, R. Fereidonnejad, M. Naseri, N. Shaburova, D. Mikhailov, S. Uporov and E. Trofimov, Synthesis and characterization of novel high entropy Heusler intermetallics, Intermetallics, 2023, 159, 107917 CrossRef.
- M. Salaheldeen, A. Wederni, M. Ipatov, V. Zhukova and A. Zhukov, Preparation and Magneto-Structural Investigation of High-Ordered (L21 Structure) Co2MnGe Microwires, Processes, 2023, 11(4), 1138 CrossRef CAS.
- A. Kumar, T. Chandel and T. N. Diwaker, Predicting the magnetism, structural, thermodynamic and electronic properties of new co-based Heuslers: first principle perspective, Philos. Mag., 2020, 100(21), 2721–2734 CrossRef CAS.
- K. Elphick, W. Frost, M. Samiepour, T. Kubota, K. Takanashi, H. Sukegawa, S. Mitani and A. Hirohata, Heusler alloys for spintronic devices: review on recent development and future perspectives, Sci. Technol. Adv. Mater., 2021, 22(1), 235–271 CrossRef CAS PubMed.
- S. Kumar, S. L. Gupta, D. Kumar and K. S. Vinayak, Structural properties of novel potential spintronic material and Full-Heusler alloy Co2NbGe: First Principles investigation, Mater. Today: Proc., 2022, 1–12 Search PubMed.
- J. D. D. Enamullah, K. G. Suresh and A. Alam, Half-metallic Co-based quaternary Heusler alloys for spintronics: Defect-and pressure-induced transitions and properties, Phys. Rev. B, 2016, 94(18), 184102 CrossRef.
- C. J. Palmstrøm, Heusler compounds and spintronics, Prog. Cryst. Growth Charact. Mater., 2016, 62(2), 371–397 CrossRef.
- A. Candan, A study on magnetic, electronic, elastic and vibrational properties of Ir2MnAl Heusler alloy for spintronic applications, Mater. Res. Express, 2019, 6(9), 096571 CrossRef CAS.
- K. Hu, R. Xie, C. Shen, H. Peng, H. Liu and H. Zhang, High-throughput design of Co-based magnetic Heusler compounds, Acta Mater., 2023, 259, 119255 CrossRef CAS.
- S. Tavares, K. Yang and M. A. Meyers, Heusler alloys: Past, properties, new alloys, and prospects, Prog. Mater. Sci., 2023, 132, 101017 CrossRef CAS.
- S. Parkin, Magnetic Race-Track—a Novel Storage Class Spintronic Memory, Int. J. Mod. Phys. B, 2008, 22(01n02), 117–118 CrossRef.
- S. Yousuf and D. C. Gupta, Insight into half-metallicity, spin-polarization and mechanical properties of L21 structured MnY2Z (Z= Al, Si, Ga, Ge, Sn, Sb) Heusler alloys, J. Alloys Compd., 2018, 735, 1245–1252 CrossRef CAS.
- B. Hillebrands and C. Felser, High-spin polarization of Heusler alloys, J. Phys. D: Appl. Phys., 2006, 39(5), E01 CrossRef.
- A. Kumar, T. Chandel and N. Thakur, Robust stability, half metallic Ferromagnetism and structural properties of Co2RhSi, and Co2RuSi Heusler compounds-A first principles approach, Mater. Today: Proc., 2022, 66, 3292–3297 CAS.
- S. L. Gupta, S. Kumar, T. S. S. Anupam and S. Panwar, Diwaker. A newly proposed full Heusler alloy Ir2VZ (Z= Sn, In) suitable for high-temperature thermoelectric applications: A DFT approach, Mod. Phys. Lett. B, 2024, 38(09), 2450030 CrossRef CAS.
- Q. Gao, I. Opahle and H. Zhang, High-throughput screening for spin-gapless semiconductors in quaternary Heusler compounds, Phys. Rev. Mater., 2019, 3(2), 024410 CrossRef CAS.
- B. Gurunani and D. C. Gupta, Tailoring the intrinsic magneto-electronic, mechanical, thermo-physical and thermoelectric response of cobalt-based Heusler alloys: an ab initio insight, RSC Adv., 2023, 13(43), 29959–29974 RSC.
- A. A. Santos and P. D. Borges, Ab initio investigation of Co–Ta–Sn Heusler alloys for thermoelectric applications, Comput. Theor. Chem., 2023, 1229, 114301 CrossRef CAS.
- H. Alam and S. Ramakrishna, A, review on the enhancement of figure of merit from bulk to nano-thermoelectric materials, Nano energy, 2013, 2(2), 190–212 CrossRef CAS.
- S. A. Khandy, I. Islam, D. C. Gupta and A. Laref, Full Heusler alloys (Co2TaSi and Co2TaGe) as potential spintronic materials with tunable band profiles, J. Solid State Chem., 2019, 270, 173–179 CrossRef CAS.
- R. A. De Groot, F. M. Mueller, P. V. van Engen and K. H. Buschow, New class of materials: half-metallic ferromagnets, Phys. Rev. Lett., 1983, 50(25), 2024 CrossRef CAS.
- H. Luo, G. Liu, F. Meng, J. Li, E. Liu and G. Wu, Half-metallicity in Fe-based Heusler alloys Fe2TiZ (Z= Ga, Ge, As, In, Sn and Sb), J. Magn. Magn. Mater., 2012, 324(20), 3295–3299 CrossRef CAS.
- H. Z. Luo, H. W. Zhang, Z. Y. Zhu, L. Ma, S. F. Xu, G. H. Wu, X. X. Zhu, C. B. Jiang and H. B. Xu, Half-metallic properties for the Mn2FeZ (Z= Al, Ga, Si, Ge, Sb) Heusler alloys: A first-principles study, J. Appl. Phys., 2008, 103(8), 1–9 CrossRef.
- P. Giannozzi, S. Baroni, N. Bonini, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, G. L. Chiarotti, M. Cococcioni, I. Dabo and A. Dal Corso, QUANTUM ESPRESSO: a modular and open-source software project for quantum simulations of materials, J. Phys.:Condens. Matter, 2009, 21(39), 395502 CrossRef PubMed.
- N. O. Nenuwe and E. Omugbe, Electronic properties of half-Heusler compounds XCrSb (X= Fe, Ru, Os): Potential applications as spintronics and high-performance thermoelectric materials, Curr. Appl. Phys., 2023, 49, 70–77 CrossRef.
- B. Gurunani, S. Ghosh and D. C. Gupta, Comprehensive investigation of half Heusler alloy: Unveiling structural, electronic, magnetic, mechanical, thermodynamic, and transport properties, Intermetallics, 2024, 170, 108311 CrossRef CAS.
- A. Kumar, S. L. Gupta, S. Kumar and V. Kumar, Spin-polarized DFT calculations for physical properties of novel KVSb half-Heusler compound for spintronic and thermodynamic applicability, arXiv, 2024, preprint, arXiv:2406.06656, DOI:10.48550/arXiv.2406.06656.
- M. Moradi, N. Taheri and M. Rostami, Structural, electronic, magnetic and vibrational properties of half-Heusler NaZrZ (Z= P, As, Sb) compounds, Phys. Lett. A, 2018, 382(41), 3004–3011 CrossRef CAS.
- L. Damewood, B. Busemeyer, M. Shaughnessy, C. Y. Fong, L. H. Yang and C. Felser, Stabilizing and increasing the magnetic moment of half-metals: The role of Li in half-Heusler LiMn Z (Z= N, P, Si), Phys. Rev. B, 2015, 91(6), 064409 CrossRef.
- R. Ahmad and N. Mehmood, Investigation of Half-Heusler Compounds RhCrZ (Z= P, As, Sb, Sn): A First Principle Study, J. Supercond. Novel Magn., 2018, 31, 2637–2645 CrossRef CAS.
- W. Huang, X. Wang, X. Chen, W. Lu, L. Damewood and C. Y. Fong, Structural and electronic properties of half-Heusler alloys PtXBi (with X= Mn, Fe, Co and Ni) calculated from first principles, J. Magn. Magn. Mater., 2015, 377, 252–258 CrossRef CAS.
- M. A. Behbahani, M. Moradi, M. Rostami and S. Davatolhagh, First principle study of structural, electronic and magnetic properties of half-Heusler IrCrZ (Z= Ge, As, sn and sb) compounds, J. Phys. Chem. Solids, 2016, 92, 85–93 CrossRef.
- Y. Xue, Z. Shen, Z. Wu and C. Song, Theoretical prediction of Curie temperature in two-dimensional ferromagnetic monolayer, J. Appl. Phys., 2022, 132(5), 1–8 CrossRef.
- V. V. Marchenkov and V. Y. Irkhin, Half-metallic ferromagnets, spin gapless semiconductors, and topological semimetals based on Heusler alloys: Theory and experiment, Phys. Met. Metallogr., 2021, 122, 1133–1157 CrossRef CAS.
- G. Saravanan, V. Asvini, R. K. Kalaiezhily, I. M. Parveen, and K. Ravichandran. Structural and magnetic characterization of Fe2CrSi Heusler alloy nanoparticles as spin injectors and spin based sensors, in AIP Conference Proceedings, AIP Publishing, 2018, vol. 1953, No. 1 Search PubMed.
- K. V. Larionov, J. J. Pais Pereda, S. Li, S. Sakai and P. B. Sorokin, Half-metallic Heusler alloy/MoS2 based magnetic tunnel junction, ACS Appl. Mater. Interfaces, 2022, 14(49), 55167–55173 CrossRef CAS PubMed.
- Y. Kawasugi, T. Ujino and H. Tada, Room-temperature magnetoresistance in organic spin-valves based on a Co2MnSi Heusler alloy, Org. Electron., 2013, 14(12), 3186–3189 CrossRef CAS.
- M. Saleem and M. Shakil, Determination of tunnelling magneto resistance of magnetic tunnel junction designed using Co2TiAl Heusler alloy with MgO spacer layer, Phys. B, 2023, 649, 414458 CrossRef CAS.
- A. Hirohata, W. Frost, M. Samiepour and J. Y. Kim, Perpendicular magnetic anisotropy in Heusler alloy films and their magnetoresistive junctions, Materials, 2018, 11(1), 105 CrossRef PubMed.
- M. Marathe and H. C. Herper, Exploration of all-3 d Heusler alloys for permanent magnets: Ab initio based high-throughput study, Phys. Rev. B, 2023, 107(17), 174402 CrossRef CAS.
- P. Blaha, K. Schwarz, F. Tran, R. Laskowski, G. K. Madsen and L. D. Marks, WIEN2k: An APW+ lo program for calculating the properties of solids, J. Chem. Phys., 2020, 152(7), 1–6 CrossRef PubMed.
- P. Blaha, K. Schwarz, G. K. H. Medsen, D. Kvasnicka and J. Luitz, WIEN2k, An Augmented Plane Wave Plus Local Orbitals Program for Calculating Crystal Properties, Vienna University of Technology, Vienna, Austria, 2001 Search PubMed.
- P. Nayak and D. C. Gupta, Exploring the multifaceted properties: electronic, magnetic, Curie temperature, elastic, thermal, and thermoelectric characteristics of gadolinium-filled PtSb3 skutterudite, RSC Adv., 2024, 14(25), 17364–17379 RSC.
- B. Gurunani and D. C. Gupta, Half-metallicity and thermoelectric performance: A multifaceted investigation of Zr-based half-Heusler alloys, Mater. Sci. Eng., B, 2025, 311, 117783 CrossRef CAS.
- K. Schwarz and P. Blaha, Solid state calculations using WIEN2k, Comput. Mater. Sci., 2003, 28(2), 259–273 CrossRef CAS.
- B. Gurunani and D. C. Gupta, A holistic approach to understanding structural, magneto-electronic, thermoelectric, and thermodynamic properties of RhMnZ (Z= Si, Ge) half Heusler alloys, Phys. Chem. Chem. Phys., 2024, 26(48), 30002–30017 RSC.
- K. Schwarz, P. Blaha and S. B. Trickey, Electronic structure of solids with WIEN2k, Mol. Phys., 2010, 108(21–23), 3147–3166 CrossRef CAS.
- G. K. Madsen, P. Blaha, K. Schwarz, E. Sjöstedt and L. Nordström, Efficient linearization of the augmented plane-wave method, Phys. Rev. B, 2001, 64(19), 195134 CrossRef.
- H. J. Monkhorst and J. D. Pack, Special points for Brillouin-zone integrations, Phys. Rev. B, 1976, 13(12), 5188 CrossRef.
- T. Lin, Q. Gao, J. Zhong, S. Yu and G. Liu, Structural, Mechanical, and Thermoelectric Properties of Quaternary Heusler Compounds CuCoZrZ (Z= Sn, Pb): A First-Principles Investigation, Phys. Status Solidi B, 2025, 2400278 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2025 |