DOI:
10.1039/D4RA08271K
(Review Article)
RSC Adv., 2025,
15, 3607-3645
Recent advances in the synthesis, reaction, and bio-evaluation potential of purines as precursor pharmacophores in chemical reactions: a review
Received
21st November 2024
, Accepted 22nd January 2025
First published on 4th February 2025
Abstract
Purines are nitrogenous heterocyclic compounds characterized by the presence of two fused rings: pyrimidine and imidazole. Their significance is underscored by their widespread occurrence in natural products as the metabolic processes of all living organisms heavily rely on purines and their synthetic derivatives. Furthermore, purines exhibit considerable bioactivity, highlighting their importance in biological systems. Given their unique structural characteristics and ability to yield a diverse array of bioactive molecules, purines have attracted substantial attention from researchers. This review illustrates the recent methods for the synthesis of purines from diaminomaleonitrile, urea derivatives, imidazole, and pyrimidine derivatives reported from 2019 to 2024. Additionally, it elucidates the various chemical modifications applied to the purine nucleus, including benzoylation, alkylation, halogenation, amination, selenylation, thiolation, condensation, diazotization, coupling reactions, and other miscellaneous reactions. Moreover, this review discusses several biological evaluations, including the mechanisms of action of purine derivatives as anticancer, antimicrobial, anti-inflammatory, antiviral, antioxidant, and anti-Alzheimer agents. This review aims to assist researchers in synthetic organic and medicinal chemistry toward the development and enhancement of novel methodologies for the synthesis of new purine molecules while supporting biologists in the identification of new targets for bio-evaluation.
 Ahmed Ragab | Ahmed Ragab is recognized as a prominent figure in the field of organic chemistry, particularly in pharmaceutical organic chemistry. He serves as an associate professor of organic chemistry in the Chemistry Department at the Faculty of Science, Al-Azhar University, Cairo, Egypt. Currently, he is on secondment to the Chemistry Department, Faculty of Science, Galala University, where he continues his academic pursuits. Ahmed obtained his bachelor's degree in special chemistry from the Faculty of Science. Subsequently, he earned both a master's degree and a PhD in organic chemistry, specializing in the synthesis of heterocyclic organic compounds, from Al-Azhar University, Cairo. His master's and doctoral research were conducted under the supervision of Professor Dr Ahmed Shehab El-Sherief, Professor Dr Yousry Ammar, and Professor Dr Yahya M. Abdelfatah. His research interests are focused on the synthesis of novel heterocyclic compounds with significant biological activity, particularly drug derivatives and natural products, along with the evaluation of their biological activities as antifungal, antimicrobial, and anticancer agents. He also engages in organic process development, drug design, as well as medicinal and pharmaceutical chemistry. In the past five years, his work has concentrated on synthesizing new hybrid bioactive heterocyclic compounds for biological evaluation, covering areas such as antimicrobial, anticancer, anti-inflammatory, and antidiabetic activities, all based on the active core of the synthesized compounds. Furthermore, he is involved in the design of new dyes and sensors. His research has included many computational studies, such as density functional theory (DFT) and molecular docking simulations, and he has collaborated with various research teams. In 2023, he contributed to the modification of novel polymeric materials by designing new polymer–drug conjugations at the Department for Biomaterials Research, Polymer Institute, Slovak Academy of Sciences, Bratislava, Slovakia. |
1. Introduction
Purine (imidazo[4,5-d]pyrimidine) is a heterocyclic aromatic system composed of fused pyrimidine and imidazole rings. It is found in four tautomeric N–H forms. Purine tautomer stability increases with increasing aromaticity, and the order is as follows: 9-H > 7-H > 3-H > 1-H. It has been reported that deprotonated NH-pyrimidine (H9 and H7) displays higher aromaticity, while its aromaticity decreases in imidazole (H1 and H3).1 These results were confirmed by the harmonic oscillator model of aromaticity (HOMA). Pyrimidine without NH showed an HOMA of 0.971, and imidazole showed an HOMA of 0.742. The structure of purine serves as a basis for synthesizing many biologically significant compounds, including guanine, adenine, isoguanine, xanthine, caffeine, hypoxanthine, and uric acid. Moreover, adenine and guanine are important because they are the fundamental building blocks of DNA, RNA, and ATP systems, which are vital to life processes (Fig. 1).1
 |
| | Fig. 1 Structure of purine and its analogues. | |
Purine is a nine-atom compound with four nitrogen atoms at positions 1, 3, 7, and 9. Numbering in purines starts with nitrogen 1 of the six-membered ring and moves in an anticlockwise direction, and it continues on the imidazole ring but in a clockwise manner.1 Additionally, purines are essential components of nucleic acids and play a role as energy cofactors for coenzymes involved in redox processes. They are also involved in several intracellular signal transduction processes and function as direct neurotransmitters.2 Purines have received significant research attention owing to their distinct properties and their value as a valuable source of various bioactive molecules in medicinal chemistry.3 Moreover, the importance of purine derivatives originates from their abundant presence in natural products and synthetic compounds, as well as their potential for bioactivity. The metabolism of all living organisms heavily depends on purine and purine bases, such as adenine, guanine, and xanthine (Fig. 1).4
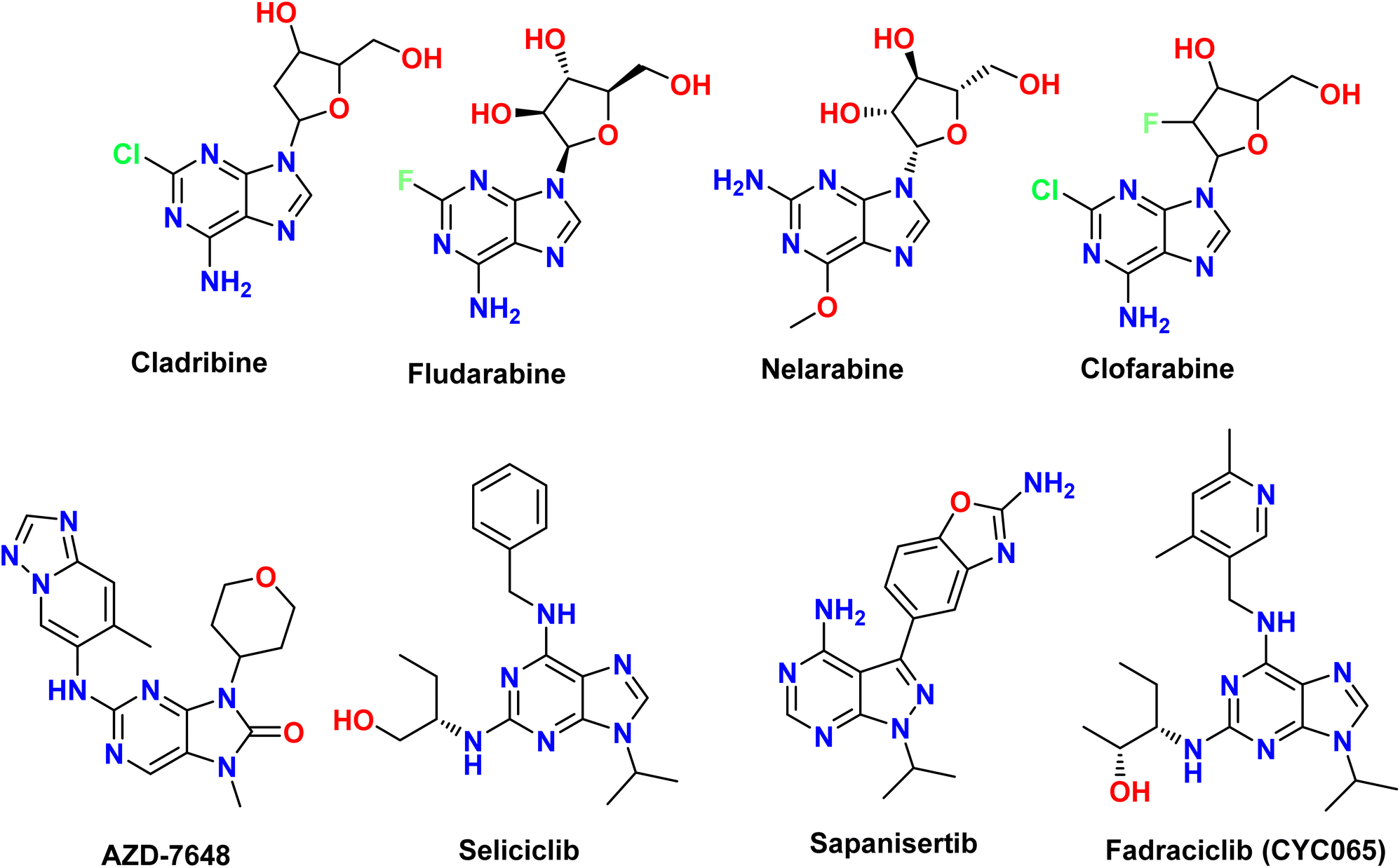
Furthermore, the purine scaffold is has been synthesized and analyzed to develop beneficial drugs with broad biological activity, such as anti-cancer drugs with different targets (m-TOR, EGFR, FGFR, VEGFR, PI3Kα, and B-RafV600E), anti-bacterial (standard strains, clinical isolates, and Helicobacter pylori), anti-inflammatory (JAK2/BRD4, COX-2, and 15-LOX), anti-fungal, anti-oxidant, anti-tuberculosis, anti-HSV, anti-diabetic, anti-viral, and anti-Alzheimer's (AChE and BChE) activities.5–15 Additionally, many purine-based medications, including 6-thio-purine (mercaptopurine) and derivatives, cladribine, fludarabine, nelarabine, clofarabine, fadraciclib (CYC065), seliciclib, sapanisertib, and AZD-7648 (Fig. 2) are used in clinical trials.16,17 Most of them are used to treat cancers and inflammation, and their mode of action involves the impairment of the ability to synthesize nucleic acids or the inhibition of important metabolic enzymes.6,18 Moreover, numerous review articles have previously addressed the synthesis, reactions, and bio-evaluation of the purine scaffold in various contexts, including the utilization of purine-containing compounds as anticancer agents19,20 and cancer-targeting kinase inhibitors.21 Furthermore, the role of purines in inflammatory responses, encompassing their release, metabolism, and signaling, has been extensively explored.22 Purine nucleotides have also been identified as signaling molecules23 and promising scaffolds in drug discovery.24 Additionally, the chemistry and biosynthetic pathways of purines, as well as related reactions, biomedical prospects, and clinical applications, are discussed.25 Furthermore, the synthetic routes and pharmaceutical activities of purine derivatives have been examined,26 alongside their neurotoxic effects.27 However, the advantage of this review over the others is that it presents several examples from recent studies, newer synthetic methods, and some bio-evaluation from the perspective of SAR studies in a simple way.
 |
| | Fig. 2 Structures of purine-based FDA-approved drugs and those under clinical trials. | |
Overall, this review article provides a comprehensive overview and summary of various methods employed for synthesizing purine derivatives over the past five years (2019–2024). In addition, this review article is one of my continuous efforts to gain insights into the design, synthesis, and bio-evaluation of heterocyclic scaffolds that are designed to modify and develop the bio-evaluation based on their structure or based on a hybridization approach for more than one nucleus to obtain one bioactive molecule, as described in my articles with my group.28–32 This review highlights numerous approaches and reagents utilized in these syntheses, including 2,3-diaminomaleonitrile, urea derivatives, imidazole derivatives, and pyrimidine derivatives. Furthermore, the article details an array of reactions involving purine derivatives, such as benzoylation, alkylation (mono-, di-, and tri-), halogenation (bromination), amination (nucleophilic substitution reactions), selenylation, thiolation, condensation, and cyclization, which facilitate the formation of new heterocyclic rings. Additional reactions discussed include diazotization and coupling reactions, among others. In conclusion, the article presents an overview of the biological evaluation of purine derivatives, which are categorized according to their activities, including anticancer, antimicrobial, anti-inflammatory, antiviral, antioxidant, and anti-Alzheimer effects. It is anticipated that this review can guide future researchers in exploring rational approaches to the application of purine scaffolds as pharmaceuticals targeting various biological pathways in clinical trials.
2. Synthesis of purine derivatives
2.1. From diaminomaleonitrile and urea derivatives
Bizzarri et al. illustrated the synthesis of amino-acid-decorated purines 4a–f, which were obtained as the main products in the multicomponent reaction between 2,3-diaminomaleonitrile (DAMN) 1, trimethyl orthoacetate 2, and α-amino acid derivatives 3a–f in acetonitrile containing triethyl amine (TEA) under reflux conditions and photon irradiation (290–315 nm) for 15 h. This way, they combined thermal and photochemical conditions to improve the complexity of the reaction pathway.33
Moreover, the reaction can be described by the photoisomerization of diaminomaleonitrile (DAMN) to diaminofumaronitrile (DAFN), followed by the cyclization of diaminofumaronitrile (DAFN) to amino imidazole carbonitrile (AICN) 7 (ref. 34) via an unstable azetidine intermediate 5.35 The azetenes 5 undergo C–C bond cleavage and ultimate rearrangement to give N-heterocyclic carbene 6, which tautomerizes readily to form AICN 7. Additionally, amino imidazole carbonitrile (AICN) reacts with trimethyl orthoacetate 2 and produces the imidazole imino ether derivative 8, which subsequently reacts with amino acid derivatives to give the desired purine derivatives, as shown in Scheme 1.33
 |
| | Scheme 1 Synthesis of 6-imino-6,9-dihydro-1H-purine derivatives 4a–f decorated with amino acids. | |
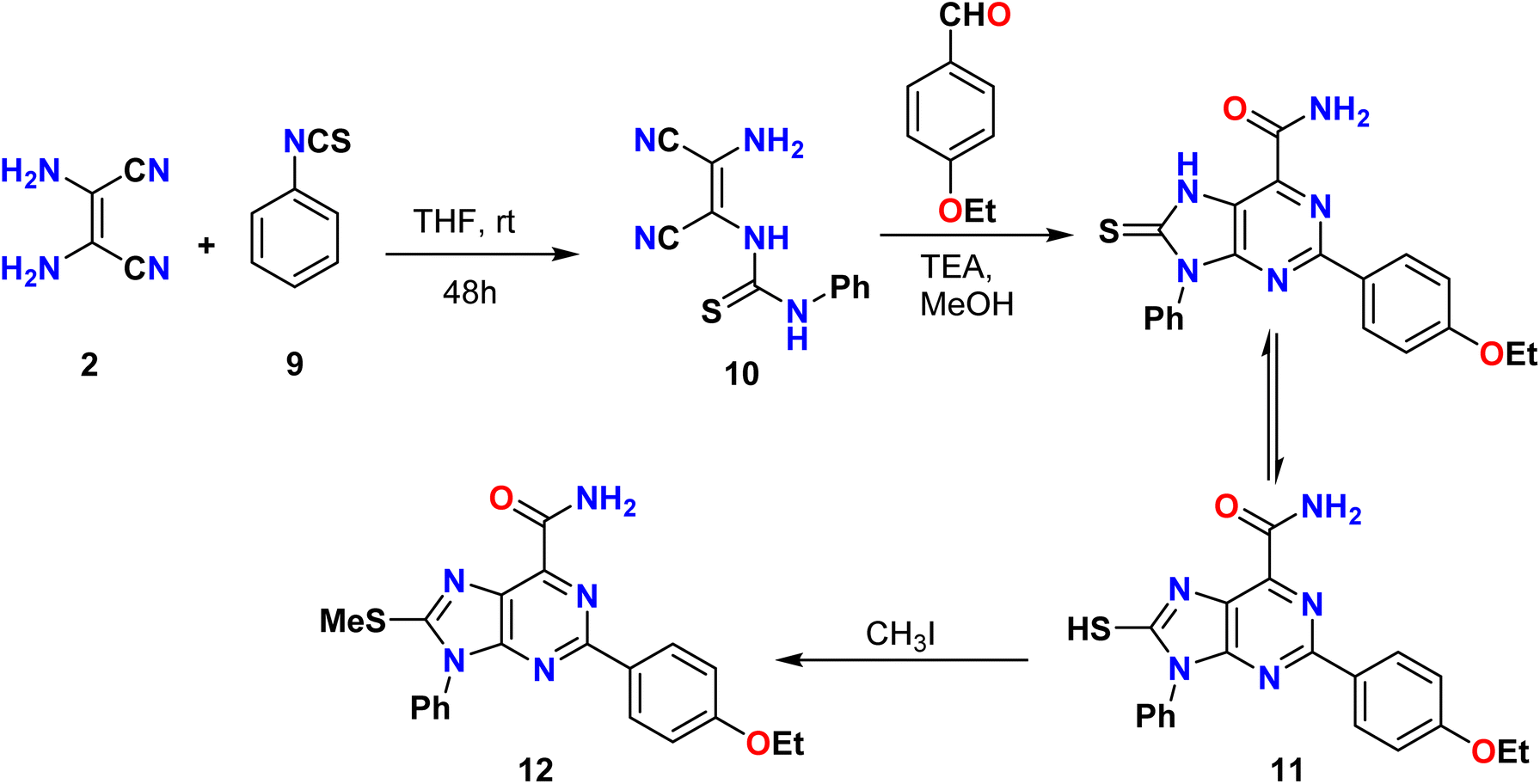
Huang et al. described the synthesis of a new 8-(methylthio)-9-phenyl-9H-purine-6-carboxamide derivative 12 by treating phenyl isothiocyanate 9 with diaminomaleonitrile (2) in the presence of THF as the solvent. The as-formed thiourea derivative then reacted with 4-ethoxybenzaldehyde to provide the 8-mercaptopurine-6-carboxamide derivative 11 as the desired product. The reaction between 11 and iodo-methane occurred exclusively at the relatively soft sulfur atom, yielding the S-alkylation product 12 (Scheme 2).36
 |
| | Scheme 2 Synthesis of 8-(methylthio)-9-phenyl-9H-purine-6-carboxamide derivatives 11 and 12 from diaminomaleonitrile. | |
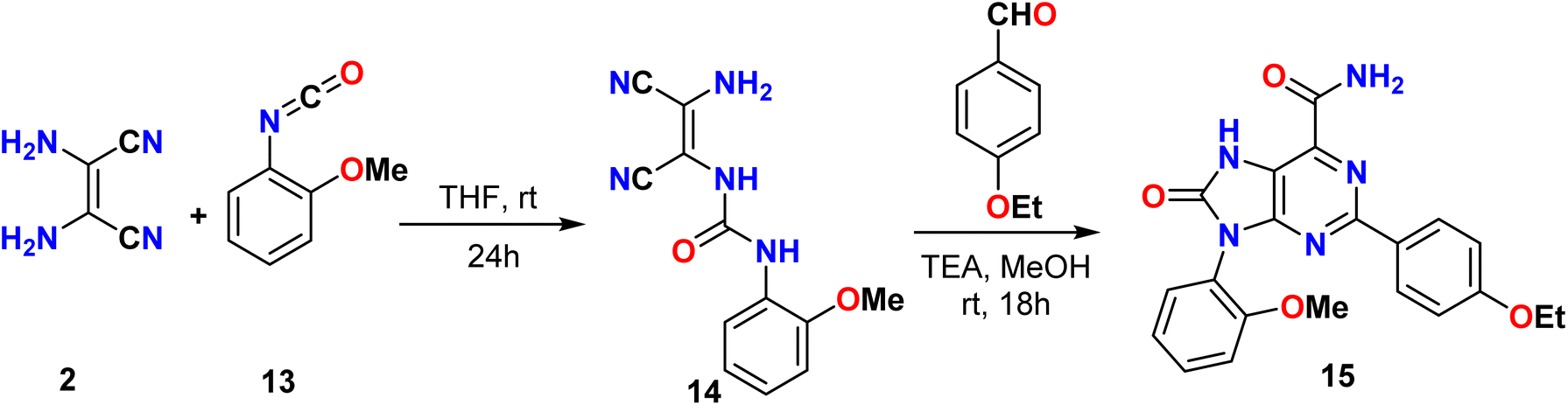
In the same way, Tseng et al. reported a synthetic method for the 2,9-(substituted-phenyl)-8-oxo-7H-purine-6-carboxamide derivative 15 based on the condensation reaction between 2-methoxyphenyl isocyanate 13 and diaminomaleonitrile (DAMN) 2, which yielded a urea derivative 14. This urea compound was then treated with 4-ethoxybenzaldehyde to synthesize a series of purine derivatives 15 that could be used as dual-target anticancer agents, as illustrated in Scheme 3.37
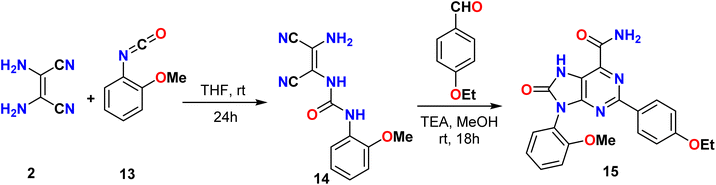 |
| | Scheme 3 Synthesis of 2,9-(substituted-phenyl)-8-oxo-7H-purine-6-carboxamide derivative 15. | |
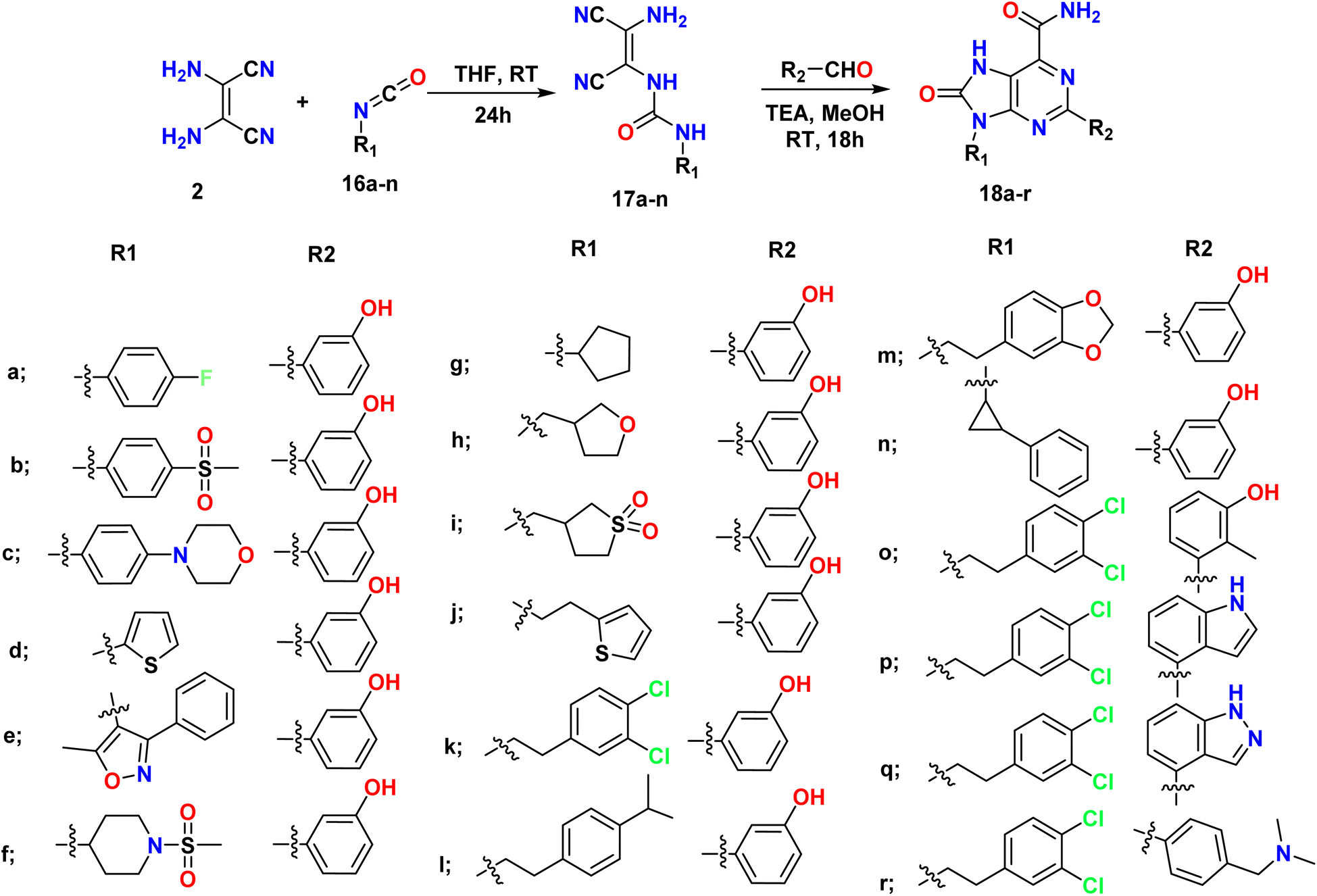
Mazzucato et al., synthesized 7H-purin-8(9H)-one derivatives 18a–r in two steps; the first step involved the reaction of 2,3-diaminomaleonitrile (DAMN) 2 and commercial isocyanate 16a–n to afford ureic intermediates 17a–n with the first substituent R1. The second step involved the formation of a core and the addition of a second substituent (R2) through the reaction of the intermediates with different aldehydes in the presence of TEA and catalytic quantities of I2, as shown in Scheme 4.38
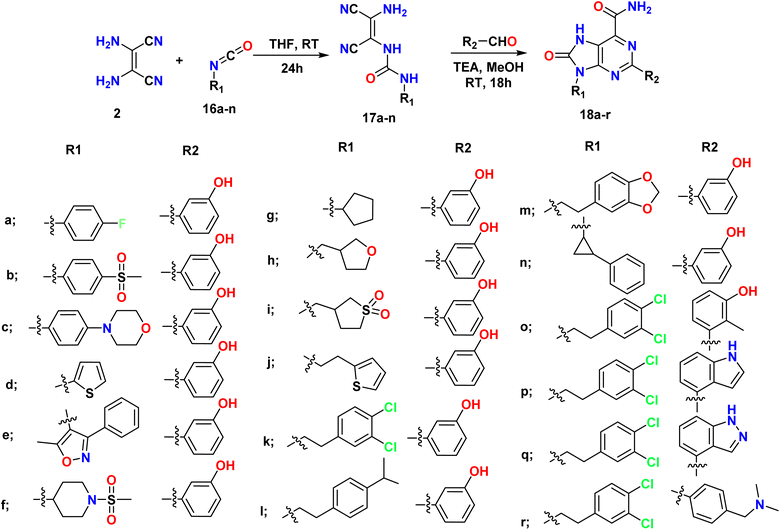 |
| | Scheme 4 Synthesis of 7H-purin-8(9H)-one from alkyl/aryl isocyanate using 2,3-diaminomaleonitrile (DAMN) and aldehyde. | |
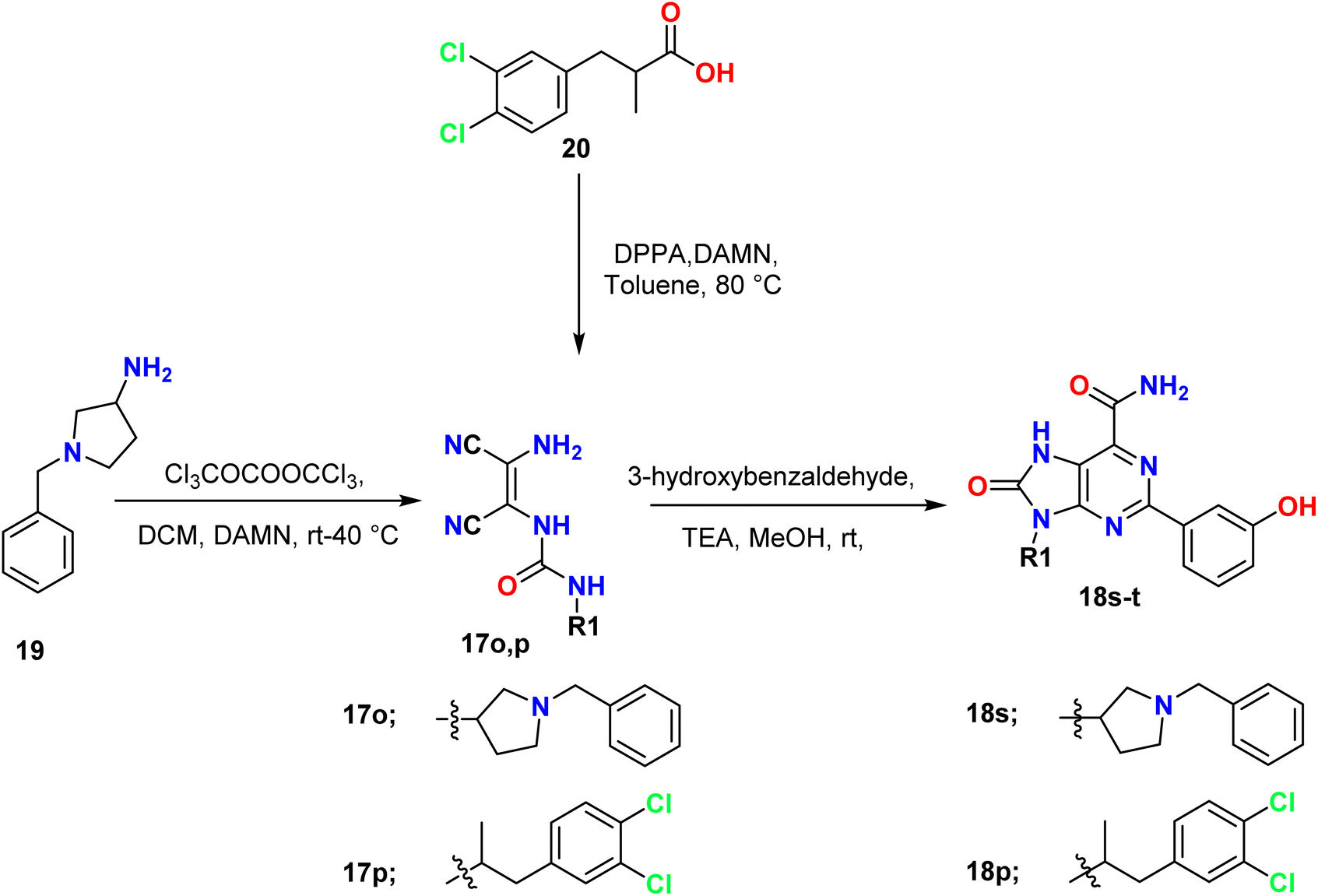
Moreover, 1-benzyl-3-isocyanatopyrrolidine was prepared by the reaction of 1-benzylpyrrolidin-3-amine 19 with triphosgene [O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C(OCCl3)2]; then the obtained isocyanate was reacted in situ with diaminomaleonitrile 1 (DAMN) to afford the corresponding urea derivative 17o, which when treated with 3-hydroxybenzaldehyde and TEA gave the final product 18s. Additionally, 2-benzylpropionic acid 20 was converted to an isocyanate (R–NCO) through Curtius rearrangement by using diphenylphosphoryl azide (DPPA) to convert the acid to azide and further to isocyanate. R–NCO then reacted in situ with diaminomaleonitrile (DAMN) 2 to give the urea derivative 17p, which was treated with 3-hydroxybenzaldehyde to form the 2,9-diaryl-8-oxo-7H-purine-6-carboxamide derivative 18t (Scheme 5).38
C(OCCl3)2]; then the obtained isocyanate was reacted in situ with diaminomaleonitrile 1 (DAMN) to afford the corresponding urea derivative 17o, which when treated with 3-hydroxybenzaldehyde and TEA gave the final product 18s. Additionally, 2-benzylpropionic acid 20 was converted to an isocyanate (R–NCO) through Curtius rearrangement by using diphenylphosphoryl azide (DPPA) to convert the acid to azide and further to isocyanate. R–NCO then reacted in situ with diaminomaleonitrile (DAMN) 2 to give the urea derivative 17p, which was treated with 3-hydroxybenzaldehyde to form the 2,9-diaryl-8-oxo-7H-purine-6-carboxamide derivative 18t (Scheme 5).38
 |
| | Scheme 5 Synthesis of 2,9-diaryl-8-oxo-7H-purine-6-carboxamide derivative 18s–t. | |
Bettencourt et al. synthesized 6-cyanopurine derivatives 23a–e containing a phenolic moiety through the reaction of O-alkylformamidoximes 21a, b [O-benzylformamidoxime 21a and O-methylformamidoxime 21b] with phenolic aldehyde and triethyl amine TEA (cat.) in an ether solution for 15 minutes on an ice bath under stirring. The reaction proceeded via the formation of amidoxime 22a–f by intramolecular cyclization and the elimination of an alcohol molecule. The elimination of methanol resulted in a simpler isolation process than benzoyl alcohol due to its high boiling point (Scheme 6).39
 |
| | Scheme 6 Synthesis of 6-cyanopurine derivatives 23a–e containing a phenolic moiety. | |
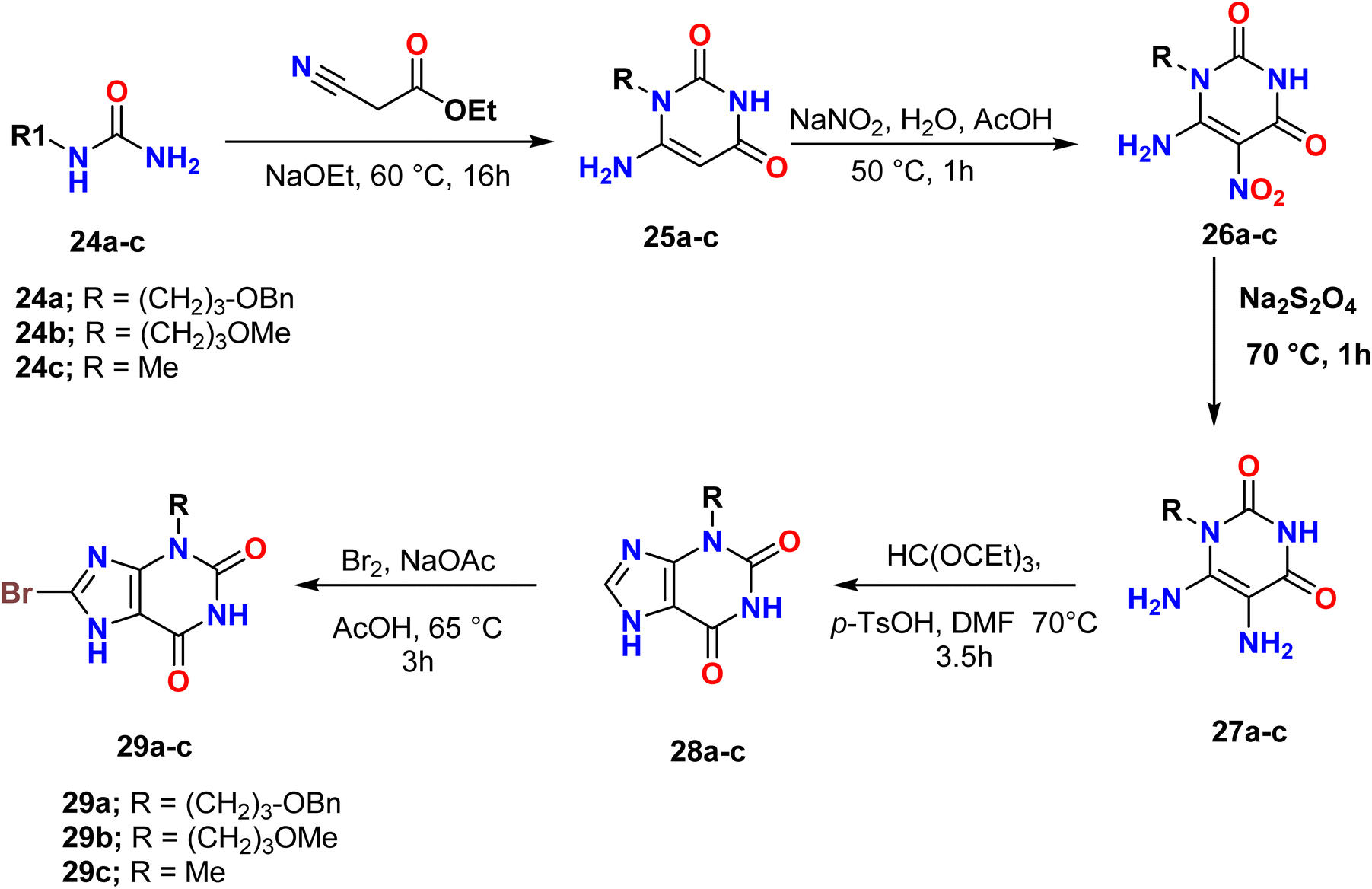
Pretze et al. reported the synthesis of 3-substituted-8-bromo-1H-purine-2,6(3H,7H)-diones 29a–c from urea derivatives. The alkylated urea derivatives 24a–c were prepared from primary amines using sodium cyanate. Additionally, the asymmetrically alkylated urea derivatives 24a–c reacted with ethyl 2-cyanoacetate in the presence of sodium ethoxide to afford N-substituted uracil derivatives 25a–c, which were subsequently treated with sodium nitrite (NaNO2) to produce the nitroso-uracil derivatives 26a–c under acidic conditions. These derivatives then underwent a reduction reaction with sodium dithionite to give 5,6-diamino-pyrimidine-2,4(1H,3H)-diones 27a–c. Moreover, the treatment of the diamino-pyrimidine-2,4-ones with triethyl orthoformate in the presence of toluenesulfonic acid (p-TsOH) produced 3-substituted-1H-purine-2,6(3H,7H)-diones 28a–c, which were then brominated using bromine for 3 hours at 65 °C to give the desired purine derivatives (Scheme 7).40
 |
| | Scheme 7 Synthesis of brominated purine derivatives starting from alkylated urea. | |
2.2. From imidazole derivatives
Gonçalves et al. reported the synthesis 2-amino-6-cyano-purine 35 by treating 5-amino-4-cyanoformimidoyl imidazoles 30 with cyanamide in acetic acid and a small volume of DMSO, which afforded the N-cyanoimidoyl cyanide intermediates 33; these underwent intramolecular cyclization and finally generated 2-amino-6-cyano-purine 35. The reaction mechanism involved the attack of the cyanamide on the imino group of 4-cyanoformimidoyl, leading to the loss of an ammonia molecule (NH3) and the formation of a new intermediate 34. Finally, the amino group of imidazole at C5 attacks the nitrile group and the subsequent proton transfer affords the desired product 35 (Scheme 8).41
 |
| | Scheme 8 Synthesis of 2-amino-6-cyano-purine 35 from 5-amino-4-cyanoformimidoyl imidazoles 30 using cyanamide. | |
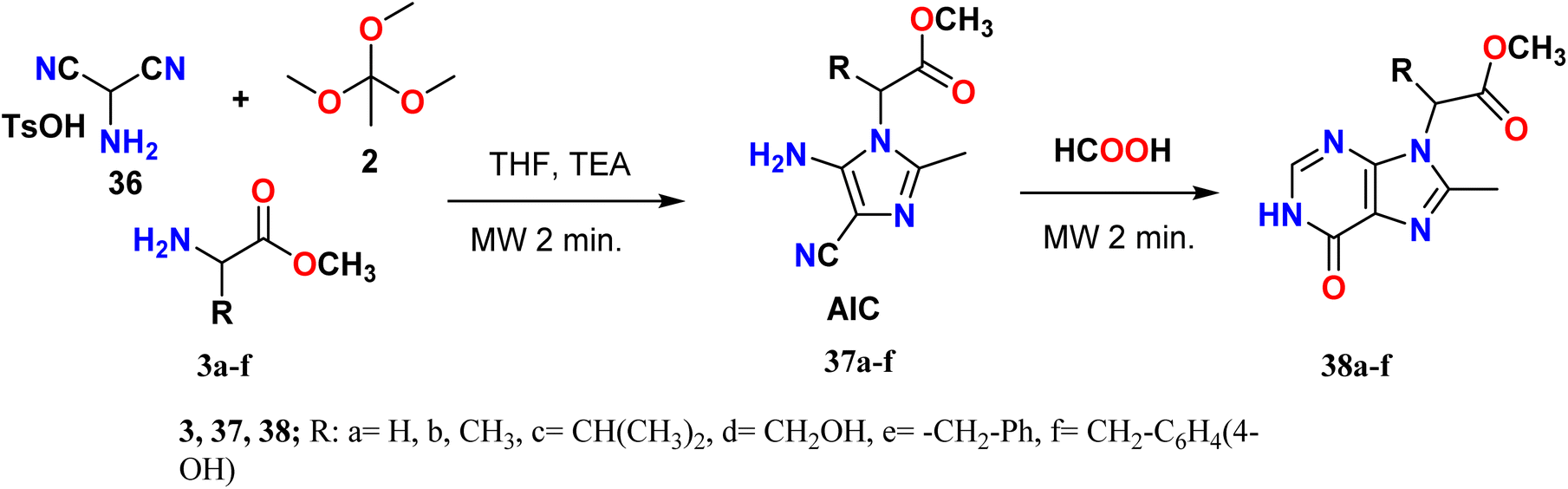
Bizzarri et al. synthesized 8,9-disubstituted-1H-purin-6-one derivatives 38a–f via a one-pot multicomponent reaction using microwave irradiation (250 W, 250 psi, 2.0 min at 200 °C). First, the enaminonitrile–imidazole derivatives 37a–f (AIC) were prepared by the condensation of aminomalononitrile p-toluenesulfonate (AMNS) 36, triethyl orthoacetate (TOA) 2 and methyl esters of amino acids 3a–f (glycine, alanine, valine, serine, phenylalanine, and tyrosine). Eventually, the amino group of AIC reacted with formic acid, leading to the loss of a water molecule under MW irradiation, and the cyano group hydrolyzed to amide (CONH2) and cyclized to give compound 38 (Scheme 9).42
 |
| | Scheme 9 Synthesis of 8,9-disubstituted-1H-purin-6-one derivatives 38a–f from enaminonitrile–imidazole using microwave irradiation. | |
Tber et al. described a multi-step method for synthesizing 2-(trifluoromethyl)pyrido[1,2-e]purine derivatives from 2-aminopyridine. First, they used a desilylative Strecker–Ugi type multicomponent reaction to prepare 3-aminoimidazo[1,2-a]pyridine-2-carboxylate 40a. This involved treating 2-aminopyridine with ethyl glyoxylate in the presence of trimethylsilyl cyanide (TMSCN) as a source of cyanide and 1,4-diazabicyclo[2.2.2]octane (DABCO) as a strong base under microwave irradiation. Then, they heated the ester imidazole with an ammonia solution at 70 °C for 48 hours to produce the 3-aminoimidazo[1,2-a]pyridine-2-carboxamide derivative 42a. This compound was then reacted with 2,2,2-trifluoroacetamide (10 equiv.) under argon at 160 °C for 24 hours to yield 2-(trifluoromethyl)pyrido[1,2-e]purin-4-ol 43a. Finally, this compound was treated with thionyl chloride to obtain the dehydroxychlorinated pyrido[1,2-e]purin-4-ol derivatives 44a–c. In the same way, the authors also described the synthesis of imidazo[1,2-a]pyridine-2-carboxylates 40b, c from 2-aminopyrimidine and ethyl bromopyruvate. The product was then nitrated, and the ester group was converted to an amide. Finally, the nitro group was reduced to afford 3-aminoimidazo[1,2-a]pyridine-2-carboxamides 42b, c (Scheme 10).43
 |
| | Scheme 10 Synthesis of 2-(trifluoromethyl)pyrido[1,2-e]purine derivatives 43 and 44 from 2-aminopyridine derivatives. | |
2.3. From pyrimidine derivatives
Verma et al. described a new strategy for the synthesis of 1,3-dimethyl-2,6-dioxo-1H-purine derivatives containing the acetonitrile group. The reaction involved treating 1,3-dimethyl-5,6-diaminopyrimidine 45 with ethyl 2-cyanoacetimidate 46 in the presence of HCl under reflux conditions. Additionally, these materials were used as the starting material to prepare new purine derivatives attached to a pyridine moiety via a two-step reaction. The first step involved the Claisen–Schmidt condensation of 1,3-dimethyl-8-(acetonitrilyl)-3,9-dihydro-1H-purine-2,6-dione 47 with substituted benzaldehyde in pyridine, which acted as a basic medium, to afford acrylonitrile-purine derivatives 48a–d. These were then reacted with malononitrile in ethanol and in the presence of a few drops of pyridine to afford 4-amino-5-(2,6-dioxo-1H-purin-8-yl)pyridine-3-carbonitriles 49a–d (Scheme 11).44
 |
| | Scheme 11 Synthesis of 1,3-dimethyl-2,6-dioxo-1H-purine derivatives 49a–d from diaminopyrimidine. | |
El-Kalyoubi et al. described the synthesis of N-substituted-8-purines 54a–c by the cyclo-condensation of 5,6-diaminouracils 50a–c with an oxazolone derivative 51 in acetic acid on a water bath for 1 hour. The mechanistic pathway illustrated that the oxazolone derivative 51 was protonated at the carbonyl moiety, and the C5 amino group of the uracil derivatives 54a–c attached with the carbonyl, which led to ring opening; then, the second amino group C6 attached with the amidic adjacent carbonyl, leading to the formation of imidazopyridine derivatives (Scheme 12).45
 |
| | Scheme 12 Synthesis of N-substituted-8-purine derivatives 54a–c via the cyclo-condensation of 5,6-diaminouracils 50a–c with the oxazolone derivative 51. | |
Chakraborty et al. explored the synthesis of 8-substituted purines 56a–d and 8,9-disubstituted purines 57a–d via an acceptor-less dehydrogenative coupling reaction of benzyl alcohol with 4,5-diaminopyrimidine and Ni(II)-catalyst [Ni(MeTAA)] under aerobic conditions in toluene and potassium tert-butoxide (KOtBu), and the reaction was dependent on their molar ratio.1 The reaction required prolonged time rather than the formation of benzimidazole, and the yield obtained ranged from moderate to good (Scheme 13).
 |
| | Scheme 13 Synthesis of 8-substituted purines 56a–d and 8,9-disubstituted purines 57a–d via acceptorless dehydrogenative coupling (ADC). | |
El-Kalyoubi et al. achieved purine derivatives through the formation of Schiff bases followed by aza-Michael addition. The substituted purines were obtained in a high yield by subjecting 1,3-dimethyl-5,6-diaminouracil 58 to heat in the presence of p-bromoacetophenone 59 in DMF (1 mL). Although most of their synthesized derivatives were screened against four cancer cell lines (HepG2, Huh7, MCF7, and A549), the anti-cancer activity of 8-(4-bromophenyl)-1H-purine-2,6-dione 60 (ref. 46) (Scheme 14) was not evaluated.
 |
| | Scheme 14 Synthesis of 8-(4-bromophenyl)-1H-purine-2,6-dione 60 from 1,3-dimethyl-5,6-diaminouracil 58 and acetophenone derivative. | |
Doganc et al. described the synthesis of 2-chloro-8-(substituted phenyl)-9H-purine via two steps; the first step involved the formation of Schiff bases 62a, b from 2-chloropyrimidine-4,5-diamine 61 and benzaldehyde derivatives, especially those with 4-fluoro or 3,4-dimethoxy phenyl substituents, under reflux conditions for 1 hour in ethanol. Moreover, the as-formed Schiff base was treated with N-bromosuccinamide in chloroform under reflux, and the product was separated from the solvent under reduced pressure as two isomers N9 and N7. Additionally, they reported that the reaction of 4,5-diaminopyrimidine with 4-fluorobenzoyl chloride afforded 6-chloro-8-(4-fluorophenyl)-3H-purine 64 as the sole product. The structures of the designed purine derivatives were confirmed by 1H–1H NOE (nuclear Overhauser effect spectroscopy, NOESY), 1H–13C/15N HMBC (heteronuclear multiple bond correlation) methods and X-ray crystallographic data (Scheme 15).2
 |
| | Scheme 15 Synthesis of 2-chloro-8-(substituted phenyl)-9H-purine derivatives 63 and 64 from a Schiff base, N-bromosuccinimide, and benzoyl chloride derivatives. | |
Fedotov et al. synthesized benzimidazopurines 67a–e as poly-condensed purine derivatives through many pathways. Several trials of treating nitrobenzimidazo-pyrimidines 65a–e with sodium dithionite and catalytic hydrogenation in different reaction media did not result in the desired diamines. Therefore, the starting material was isolated in all cases. Additionally, the target compounds were produced by the reduction of the nitro group using iron dust in a mixture of triethyl orthoformate and acetic acid; the reduction was followed by the aromatization of the pyrimidine cycle, leading to the formation of tetracyclic benzimidazopurines 67a–e with yields up to 85%. The mechanistic studies illustrated that the reaction proceeded through hydroxylamine B formation, followed by the aromatization of the pyrimidine ring C and then the final product. Finally, they proposed that metals in acidic media reduce nitroamines more efficiently than heterogeneous hydrogenation, which is uncommon (Scheme 16).47
 |
| | Scheme 16 Synthesis of poly-condensed purine derivatives 67a–e from nitro-benzimidazopyrimidines 65a–e via reduction and cyclization. | |
Lei et al. prepared 9-heterocyclyl-substituted 9H-purine derivatives 71a–l by the reduction of 5-nitropyrimidine derivatives 68 using palladium on carbon (Pd/C) (5%) to afford the corresponding amines. Additionally, the intermediate amines 68 were treated with phenyl isothiocyanate or 4-fluorophenyl isothiocyanate to generate 9H-purine-2,8-diamines 69a–i, some of which contained the tert-butoxycarbonyl protecting group (BOC group), which was removed using trifluoroacetic acid to generate 70a–f; these were subsequently reacted with sulfonyl chloride and glycolic acid or ethyl bromoacetate to give compounds 71a–l (Schemes 17 and 18).48
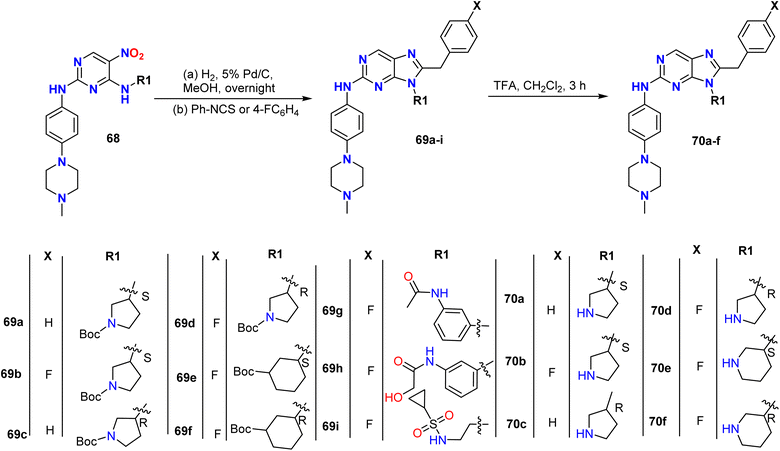 |
| | Scheme 17 Synthesis of 9-heterocyclyl substituted 9H-purine derivatives 69 and 70. | |
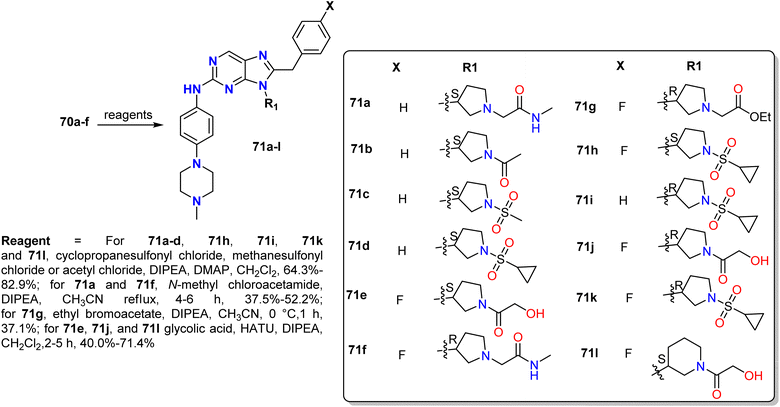 |
| | Scheme 18 Synthesis of 9-heterocyclyl substituted 9H-purine derivatives 71a–l. | |
Lorente-Macías et al. explored 6-alkoxy purine as a Jurkat-selective proapoptotic agent. The 6-alkoxy purine derivatives were prepared in two steps; reacting dichloropyrimidines with various alkyl amines in the first step yielded 6-chloro diaminopyrimidine derivatives 73. Subsequently, the chloropyrimidines were displaced at C6 with suitable alcohols under strong basic conditions in the presence of N,N-dimethylamide. This synthetic process led to the production of purine analogs 74a–c. The reaction was controlled by the type of N,N-dimethylamide used as the solvent and reagent. For example, using dimethylformamide (DMF) facilitated the synthesis of 6,9-disubstituted purines with nonsubstituted C8 (route A). On the other hand, the presence of sterically hindered N,N-dimethylamides like dimethylbenzamide (DMB) resulted in the synthesis of tri-substituted purines 75a–e (route B), where the alcohols (benzyl alcohol and 4-bromobenzyl alcohol) generated fragments at both C6 and C8. Additionally, microwave irradiation at higher temperatures was used for the 2,5-diaminopyrimidine core due to its lower reactivity than the 5-amino-2-chloropyrimidine core (Scheme 19).49
 |
| | Scheme 19 Synthesis of 6-alkoxy purine derivatives 74a–c and 75a–e from dichloropyrimidines. | |
Polat et al. synthesized a series of 6,8,9-trisubstituted purine derivatives 79 starting from 4,6-dichloro-5-nitropyrimidine 76. 5-Nitropyrimidine 76 was reduced in the presence of tin(II) chloride to produce 6-dichloropyrimidin-5-amine 72a, then the chlorine atom at C4 of pyrimidine was substituted with cyclopentyl amine in the presence of triethyl amine via a nucleophilic substitution reaction to afford 6-chloro-N4-cyclopentylpyrimidine-4,5-diamine 77, which was subsequently reacted with substituted benzaldehyde under p-TSA catalysis and cyclized to obtain 6-chloro-8,9-disubstituted 7H-purine derivatives 78a–f. Finally, the tri-substituted purines 79a–r′ were obtained by the reaction of 6-chloropurine derivatives 78a–f with appropriate N-substituted piperazines (Scheme 20).50
 |
| | Scheme 20 Synthesis of di- and tri-substituted purines from 4,6-dichloro-5-nitropyrimidine. | |
In the same way, Kucukdumlu et al. reported the synthesis of 5-amino-4,6-dichloro pyrimidine 72a by the reduction of 4,6-dichloro-5-nitro-pyrimidine 76 using SnCl2, followed by amination with benzylamine to give 4-(4-substituted benzyl)pyrimidines 80a, b. The 6-chloro purines 81a, b were prepared by the condensation of compounds 80a, b with triethyl orthoformate in the presence of toluenesulfonic acid to afford intermediates 81a, b, which underwent amination to afford 6,9-disubstituted purine derivatives 82a–t (Scheme 21).51
 |
| | Scheme 21 Synthesis of 6,9-disubstituted purine derivatives 82a, b. | |
Orduña et al. illustrated the synthesis of new substituted purine derivatives containing amino groups using a low-power microwave. They synthesized a 5,6-diamino-pyrimidine derivative 83 from 4,6-dichloropyrimidin-5-amine 72a via amination using 3-nitro aniline under MWI at 150 °C for 10 minutes, which afforded the corresponding 9-substituted purine derivative 84 in a good yield (67%). Additionally, the second amination process was performed with p-methoxy benzylamine under MWI at 100 °C for 1 hour, and finally, the nitro group of the 6,9-disubstituted purine derivative 85 was treated with SnCl2 in a mixture of ethanol and ethyl acetate under reflux conditions for 2 h to afford the purine derivative 86 containing an amino group (Scheme 22).52
 |
| | Scheme 22 Synthesis of 6,9-disubstituted purine derivative 86 from 4,6-dichloropyrimidin-5-amine 72a. | |
Bigonah-Rasti et al. synthesized a triazolo[5,1-f]purine derivative 91 starting from 5-amino-3-(methylthio)-1H-1,2,4-triazole 87, which was used as a bi-nucleophile with the 5-dichloropyrimidine derivative 88 under heating conditions and TEA as the catalyst to afford the 5-bromo-2-chloro-6-methyl-pyrimidin-4-amine derivative 89. Additionally, the tricyclic heterocyclic core was alkoxylated using several aliphatic alcohols in KOH. The reaction then progressed through two consecutive aromatic nucleophilic substitutions (SNAr), which involved intramolecular cyclocondensation and the formation of a non-isolated adduct intermediate, leading to the elimination of HBr and MeSH in two successive steps (Scheme 23).53
 |
| | Scheme 23 Synthesis of tricyclic 2-alkoxy-4H-[1,2,4]triazolo[5,1-f]purine derivatives. | |
3. Reactions of purine derivatives
The reaction of purines can proceed via more than one functional group and therefore, can be classified as follows.
3.1. Benzoylation
Attia et al. described the formation of a new guanosine monophosphate 93 with an amide linker by modifying the amino group of guanosine monophosphate 92 using benzoyl chloride (Scheme 24).54
 |
| | Scheme 24 Benzoylation of the 2-amino-9-alkyl purine derivative. | |
3.2. Alkylation
The alkylation of purine derivatives can be of three types based on the substitution of the nitrogen atom and therefore, can be classified as mono, di, and tri-substituted purines. Wang et al. synthesized new 9-substituted-9H-purine derivatives 97a–h, 98a–f, and 99 through the reaction of 6-chloro-9H-purine 94 with 4-nitrobenzyl bromide to afford 6-chloro-9-(4-nitrobenzyl)-9H-purine 95, which underwent a reduction reaction in the presence of Fe powder and ammonium chloride (NH4Cl) to give the corresponding amino derivative 96 (Scheme 25).
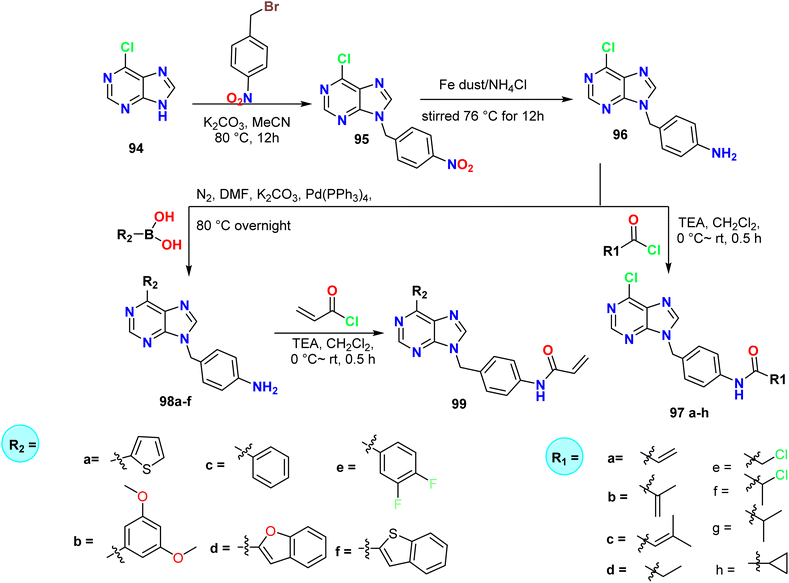 |
| | Scheme 25 Synthesis of 9-substituted-9H-purine derivatives 97a–h, 98a–f, and 99a–f through alkylation reactions. | |
Additionally, amide condensation reactions with Michael receptors, such as acryloyl chloride and analogs, were carried out using the amine derivative in order to obtain the desired products 97a–h. Moreover, compound 96 was first exposed to a Suzuki coupling reaction with compounds containing boronic acid, which produced disubstituted purine intermediates 98a–f, which subsequently underwent an amide condensation process with acryloyl chloride to produce the target compound 99 (Scheme 25).55
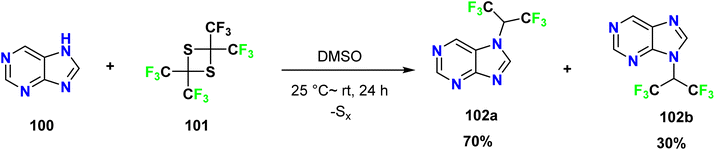
Petrov et al. discovered that purine 100 could react with tetrakis(trifluoromethyl)-1,3-dithietane 101 in DMSO in the absence of a catalyst at room temperature, affording 7- and 9-(hexafluoroisopropyl)purines (102a and 102b) in the ratio of 70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :30% (Scheme 26).56
:30% (Scheme 26).56
 |
| | Scheme 26 Synthesis of 7- and 9-(hexafluoroisopropyl)purine derivatives 102a and 102b. | |
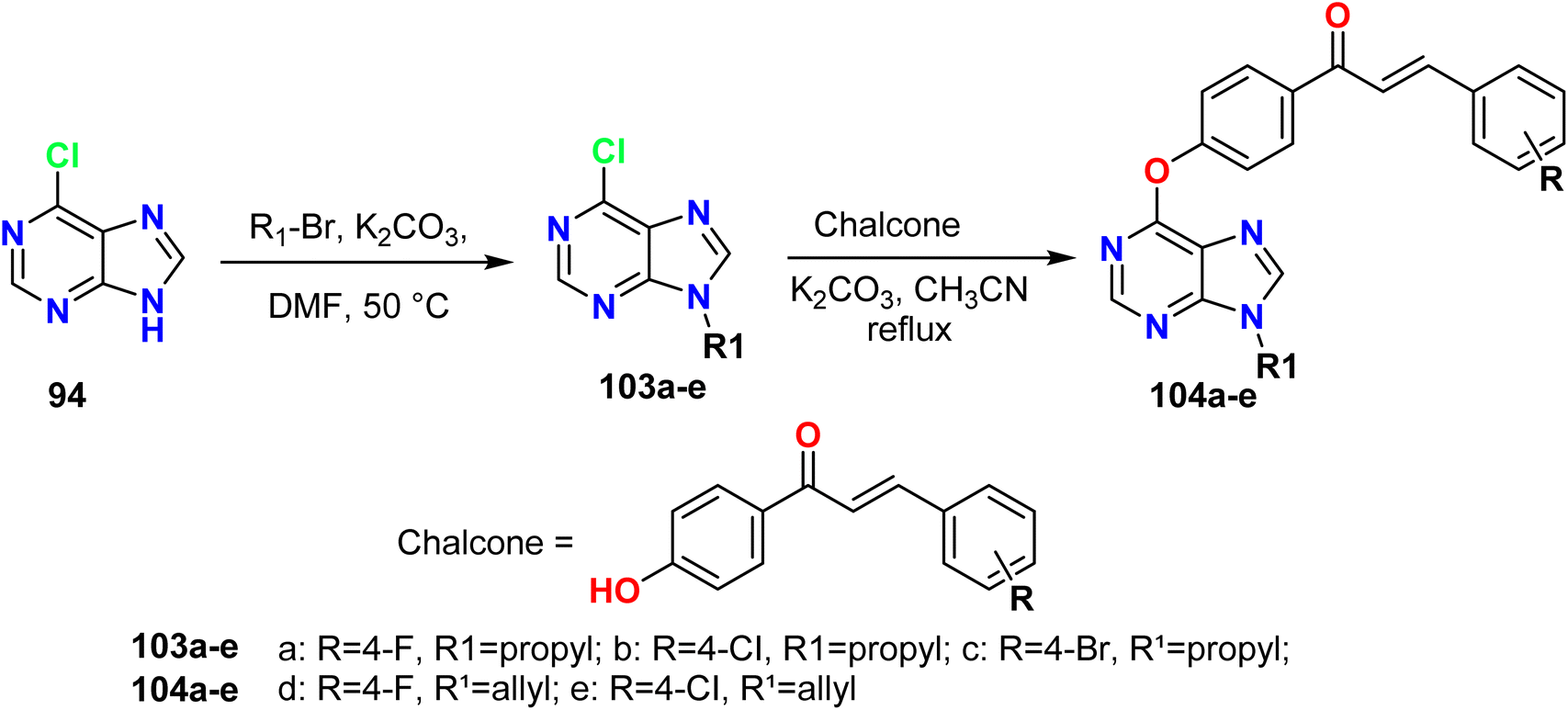
Liu et al. outlined the synthesis of 9-substituted purine derivatives 103a–e from 6-chloro-9H-purine 94 and 1-bromopropane in the presence of K2CO3; it was subsequently treated with chalcones containing the phenolic hydroxyl group to afford the corresponding 6,9-bis substituted purine derivatives 104a–e (Scheme 27).57
 |
| | Scheme 27 Synthesis of 6,9-disubstituted purine derivatives 104a–e from 6-chloro-9H-purine. | |
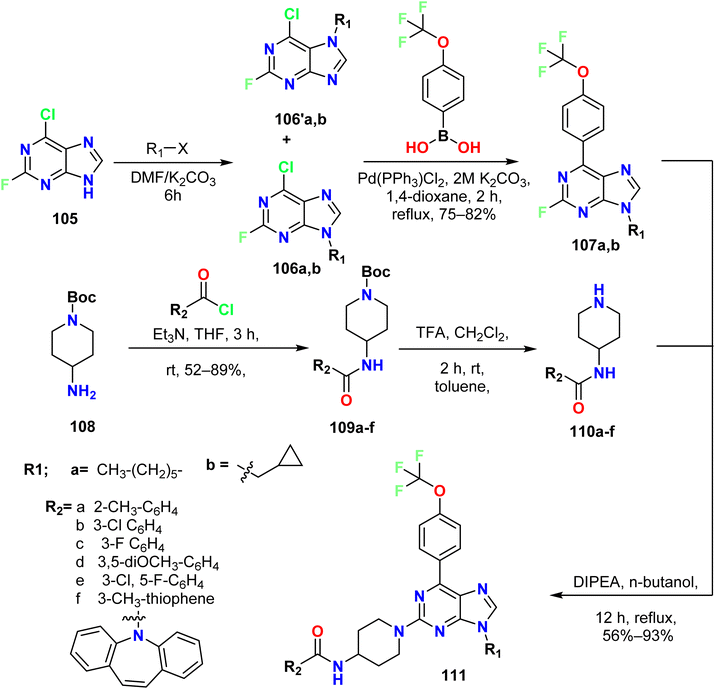
Villegas et al. described the synthesis of 2,6,9-trisubstituted purine derivatives 111 from 6-chloro-2-fluoro-9H-purine 105. Firstly, the alkylation of 6-chloro-2-fluoro-9H-purine 105 was carried out by treating it with alkyl halide in the presence of K2CO3 in DMF at room temperature to give a mixture of N7 and N9 alkylated purine regio-isomers 106a–b and 106′a, b. The second alkylation process on N6 of the purine was performed by a Suzuki reaction using trifluoromethoxyphenylboronic acid to afford the 6,9-diaryl purine derivatives 107a–b (Scheme 28).
 |
| | Scheme 28 Synthesis of 2,6,9-trisubstituted purine derivatives via three-step alkylation. | |
At the same time, different piperidine amide derivatives 110a–f were produced by reacting tert-butyl-4-aminopiperidin-1-carboxylate 108 with different acyl chlorides and Et3N. Moreover, the salts of piperidine amido derivatives 110a–f were prepared by treating amino piperidine derivatives 109a–f with trifluoroacetic acid (TFA) in methylene chloride at room temperature. Finally, the diaryl purines were reacted with salts of piperidine amide in the presence of N,N-diisopropylethylamine (DIPEA) in butanol to afford the triaryl purine derivatives 111 (Scheme 28).58
Popov et al. synthesized the diazido derivative 114a from the reaction of 2-bromoethyl-6-chloropurine 113a–c with sodium azide, and the product was obtained in two different tautomeric forms. Moreover, this compound was subjected to Cu(I)-catalyzed azide–alkyne cycloaddition (CuAAC) with sodium azide in acetone, yielding the corresponding 115. The authors also outlined a strategy for the synthesis of 6-substituted bis-purines 118 connected via different spacers. Initially, 6-alkyl purine was reacted with sodium azide to afford mono-azide derivatives 116, which were subsequently reacted with bis-alkynes 117a–c to obtain the target 6-substituted bis-purine 118 and mono-purine derivatives 119 via a CuAAC reaction of the heterocycle. This reaction was carried out, using different catalytic systems and reaction conditions, such as ultrasound irradiation to shorten the reaction time and optimize the synthesis of both mono- and bis-purine compounds (Scheme 29).59
 |
| | Scheme 29 Synthesis of 6-substituted bis-purines 118 and mono-purine derivatives 119. | |
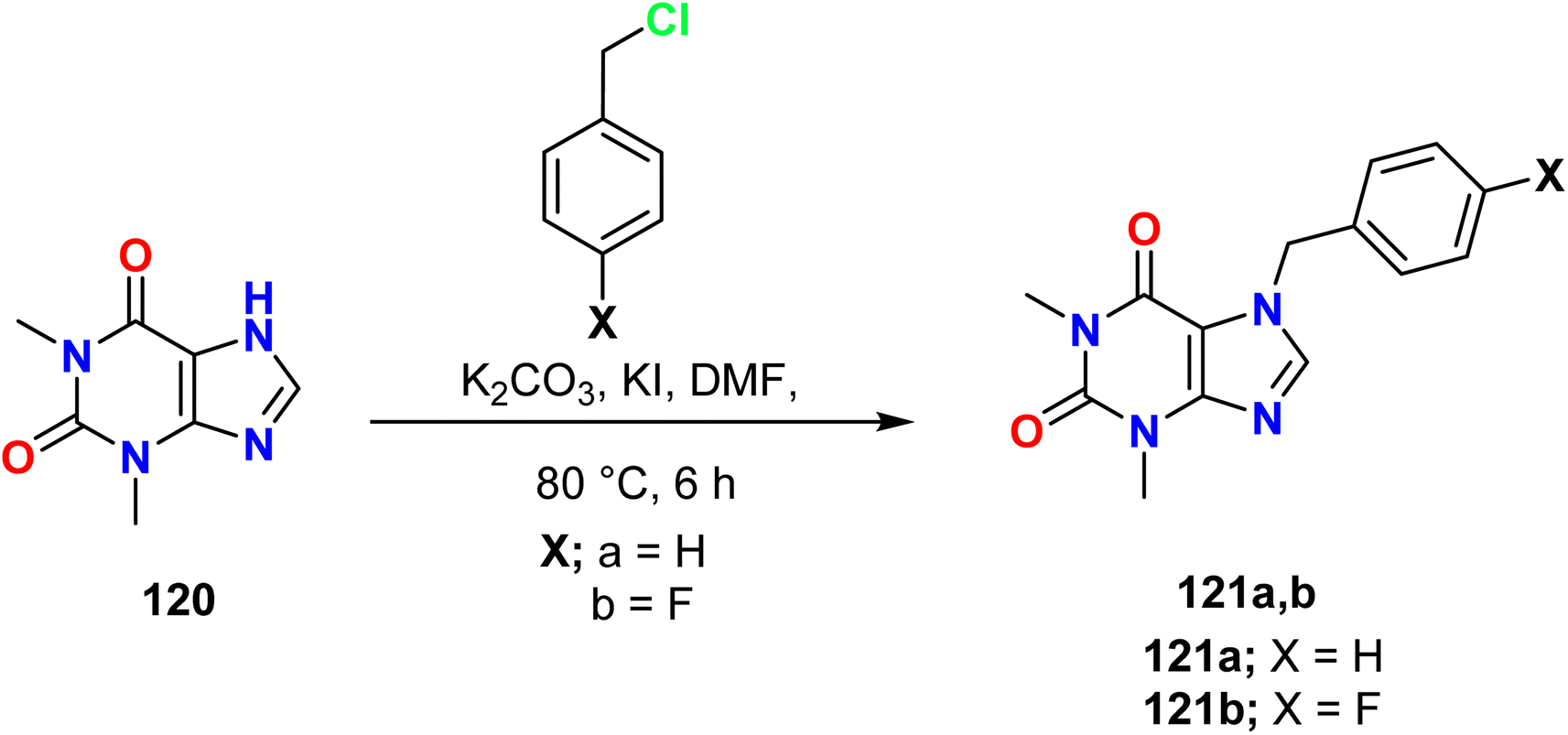
Mohamed et al. showed the alkylation of theophylline 120 at N7 using benzyl chloride (H and 4-F) via a nucleophilic substitution reaction (SN2) to afford 7-(4-substituted benzyl)-3,7-dihydro-1H-purine-2,6-dione derivatives 121a, b. The reaction was carried out in the presence of a mixture of potassium carbonate and potassium iodide (K2CO3 and KI) in dimethylformamide (DMF) at 80 °C for 6 hours (Scheme 30).7
 |
| | Scheme 30 Alkylation of theophylline using benzyl chloride derivatives. | |
3.3. Halogenation (bromination)
Mohamed et al. reported the bromination of 1,3-dimethyl-7-(alkyl)-3,7-dihydro-1H-purine-2,6-dione derivatives 121a, b using N-bromosuccinimide (NBS) in DMF under reflux conditions at 90 °C for 8 h, and bromination occurred at the C8 position (Scheme 31).7
 |
| | Scheme 31 Bromination of theophylline using N-bromosuccinimide. | |
Konduri et al. described the bromination of the 3-substituted purine derivative 120 by another method. The reaction of the 3-methyl-3,7-dihydro-1H-purine-2,6-dione derivative 120 with bromine in acetic acid provided the 8-bromo-3-methyl-1H-purine-2,6-dione derivative 123 was 92% yield.60 Additionally, Rad et al. tried to synthesize bromo-caffeine as a tri-substituted purine 124 using various methods, but the N-bromosuccinimide (NBS) in dichloromethane (CH2Cl2) and water at room temperature was preferred due to the purity of the product 125 (Scheme 32).61
 |
| | Scheme 32 Synthesis of 8-bromo-1H-purine-2,6-dione. | |
3.4. Amination (nu substitution reaction)
Konduri et al. aminated 8-bromo-1,3-disubstituted-1H-purine-2,6-dione derivative 126 using 1-BOC-piperazine in the presence of sodium carbonate in DMF solvent, successfully incorporating the piperazine group and forming compound 127. The BOC group was cleaved using methanolic HCl in an acid-mediated reaction, resulting in the formation of the important intermediate amine 128 (Scheme 33).60
 |
| | Scheme 33 Synthesis of the 1-alkyl-3-methyl-8-(piperazin-1-yl)-1H-purine-2,6-dione derivative. | |
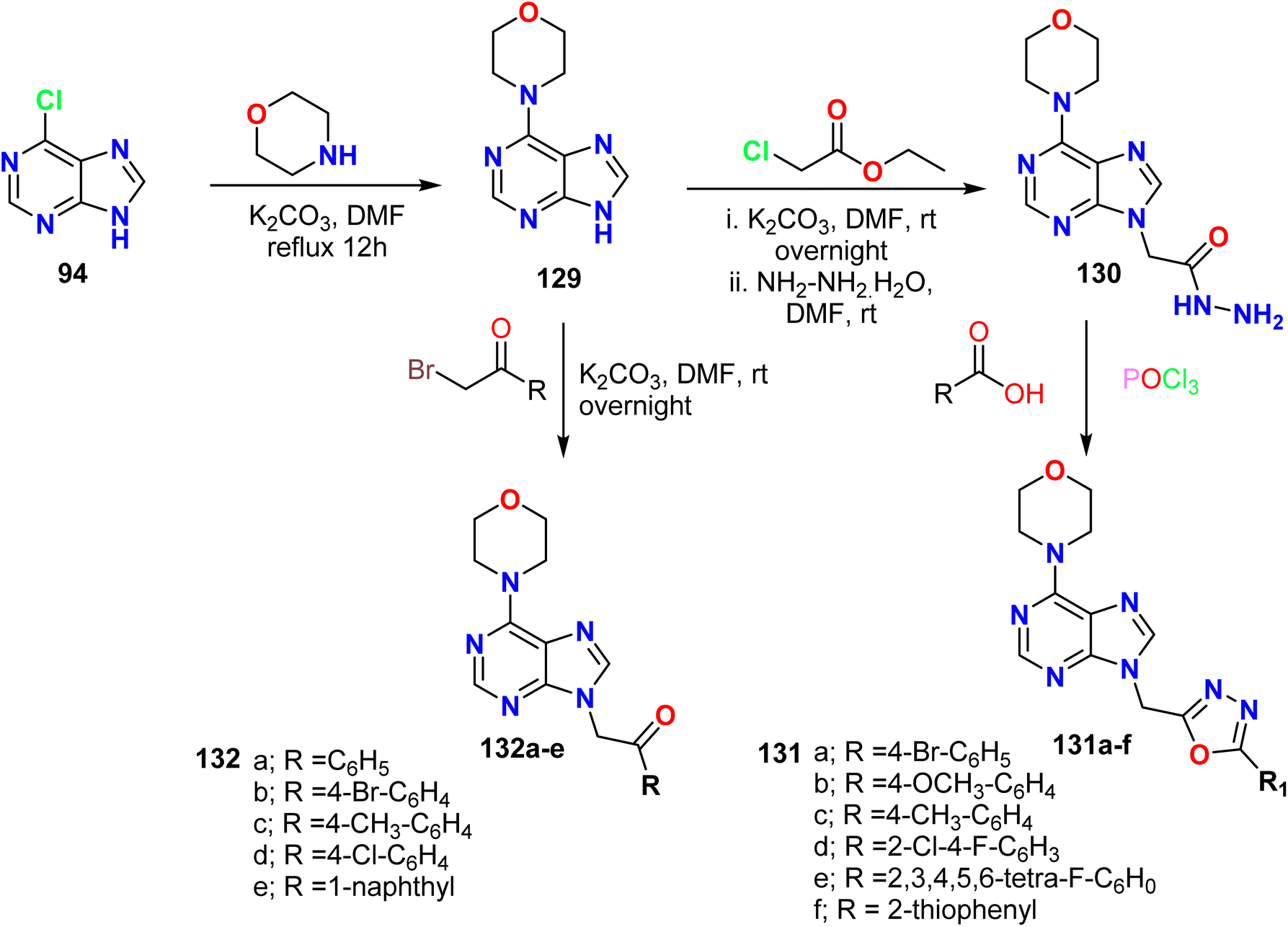
Nadaf et al. described the reaction of 6-chloro-9H-purine 94 with morpholine to form the 4-(9H-purin-6-yl)morpholine 129, which was then alkylated using different phenyl bromides to obtain the 6,9-disubstituted purine derivatives 132. On the other hand, alkylating 4-(9H-purin-6-yl)morpholine 129 with ethyl chloroacetate afforded an alkylated purine with an ester group, which was treated with hydrazine hydrate to form a new hydrazine derivative 130. The hydrazine derivative reacted with various aromatic acids in the presence of phosphorous oxychloride to afford purines with alkylated oxadiazoles 131a–f (Scheme 34).62
 |
| | Scheme 34 Amination of 6-chloro-9H-purine with morpholine and subsequent alkylation. | |
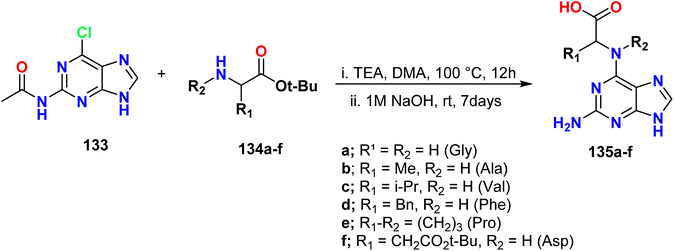
Krasnov et al. described the synthesis of novel purine derivatives with amino acids attached to C6. The process involved the treatment of N-(6-chloro-9H-purin-2-yl)acetamide 133 with tert-butyl esters of amino acids 134a–f in dimethylacetamide (DMA) as the solvent and triethylamine (TEA) as the catalyst at 100 °C under reflux conditions. The products were obtained with yields ranging from 32% to 83%. Subsequently, alkaline hydrolysis was performed to remove the N-acetyl and ester groups, resulting in the formation of the desired products 135a–f (Scheme 35).63
 |
| | Scheme 35 Synthesis of purine conjugates with natural amino acids. | |
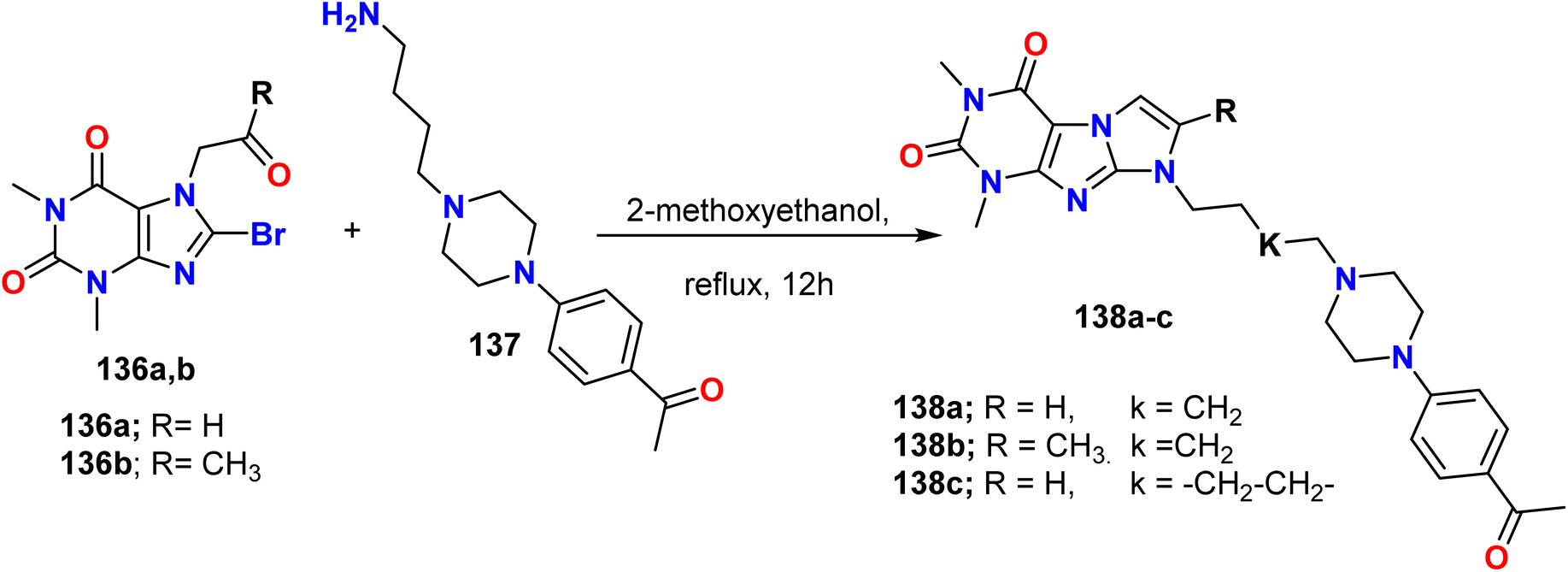
Zagórska et al. reported the synthesis of 1H-imidazo[2,1-f]purine derivatives 138a–c from 8-bromotheophylline derivatives 136a, b via a reaction with N-(aminoalkyl)-4-acetylphenylpiperazine 137 followed by cyclo-condensation. First, 8-bromotheophylline was aminated, and then the 8-amino derivatives spontaneously cyclized through the reaction of NH at C8 with the carbonyl group at N9, resulting in the loss of a water molecule under the reaction conditions (Scheme 36).64
 |
| | Scheme 36 Synthesis of 1H-imidazo[2,1-f]purine derivatives 138a–c. | |
On the other hand, Mohamed et al. reported the amination of 8-bromo-7-(alkyl)-1H-purine-2,6-dione derivatives 122a, b using 3-iminosaccharin and sulfathiazole in DMF and in the presence of a catalytic amount of dimethylaminopyridine (DMAP) under reflux conditions to afford 139 and 140, respectively. Moreover, compounds 141, 142, and 143 were readily obtained by the reaction of the 8-bromo-7-(alkyl)-1H-purine derivative 122a with amine derivatives using potassium carbonate in DMF. The authors also described that in order to generate the activity of 2-chloroethylamine–HCl in a mild basic condition, it was necessary to use triethyl amine in DMF to react with 122a; then the reaction was terminated by the addition of potassium carbonate to give 142. For the reaction involving 6-aminouracil and 122a, it was necessary to adjust the pH of the medium in order to facilitate the nucleophilic attack using the primary amine. To achieve this, sodium acetate was used as a base in acetic acid. This prevented the de-protonation of uracil NHs and resulted in the formation of compound 143 (Scheme 37).7
 |
| | Scheme 37 Amination of 1,3-dimethyl-7-(alkyl)-3,7-dihydro-1H-purine-2,6-dione derivatives 122a, b with appropriate amines. | |
3.5. Selenylation and thiolation
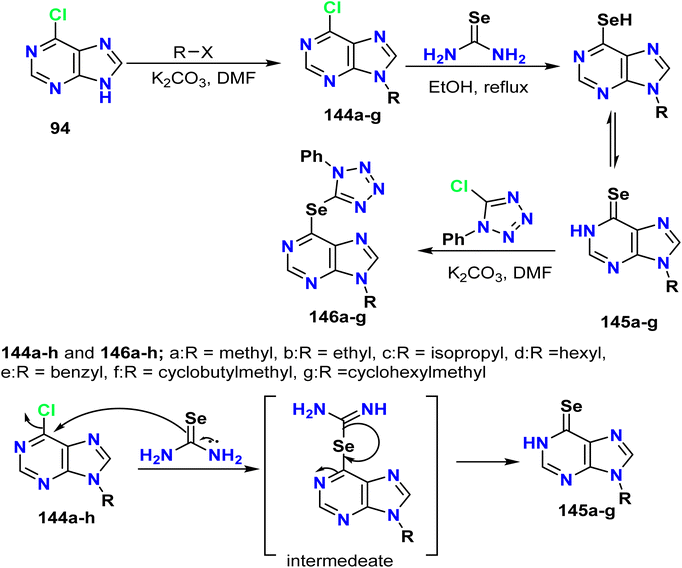
Dilek et al. reported the generation of selenotetrazole-purine derivatives 146a–g through a three-step synthetic process. Initially, 6-chloropurine was reacted with specific alkyl halides in DMF and K2CO3 to afford 6-chloro-9-alkyl-9H-purines 144a–g. Additionally, selenopurines 145a–g were prepared by reacting compounds 144a–g with selenourea in absolute ethanol at room temperature by the Mautner method. The product yields ranged from 30–88%. Compounds 145a–g were allowed to react with 5-chloro-1-phenyl-1H-tetrazole to give the desired products 146a–g (Scheme 38).65
 |
| | Scheme 38 Synthesis of selenotetrazole purine derivatives 146a–g. | |
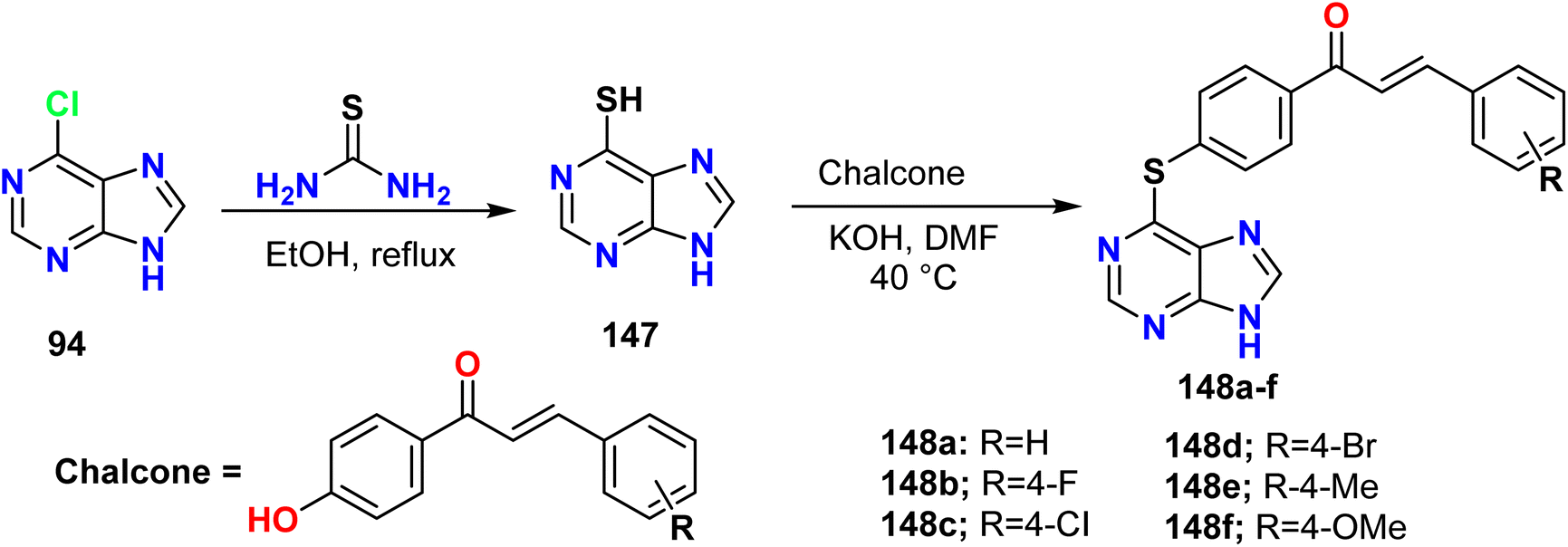
Liu et al. outlined the reaction of 6-chloro-9H-purine 94 and thiourea under reflux conditions to afford the corresponding 9H-purine-6-thiol 147, which reacted with the potassium salt of the phenolate hydroxyl group of a chalcone to afford 6-thioalkyl purine derivatives 148a–f (Scheme 39).57
 |
| | Scheme 39 Synthesis of target 9H-purine-6-thiol 147 and 6-thioalkyl purine derivatives 148a–f. | |
3.6. Condensation and cyclization to form new heterocyclic rings
Singh et al. synthesized new Schiff bases 150 and 151 by reacting 2,6-diaminopurine with 2-hydroxy-3-methoxybenzaldehyde and chromone-3-carboxaldehyde. The reaction was carried out in methanol without any catalyst under reflux and stirring conditions for 6 hours. The authors used these two Schiff bases as the starting material to prepare new copper, zinc, and cobalt metal complexes with targeted DNA-binding activity (Scheme 40).66
 |
| | Scheme 40 Synthesis of new Schiff bases by reacting 2,6-diaminopurine 149 with formyl derivatives. | |
Furthermore, Afifi et al. reported the synthesis of novel hydrazone derivatives based on the purine scaffold. This was achieved by treating 8-hydrazinyl-1H-purine-2,6-dione 152 with acetophenone derivatives. Subsequently, the resulting compounds underwent a Vilsmeier reaction using phosphorus oxychloride in DMF, leading to the formation of purine-pyrazolecarbaldehyde derivatives 154a–d. Furthermore, the pyrazolecarbaldehyde derivatives were subjected to Knoevenagel condensation with the activated methylene of 2-thioxo-thiazole derivatives under basic conditions in dioxane, which resulted in the formation of the corresponding purine derivatives 155 and 156 (Scheme 41).67
 |
| | Scheme 41 Synthesis of new Schiff bases based on purine derivatives. | |
3.7. Diazotization and coupling reaction
Khalifa et al. illustrated the synthesis of the 8-diazonium purine salt 158 via the reaction of 8-aminopurine 157 with nitrous acid; 158 was then coupled with many aryl (phenol, 1-naphthol, and 2-naphthol) and/or hetaryl (3-methyl-6-oxo-thieno[2,3-b]pyridine-5-carbonitrile) compounds to afford the diazenyl-purine derivatives 159, 160, 161, and 162 (Scheme 42).68
 |
| | Scheme 42 Synthesis of 8-diazonium purine and its coupling reactions with different reagents. | |
Furthermore, the diazonium salt of the aminopurine derivative 158 and the 2-phenylthiocarbamide derivative 163 underwent a Japp–Klingemann reaction and afforded a cyano-purine hydrazone derivative 164 by losing the acetyl group. Additionally, the cyclization of the cyano-purine 164 in a bromine solution (Br2 in ethyl acetate) in pyridine produced the benzothiazolyl acetonitrile derivative 165 (Scheme 43).68
 |
| | Scheme 43 Synthesis of the benzothiazolyl acetonitrile derivative 165. | |
3.8. Miscellaneous reactions
Khalifa described the treatment of 8-amino purine 157 with active α-halo carbonyl compounds, such as phenacyl chloride, chloroacetone, and ethyl chloroacetate, in the presence of anhydrous potassium carbonate; the subsequent cyclization process resulted in imidazopurine derivatives 166a, b and 167 (Scheme 44).68
 |
| | Scheme 44 Synthesis of imidazopurine derivatives 166a, b and 167. | |
Furthermore, to synthesize heterocyclic nucleus hybrid with purine nucleus, initially, the purine derivatives 49a–d containing enaminonitrile–pyridine at C8 were used as starting material for the synthesis of new purines containing heterocyclic fragments at the same carbon, such as pyrido[4,3-d]pyrimidine 168a–c, pyrazolo[4,3-c]pyridine 169a–c, 2-thioxopyrido[4,3-d]pyrimidine 170a–c, 2-oxopyrido[4,3-d]pyrimidine 171a–c, and 2-oxo-2H-pyrano[2,3-b][1,6]naphthyridine-3-carbonitrile 172a–c, by reaction with formic acid, hydrazine hydrate, urea, thiourea, and ethyl cyanoacetate, respectively. All these derivatives were screened for antimicrobial, antiproliferative, and antioxidant activity (Scheme 45).44
 |
| | Scheme 45 Synthesis of many heterocyclic cores with purine as the base nucleus. | |
Hassan et al. described the synthesis of chiral-carbon-containing purine derivatives through a one-pot double Mannich-type reaction. The reaction was accomplished by treating the purine derivative 173 with various secondary amines in the presence of an excess amount of formaldehyde (nonenolizable aldehyde) under reflux conditions to afford 1,3,5-thiadiazino[2,3-f]purine derivatives 175a–e. This reaction proceed by double Mannich reaction that involved attach the formaldehyde and led to loss of water molecule and afford methylamine carbocation derivative 174b, which subsequently reacted with secondary amine and thiocarbonyl tautomerized to give thio group which N-methyl amine derivative 174e and loss the second water molecule leading to formation new thio-heterocyclic ring Scheme 46.69
 |
| | Scheme 46 Formation of new purines attached to chiral-carbon-containing heterocyclic scaffolds and the mechanistic pathway. | |
Husseiny et al. reported the synthesis of new thiazepinopurines via the reaction of 7H-purine-6-thiol 147 with arylidenes of malononitrile or ethyl cyanoacetate in a basic medium. The reaction of 7H-purine-6-thiol 147 with arylidenes of malononitrile afforded 9H-[1,4]thiazepino[4,3,2-gh]purine-8-carbonitrile 176a–d as the sole product. On the other hand, treating 7H-purine-6-thiol 147 with arylidenes of ethyl cyanoacetate under reflux conditions revealed two pathways based on the catalyst (trimethylamine or sodium ethoxide) to furnish 2-aminocarboxylate 177a–c or hydroxyl carbonitrile derivatives 178a–c. The reaction proceeded via the Michael addition of purine thiol to furnish an olefinic bond in arylidenes, followed by nucleophilic addition to give the desired product (Scheme 47).70
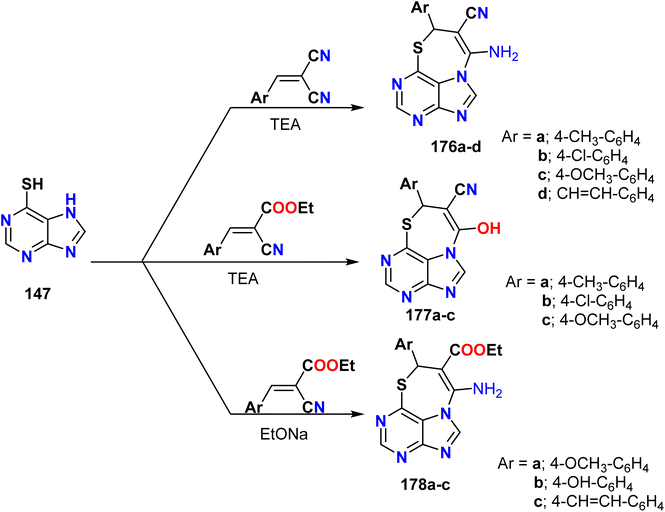 |
| | Scheme 47 Synthesis of the new thiazepinopurine derivatives 176–178. | |
4. Biological evaluation of purine derivatives
4.1. Anti-cancer activity
Purine derivatives exhibit anticancer activity with different modes of action, but the general mode of action of drugs containing the purine scaffold involves hampering the synthesis of nucleic acids or inhibiting critical enzymes involved in cellular metabolism. It is well-documented that purine analogs have specific targeting abilities and anticancer behavior. For example, tioguanine, also known as 6-thioguanine, is a leukemia drug that has been approved by the FDA. Cytosine methylation in human cells is reduced because it inhibits cytosine residue methylation. Similarly, clofarabine, an FDA-approved medicine based on second-generation purines, exhibits anti-cancer properties by inhibiting ribonucleotide reductase and stopping the elongation of DNA chains.71,72
Lei et al. screened the 9-heterocyclyl substituted 9H-purine derivatives 71a–l for anti-proliferation activity against four cell lines (HCC827, H1975, A549, and A431) by using the MTT assay, and the results showed their potency against the human lung cancer cell line (HCC827) rather than other tested cells (Fig. 3).48 Additionally, 71i had an IC50 value of 0.00088 μM against HCC827 cells, but for H1975 cells, it was 0.20 μM. Notably, four derivatives 71d, 71i, 71k, and 71l exhibited very strong anti-proliferation and EGFRL858R/T790M/C797S inhibition activities. 1i demonstrated anti-proliferative activity against HCC827 and H1975 cell lines with IC50 values of 0.00088 and 0.20 μM, respectively. Additionally, the most active derivative 71i suppressed EGFRL858R/T790M/C797S with an IC50 of 18 nM. The derivatives 71d, 71i, 71k, and 71l induced apoptosis at the G0/G1 cell cycle phase, and their percentage of apoptosis in HCC827 cells were 57.74, 56.91, 60.08, and 55.90%, respectively, at 1 μM concentration, respectively. The results of mutant EGFR (epidermal growth factor receptor) were compared with the positive control osimertinib, which displayed an IC50 value of 110 ± 6.0 nM. The activity was assessed using a western blot assay to better elucidate the mechanism of their anti-proliferative action. Compounds 71d, 71i, and 71k inhibited EGFR phosphorylation in H1975, HCC827, and A549 cell lines in a concentration-dependent manner (Fig. 3).48
 |
| | Fig. 3 Structures of the most active 9-heterocyclyl substituted 9H-purine derivatives and their values against the EGFRL858R/T790M/C797S compared to osimertinib as a positive control. | |
Polat et al. reported the cytotoxicity of tri-substituted purines against different cell lines, including HUH7 (liver), MCF7 (breast), and HCT116 (colon), based on the SRB assay. SAR study demonstrated that substitution with different phenyl moieties at C8 of purine affects the activity; for example, introducing trifluoromethyl phenyl (79j, 79k, 79l), 4-methoxy phenyl (79s), and 4-fluoro phenyl (79z) exhibited IC50 values less than 10 μM against liver cell lines (HUH7). Furthermore, the most potent purine derivatives were also tested using the NCI-SRB assay on drug-sensitive hepatocellular carcinoma (HCC) cell lines, including SNU475, SNU387, HepG2, SNU423, Hep3B, SNU182, SNU449, Mahlavu, Huh7, SNU398, FOCUS, and PLC. Compound 79j had significant anticancer activity with IC50 values ranging between (2.9–9.3 μM) against Huh7, FOCUS, SNU475, SNU182, HepG2, and Hep3B cells compared with the positive control fludarabine. In the same way, the substituted purines 79l and 79s showed more significant activity than fludarabine against the drug-sensitive HepG2 and Hep3B cells with low IC50 values (Fig. 4).50
 |
| | Fig. 4 Structure of the most active tri-substituted purines with cytotoxicity against hepatocellular carcinoma (HCC) cell lines. | |
Moreover, Verma et al. evaluated the anticancer activity of purine derivative 168b against four cell lines. The IC50 (μM) values against the MCF-7, A549, HeLa, and Panc-1 cancer cell lines were 0.8 ± 0.61, 1.0 ± 0.3, 1.2 ± 0.7, and 0.90 ± 0.71 μM compared with those of doxorubicin (0.92 ± 0.50, 1.02 ± 0.80, 1.02 ± 0.72, and 1.41 ± 0.58 μM), respectively, indicating its strong cytotoxic action. Additionally, compounds 168c and 172c showed remarkable radical-scavenging action at concentrations between 25 and 100 μg mL−1, with ED50 values of 3.39 ± 0.3 and 4.27 ± 0.5 μM, respectively, comparable to butylated hydroxyanisole (BHA) 5.51 ± 0.3 μM (Scheme 45 and Fig. 5).44
 |
| | Fig. 5 Structure of 8-substituted purine derivatives attached to heterocyclic scaffolds as anticancer agents. | |
Kucukdumlu et al. screened the designed di-substituted purines against Huh7 (liver), HCT116 (colon), and MCF7 (breast) cell lines. The results exhibited broad activity against the tested cells with IC50 values ranging from 0.05 to 21.8 μM, except for the compound derived from 4-chlorobenzylamine using 1-((4-trifluoromethyl)phenyl)piperazine, which displayed weak activity (Scheme 21). Additionally, compounds 82f and 82p exhibited significant activity against all cell lines, with IC50 values of (0.13 ± 0.12 and 0.08 ± 0.06 μM), (0.42 ± 0.08 and 0.04 ± 0.004 μM), and (0.4 and 0.05 μM) against Huh7, HCT116, and MCF7, respectively, compared with those of 5-FU (30.6 ± 1.8, 4.1 ± 0.3, and 3.5 ± 0.7 μM) and cladribine (0.9 ± 0.7, <0.1, and 2.4 ± 2.4 μM) against the same cell lines. Several liver cancer cell (HCC) lines, including Huh7, HepG2, Mahlavu, and FOCUS, were also used to test the cytotoxic activity of purine analogs. Compounds 82f and 82s demonstrated high cytotoxic activity against Huh7 cells, with IC50 values in the 0.08–0.13 μM range, which were comparable to CPT and superior to cladribine, fludarabine, and 5-FU (Scheme 21 and Fig. 6).51
 |
| | Fig. 6 The most active 6,8-disubstituted purine derivatives 82f, 82p, and 82s against hepatocellular carcinoma (HCC) cell lines: (Huh7, HepG2, MAHLAVU, and FOCUS). | |
Attiogbe et al. reported the cytotoxicity of 179 against different cell lines, and the results exhibited strong activity against the growth of melanoma, breast, and lung cancer cells; it displayed more sensitivity to HCC827 and H1975 lung cancer cells and T47D breast cancer cells. Surprisingly, the in vivo results confirmed that 179 inhibited the proliferation of HCC827 and H1975 tumors. Compound 179 inhibits the EGFR pathway by downregulating p-EGFR, p-Akt, and p-MAPK, as confirmed by western blotting analysis. Apoptosis and cell cycle analyses were conducted on HCC827 and H1975 cell lines. The results showed that the purine derivative 179 could enhance apoptosis in both early- and late-stage cancer compared with the control. Additionally, purine derivative 179 was found to induce cell cycle arrest at the S phase (Fig. 7).18
 |
| | Fig. 7 Structure of the 2,8,9-trisubstituted purine (179). | |
Husseiny et al. screened the synthesized thiazepinopurine derivatives against three cancer cell lines (HepG2, MCF-7, and PC-3) and one normal cell line (WI38). The IC50 of the thiazepinopurines exhibited broad activity against the tested cell lines. A significant antiproliferative effect was observed for compounds 176b and 176c, whose IC50 values ranged from 5.52 to 17.09 μM compared with roscovitine (9.32 to 13.82 μM). Moreover, both derivatives exhibited good selectivity indexes, and compound 176b showed the best selectivity index. Compound 176b showed CDK2 inhibitory potential with an IC50 0.219 μM. Furthermore, 176b was found to arrest MCF-7 cells at the S phase and induce apoptosis by suppressing Bcl-2 expression, as well as increasing the expression of Bax and caspases-8 and 9. The SAR study is summarized in Fig. 8.70
 |
| | Fig. 8 Illustration of the SAR study of thiazepinopurine derivatives using cancer cell lines (HepG2, MCF7, and PC-3). | |
Rad et al. synthesized new 8-((1-alkyl-1H-1,2,3-triazol-4-yl) piperazin-1-yl)-1H-purine-2,6-dione 180a–o and screened the designed derivatives against two cancer cell lines, including A-375 (malignant melanoma) and MCF7 (common breast cancer), using the MTT assay. The results revealed that compounds 180i, 180j, and 180k demonstrated IC50 values of (371 ± 2.5, 323 ± 2.6, and 367 ± 3.4 μM) and (260 ± 2.2, 245 ± 2.3, and 175 ± 3.2 μM) against A-375 and MCF7, respectively. Additionally, screening the synthesized compounds against the normal cell line HEK-293 indicated non-toxic properties with IC50 values above that of methotrexate (MTX) (IC50 = 199 ± 2.4 μM). Based on the SAR study, the derivatives could be summarized into four categories: (1) compounds with simple aryl–alkyl substituents, such as 180a and 180c–180f; (2) compounds with alkyl–ester moieties; (3) compounds with alkyl–imide moieties, such as 180l–180o; and finally, (4) compounds containing normal alkyl chain substituents 180h–180k. While the first three groups exhibited no effect on cancer cells, compounds 180h–180k responded most favorably to both cancer cell lines (Fig. 9).61
 |
| | Fig. 9 Structure of 8-((1-alkyl-1H-1,2,3-triazol-4-yl)piperazin-1-yl)-1H-purine-2,6-dione derivatives 180a–o. | |
Wang et al. reported that compound 99b showed the greatest efficacy in inhibiting enzyme activity and displaying anti-proliferative effects against the cancer cell lines. Compound 99b exhibited kinase activity against fibroblast growth factor receptors (FGFRs), especially FGFR1 (IC50 = 0.20 ± 0.02 nM) and FGFR4 (IC50 = 0.40 ± 0.03 nM), compared with fexagratinib (AZD4547), showing an IC50 value of 0.95 ± 0.03 nM for FGFR1 and 369.46 ± 26.94 nM for FGFR4, which illustrates that this compound formed irreversible covalent bonds with FGFR1 and FGFR4 proteins, thus effectively inhibiting their enzyme activity. Furthermore, it regulated the FGFR-mediated signaling pathway and the mitochondrial apoptotic pathway, promoting cancer cell apoptosis and hindering cancer cell invasion and metastasis. Moreover, 99b demonstrated good metabolic stability and showed significant anti-tumor activity in the MDA-MB-231 xenograft tumor mice model (Fig. 10).55
 |
| | Fig. 10 Structure of compound 99b with inhibitory activity against fibroblast growth factor receptors (FGFRs). | |
4.2. Anti-microbial activity
Verma et al. revealed the antimicrobial activity of 1,3-dimethyl-2,6-dioxo-1H-purine derivatives. The SAR study of antimicrobial activity revealed that electron-withdrawing groups, such as nitro and chloro, enhanced the activity greater than electron-donating groups because the hydrophobic nature of nitro and halogen groups changes the physicochemical properties of these compounds to transect microbial membranes. The 8-{7-(4-chlorophenyl)-4-oxopyrido[4,3-d]pyrimidin-8-yl}-1,3-dimethyl-1H-purine-2,6-dione derivative 168a showed strong action against Gram-negative bacteria, whereas 8-{6-(4-nitrophenyl)-1H-pyrazolo[4,3-c]pyridin-7-yl}-1H-purine-2,6-dione 169b showed greatest activity against Gram-positive bacteria. Compound 168a exhibited the most potent MIC value of 1.5 μg mL−1 against Escherichia coli and Klebsiella pneumoniae compared with ciprofloxacin (3.12 and 1.5 μg mL−1). Similarly, compound 169b showed MIC values of 12.5 and 25 μg mL−1 against Staphylococcus aureus and Bacillus subtilis, which are greater than the MIC values of ciprofloxacin (3.12 and 1.5 μg mL−1), respectively. As for antifungal activity, purine derivative 171b showed MIC values of 3.12, 1.5, 1.5, and 3.12 μg mL−1 compared to fluconazole (MIC = 1.5, 3.12, 1.5, 3.12 μg mL−1) against Aspergillus niger, Aspergillus oryzae, Candida albicans, and Penicillium chrysogenum, respectively (Fig. 11).44
 |
| | Fig. 11 Structure of 8-purine derivatives with heterocyclic fragments. | |
Hu et al. evaluated the antimicrobial activity of purine azole derivatives against five Gram-positive and six Gram-negative isolates. A purine nucleus hybrid with metronidazole compound 181 exhibited significant inhibitory activity, with a MIC value of 6 μM, which is nearly four times higher than that of norfloxacin (MIC = 25 μM). Additionally, compound 181 displayed no toxic activity against normal mammalian cells (RAW 264.7). Compound 181 could disrupt the MRSA cell membrane and also get inserted into the MRSA DNA to generate a stable complex known as compound 181-MRSA DNA. This complex could inhibit the replication and proliferation of MRSA. Finally, it was found that compound 181 exhibited antimicrobial activity via a dual-targeted inhibitory action, that is, damaging the MRSA cell membranes and causing changes in the DNA double helix (Fig. 12).73
 |
| | Fig. 12 Purine-metronidazole derivative 181 as a dual-targeting MSRA inhibitor. | |
Paulsen et al. conducted a screening to investigate the antimicrobial potential of synthesized 6-(hydroxyimino)-9-methyl-1H-purin-7-ium derivatives. The study identified two derivatives 182 and 183, which exhibited antimicrobial activity against various pathogenic bacteria and protozoa. Notably, compounds 182 and 183, characterized by larger lipophilic side chains, demonstrated good to very good activity against all microorganisms, except for E. coli. Furthermore, these compounds displayed the highest potency, with IC50 (μM) values of 1.89 and 1.84 μM against S. aureus, respectively. Additionally, compounds 182 and 183 exhibited IC50 (μM) values of 0.54 and 0.53 μM against T. cruzi, and an IC50 value of 0.50 μM against T. rhodesiense. Importantly, these derivatives demonstrated non-toxic activity against mammalian MRC-5sv2 (human lung fibroblast) cells and exhibited the ability to reduce bacterial biofilm formation by varying percentages. Compounds 182 and 183 demonstrated a significant 90% decrease in S. epidermidis biofilm formation at concentrations of 125 and 63 μM, respectively. In contrast, at the same concentrations (125 and 63 μM), the bacterial growth was reduced by 50% and 15%, respectively (Fig. 13).74
 |
| | Fig. 13 Structure of the most active 6-(hydroxyimino)-9-methyl-1H-purin-7-ium derivatives. | |
Nadaf et al. synthesized 9-alkyl-6-(4-(4-propoxyphenyl)piperazin-1-yl)-9H-purine derivatives and tested their antituberculosis (antimycobacterial), antibacterial, and antifungal activities. As for antimycobacterial activity, compounds 184a, 184b, 184c, and 184d showed MIC values of 6.2, 3.125, 3.125, and 6.25 μg mL−1, respectively, while other derivatives showed MIC values of 12.5 μg mL−1. Similarly, ciprofloxacin, pyrazinamide, and streptomycin revealed MIC values of 3.125, 3.125, and 6.25 μg mL−1, respectively. To test the antimicrobial activity, the designed derivatives were evaluated against Staphylococcus aureus, Bacillus subtilis, Escherichia coli, and Pseudomonas aeruginosa. Compounds 184a–f revealed potent antibacterial activity against the tested strains, with MIC values ranging from 2–4 μg mL−1, while other derivatives 184g–j displayed MIC values between 8 and 64 μg mL−1, displaying comparable activity to tetracycline or higher (MIC = 2–4 μg mL−1). As for antifungal activity, the designed purine derivatives were screened against Aspergillus flavus, Trichoderma harzianum, Penicillium chrysogenum, and Candida albicans, and the same results were observed. Compounds 184a–f showed good antifungal activity against the tested strains, with MIC values ranging between 1 and 8 μg mL−1. For compounds 184g–j, the MIC ranged from 16 to 64 μg mL−1 compared to nystatin and fluconazole (MIC = 2–4 μg mL−1). Finally, the SAR study illustrated that the increased length of the alkyl chain decreased the activity of the compound (Fig. 14).75
 |
| | Fig. 14 New 6,9-disubstituted purine derivatives exhibited antimycobacterial, antibacterial, and antifungal activity. | |
4.3. Anti-inflammatory activity
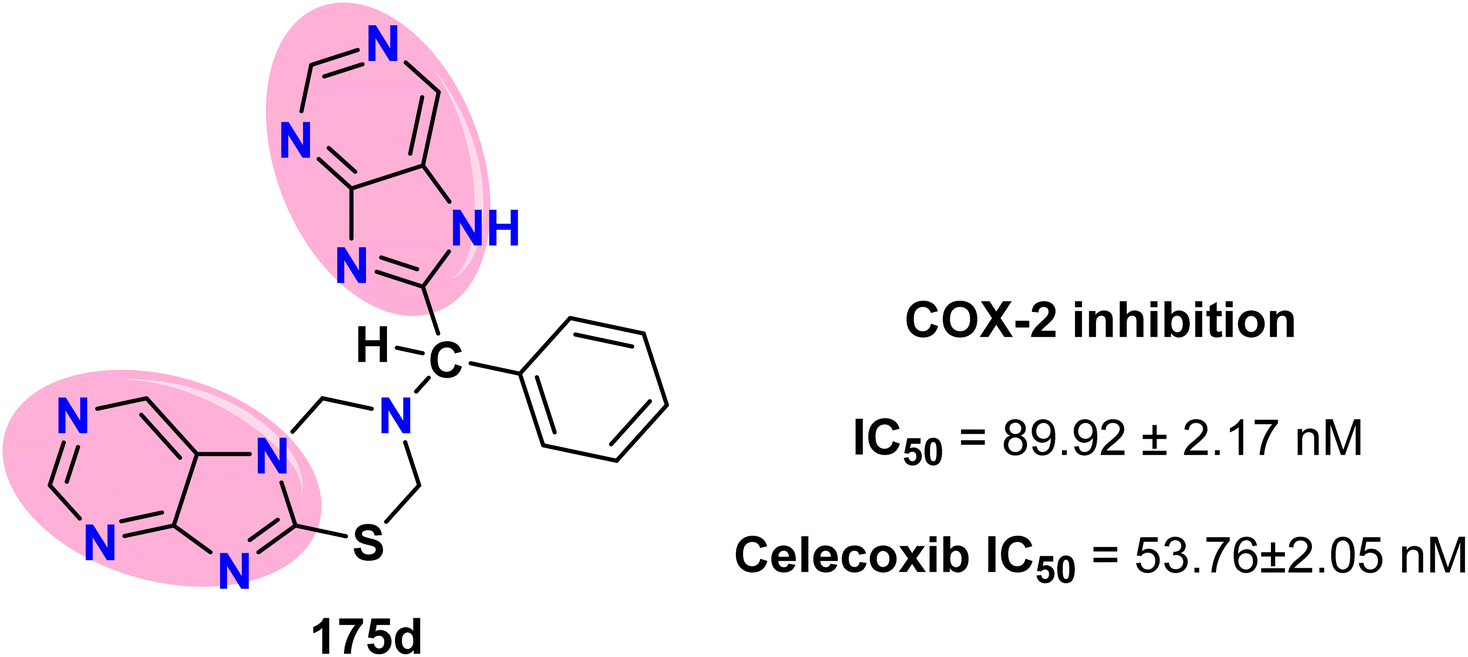
Hassan et al. demonstrated that the [1,3,5]thiadiazino[2,3-f]purine derivative 175d exhibited anti-inflammatory activity by inhibiting the COX-2 enzyme. The IC50 value of this purine derivative was 89.92 ± 2.17 nM, whereas the IC50 value of Celecoxib was 53.76 ± 2.05 nM (Scheme 46 and Fig. 15).69
 |
| | Fig. 15 Structure of the 1,3,5-thiadiazino[2,3-f]purine derivative 175d, which acts as a COX-2 inhibitor. | |
Similarly, lipoxygenases (Lox) are a class of oxidative enzymes that contain a non-heme iron atom in their active site. Lox regulates inflammatory responses by producing leukotrienes as pro-inflammatory mediators and lipoxins as anti-inflammatory mediators.76 Afifi et al. tested the anti-inflammatory action of the designed purine-pyrazole derivatives 154a–d, 155a–d, and 156a–d against 15-LOX. The IC50 of all tested compounds ranged from 1.96 to 6.12 μm, indicating potent 15-LOX inhibition comparable to zileuton (IC50 = 3.98 μM), quercetin (IC50 = 6.87 μM) and meclofenamate sodium (IC50 = 5.64 μM). Purine derivatives 156a exhibited the highest potency, while 154d showed the least activity. The SAR study elucidated that the test pyrazole-carboxaldehydes 154a–d had moderate activity. A general observation was that the derivatives obtained from condensation with rhodanine derivatives 156a, 156b, 156d, 155a, 155b, and 155d were more potent than the starting pyrazole carboxaldehydes. Moreover, purine derivatives containing acetic acid as a side chain (155a, 155b, and 155d) exhibited greater inhibitory activity against 15-LOX in comparison with their benzene sulphonamide counterparts 156a, 156b, and 156d (Scheme 41 and Fig. 16).67
 |
| | Fig. 16 Structure of the purine analog with the most potent 15-LOX inhibitory action. | |
4.4. Anti-viral activity
Mohammed et al. synthesized a series of tricyclic penciclovir (PCV) derivatives that showed promising antiviral activity, especially against the herpes simplex virus (HSV). Among the synthesized derivatives, the 9H-imidazo[1,2-a]purin-9-one derivative 185 exhibited potent antiviral activity, with EC50 = 1.5, 0.8, and 0.8 μM comparable to cidofovir (CDV) EC50 = 2.0, 2.0, and 5.0 μM and aciclovir (ACV) EC50 = 0.9, 0.9, and 100 μM against HSV-1, HSV-2, and HSV-1 TK + VZV Oka strains, respectively (Fig. 17).77
 |
| | Fig. 17 Structure of purine derivatives with antiviral activity. | |
Furthermore, Deng et al. described the synthesis of new purine hybrids with quinazoline and then evaluated their activity against the tobacco mosaic virus. In the antiviral bioassays against plant viruses, the in vivo antiviral activity of some purine-quinazoline derivatives against tobacco mosaic virus (TMV) was higher than that of the commercial drug ribavirin. To our surprise, compound 186 containing 8-fluro quinazoline (500 mg L−1) showed curative activity and protective inhibitory action up to 65.2% and 60.2%, respectively, higher than those of ribavirin (50.1% and 57.2%). Similarly, the in vivo study of TMV showed that the EC50 value of compound 186 (162.3 mg L−1) was greater than that of ribavirin (314.45 mg L−1), indicating that the compound had a significant therapeutic effect (Fig. 17).78
4.5. Anti-oxidant activity
Mangasuli et al. synthesized new coumarin–purine hybrids and screened them for in vitro antioxidant activity using DPPH. By substituting OH at C7 of the coumarin ring, the most active derivative with excellent DPPH scavenging properties was obtained. Compound 187 exhibited radical scavenging activity (%) values of 30.09 ± 1.16%, 43.90 ± 2.39%, 62.51 ± 0.92%, 75.24 ± 1.78%, and 87.04 ± 1.54% at concentrations of 20, 40, 60, 80, and 100 μg mL−1, respectively, compared to ascorbic acid, which showed values of 40.36 ± 0.75%, 58.60 ± 0.916%, 75.40 ± 2.12, 85.23 ± 1.21%, and 95.15 ± 1.91% at the same concentrations, respectively (Fig. 18).79
 |
| | Fig. 18 Structure of the 9-(coumarin)-purine derivative 187 with antioxidant activity. | |
Valdes et al. modified some purine derivatives (adenosine) and evaluated their antioxidant activity using DPPH and ABTS assays. Compounds 188, 189, and 190 had a powerful antioxidant effect compared with the reference compound ascorbic acid. As for DPPH, the purine derivatives 188, 189, and 190 demonstrated IC50 values of 77.25 ± 1.5, >100, and 17.12 ± 1.9 μg mL−1, respectively, much higher than that of ascorbic acid (IC50 = 1.5 ± 0.2 μg mL−1). On the other hand, these derivatives revealed IC50 values of 7.25 ± 1.5, 83.24 ± 2.2, and 20.05 ± 2.5 μg mL−1, respectively, which were much lower compared to that of ascorbic acid (IC50 = 27.62 ± 3.5 μg mL−1) in the ABTS assay (Fig. 19).80
 |
| | Fig. 19 Structures of some modified adenosine purine analogs exhibiting anti-oxidation potential. | |
4.6. Anti-Alzheimer activity
Sharma et al. synthesized three series of caffeine-based triazoles starting from a 1,3-dimethyl-1H-purine-2,6-dione derivative. These three series differed only in the linker substituted at N9 of the purine ring: compounds with methylene are denoted as 191a–l, while those with alkylmethyl acetate are 192a–o and compounds with N-(alkyl)acetamide are 193a–d. All the synthesized derivatives were screened in vitro against AChE and BChE at 10 μM and exhibited good to potent inhibitory activity. Compounds with high inhibitory activity were further selected for IC50 value determination. The results showed that the purine derivative 192d showed promising activity with dual potential against AChE and BChE, exhibiting IC50 values of 1.43 and 10.9 μM, respectively. Cryptolepine IC50 = 0.26 ± 0.13 μM and rivastigmine IC50 = 40.57 ± 0.69 μM were used as positive controls for AChE, while β-secretase inhibitor-IV was used as the positive control for BChE (IC50 = 0.74 ± 0.19 μM) (Fig. 20).5
 |
| | Fig. 20 Structure of caffeine-based triazoles as AChE and BChE inhibitors. | |
5. Conclusion
In drug development, purine is a promising lead molecule due to its ability to interact with various essential biomolecules involved in metabolic processes. Consequently, synthesizing novel derivatives and structural modifications of purine has become a significant area of research because it is considered a building block of nucleic acids and implicated in cellular energy transfer, and signaling pathways, many drug modifications, modified nucleosides, etc. This review focuses on the recently developed synthetic methods, chemical modifications of purine scaffolds, and some pharmacological activities, including their structure–activity relationships and modes of action. It hopes to pave the way for researchers to develop purine nuclei and explore the effect of different substituents in many biological evaluations. Finally, this review is expected to serve as a valuable reference for researchers involved in the investigation of novel purine scaffolds.
Abbreviations
| HOMA | Harmonic oscillator model of aromaticity |
| DNA | Deoxyribonucleic acid |
| RNA | Ribonucleic acid |
| ATP | Adenosine triphosphate |
| BChE | Butyrylcholinesterase |
| AChE | Acetylcholinesterase |
| mTOR | Mammalian target of rapamycin |
| VEGFR | Vascular endothelial growth factor receptor |
| VEGF | Vascular endothelial growth factor |
| PI3K | Phosphoinositide-3 kinases |
| BRAF | V-Raf murine sarcoma viral oncogene homolog B1 |
| JAK2 | Janus kinase 2 |
| BRD4 | Bromodomain-containing protein 4 |
| DAMN | 2,3-Diaminomaleonitrile |
| TEA | Triethyl amine |
| DAFN | Diaminofumaronitrile |
| AICN | Amino imidazole carbonitrile |
| THF | Tetrahydrofuran |
| DPPA | Diphenylphosphoryl azide |
| p-TsOH | Toluenesulfonic acid |
| DMSO | Dimethyl sulfoxide |
| AMNS | Aminomalononitrile p-toluenesulfonate |
| TOA | Triethyl orthoacetate |
| TOF | Triethyl orthoformate |
| TMSCN | Trimethylsilyl cyanide |
| DABCO | Diazabicyclo[2.2.2]octane |
| KOtBu | Potassium tert-butoxide |
| ADC | Acceptorless dehydrogenative coupling |
| DMF | Dimethylformamide |
| NOESY | Nuclear Overhauser effect spectroscopy |
| HMBC | Heteronuclear multiple bond correlation |
| DMB | Dimethylbenzamide |
| MWI | Microwave irradiation |
| SNAr | Aromatic nucleophilic substitutions |
| KOH | Potassium hydroxide |
| TFA | Trifluoroacetic acid |
| DIPEA | N,N-Diisopropylethylamine |
| CuAAC | Cu(I)-catalyzed azide–alkyne cycloaddition |
| NBS | N-Bromosuccinimide |
| DMAP | Dimethylaminopyridine |
| FDA | Food and Drug Administration |
| MTT | 3-[4,5-Dimethylthiazol-2-yl]-2,5-diphenyl tetrazolium bromide |
| IC50 | Half-maximal inhibitory concentration |
| EC50 | Half maximal effective concentration |
| EGFR | Epidermal growth factor receptor |
| SRB | Sulforhodamine B |
| SAR | Structure–activity relationship |
| HCC | Hepatocellular carcinoma |
| BHA | Butylated hydroxyanisole |
| 5-FU | 5-Fluorouracil |
| CPT | Camptothecin |
| WI38 | Human fetal lung fibroblast cells and its normal cell line |
| CDK2 | Cyclin-dependent kinase 2 |
| HEK-293 | Human embryonic kidney cells |
| MTX | Methotrexate |
| FGFRs | Fibroblast growth factor receptors |
| MIC | Minimum inhibitory concentration |
| MSRA | Methicillin-resistant Staphylococcus aureus |
| RAW 264.7 | Normal mammalian cells and macrophage cell line |
| COX-2 | Cyclooxygenase-2 |
| 15-LOX | 15-Lipoxygenase |
| HSV | Herpes simplex virus |
| CDV | Cidofovir |
| ACV | Aciclovir |
| TMV | Tobacco mosaic virus |
| DPPH | 2,2-Diphenyl-1-picrylhydrazyl |
| ABTS | 2,2-Azino-bis-3-ethylbenzothiazoline-6-sulphonic acid |
Data availability
All data will be made available on request.
Conflicts of interest
The author declares no conflicts of interest.
References
- G. Chakraborty, R. Mondal, A. K. Guin and N. D. Paul, Org. Biomol. Chem., 2021, 19, 7217–7233 RSC.
- F. Doganc, A. S. Aydin, E. Şahin and H. Göker, J. Mol. Struct., 2023, 1272, 134200 CrossRef CAS.
- N. Abad, S. Buhlak, M. Hajji, S. Saffour, J. Akachar, Y. Kesgun, H. Al-Ghulikah, E. Hanashalshahaby, H. Turkez and A. Mardinoglu, J. Mol. Struct., 2024, 1311, 138400 CrossRef CAS.
- T. Cheviet, I. Lefebvre-Tournier, S. Wein and S. Peyrottes, J. Med. Chem., 2019, 62, 8365–8391 CrossRef CAS PubMed.
- M. Sharma, A. Sharma, V. K. Nuthakki, S. Bhatt, U. Nandi and S. B. Bharate, Drug Dev. Res., 2022, 83, 1803–1821 CrossRef CAS PubMed.
- A. Chaurasiya, S. K. Wahan, C. Sahu and P. A. Chawla, J. Mol. Struct., 2023, 1274, 134308 CrossRef CAS.
- A. R. Mohamed, A. M. El Kerdawy, R. F. George, H. H. Georgey and N. M. A. Gawad, Bioorg. Chem., 2021, 107, 104569 CrossRef CAS PubMed.
- Y. Guo, Y. Zou, Y. Chen, D. Deng, Z. Zhang, K. Liu, M. Tang, T. Yang, S. Fu, C. Zhang, W. Si, Z. Ma, S. Zhang, B. Peng, D. Xu and L. Chen, Bioorg. Chem., 2023, 132, 106386 CrossRef CAS PubMed.
- M. A. Bhat, B. Tüzün, N. A. Alsaif, A. A. Khan and A. M. Naglah, J. Mol. Struct., 2022, 1257, 132600 CrossRef CAS.
- Y. Y. Yang, W. L. Wang, X. T. Hu, X. Chen, Y. Ni, Y. H. Lei, Q. Y. Qiu, L. Y. Tao, T. W. Luo and N. Y. Wang, Bioorg. Chem., 2023, 132, 106356 CrossRef CAS PubMed.
- A. K. Maddheshiya, M. Kumar, A. Tufail, P. S. Yadav, Y. Deswal, N. Yadav, T. P. Yadav and A. Dubey, ACS Appl. Bio Mater., 2024, 7(9), 5906–5924 CrossRef CAS PubMed.
- P. Salas-Ambrosio, S. Vexler, R. P S, I. A. Chen and H. D. Maynard, ACS Bio Med Chem Au, 2023, 3, 189–200 CrossRef CAS PubMed.
- M. Ali, E. N. Sholkamy, A. S. Alobaidi, M. K. Al-Muhanna and A. Barakat, ACS Omega, 2023, 8, 47304–47312 CrossRef CAS PubMed.
- F. He, J. Shi, Y. Wang, S. Wang, J. Chen, X. Gan, B. Song and D. Hu, J. Agric. Food Chem., 2019, 67, 8459–8467 CrossRef CAS PubMed.
- I. Schino, M. Cantore, M. de Candia, C. D. Altomare, C. Maria, J. Barros, V. Cachatra, P. Calado, K. Shimizu, A. A. Freitas, M. C. Oliveira, M. J. Ferreira, J. N. C. Lopes, N. A. Colabufo and A. P. Rauter, Pharmaceuticals, 2023, 16(1), 54 CrossRef CAS PubMed.
- J. Liu, S. Hong, J. Yang, X. Zhang, Y. Wang, H. Wang, J. Peng and L. Hong, J. Ovarian Res., 2022, 15, 93 CrossRef CAS PubMed.
- A. Chaurasiya, C. Sahu, S. K. Wahan and P. A. Chawla, J. Mol. Struct., 2023, 1280, 134967 CrossRef CAS.
- M. K. I. Attiogbe, H. y. Zhao, J. Wang, T. t. Huang, P. p. Yan, Y. n. Liu, W. Li, L. Cao, S. q. Zhang and Y. x. Cao, Life Sci., 2024, 336, 122308 CrossRef CAS PubMed.
- T. Manna, S. Maji, M. Maity, B. Debnath, S. Panda, S. A. Khan, R. Nath and M. J. Akhtar, Mol. Divers., 2024, 29(1), 81–848, DOI:10.1007/s11030-024-10870-4.
- D. D. Umesh, P. K. Nagaraj and L. K. Karadigere, Int. J. Pharm. Invest., 2025, 15, 19–29 CrossRef.
- S. Sharma, S. Mehndiratta, S. Kumar, J. Singh, P. M. S. Bedi and K. Nepali, Recent Pat. Anti-Cancer Drug Discov., 2015, 10, 308–341 CrossRef CAS PubMed.
- J. Linden, F. Koch-Nolte and G. Dahl, Annu. Rev. Immunol., 2019, 37, 325–347 CrossRef CAS PubMed.
- G. Burnstock, Neuropharmacology, 1997, 36, 1127–1139 CrossRef CAS PubMed.
- N. Rana, P. Grover and H. Singh, Curr. Top. Med. Chem., 2024, 24, 541–579 CrossRef CAS PubMed.
- A. A. Abu-Hashem, O. Hakami, M. El-Shazly, H. A. S. El-Nashar and M. N. M. Yousif, Chem. Biodivers., 2024, 21, e202400050 CrossRef CAS PubMed.
- S. Benkirane, H. Misbahi, M. Boudkhili, Y. K. Rodi, N. K. Sebbar and E. M. Essassi, Curr. Org. Chem., 2023, 27, 1683–1696 CrossRef CAS.
- B. D. Cheson, D. A. Vena, F. M. Foss and J. M. Sorensen, J. Clin. Oncol., 2024, 12, 2216–2228 CrossRef PubMed.
- A. Ragab, S. A. Fouad, Y. A. Ammar, D. S. Aboul-Magd and M. S. Abusaif, Antibiotics, 2023, 12, 128 CrossRef CAS PubMed.
- A. Ragab, M. S. Abusaif, N. A. Gohar, D. S. Aboul-Magd, E. A. Fayed and Y. A. Ammar, Bioorg. Chem., 2023, 131, 106307 CrossRef CAS PubMed.
- A. Ragab, M. S. Abusaif, D. S. A. Magd and M. M. S. Wassel, Drug Dev. Res., 2022, 83, 1305–1330 CrossRef CAS PubMed.
- M. M. Abdelgalil, Y. A. Ammar, G. A. M. E. Ali, A. K. Ali and A. Ragab, J. Mol. Struct., 2023, 1274, 134443 CrossRef CAS.
- R. Ayman, M. S. Abusaif, A. M. Radwan, A. M. Elmetwally and A. Ragab, Eur. J. Med. Chem., 2023, 249, 115138 CrossRef CAS PubMed.
- B. M. Bizzarri, A. Fanelli, S. Cesarini and R. Saladino, Eur. J. Org. Chem., 2022, 2022, e202200598 CrossRef CAS.
- I. Reva, H. Rostkowska and L. Lapinski, Photochem, 2022, 2, 448–462 CrossRef CAS.
- N. J. Green, J. Xu and J. D. Sutherland, J. Am. Chem. Soc., 2021, 143, 7219–7236 CrossRef CAS PubMed.
- M.-R. Huang, Y.-L. Hsu, T.-C. Lin, T.-J. Cheng, L.-W. Li, Y.-W. Tseng, Y. Chou, J.-H. Liu, S.-H. Pan, J.-M. Fang and C.-H. Wong, Eur. J. Med. Chem., 2019, 181, 111551 CrossRef CAS PubMed.
- Y.-W. Tseng, T.-J. Yang, Y.-L. Hsu, J.-H. Liu, Y.-C. Tseng, T.-W. Hsu, Y. Lu, S.-H. Pan, T.-J. R. Cheng and J.-M. Fang, Eur. J. Med. Chem., 2024, 265, 116042 CrossRef CAS PubMed.
- R. Mazzucato, M. Roberti, A. M. Capelli, F. Rancati, M. Biagetti, C. Fiorelli, P. Bruno, P. Ronchi, S. Bertolini, M. Corsi and D. Pala, Eur. J. Med. Chem., 2023, 254, 115331 CrossRef CAS PubMed.
- A. P. Bettencourt, M. Castro, J. P. Silva, F. Fernandes, O. P. Coutinho, M. J. Sousa, M. F. Proença and F. M. Areias, Med. Chem., 2019, 15, 341–351 CrossRef CAS PubMed.
- M. Pretze, C. Neuber, E. Kinski, B. Belter, M. Köckerling, A. Caflisch, J. Steinbach, J. Pietzsch and C. Mamat, Org. Biomol. Chem., 2020, 18, 3104–3116 RSC.
- J. M. Gonçalves, J. N. D. Gonçalves, A. S. Pêra, N. R. Senhorães, A. R. O. Rodrigues, R. Oliveira, P. J. G. Coutinho, E. M. S. Castanheira and A. M. Dias, Eur. J. Org. Chem., 2023, 26, e202300176 CrossRef.
- B. M. Bizzarri, A. Fanelli, L. Botta, M. De Angelis, A. T. Palamara, L. Nencioni and R. Saladino, RSC Adv., 2021, 11, 30020–30029 RSC.
- Z. Tber, N. G. Biteau, L. Agrofoglio, J. Cros, S. Goffinont, B. Castaing, C. Nicolas and V. Roy, Eur. J. Org. Chem., 2019, 2019, 5756–5767 CrossRef CAS.
- V. A. Verma, B. Halu, A. R. Saundane and R. S. Meti, Polycycl. Aromat. Compd., 2022, 42, 3694–3716 CrossRef CAS.
- S. El-Kalyoubi, F. Agili, W. A. Zordok and A. S. A. El-Sayed, Int. J. Mol. Sci., 2021, 22, 1–36 Search PubMed.
- S. El-Kalyoubi, F. Agili, I. Adel and M. A. Tantawy, Arab. J. Chem., 2022, 15, 103669 CrossRef CAS.
- V. V. Fedotov, E. N. Ulomsky, K. V. Savateev, E. M. Mukhin, D. A. Gazizov, E. B. Gorbunov and V. L. Rusinov, Synthesis, 2020, 52, 3622–3631 CrossRef CAS.
- H. Lei, S. Fan, H. Zhang, Y. J. Liu, Y. Y. Hei, J. J. Zhang, A. Q. Zheng, M. Xin and S. Q. Zhang, Eur. J. Med. Chem., 2020, 186, 111888 CrossRef CAS PubMed.
- Á. Lorente-Macías, I. Iañez, M. C. Jiménez-López, M. Benítez-Quesada, S. Torres-Rusillo, J. J. Díaz-Mochón, I. J. Molina and M. J. P. de las Infantas, Arch. Pharm., 2021, 354, 2100095 CrossRef PubMed.
- M. F. Polat, I. D. Şahin, P. Kul, R. C. Atalay and M. Tuncbilek, Bioorg. Med. Chem. Lett., 2024, 106, 129775 CrossRef PubMed.
- A. Kucukdumlu, M. Tuncbilek, E. B. Guven and R. C. Atalay, Acta Chim. Slov., 2020, 67, 70–82 CrossRef CAS PubMed.
- J. M. Orduña, N. del Río and M. J. Pérez-Pérez, Org. Biomol. Chem., 2023, 21, 5457–5468 RSC.
- S. Bigonah-Rasti, S. Sheikhi-Mohammareh, K. Saadat and A. Shiri, Polycycl. Aromat. Compd., 2022, 42, 2644–2654 CrossRef CAS.
- R. T. Attia, M. A. Ewida, E. Khaled, S. A. Fahmy and I. M. Fawzy, ACS Omega, 2023, 8, 37864–37881 CrossRef CAS PubMed.
- Y. Wang, Y. Pan, Z. Lv and S. Gou, Eur. J. Med. Chem., 2024, 271, 116415 CrossRef CAS PubMed.
- V. Petrov, R. J. Dooley, A. A. Marchione, E. L. Diaz, B. S. Clem and W. Marshall, Beilstein J. Org. Chem., 2020, 16, 2739–2748 CrossRef CAS PubMed.
- Y. Fu, D. Liu, H. Zeng, X. Ren, B. Song, D. Hu and X. Gan, RSC Adv., 2020, 10, 24483–24490 RSC.
- A. Villegas, R. Satheeshkumar, A. Ballesteros-Casallas, M. Paulino, A. Castro, C. Espinosa-Bustos and C. O. Salas, J. Heterocycl. Chem., 2022, 59, 97–111 CrossRef CAS.
- A. Bistrović Popov, A. B. Popov, R. Vianelo, P. Grbčić, M. Sedić, S. K. Pavelić and S. Raić-Malić, Molecules, 2021, 26, 3334 CrossRef PubMed.
- S. Konduri, J. Prashanth, V. S. Krishna, D. Sriram, J. N. Behera, D. Siegel and K. P. Rao, Bioorg. Med. Chem. Lett., 2020, 30, 127512 CrossRef CAS PubMed.
- M. N. S. Rad, S. Behrouz, K. Shahbazkhani, M. Behrouz, E. Zarenezhad and A. Ghanbariasad, RSC Adv., 2023, 13, 24656–24673 RSC.
- A. Q. A. Nadaf, S. Dixit, M. Yaseen, S. Mantur, M. S. Najare, S. Joshi, S. Vootla and I. A. M. Khazi, ChemistrySelect, 2020, 5, 8635–8643 CrossRef CAS.
- V. P. Krasnov, G. L. Levit, V. V. Musiyak, D. A. Gruzdev and V. N. Charushin, Pure Appl. Chem., 2020, 92, 1277–1295 CrossRef CAS.
- A. Zagórska, A. Czopek, A. Jaromin, M. Mielczarek-Puta, M. Struga, D. Stary and M. Bajda, Materials, 2021, 14(15), 4156 CrossRef PubMed.
- G. Dilek, I. O. Tekin, B. Coban, A. Disli and Z. Gercek, Med. Chem. Res., 2021, 30, 84–97 CrossRef CAS.
- A. Singh, S. K. Maiti, H. P. Gogoi and P. Barman, Polyhedron, 2023, 230, 116244 CrossRef CAS.
- O. S. Afifi, O. G. Shaaban, H. A. A. El Razik, S. E.-D. A. Shams El-Dine, F. A. Ashour, A. A. El-Tombary and M. M. Abu-Serie, Bioorg. Chem., 2019, 87, 821–837 CrossRef CAS PubMed.
- M. E. Khalifa, J. Mol. Struct., 2021, 1229, 129843 CrossRef CAS.
- A. Y. Hassan, E. S. Abou-Amra and S. A. El-Sebaey, J. Mol. Struct., 2023, 1278, 134930 CrossRef CAS.
- E. M. Husseiny, H. S. Abulkhair, A. Saleh, N. Altwaijry, R. A. Zidan and F. G. Abdulrahman, Bioorg. Chem., 2023, 140, 106789 CrossRef CAS PubMed.
- W. C. Hahn, J. S. Bader, T. P. Braun, A. Califano, P. A. Clemons, B. J. Druker, A. J. Ewald, H. Fu, S. Jagu and C. J. Kemp, Cell, 2021, 184, 1142–1155 CrossRef CAS PubMed.
- A. Chaurasiya, S.
K. Wahan, C. Sahu and P. A. Chawla, J. Mol. Struct., 2023, 1274, 134308 CrossRef CAS.
- Y. Hu, S. Hu, G. Pan, D. Wu, T. Wang, C. Yu, M. F. Ansari, R. R. Y. Bheemanaboina, Y. Cheng, L. Bai, C. Zhou and J. Zhang, Bioorg. Chem., 2021, 114, 105096 CrossRef CAS PubMed.
- B. Paulsen, K. A. Fredriksen, D. Petersen, L. Maes, A. Matheeussen, A. O. Naemi, A. A. Scheie, R. Simm, R. Ma, B. Wan, S. Franzblau and L. L. Gundersen, Bioorg. Med. Chem., 2019, 27, 620–629 CrossRef CAS PubMed.
- A. Q. A. Nadaf, M. S. Najare, M. Garbhagudi, S. Mantur, M. G. Sunagar, S. Gaonkar, S. Joshi and I. A. M. Khazi, Chem. Biodivers., 2020, 17(5), e2000053 CrossRef CAS PubMed.
- A. Ohler, P. E. Taylor, J. A. Bledsoe, A. T. Iavarone, N. C. Gilbert and A. R. Offenbacher, ACS Catal., 2024, 14, 5444–5457 CrossRef CAS PubMed.
- A. F. Mohammed, G. Andrei, A. M. Hayallah, S. G. Abdel-Moty, R. Snoeck and C. Simons, Bioorg. Med. Chem., 2019, 27, 1023–1033 CrossRef CAS PubMed.
- Y. Deng, M. Chen, J. Yi and Y. Zheng, Phytochem. Lett., 2024, 59, 10–14 CrossRef CAS.
- S. N. Mangasuli, K. M. Hosamani and P. B. Managutti, Heliyon, 2019, 5, e01131 CrossRef PubMed.
- F. Valdes, N. Brown, A. Morales-Bayuelo, L. Prent-Peñaloza and M. Gutierrez, Antioxidants, 2029, 8(10), 468 CrossRef PubMed.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab
*ab