
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Contrast-enhanced imaging of carbon fiber composites using hafnium oxide nanocrystals†
Eline
Goossens
 ab,
Ives
De Baere
c,
Yuriy
Sinchuk
d,
Evert
Dhaene
a,
John
De Vos
a,
Pauline
Rooms
a,
Matthieu N.
Boone
e,
Jonathan
De Roo
b,
Isabel
Van Driessche
a,
Wim
Van Paepegem
c and
Klaartje
De Buysser
*a
ab,
Ives
De Baere
c,
Yuriy
Sinchuk
d,
Evert
Dhaene
a,
John
De Vos
a,
Pauline
Rooms
a,
Matthieu N.
Boone
e,
Jonathan
De Roo
b,
Isabel
Van Driessche
a,
Wim
Van Paepegem
c and
Klaartje
De Buysser
*a
aDepartment of Chemistry, Ghent University, 9000 Ghent, Belgium. E-mail: Klaartje.DeBuysser@UGent.be
bDepartment of Chemistry, University of Basel, 4058 Basel, Switzerland
cDepartment of Materials, Textiles and Chemical Engineering, Ghent University, 9052 Zwijnaarde, Belgium
dLaboratory of Mechanics Paris-Saclay, 91192 Gif-sur-Yvette Cedex, France
eCenter for X-ray Tomography, Department of Physics and Astronomy, Ghent University, 9000 Ghent, Belgium
First published on 28th March 2025
Abstract
Carbon fiber-reinforced polymers (CFRPs) are of utmost importance in high-performance structural applications due to their unique combination of light-weight, yet high-strength behaviour. However, their behaviour and damage mechanisms are not fully understood. Damage propagation models need to take into account the type, geometry and orientation of a multitude of failure mechanisms and defects. Micro-Computed Tomography (micro-CT) data could provide the necessary input for these models, but the poor contrast between the carbon fibers and the polymer matrix does not allow automatic geometry extraction needed as input. To overcome this issue, hafnium oxide nanocrystals (HfO2 NCs) were introduced as CT contrast agents to the polymer matrix to provide the required contrast. To ensure a homogeneous and stable dispersion of the NCs in the epoxy resin, a bisphosphonic acid ligand consisting of ethylene glycol oligomer segments was used. The NCs did not significantly alter important parameters such as matrix stiffness, viscosity, glass transition temperature and curing time, enabling them to be implemented without having to alter current composite resin infusion methods. NC-doped CFRPs with 5 and 10 m% of HfO2 NCs added showed a drastic improvement in CT contrast, allowing for segmentation of the carbon fiber tows and visualization of micrometer-scale cracks. The contrast-enhanced NC-doped composites thereby enable the validation of damage models by accurate micro-CT data.
Introduction
Fiber-reinforced polymers (FRPs) are the dominant composite material used in applications such as aerospace,1,2 automotive,3,4 wind energy5,6 and more.7–9 Their high corrosion resistance and specific strength and stiffness make them of interest for weight-critical structural applications. In structural applications, FRPs typically consist of a thermoset (cross-linked) polymer matrix because of its improved thermal and chemical stability, as well as fiber bonding properties, compared with thermoplastic (non-crosslinked) matrices.10,11 The polymer is reinforced with micrometer-sized fibers, typically carbon (CFRPs) or glass fibers (GFRPs). The resulting material is heterogeneous and anisotropic in nature, making its behaviour different from traditional structural materials such as steel and convoluting the prediction of damage. The type, geometry and orientation of a multitude of failure mechanisms and defects (i.e. delamination, matrix cracking, fiber failure, fiber–matrix debonding, inclusions and voids)12,13 need to be taken into account. Furthermore, the interactions between these defects are of interest, as they often lay the basis for final composite failure.Micro-Computed Tomography (micro-CT) has become paramount in the development of damage and failure prediction models, as it is the only non-destructive evaluation technique that gives a high-resolution three-dimensional (3D) representation of the scanned object with a minimum of sample preparation.14–17 By sending X-rays through the sample, its internal structure is revealed with (sub-)micrometer resolution. Micro-CT analysis is therefore used in composite research for the analysis and identification of damage and microstructures.12 It has been applied for crack visualization,18,19 damage due to loading,20–22 impact,23,24 fatigue25,26 and to determine manufacturing defects such as porosity27,28 and fiber misalignment.29,30 With time-lapse 3D imaging even the in situ propagation of damage effects during loading can be visualized.18,31 CT relies on differences in X-ray attenuation to generate a tomographic image. The aforementioned studies are thus limited to metal and ceramic matrix composites or synchrotron sources.12 Synchrotron CT scans can obtain better contrast-to-noise and are therefore often used, but come at the downside of large and expensive facilities with limited accessibility. Crucial research is therefore slowed down or restricted because synchrotron sources are time-consuming and can only scan small samples. For lab-based sources, the current state-of-the-art to visualize cracks in polymer matrices utilizes iodine-based dye penetrants. However, this approach can only visualize the cracks that are exposed to the penetrant, i.e. those that are directly connected to the external surface.12,15,32 Dye penetrants also affect the growth of cracks and significantly stiffen the tows, which makes the mechanical performance after staining unreliable.32–34
Simulations of composite behavior and its damage propagation are primarily done via finite element modelling (FEM), which subdivides a highly complex system into a mesh of smaller, manageable elements whose behaviour is more readily understood.35 Significant improvements have been obtained with models based on micro-CT images,36–41 but the quality of the final model strongly depends on the input CT image quality.42 The image-based mesh is constructed from the micro-CT scan via segmentation, a post-processing technique which virtually separates different elements based on their difference in greyscale value (i.e. contrast).17 The segmentation efficiency is mainly dependent on signal-to-noise and contrast,43 and the latter is largely material-dependent. CT analysis of GFRPs can readily extract the glass fibers from the polymer matrix. CFRPs on the other hand have poor contrast between carbon fibers and the polymer matrix, making automatic segmentation of single fibers or tows from the undoped matrix a challenging problem. CFRPs thus rely on contrast enhancement for segmentation of CT images. Besides the aforementioned dye penetrants to visualize surface-accessible cracks, contrast enhancement of the boundaries between tows and resin has been done using nanometer-sized coatings on the tows, typically gold or copper, or using resin additives.32,44 The addition of high-density particles which improve the contrast of the matrix is the only option allowing visualization of cracks throughout the complete matrix and making automatic segmentation possible even to the individual fiber level. Djukic et al. introduced Al2O3 and BaSO4 nano- and microparticles to epoxy; however, the particles were not uniformly distributed and did not result in a stable nanocomposite mixture.44 An unstable particle-based contrast agent creates problematic local artefacts in micro-CT, rather than providing homogeneous contrast enhancement. Furthermore, agglomerated particles have a large, typically negative, effect on the properties of the matrix, such as an increase in viscosity of the resin which complicates the manufacturing process. The issues with gravitational sinking and/or agglomeration of the nanoparticles mean that nanoparticle-based contrast agents in polymer nanocomposites are limited and there are no homogeneous dispersions available for fiber-reinforced polymers. High-atomic number nanoparticles (NPs) such as Au,45,46 TaOx,47,48 WOx,49,50 Bi2S3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 51,52 and HfO253–56 have been used in clinical settings as CT contrast agents. Recently, we used HfO2 nanocrystals (NCs) as CT contrast agents in a polymethylmethacrylate polymer resin for the casting of biological specimens.57 In this study, we demonstrate the potential of HfO2 NCs as CT contrast agents for CFRPs. By careful surface chemistry design, we obtain a homogeneous and stable dispersion in the epoxy resin using of a bidentate phosphonic acid ligand. The HfO2 NCs remain stable during and after curing, and significantly improve the CT contrast of the composites. Because of the homogeneous distribution, mechanical properties of the NC-doped matrix, such as matrix stiffness, viscosity, glass transition temperature and curing time, are not negatively affected. Current existing resin infusion methods can be used without adaptation. The NC-doped CFRPs demonstrate significant contrast enhancement, which increases with increasing HfO2 NCs added. The CT contrast allows for straightforward and accurate segmentation of the carbon fiber tows, opening the door to modeling of carbon fiber composites with automatic segmentation using lab-based CT. These damage and behaviour models lead to a better understanding of composite behaviour, an important step towards the elimination of unexpected composite failure.
51,52 and HfO253–56 have been used in clinical settings as CT contrast agents. Recently, we used HfO2 nanocrystals (NCs) as CT contrast agents in a polymethylmethacrylate polymer resin for the casting of biological specimens.57 In this study, we demonstrate the potential of HfO2 NCs as CT contrast agents for CFRPs. By careful surface chemistry design, we obtain a homogeneous and stable dispersion in the epoxy resin using of a bidentate phosphonic acid ligand. The HfO2 NCs remain stable during and after curing, and significantly improve the CT contrast of the composites. Because of the homogeneous distribution, mechanical properties of the NC-doped matrix, such as matrix stiffness, viscosity, glass transition temperature and curing time, are not negatively affected. Current existing resin infusion methods can be used without adaptation. The NC-doped CFRPs demonstrate significant contrast enhancement, which increases with increasing HfO2 NCs added. The CT contrast allows for straightforward and accurate segmentation of the carbon fiber tows, opening the door to modeling of carbon fiber composites with automatic segmentation using lab-based CT. These damage and behaviour models lead to a better understanding of composite behaviour, an important step towards the elimination of unexpected composite failure.
Results and discussion
Synthesis of HfO2 nanocrystals
Hafnium oxide nanocrystals were synthesized via a microwave-assisted solvothermal synthesis, starting from HfCl4·2THF in benzyl alcohol as shown in Fig. 1A.58,59 We recently scaled up this reaction, resulting in multiple grams of HfO2 NCs per synthesis,57 and we have shown that the reaction time can be reduced to 1 hour.60 The bottleneck for large-scale NC production is currently the post-synthetic purification, which requires multiple time-consuming centrifugation steps. We present a work-up protocol optimized for gram-scale syntheses, without the need for centrifugation. After synthesis the reaction mixture is split up into an aqueous phase containing the NCs and an organic phase, which can be separated using a separation funnel. From the observation that particles agglomerate to clusters larger than 1 μm when an appropriate non-solvent is added, further purification of excess chloride and ligands is possible using por5 fritted filters. The filtrate is mostly NC-free, while the agglomerated NCs remain on the filter. The resulting NCs are ellipsoidal, with a major axis of 8.5 ± 5.5 nm and a minor axis of 5.2 ± 2.5 nm (μ ± 3σ) as determined via transmission electron microscopy (TEM) (Fig. 1B). Powder X-ray diffraction (PXRD) confirmed their monoclinic (P21/c) crystal structure (Fig. 1C). After synthesis, the resulting particles are de-aggregated using a mixture of fatty acid in the presence of a base to functionalize the surface.58 Typically the NC surface is functionalized with oleic acid, providing excellent stability in nonpolar solvents. The final NC stability is not affected by the filter steps, as evidenced by dynamic light scattering (DLS). In chloroform, the suspension of HfO2/oleate has a Z-average value of 17 ± 6 nm (μ ± 3σ)(Fig. 1D). The appropriate non-solvent is used to precipitate the NCs and wash them on the fritted filter. The NCs are washed with the same nonsolvent in order to remove all excess (free) ligand. Broadened 1H nuclear magnetic resonance (NMR) peaks are indicative of bound ligands and show good purification (Fig. S1†).61–63 | ||
| Fig. 1 (A) Synthesis reaction scheme of HfO2 NCs starting from HfCl4·2THF in benzyl alcohol. (B) TEM image of the synthesized NCs. The size distribution and a zoom of a single NC is shown in the top and bottom left corner, respectively. (C) XRD spectrum (grey) with a monoclinic reference spectrum (black, COD ID 9013470). (D) Volume size distribution DLS measurement of HfO2/oleate in chloroform. | ||
Influence of the surface chemistry
High-resolution (micro-) CT can obtain resolutions in the sub-micrometer range.64,65 The HfO2 NCs should therefore be homogeneously dispersed and remain stable in the composite resin during curing. Micrometer-sized NC agglomerates are detected in the micro-CT scans, complicating or even eliminating the possibility of automatic segmentation. The most common thermoset matrices for FRPs are epoxy, polyester, vinylester, phenolic and polyurethane.10 Though our approach can be expanded to different resins by optimizing the surface chemistry to match the resin of choice, we chose to focus on epoxy as it is considered the most widely utilized resin for high-performance structural applications.66 Although it is more expensive, epoxy outperforms other resins in terms of mechanical properties (e.g. tensile strength) and durability (corrosion and moisture resistance).66 We used a commercially available epoxy (Epikote RIMR135) with a diamine curing agent (Epikure RIMH137) widely used for composite synthesis.67–71 The stability of NCs is largely determined by the ligands capping the NC surface,72,73 especially in challenging environments such as polymer resins.74–76 Matrix-compatible ligands improve the enthalpic compatibility and have been shown to give great colloidal stability.77,78 To assess the influence of ligand binding groups and tails on the stability in the resin, three different binding groups with a high binding affinity for the metal oxide surface were chosen: carboxylic acids,79–81 phosphonic acids82–86 and bisphosphonic acids87–89 (Fig. 2A). The colloidal stability of the ligand-functionalized NCs in the resin was determined by DLS measurements. Lower solvodynamic diameters indicate well-dispersed NCs. An overview of all 1H NMR spectra confirming the successful exchange and the corresponding volume distributions in the resin according to DLS is given in Fig. S2.† A selection of those volume distributions in Fig. 2B highlights two noticeable trends. First, carboxylic acids perform worse overall than phosphonic acids and bisphosphonic acids, indicating that the binding group strength plays a role in the overall colloidal stability. Second, the polarity of the ligand tail is the determining factor for the colloidal stability in the resin. Ligands with repeating polyethyleneglycol (PEG) units have better stability than aliphatic chains. The best colloidal stability in the resin, confirmed by DLS, was obtained with (bis)phosphonic acids having PEG-units as organic tail. Based on the Z-average value, which describes the intensity-weighted average particle size and is most sensitive to agglomeration, bisPA-mPEG[6] (μ ± 3σ = 23 ± 6 nm) and PA-mPEG[2] (24 ± 3 nm) have superior stability to the other ligands. Selected mixed ligand shells, combining polar and nonpolar tails, that were tested do not outperform a fully (bis)phosphonic acid shell (Fig. S2†). | ||
| Fig. 2 (A) Overview of the ligand library separated by binding group and polarity. (B) Volume size distribution DLS data of HfO2 NCs characterized with several ligands. (C) TEM slice of 5 m% HfO2/BisPA-mPEG[6] in epoxy shows the homogeneous distribution. | ||
Solely based on DLS data one might expect that the bisphosphonic acid only slightly outperforms the phosphonic acid in terms of stability in the resin. After curing, however, clear visual turbidity in the phosphonic samples could be observed whereas this was completely absent in the bisphosphonic acid samples, shown in Fig. S3A.† The turbidity is linked to the presence of large NC agglomerates, evidenced by TEM images of the nanocomposite samples (Fig. S3D†). The epoxy resin is poly(bisphenol-A-co-epichlorhydrin)-based and crosslinks upon addition of a combination of diamine curing agents. We found that the latter reacts with the phosphonic acid on the NC surface and strips it during curing, leaving the NCs destabilized in the resin. 31P NMR spectra of the phosphonic acid ligand-functionalized NCs originally showed a broad peak, indicative of bound ligands. After addition of the curing agent, a sharp peak of free ligand appeared within minutes, proving that ligands are actively stripped from the surface (Fig. S3B†). Bisphosphonic acids on the other hand have a higher binding affinity to the HfO2 surface due to their multidentate binding mode (evidenced by the lower ligand density, see ESI†) and do not get stripped from the surface during curing. A control experiment using an excess of curing agent revealed that ligand removal for the bisphosphonic acid was only observed after 48 h, but as the epoxy resin itself will be cured in a few hours, the particles are set in place before they can agglomerate. TEM slices of cured composites showed that no agglomerations larger than 50 nm were present in the nanocomposite sample of HfO2/bisPA-mPEG (Fig. 2C). In addition, the NCs were stable in the uncured resin mixture up to a week, demonstrated using DLS in Fig. S4.† Bisphosphonic acids will therefore be the preferred ligand for other resins which contain materials that could compete with the surface ligands, e.g. polyesters or vinylesters.
Ligand synthesis and exchange
In the application of CT contrast-enhanced CFRPs, the NC dispersion is required to be both homogeneous and stable to create homogeneous contrast and have a minimal influence on the mechanical properties. After evaluating the surface chemistry, the bisphosphonic acid bisPA-mPEG[6] was shown to result in such a dispersion. Having thus found the optimum ligand, we sought to improve its synthesis and post-synthetic ligand exchange. We based our synthesis of bisPA-mPEG[6] on the synthetic strategy for alkylbisphosphonic acids published by Egorov et al.90 This one-pot synthesis started from the corresponding acid (Fig. 3A) and received higher purity (97% based on 31P NMR) than the commercially available bisPA-mPEG[6]. The final ligand was fully characterized with MS and NMR spectroscopy, see Fig. S5–8.† The synthesized bisPA-mPEG[6] could be successfully exchanged on the HfO2 NC surface as confirmed in Fig. 3B by the broadened 1H and 31P NMR resonances. The functionalized NCs had a comparable ligand density (1.26 ligands per nm2) to the commercial equivalent (1.28 ligands per nm2) as determined via thermogravimetric analysis (TGA),57 see Fig. 3C. Their stability in the epoxy resin slightly outperformed the NCs functionalized with the commercial equivalent (Fig. S9†), likely due the higher quality compared with the commercial product. The synthesis can also be applied starting from more economical carboxylic acids with shorter PEG-chains and thus allows for the creation of bisphosphonic acid ligands with a variety of ligand tail lengths or types better suited for other resins. Shorter bisphosphonic acids provide also good colloidal stability in epoxy (Fig. S2†). To demonstrate this, we synthesized bisPA-mPEG[2], again with higher purity (96% based on 31P NMR) than the commercially available alternative, and fully characterized with MS and NMR spectroscopy, shown in Fig. S10–S13.† The synthesized HfO2/bisPA-mPEG[2] NC stability in the resin was also slightly better than the commercial ligand, see Fig. S14.† | ||
| Fig. 3 (A) Reaction scheme of one-pot synthesis of bisPA-mPEG[6] starting from the carboxylic acid. (B) 1H NMR spectrum of HfO2/BisPA-mPEG[6] NCs in CDCl3. The inset shows the 31P NMR. (C) TGA data show the mass loss of the ligands attached to the surface. Measurement was done from 25 °C to 800 °C, followed by a 30-minute isothermal step at 800 °C. | ||
The NC surface is typically stabilized by oleate ligands after synthesis (Fig. S1†). Bisphosphonic acids can be exchanged on the metal oxide surface from oleate in an X-for-X ligand exchange91 due to their higher binding affinity.88 Following our previously reported protocol,57 we simplified the post-synthetic surface modification. The bisphosphonic acid was added directly after synthesis, eliminating the need for the oleate in-between step. Fig. S15† shows the reaction scheme for the indirect and direct exchange. The direct exchange was achieved using triethyl amine as a base to drive the reaction equilibrium to a full exchange, see Fig. S16.† A photometric chloride test confirmed the removal of all adsorbed chloride on the NC surface after the exchange (calculations in ESI†).
NC-doped epoxy mechanical properties
In order to construct accurate damage propagation models, the presence of the NCs should not negatively affect the composite's mechanical properties, which typically takes place when large agglomerates are present. NPs influence the local stress state of the surrounding polymer matrix, creating a radial zone which substantially differs from the properties of the bulk material.92–94 An uneven dispersion results in heterogeneity and structural defects.81,95,96 Furthermore, examples with carbon nanotubes (CNTs) are known to cause drastic viscosity increases when mixing them in a polymer resin,97–100 thereby limiting their application potential. Using HfO2/bisPA-mPEG[6], we achieved a homogeneous and stable dispersion in the resin and could evaluate nanocomposite samples with 0, 5 and 10 m% (ligand weight subtracted) of NCs added. To ensure that matrix is barely affected by the presence of the NCs, we measured a set of properties which influence polymer performance: matrix stiffness, insoluble fraction, water absorption, degradation temperature, curing time, viscosity and glass transition temperature.The Young's modulus was calculated from the stress–strain curve measured by a tensile test, shown in Fig. 4A. With increasing mass percentage of NCs added, the modulus increased, as has been observed before with several (metal oxide) nanoparticles.93,95,101 Minor improvements in ultimate force and ultimate stress were observed as well (values in Table S1†). Due to their brittle nature, epoxies have a low elongation at break.66 The presence of the NCs slightly improves the total elongation; similar effects have often been attributed to crack bridging and deflection, though typically for larger (>200 nm) NPs, or increased flexibility (necking) of the NCs.102,103 As the carbon fibers are the main load-bearing constituents, this minor matrix improvement has a negligible effect on the overall strength of the structure. Fig. 4B shows the evolution in insoluble fraction, the fraction of material that has formed a three-dimensional crosslinked network. The reference (0 m% NCs) sample was completely cured (μ ± σ = 100 ± 0%). While the insoluble fraction remains high, it decreased with m% of NCs added (97.3 ± 1.8% for 5 m% and 96.1 ± 0.4% for 10 m%). We hypothesize this was caused by the ligands on the NC surface interacting with the diamine curing agent, lowering the crosslink density, i.e. the insoluble fraction.
 | ||
| Fig. 4 Nanocomposite properties of epoxy with 0, 5 and 10 m% HfO2/BisPA-mpPEG[6] added. (A) Stress–strain measurements; the inset shows the Young's modulus calculated from the data. An example of necking on a dogbone-shaped sample is drawn. (B) The insoluble fractions and (C) water absorption after 7 days. (D) TGA data shown as mass loss in function of temperature. The degradation temperatures (defined at 5 m% sample loss) are given. (E) The curing time defined by the crossover of the storage modulus G′ and loss modulus G′′. Moduli are measured at 80 °C in function of time. (F) Viscosity (average of 0.1–100 Hz) and viscosity in function of increasing temperature. (G) DSC data plotted as heat flow in function of temperature; the glass transition temperatures are indicated. | ||
Water absorption is an issue in high-performance materials. Specifically in epoxies, it has a plasticizing effect (reduced Tg and tensile properties) and leads to polymer chain scission.104–108 These drawbacks are also cause for concern in epoxy/(carbon) fiber composites, even leading to debonding of the polymer from the fibers.106,109,110 Addition of nanofillers (nanoclay, nanotubes, nanoparticles) has been shown to slightly reduce the water uptake with increasing mass percentage, often accredited to the tortuosity path created by the nanofillers,105,111,112 though concerns have been raised about some nanofillers increasing the water absorption.113,114 For our samples, we found a small decrease in water absorption with increasing mass percentage of HfO2 NCs added (Fig. 4C).
The maximum temperature at which a polymer can be used should not be significantly affected by the NCs. This degradation temperature, defined as the temperature at which 5 m% mass is lost, was determined via TGA measurements (Fig. 4D). It was largely unaffected by the presence of the NCs and decreased a few degrees Celsius with increasing mass percentage. Similarly, the NC-doped resin should not have an accelerated curing time. The curing times of the three formulations were determined by the crossover of the storage modulus G′ and loss modulus G′′ at 80 °C, see Fig. 4E. For all samples this crossover occurred around ca. 7 kPa, indicating similar homogeneities of all the mixed resins (Fig. S17†). A slightly faster (4 min) or slower (5 min) gelation was observed for 10 m% and 5 m%, respectively, compared with the commercial formulation. As the curing process is >24 h, no significant differences are expected.
A drastic increase in resin viscosity would impede the infusion process of the carbon fiber fabric. However, contrary to the large viscosity increases observed with CNTs, the HfO2 NCs only showed a minimal viscosity increase which did not affect the ability for resin infusion. If needed, the viscosity increase can also be combatted by a small temperature increase (ΔT ≈ 5 °C), rapidly dropping even below the value for 0 m% at room temperature, as demonstrated in Fig. 4F. Finally, the glass transition temperature (Tg) was determined by DSC measurements, and is the temperature at which the materials’ physical properties will start to change. A small, but non-significant decrease in Tg was found, attributed to the plasticizing effect of the NCs (Fig. 4G). Overall the NCs had a slightly positive to negligible effect on the final nanocomposite's mechanical properties. Importantly, the NCs can no longer migrate or agglomerate after curing. The minimal impact on the mechanical properties is therefore maintained even under varying environmental conditions, as confirmed in Fig. S18.†
Proof-of-concept CT
Contrast enhancement using the HfO2 NCs was evaluated first by doping the epoxy resin, ranging from 0 to 10 m% NCs added (mass does not include the ligands), and adding a single carbon fiber bundle to each sample. Small fragments were cut after curing and analyzed via micro-CT using a peak tube potential of 90 kV and a 173 μm3 voxel size.A signal-to-noise plot, shown in Fig. S19,† was constructed by comparing the ratio of the attenuation values of the background with those of the polymer matrix. The plot shows a linear increase of CT contrast with m% NCs added. Nonetheless, limited contrast improvement was observed between 0, 1 and 2.5 m%, as can be seen in Fig. 5A. This was attributed to the difference in X-ray attenuation of the carbon fibers and the polymer matrix. The latter attenuated slightly less than the carbon fibers when no NCs were added, as can be seen in Fig. 5B which plots the linear attenuation coefficients. Upon improving the attenuation of the polymer matrix by adding the HfO2 NCs, the contrast between matrix and fibers decreased before the attenuation of the matrix overtook the fibers and the contrast improved. The attenuation coefficient of the fibers appeared to increase as well with increasing m% NCs; this artefact is due to the partial volume effect averaging the greyscales of the fibers with the polymer within voxels, i.e. the actual attenuation difference will be higher than depicted here.
 | ||
| Fig. 5 Contrast enhancement of HfO2 NC-doped CFRPs with increasing m% of NCs added. (A) Axial view of slices containing one fiber bundle, (B) linear attenuation coefficient of the fibers and the polymer matrix shows the cross-over in contrast, (C) 3D volume renders of samples with increasing m%. From 5 m% on it was possible to automatically extract the fiber bundle from the polymer matrix. | ||
From 5 m% on, the carbon fiber bundle was both visually clearly distinguishable and could be automatically extracted, Fig. 5C. We opted for seeded region-growing based segmentation for its speed, high accuracy and minimal manual input needed.115 The segmentation starts from a manually placed seed and grows automatically to regions of the same contrast, continuously evaluating neighbouring voxels's greyscales. When a user pre-determined contrast difference is encountered it is indicated as the edge of this region.16,17 In the range of 5 to 10 m% concentration the obtained segmentation was more exact and detailed with increasing HfO2 NCs. No agglomerations were found in any of the samples throughout the entire 3D volume due to the stability provided by the bisPA-mPEG[6] ligand. Fig. S20† shows the importance of choosing a well-suited contrast agent by comparing the X-ray contrast of HfO2 (high Z and high density) and ZrO2 NCs (lower Z and density). To the contrary, the negligible difference in tensile curves indicates that the material type is of lesser importance for the mechanical properties.
Contrast-enhanced CT visualization of CFRPs
Having determined the minimum amount of NCs needed for automatic extraction of the fiber bundles in the previous section, NC-doped CFRPs were manufactured with 5 and 10 m% of HfO2/BisPA-mPEG[6]. Laminates consisting of six layers of woven carbon fiber fabric were made using vacuum-assisted resin infusion (VARI). Note that since the presence of the NCs does not influence the resin properties, composite manufacturing techniques can be used without having to adapt them. This method can thus be expanded to other (non-)continuous FRPs and processes such as hand lay-up, resin transfer molding, pultrusion, etc. The infusion is not hindered after addition of the NCs due to the minimal viscosity increase (Fig. 4F). Gravimetric analysis and radiography of the cured nanocomposite present in the inlet and outlet tube connected to the infusion setup showed that the amount of NCs entering and exiting the fiber stack was equal, confirming the absence of any possible filtering of the NCs caused by the densely packed carbon fibers (Fig. S21†). For woven fabrics, mesoscopic finite element modelling (FEM) starts from the representative unit cell (RUC), a small and repeatable volume which statistically represents the structure. The whole fabric is reconstructed by in-plane translations of this RUC.42 Small samples (3 × 3 mm) were diamond cut from the cured laminate and used as RUC. Fig. 6A shows a schematic of the RUC which contains fibers traversing in- and out-of-plane. The small sample size allows for high-resolution CT scans and samples were scanned using a tube potential of 90 kV and a voxel size of 3.53 μm3. Fig. 6B shows a cross-section slice of the NC-doped CFRPs compared with an undoped reference sample (videos going through the complete sample are supplied in the ESI†). The contrast between fibers and polymer matrix was significantly higher with the addition of the HfO2 NCs. The contrast-enhanced CFRPs allowed for a clear distinction between fiber bundles and the direction in- or out-of-plane. The corresponding greyscale histograms also show an increasing shift between the larger peak associated with the fibers and those with the matrix, as well as a shift away from the background values. | ||
| Fig. 6 (A) Schematic showing the point-of-view of the micro-CT scan slices on the woven fabric. Weft and warp directions are indicated. (B) Cross-section slices of micro-CT scans demonstrate the CT contrast improvement of the CFRPs with, from left to right, 0, 5 and 10 m% HfO2/BisPA-mPEG[6] NCs added to the polymer matrix. (C) 3D volume renders of the 5 and 10 m% samples. The segmented matrix is highlighted in blue. (D) The automatic segmentation on the micro-CT scans is increasingly more accurate with HfO2 NCs added. (E) Greyscale histograms with Gaussians fitted to the fibers and polymer matrix contributions. For the 0 m% sample the matrix cannot be distinguished from the fibers. | ||
Using the same region-growing approach as described above, we attempted to automatically segment the fiber bundles from the polymer matrix. In Fig. 6C and D the segmented region is highlighted. For the undoped reference CFRP, the method failed to segment the tows region. With 5 m% of NCs added, the automatic segmentation could extract the polymer-rich zones between fiber bundles and differentiate the polymer from the fibers. Increasing the amount of NCs to 10 m% made the segmentation even more detailed. It could detect the different fiber bundles both in- and out-of-plane. A more accurate segmentation was achieved using a deep learning approach, which is comparatively more resource-intensive in terms of computational resources and requires manual annotation. Deep learning models, a subset of machine learning, are trained by labeled data sets and use neural networks to understand complex patterns.
We constructed a training set by preprocessing and manually annotating the 5 m% and 10 m% scans. The training dataset included information from only 20 slices of each CT image (10 warp and 10 weft). This is about 1.3% of the total number of inferred slices. The resulting annotation was converted into the boundary surface mesh of tows, which was then voxelized, i.e. converted into images, as shown in Fig. S22.† For woven composites, the multiclass segmentation needs to distinguish between the different directions of the fibers (warp or weft, Fig. 6A) and the matrix. This was successfully converted to a binary segmentation using the identification of the tow cross-section regions (is or is not a cross-section). Once trained, the neural network yielded highly accurate results in both the warp and weft direction, achieving voxel-wise accuracies of >96% for all samples (Table 1). The distinction was made between the ability to segment tows from matrix or the more complicated task of distinguishing the fiber bundle directions (warp or weft) within the tows as well. The network was trained both with the original dataset using high-performance GPUs, as well as with a reduced input dataset using downscaled images. Despite the use of limited computational resources, the latter obtained a similar voxel-wise accuracy with approximately 8 times less training time needed (Table S3†). Fig. S23† depicts the evolution of both the original and reduced training processes in terms of the difference between predicted output and actual value (loss function) and Fig. 7 shows the final segmentation results.
 | ||
| Fig. 7 Deep learning segmentation results on NC-doped fibres with 5 m% (top) and 10 m% (bottom) NCs added. (A) Input images and (B) manual segmentation compared with the (C) reduced and (D) original resolution datasets. | ||
| Tows/matrix | Warp/weft/matrix | |||
|---|---|---|---|---|
| 5 m% | 10 m% | 5 m% | 10 m% | |
| Original | 97.0 | 98.4 | 95.7 | 96.3 |
| Reduced | 97.2 | 98.4 | 96.0 | 96.7 |
Detection of micro-cracks
In polymer matrix composites, crack visualization becomes inherently difficult due to the low contrast difference between the crack and the resin.15 The smallest detectable crack opening is believed to be dependent on both the spatial resolution of the scan (in turn dependent on many factors such as field of view, X-ray source, pixel size, spot size, magnification, scintillator properties) and the contrast difference.14 The higher the contrast, the smaller the crack that can be visualized at a given resolution.15,31 Fatigue studies predicting composite end-of-life rely heavily on the detection of short cracks, of which the growth is significantly influenced by the microstructure.19,116,117After introduction and confirmation of damage to the NC-doped CFRP samples, they were re-scanned in CT using the same aforementioned settings (Fig. 8A). The extent of the damage was of lesser concern, as long as microcracks were present, which was verified via scanning electron microscopy, (Fig. 8B). Region growing was applied to segment the cracks present in the damaged samples. With a voxel size of 3.53 μm3, microcracks of a few micrometers will be below the resolution limit. Nonetheless a trend was observed with increasing masspercentage of nanocrystals added. For the reference sample, most larger cracks were detected. However cracks with sizes ca. <10 μm that extended into the polymer matrix could not be distinguished by the segmentation. Fig. 8C shows the segmented region; cracks that could not be extracted are highlighted. With 5 m% of HfO2 NCs added, the segmentation also encompasses cracks with sizes below 10 μm, though from ca. 6 μm and below some remain undetected (Fig. 8B). Region growing on the 10 m% damaged sample was able to detect almost all cracks, even with sizes of 2 to 3 μm which border the resolution limit (Fig. 8C). Extra micro-CT cross-section slices of segmented micro-cracks are shown in Fig. S24–26.†
 | ||
| Fig. 8 Increasingly accurate segmentation of microcracks in NC-doped CFRPs with, from top to bottom, 0, 5 and 10 m% of HfO2 NCs added. (A) 3D volume render of the micro-CT scans of the damaged samples. (B) SEM images confirming the presence of microcracks. (C) Cross-section slices with region growing based segmentation (highlighted in blue). A selection of cracks are measured and indicated. | ||
Conclusion
In order to avoid unexpected composite damage and failure, it is necessary to understand the material's behaviour. In situ visualization using lab-based micro-CT can provide valuable input, but the poor contrast between carbon fibers and polymer matrix complicates segmentation. We presented a new method to visualize carbon fiber-reinforced polymers, using contrast-enhanced computed tomography with HfO2 NCs as CT contrast agents. The post-synthetic work-up of the gram-scale NC syntheses was optimized and the HfO2 NCs were added to the epoxy polymer resin. We have evaluated common ligand-binding groups with a high binding affinity and found the stability in the epoxy resin to be dependent both on the binding group and the organic ligand tail. Using a PEGylated bisphosphonic acid (bisPA-mPEG[6]) resulted in excellent colloidal stability in the resin as the NCs remained stable during and after curing. A new synthetic strategy to the bisPA-mPEG[6] ligand was introduced, as well as a single-step post-synthetic ligand exchange on the NC surface. Mechanical properties such as matrix stiffness, insoluble fraction, water absorption, degradation temperature, curing time, resin viscosity and glass transition temperature of the HfO2/bisPA-mPEG[6] NC-doped epoxy were evaluated. Due to their homogeneous dispersion, the NCs did not negatively affect the polymer and have only a minor influence; manufacturing of CFRPs was also not affected. The proof-of-concept CT analysis showed that a minimum of 5 m% of HfO2 NCs yields drastic contrast improvement between the carbon fibers and the polymer matrix, allowing for automatic segmentation techniques. Deep learning segmentation allowed for voxel-wise accuracies of more than 96% with reduced computational effort and time. Finally, damaged laminates were found to have an improved visualization and detection of microcracks with increasing m% of NCs added. The contrast-enhanced NC-doped CFRPs allowed for automatic extraction of geometry and damage features, creating a better understanding of composite behaviour through more accurate modeling, which can reduce unexpected failure.Experimental
Materials
Hafnium(IV) chloride (98%), zirconium(IV) chloride tetrahydrofuran complex (1:2, 99%), oleic acid (90%), oleylamine (70%), triethylamine (≥99.5%) and chloroform (stabilized with amylenes, ≥99%) were purchased from Sigma-Aldrich. Benzyl alcohol was either purchased anhydrous (99.8%) or as ReagentPlus® (≥99%) from Sigma-Aldrich; the latter was then vacuum distilled (55 °C at 35 mTorr) and stored over sieves in a glovebox under an inert atmosphere. Tetrahydrofuran (ExtraDry, 99.5%) was purchased from Acros Organics. 2-[2-(2-Methoxyethoxy)ethoxy]acetic acid (>95.0%) was purchased from TCI Chemicals. (2-2-[2-(2-2-[2-(2-Methoxyethoxy) ethoxy] ethoxy ethoxy) ethoxy]ethoxy ethoxy) carboxylic acid (mPEG6-CH2COOH) (≥95%) was purchased from PurePEG. 6-[(2-2-(2-Methoxyethoxy) ethoxy] ethoxy hexyl)phosphonic acid (PA-hex-mPEG) (≥96%) and (2-2-[2-methoxyethoxy]-ethoxy-ethyl)phosphonic acid (PA-mPEG) (96%) were purchased from Sikémia. (2-(2-Methoxyethoxy)ethoxyx) bisphosphonic acids (bisPA-mPEG[x] with x = 2, 3, 6 or 15) were purchased from Surfactis Technologies or in-house synthesized according to the protocol below. 2-Ethylhexyl phosphonic acid (EHPA) was synthesized according to De Roo et al.84 Oleylphosphonic acid (OPA) was synthesized according to Dhaene et al.85 Marbocote 227 CEE release agent was purchased from Fatol-Kunststoffen. Epikote™ resin MGS RIMR135 and Epikure™ curing agent MGS RIMH137 were purchased from Hexion. Chemlease® 41-90 EZ was purchased from Chem-Trend. Wrightlon® 7400 nylon vacuum bagging film, Wrightlon® 5200 high-performance fluoropolymer release film and ultraweave® 606 medium weight nylon breather/bleeder were purchased from Airtech Europe SARL. Aerofilm® PP180 economy polyester peel ply and ST150 vacuum bagging gum sealant tape were purchased from EasyComposites. Solvents used for synthesis were purchased from Chemlab or Sigma-Aldrich. All deuterated solvents (CDCl3 and CD3OD) were purchased from Sigma-Aldrich or Eurisotop and stored over sieves. All manipulations were performed in air unless otherwise indicated. All chemicals were used as received unless otherwise mentioned. When required, organic solvents were dried according to the procedure described by Williams et al.118 making use of 20% m/v freshly activated 3 Å sieves for a minimum of 120 h.
Synthesis of bisPA-mPEG[6]
The synthesis was inspired by Egorov et al.90 Tris(trimethylsilyl)phosphite (P(OSiMe3)3) and catecholborane were stored in a glovebox under an inert atmosphere; the latter was stored in a freezer (−18 °C) in a glovebox. mPEG6CH2COOH was stored in a freezer (−18 °C) outside of a glovebox. 0.218 g mPEG6CH2COOH (1 eq., 0.615 mmol) was allowed to reach room temperature, after which it was weighed and dissolved in 0.5 mL THF. A two-neck flask was equipped with a septum and vacuum adapter and flushed three times with argon. The carboxylic acid was added to the flask under argon flow. In a glovebox under an inert atmosphere, a 1 mol L−1 solution of catecholborane (1.1 eq., 0.8 mmol, 0.096 g) in anhydrous THF (0.8 mL) was made. The solution was taken out of the glovebox and 0.675 mL was added to the flask, creating a solution with a yellow hue. The mixture was stirred at room temperature for 3 hours or until there was no evolution of gas. Note that hydrogen gas was being formed in this step. 0.770 g P(OSiMe3)3 (4.2 eq., 2.58 mmol, 0.862 mL) was removed from the glovebox and added to the mixture, which was left to stir for 16 hours. The mixture was hydrolyzed by adding 4 mL of dry methanol and left to stir for 4 hours. Solvents were evaporated under reduced pressure. The resulting oil was purified by dispersing in a minimal amount of methanol (ca. 0.5 mL) and precipitating with an excess of diethyl ether (ca. 10 mL) by centrifugation (8000 rcf for 4 min). This purification step was repeated five times to remove all side products. The final product was a transparent, viscous oil with 49% yield.1H NMR (400.1 MHz, CDCl3): δ 3.34–3.38 (s, 3H), δ 3.52–3.58 (m, 2H), δ 3.60–3.73 (s, 20H), δ 3.74–3.82 (s, 2H), δ 4.00–4.15 (m, 2H), δ 7.40–8.30 (s, 5H). 13C APT NMR (100.6 MHz, CDCl3): δ 59.18 (pos), δ 70.45 (neg), δ 70.53 (neg), δ 72.02 (neg), δ 74.72 (neg). 31P NMR (162.0 MHz, CDCl3): δ 17.10 (s). LC-MS calculated 500.1 for [M], found 499.1 for [M − H]−.
Synthesis of bisPA-mPEG[2]
The synthesis was inspired by Egorov et al.90 Tris(trimethylsilyl)phosphite (P(OSiMe3)3) and catecholborane were stored in a glovebox under an inert atmosphere; the latter was stored in a freezer (−18 °C). MEEAA was stored at room temperature outside of a glovebox. 0.219 g MEEAA (1 eq., 1.23 mmol) was allowed to reach room temperature, after which it was weighed and dissolved in 0.5 mL THF. A two-neck flask was equipped with a septum and vacuum adapter and flushed three times with argon. The carboxylic acid was added to the flask under argon flow. In a glovebox, a 1 mol L−1 solution of catecholborane (1.1 eq., 1.5 mmol, 0.180 g) in anhydrous THF (1.5 mL) was made. The solution was taken out of the glovebox and 1.35 mL was added to the flask, creating a solution with a yellow hue. The mixture was stirred at room temperature for 3 hours or until there was no evolution of gas. Note that hydrogen gas was being formed in this step. 1.543 g P(OSiMe3)3 (4.2 eq., 5.17 mmol, 1.728 mL) was removed from the glovebox and added to the mixture, which was left to stir for 16 hours. The mixture was hydrolyzed by adding 5 mL of dry methanol and left to stir for 4 hours. Solvents were evaporated under reduced pressure. The resulting oil was purified by dispersing in a minimal amount of methanol (ca. 0.5 mL) and precipitating with an excess of diethyl ether (ca. 10 mL) by centrifugation (8000 rcf for 4 min). This purification step was repeated five times to remove all side products. The final product was a transparent, viscous oil with 48% yield.1H NMR (400.1 MHz, CD3OD): δ 3.35–3.38 (s, 3H), δ 3.55–3.58 (m, 2H), δ 3.64–3.74 (m, 6H), δ 3.96–4.05 (t, 2H). 13C APT NMR (100.6 MHz, CD3OD): δ 59.12 (pos), δ 71.24 (neg), δ 72.09 (neg), δ 72.74 (neg), δ 75.87 (neg). 31P NMR (162.0 MHz, CD3OD): δ 17.8 (s). LC-MS calculated 324.0 for [M], found 323.0 for [M − H]−.
HfCl4·2THF synthesis
The procedure was adapted from Manxzer et al.119 and upscaled more than four times. In a glovebox under an inert atmosphere, 22 g of HfCl4 (1 eq., 0.069 mol) was added to 330 mL anhydrous dichloromethane, only partly dissolving, thereby rendering a turbid solution. Next, 22 mL (3.95 eq., 0.271 mol, 19.56 g) of anhydrous tetrahydrofuran (THF) was added in a dropwise manner and under vigorous stirring. The HfCl4 dissolved completely during addition of the THF. 220 mL anhydrous pentane was added, carefully pouring along the sides, and the solution was left in the freezer (−18 °C) for 2 hours to recrystallize. The resulting powder was filtered over a por 4 filter funnel and washed with pentane. The free-flowing white powder was dried under vacuum, resulting in a chemical yield of 89%. The complex was characterized by FTIR and 1H NMR (Fig. S27†).Gram-scale microwave-assisted HfO2 nanocrystal synthesis
The microwave synthesis was executed using a CEM Mars 6 operating at a frequency of 2.45 GHz and equipped with EasyPrep Plus vessels. Precursor preparation was executed in a glovebox under an inert atmosphere. 40 mL anhydrous benzyl alcohol (38 eq., 0.38 mol, 41.6 g) was added to the liner of the EasyPrep Plus vessel. 4.65 g HfCl4·2THF (1 eq., 0.01 mol) was carefully and slowly added to the liner under vigorous stirring. The edges of the liner were rinsed with the solution via a syringe to remove any powder stuck to the edge. Up to two or three liners could be added simultaneously to the microwave before reaching the microwave power limit required to reach 220 °C. The solution was stirred for 5 minutes, resulting in a transparent solution. The mixtures were subjected to microwave heating for 5 minutes at 80 °C (10 minutes ramp, power = 200 W), followed by 1 hour at 220 °C (20 minutes ramp, power = 800 W).Microwave-assisted ZrO2 nanocrystal synthesis
The microwave procedure was conducted using a CEM Discover SP with autosampler operating at a frequency of 2.45 GHz. Synthesis preparation was executed in a nitrogen-filled glovebox. 0.377 g (1 eq., 1 mmol) of ZrCl4·2THF was added to a 10 mL microwave vial with stirring bar. Under vigorous stirring, 4 mL (38 eq. or 48 eq., 38 mmol, 4.16 g) of anhydrous benzyl alcohol was added to the microwave vial. The microwave vial was capped, and the solution was then exited from the glovebox and stirred for 5 minutes, resulting in a transparent solution. The mixture was subjected to microwave heating for 5 minutes at 80 °C (30 W), followed by 4 hours at 220 °C (300 W) at medium stirring and PowerMax off. After synthesis, the mixture was transferred to a 15 mL plastic centrifuge tube using a Pasteur pipette. The microwave vial was rinsed with 3 mL of diethyl ether, in order to maximize the yield, after which this was also added to the centrifuge tube. After mild centrifugation (720 rcf, 2 min), three phases were observed: a transparent organic (top) phase, an aqueous, milky (middle) phase and sometimes a solid (bottom) phase of insolubles. The transparent (top) phase was removed and the milky phase was separated from the solid phase using a Pasteur pipette in a separate plastic centrifuge tube. The solid phase was discarded. Ethanol was added to the milky phase, yielding 2 mL of translucent suspension. 5 mL of diethyl ether was added and the particles were precipitated (4500 rcf, 2 min), resulting in ZrO2 nanocrystals capped with HCl.Optimized work-up protocol
The workup-protocol using centrifugation and the direct exchange was described in Goossens et al.57 The following procedure is an optimized workup protocol, described for one liner. The amount of solvents and ligands was scaled equivalently (double or triple) when two or three liners were used, respectively.After synthesis, the liners were opened and 5 mL water was added together with 200 μL aqueous HCl (37 w/w%) and left to stir for 15 minutes. The content was transferred to a separating funnel. Each liner was rinsed with 30 mL diethyl ether, which was also added to the funnel. The mixture was shaken and allowed to separate for 15 minutes. The bottom (aqueous) phase was collected in a flask and 10 mL ethanol was added to the solution. 25 mL diethyl ether was added and the precipitated NCs were filtered over a por 5 filter under vacuum with a grade 1 filter paper added. To stabilize the particles with oleate, particles were redispersed in 5 mL chloroform. Under stirring, 750 μL oleic acid was added, followed by 625 μL oleylamine. The mixture was subjected to sonication for 30 minutes. In some cases the mixture remained turbid after addition of the ligands, but would become transparent upon redispersion after the washing step. 25 mL of acetone was added and the particles were filtered over a por 5 filter with vacuum paper. Additional acetone was used to wash the particles. Chloroform can be used to remove all particles from the filter.
Ligand exchange from oleate
Ligand exchange from oleate to the bisphosphonic acids (bisPA-mPEG[x] with x = 2, 3, 6 or 15) and the phosphonic acids (PA-mPEG, PA-hex-mPEG, OPA and EHPA) was done as follows:The amount of oleate surface ligand was determined via the digital ERETIC method,120 based on the alkene resonance located around 5.3 ppm and the methyl resonance located around 0.9 ppm in dCDCl3 for 1H NMR measurements. For bisphosphonic acids and phosphonic acids, 0.5 and 1.2 equivalents were added to the nanocrystal solution, respectively, using a 100 mmol L−1 stock solution in chloroform. BisPA-mPEG[2] was added using a 100 mmol L−1 stock solution in methanol. For the bisPA-mPEG[6]/OA mixed shell only 0.25 equivalents of bisPA-mPEG[6] were added. The mixture was subjected to sonication for 30 minutes. BisPA-mPEG[x], PA-mPEG and PA-hex-mPEG were precipitated by addition of hexane (mixture of isomers), typically in a 1:5 volume ratio. OPA and EHPA were precipitated by addition of acetone, typically in a 1:5 volume ratio. After centrifugation (4500 rcf, 4 min), the particles were redispersed in a minimal volume of chloroform (ca. 100 mg mL−1). This purification procedure was repeated three times.
For the OPA/PA-mPEG mixed shell, 0.5 equivalents of OPA were added to the nanocrystal solution using a 100 mmol L−1 stock solution in chloroform, followed by sonication for 30 minutes. The nanocrystals were precipitated by addition of acetone, typically in a 1:5 volume ratio. After centrifugation (4500 rcf, 4 min), the particles were redispersed in 1 mL of chloroform. Next, 0.5 equivalents of PA-mPEG were added to the nanocrystal solution using a 100 mmol L−1 stock solution in chloroform, followed by sonication for 30 minutes. The particles were purified two more times by addition of acetone, typically in a 1:5 volume ratio, centrifugation (4500 rcf, 4 min) and redispersion in a minimal volume of chloroform using sonication.
Ligand exchange from oleate to the carboxylic acid MEEAA was done by determining the amount of oleate surface ligand as described above. 2 equivalents of carboxylic acid were added to the nanocrystal solution, followed by 30 minutes of sonication. The nanocrystals were precipitated once with hexane (mixture of isomers), typically in a 1:5 volume ratio. After centrifugation (4500 rcf, 4 min), the particles were redispersed in a minimal volume of chloroform and another 2 equivalents of carboxylic were added. This procedure was repeated three times. For the last step, the purification procedure was repeated three times.
Ligand exchange from chloride to bisPA-mPEG[6]
Direct exchange after synthesis to bisPA-mPEG[6] was done as follows:HfO2 NCs were synthesized via a small-scale microwave-assisted synthesis, starting from 0.6 mmol HfCl4·2THF in 4 mL benzyl alcohol. After synthesis, the particles were precipitated with 3 diethyl ether (720 rcf, 2 min) and redispersed in 2 mL of ethanol. 100 μmol of bisPA-mPEG[6] was added via a 500 mmol L−1 stock solution in ethanol (200 μL), followed by 90 mmol of triethylamine using a 1 mol L−1 stock solution in ethanol (90 μL). The mixture was subjected to sonication for 30 minutes, after which the mixture was transparent. The nanocrystals were precipitated by addition of diethyl ether and hexane, typically in a 1:0.5:5 volume ratio. After centrifugation (4500 rcf, 4 min), the particles were redispersed in a minimal volume of chloroform. This purification procedure was repeated two more times, but by redispersing the particles in ethanol using sonication.
Nanocrystal-doped resin formation
The appropriate amount of HfO2/bisPA-mPEG[6] was weighed, taking into account a ligand weight of 12 m%, and dispersed in acetone to yield a solution of 100 mg mL−1 (ligand weight subtracted). The nanocrystal solution was mixed manually with the epoxy resin and all acetone was evaporated using a rotary evaporator, yielding a translucent nanocomposite resin. The mixture was subjected to sonication for 30 minutes at 40 °C to fully disperse all particles.Nanocrystal-doped carbon fiber-reinforced polymer formation
Laminates were made using vacuum-assisted resin infusion (VARI). The laminate set-up is illustrated in Fig. S28.† On a clean 15 × 25 cm glass plate, a section of 6.5 × 13.5 cm was taped off and three layers of release agent (Chemlease®) were applied with a 10-minute wait time between each layer, after which the tape marking was removed. Six layers of 5 × 8 cm carbon fiber woven fabric were cut and stacked on the section coated with release agent. A peel ply fabric (5 × 10 cm) and semi-perforated release film (5 × 10 cm) was placed on top and the stacking was taped down. An inlet (ca. 30 cm) and outlet (ca. 90 cm) tube was added with plastic T-sections wrapped in bleeder fabric. A vacuum bag of 20 × 30 cm with vacuum tape along the edges was added over the stacking, not on the release agent section. Small folds above and below the inlet and outlet tubes were used. The inlet tube was clamped and the outlet tube applied to vacuum. The outlet tube was clamped as well. If this held for 24 hours without losing vacuum, the set-up was infused. The nanocrystal-doped resin was prepared as described above. For 5 m% and 10 m%, 2 and 4 grams of HfO2/bisPA-mPEG[6] (ligand weight subtracted), respectively, were mixed with 29.3 g of Epikote™ resin. 8.7 g of Epikure™ curing agent was thoroughly mixed with the resin and heated to 35 °C to counteract any viscosity increase. Next, the mixture was degassed. The outlet tube was connected to the vacuum through a resin trap and the inlet tube added to the nanocrystal–resin mixture. The set-up was infused under vacuum. After clamping the inlet and outlet tubes, the laminate was cured for 24 h at room temperature, followed by 15 h at 80 °C in a Naberthem P330 oven. After curing, the vacuum bag, tubes, peel ply and release film were removed and the laminated was released from the glass plate.Tensile testing
Stress–strain measurements were performed according to the ASTM D638 standard. A custom metal dogbone mould was made using the following dimensions (Fig. S29†): width of narrow section (W) = 6 mm, length of narrow section (L) = 57 mm, width overall (WO) = 19 mm, length overall (LO) = 195 mm, gauge length (G) = 50 mm, distance between grips (D) = 135 mm, radius of fillet (R) = 76 mm and thickness (T) = 3 mm. The mould was coated using a release agent before adding the resin. Resins were made as described above and added to the mould using a syringe and avoiding introducing air, together with a small grip to facilitate removal of the sample after curing. Samples were cured for 24 hours at room temperature and 15 hours at 80 °C.Stress–strain measurements were performed using a Tinius Olsen H10KT equipped with a 5 kN load cell and processed using Horizon software. The thickness was set as 3 mm, width as 5.85 mm and gage length as 65 mm. The tensile measurements were performed using a preload of 0.05 N and a pulling speed of 10 mm min−1 until sample failure. Tests were performed minimum in duplicate.
Water absorption
Water absorption was determined according to the ASTM D570 standard. Nanocomposite samples of approximately 50 mg (three different ones per mass%) were dried under vacuum at 100 °C for 24 hours. Afterwards they were placed in a dessicator to cool and weighed immediately upon cooling. The material was then immersed in water at room temperature for 7 days. They were removed from the water, patted dry with a lint-free cloth and weighed. The water absorption was determined according to eqn (1) where mbegin is the mass after drying and mend is the mass after submersion in water: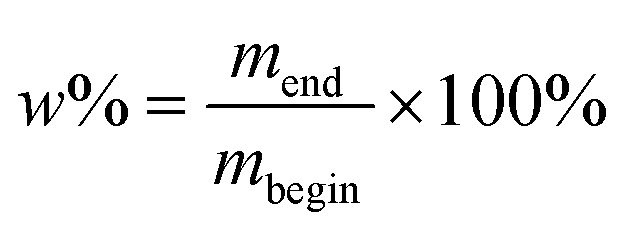 | (1) |
Insoluble fraction
To determine the appropriate solvent, samples were first submerged in different solvents and weighed before and after. The swelling degree was calculated according to eqn (1). Values are given in Table S2.† The nanocomposite samples swelled well in dichloromethane, moderately in tetrahydrofuran and barely in ethyl acetate. Therefore the first was the solvent of choice to determine the insoluble fractions.Insoluble fractions were determined by submerging the samples in dichloromethane for 24 hours. During this 24 hours the solvent was refreshed multiple times. Afterwards the samples were removed from the solvent and dried overnight in a vacuum oven at room temperature, followed by 3 hours at 150 °C. The samples were weighed again and the insoluble fraction was determined using eqn (1), where mbegin is the mass before submerging the sample and mend is the mass after drying.
DSC
Differential scanning calorimetry was performed using ca. 10 mg of nanocomposite for each measurement on a Mettler Toledo 1/700 instrument. Measurements were performed under a nitrogen atmosphere from −50 °C to 180 °C with a cooling rate of 40 °C min−1 and a heating rate of 10 °C min−1 and a 15 min stabilization step at −100 °C before measuring. The first heating run was performed to remove the thermal history of the polymer; DSC curves shown correspond to the second heating curve.Rheology
Viscosity measurements were performed on an Anton Paar rheometer equipped with a convection oven (CTD600). Samples were brought between 25 mm diameter parallel plates and the viscosity was measured from 0–100 s−1. The curing time, defined as the crossover between the storage (G′) and loss (G′′) moduli, was determined by measuring G′ and G′′ over time at 80 °C for 90 min on an Anton Paar MCR302 Rheometer equipped with a convection oven (CTD450). Samples are typically cured for 24 h at room temperature before heating to 80 °C for 15 h. Here the samples were heated immediately to 80 °C to save instrument measurement time. The samples were brought between 25 mm diameter parallel plates, with an initial gap of 1 mm and the instrument set to maintain an axial force of 0 ± 0.1 N. The samples were trimmed and a strain of 0.05% was applied onto all samples at a frequency of 10 rad s−1.TGA analysis
Thermogravimetric analysis (TGA) was done with a Netzch STA 449F3 Jupiter instrument. Powder samples were heated from 25 °C to 1200 °C at 10 °C min−1 under air flow with an isothermal step at the end of 30 min at 1200 °C. Nanocomposite samples were heated from 25 °C to 800 °C at 5 °C min−1 under airflow with an isothermal step at the end of 30 min at 800 °C. Measurements were performed in triplicate. Correction files were recorded using the same conditions as the corresponding measurement. TGA data processing was performed using the Netzch Proteus Analysis software. The lack of filtering was determined by TGA on a TA instruments TGA5500 by heating from 25 °C to 800 °C at 5 °C min−1 with an isothermal step at the end of 15 min at 800 °C.Micro-CT scans
Micro-computed tomography was done at the Ghent University Centre for X-ray Tomography (UGCT) using the HECTOR64 scanner. Samples were scanned in cone-beam mode. OCTOPUS reconstruction software version 8.9 was used to obtain a stack of reconstructed TIFF images. The proof-of-concept concentration series was scanned at a voxel size of 173 μm3, the composite laminates at a voxel size of 3.53 μm3. All were scanned at 90 kV tube potential and a power of 10 W. No hardware filter was applied. 2400 projections were taken using 1000 ms exposure time per projection.CT scan postprocessing and region growing segmentation
Datasets were processed using the VGStudio Max software. CT scans were imported as an image stack. The fibers were segmented from the surrounding matrix by creating a selection using the automatic region growing (flooding) algorithm. A seed point was manually placed in any of the 2D views after which the algorithm grew the selection, continuously expanding as long as the neighbouring voxels were within the user-specified tolerance relative to the seed. The region growing algorithm was used in static mode, i.e. voxels were added to the selection if they were connected to the region and if their greyvalues did not deviate by more than the chosen tolerance from the greyvalue of the seed point. No other constraints (such as max. radius) were applied to algorithm. The tolerance value and seed point were changed until an optimal selection was obtained. For some data, the Opening/Closing mode was applied, which eliminated gaps in the selection that was grown by expanding the selection with the user-specified number of voxels and then immediately contracting. Finally, the selection was extracted from the volume as a separate region-of-interest (ROI). The 3D volume render was exported as an image as isosurface render, displaying the surface of the selected ROI as defined by the manually set iso-level.Deep learning segmentation
The micro-CT images were preprocessed through rotation and cropping operations. The 5 m% sample had a cropped size of 672 × 768 × 768 voxels, the 10 m% sample had a cropped size of 704 × 672 × 800 voxels. Both had a voxel size of 3.53 μm3 and a greyscale depth of 16 bit. The images were annotated for the formation of the training dataset and subsequent result evaluation. Sets of tow cross-sections were manually segmented using ImageJ/Fiji.121 There were 21 tows for 5 m% and 15 tows for 10 m%. In total, cross-sections on 36 slices were manually annotated, resulting in an average of about 9 cross-sections per tow. The tow boundary mesh was generated using the interpolation technique described by Sinchuk et al.36 The meshes were converted into images (voxelized) with sizes identical to their input. Neural network training was simplified by converting the problem to binary segmentation, focusing only on identifying the tow cross-section regions. Slices transverse to the tow direction were used for training. For each image, 10 slices in the warp direction and 10 slices in the weft direction were selected for neural network training. 8 out of these 10 slices were used to form the training dataset, while the remaining 2 were used for the validation dataset. The sizes of the training and validation datasets were 32 and 8 samples, respectively. Prior to training, the source images were normalized to have a mean greyscale value of zero and a unit standard deviation. A deep learning approach based on convolutional neural networks with a 2D U-net architecture122 was employed as the segmentation method. The training process was stopped after 30 epochs ‘patience’ without a decrease in validation loss. The reduced dataset was achieved by downscaling the input images and their annotations four times in-plane and two times through the thickness, resulting in a size of 336 × 192 × 192 voxels for the 5 m% sample and 352 × 168 × 200 voxels for the 10 m% sample. The reduced training was conducted on a laptop with only 4GB of GPU memory. Table S3† summarizes the training performances for both datasets. The neural network, trained only once, was used to predict the 5 m% and 10 m% images in both the warp and weft directions (four inferences in total). The inference results were probability images with voxel values ranging from 0 to 1. A voxel was assigned to the matrix class if the sum of its warp and weft probabilities was <0.5, otherwise it was classified as a tow voxel. The warp/weft classification of the tow voxels was determined by selecting the class with the higher probability. As a postprocessing step, small isolated regions were removed followed by median filter smoothing. The volume thresholds for the small regions were set at 15k and 500 voxels for the original and reduced resolution images, respectively. The median filter kernels used were 5 × 9 × 9 for the original and 3 × 5 × 5 for the reduced resolution images. The resulting deep learning segmentations were evaluated against manual segmentations, Fig. S22.†TEM analysis
High-resolution transmission electron microscopy (HRTEM) was performed on a JEOL JEM-2200FS TEM with Cs corrector operating at 200 kV. Nanocrystals in solvents were made by drop-cast suspension on the grids. The NC-doped epoxy samples were made by microtome cutting using a Leica EM UC7 Microtome. The TEM grids used were Holey-Carbon – Cu (C200-CU) with 50 micron hole size (200 mesh). NC sizes were determined by measuring 200 particles using the polygon selection tool of ImageJ, with measurements set to fit ellipse.SEM analysis
Scanning electron microscopy (SEM) was performed using a JEOL FEG SEM JSM-7600F at an accelerating voltage of 10 kV in secondary electron mode. The samples were gold sputtered before imaging using a Bal-Tec SCD 005 Cool Sputter Coater with a sputter current of 25 mA for 40 seconds.PXRD analysis
Powder X-ray diffraction (PXRD) was performed on a Bruker D8 Advance with motorized anti-scatter screen and Autochanger and Bragg-Brentano θ–2θ geometry (goniometer radius 280 mm). The instrument uses the Cu Kα radiation (λ = 1.54184 Å) with no Kβ filter. The detector was a LynxEye XE-T Silicon strip Line detector with 192 channels. Samples were made by drop-cast suspension on a glass plate. The measurement was performed in the θ–2θ 15–60° range at a step size of 0.02° and a scan rate of 0.5° min−1.DLS analysis
Dynamic Light Scattering (DLS) measurements were recorded on a Malvern Zetasizer ZS instrument in backscattering mode (173°) at a temperature of 20 °C using a 60-second equilibration time. Nanocrystals in solvents as dispersant were measured in glass cuvettes, and nanocrystals in the epoxy Epikote RIMR135 resin as dispersant were measured in disposable plastic cuvettes. For the Epikote RIMR135 resin as dispersant, the dispersant refractive index was set as 1.550 and the viscosity as 1500 mPa s. Measurements were repeated 6 times. DLS data processing was performed using the Malvern ZS Explorer software using the general purpose analysis model.NMR analysis
Nuclear magnetic resonance (NMR) spectra were recorded at 298.15 K on a Bruker 300 instrument at 400 MHz, a Bruker Avance III spectrometer operating at a 1H frequency of 500.08 MHz and featuring a BBI probe and a Bruker UltraShield 500 spectrometer operating at a 1H frequency of 500.13 MHz. Chemical shifts (δ) are given in parts per million (ppm) and the residual solvent peak was used as an internal standard (CDCl3: δH = 7.24 ppm, δC = 77.16, CD3OD: δH = 3.31 ppm, δC = 49.00). The signal multiplicity is denoted as follows: s (singlet), d (doublet), t (triplet), quad (quadruplet), quin (quintet) and m (multiplet). All resonances were background corrected. Coupling constants are reported in Hertz (Hz). 1H, 13C and 31P spectra were acquired using the standard pulse sequences from the Bruker library: zg30, jmod (APT = attached proton test) and zgpg30 (proton decoupled), respectively. In the APT, the carbon resonances resulting from a –CH2– and quaternary –Cq– are in-phase (orientated up, denoted as pos), whereas the carbon resonances resulting from a –CH3– or –CH– are out-of-phase (orientated down, denoted as neg). Chemical shifts for 31P NMR spectra were referenced indirectly to the 1H NMR frequency of the sample with the xiref-macro in Bruker. 31P NMR spectra of NCs were processed with a line broadening to reduce noise. For the quantitative 1D 1H measurements, 64k data points were sampled with the spectral width set to 16 ppm and a relaxation delay of 30 s. Quantification was done using the Digital ERETIC method.120UV-Vis analysis
UV-Vis spectra were recorded on a PerkinElmer Lambda 365 measuring the absorbance (A) from 400 to 500 nm with a slit width of 1 nm and scan speed of 240 nm min−1.FTIR analysis
Fourier-Transform Infrared Spectroscopy (FTIR) was performed on a PerkinElmer spectrum 2 ATR-FTIR with a diamond crystal measuring 8 scans from 450 to 4000 cm−1 and using background subtraction.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the Research Foundation-Flanders (Project No. 1S11721N, 1163125N and 1S28818N), Flanders Innovation & Entrepreneurship (Project No. HBC.2022.0682), Ghent University and the University of Basel for financial support. The special research fund of the Ghent University (BOF-UGent) is acknowledged for the financial support of the UGCT Core Facility (BOF.COR.2022.008) and the Concerted Research Actions project (BOF2015/GOA/007). TEM measurements were performed at the UGent TEM Core Facility. We thank M. De Bruyne for the microtome TEM sample preparation, J. Goeman for LC-MS measurement, L. Van Bossele for PXRD measurement, H. Rijckaert for STEM measurements, J. P. Mathew for TGA measurements, T. Maiheu for assistance with the tensile tests, S. Engelen and D. Van den Eynden for assistance with the rheology measurements, I. Josipovic for micro-CT measurements, L. Sioen for preliminary experiments and E. Picavet for the inkscape fiber schematic. The TOC graphic was partly created using BioRender.com.References
- C. Soutis, Fibre reinforced composites in aircraft construction, Prog. Aerosp. Sci., 2005, 41, 143–151 CrossRef.
- S. Kesarwani, et al., Polymer composites in aviation sector, Int. J. Eng. Res., 2017, 6, 518–525 Search PubMed.
- H. Ahmad, A. Markina, M. Porotnikov and F. Ahmad, in IOP Conference Series: Materials Science and Engineering, 2020, pp. 032011.
- A. Wazeer, A. Das, C. Abeykoon, A. Sinha and A. Karmakar, Composites for electric vehicles and automotive sector: a review, Green Energy Intell. Transp., 2022, 100043 Search PubMed.
- N. Hiremath, S. Young, H. Ghossein, D. Penumadu, U. Vaidya and M. Theodore, Low cost textile-grade carbon-fiber epoxy composites for automotive and wind energy applications, Composites, Part B, 2020, 198, 108156 CrossRef CAS.
- K. Luo, L. Chen and W. Liang, Structural health monitoring of carbon fiber reinforced polymer composite laminates for offshore wind turbine blades based on dual maximum correlation coefficient method, Renewable Energy, 2022, 201, 1163–1175 CrossRef CAS.
- M. F. Petrou, D. Parler, K. A. Harries and D. C. Rizos, Strengthening of reinforced concrete bridge decks using carbon fiber-reinforced polymer composite materials, J. Bridge Eng., 2008, 13, 455–467 CrossRef.
- Y. Liu, B. Zwingmann and M. Schlaich, Carbon fiber reinforced polymer for cable structures—A review, Polymers, 2015, 7, 2078–2099 CrossRef.
- C. He, R. Yu, H. Sun and Z. Chen, Lightweight multilayer composite structure for hydrogen storage tank, Int. J. Hydrogen Energy, 2016, 41, 15812–15816 CrossRef CAS.
- J. Qureshi, A review of fibre reinforced polymer structures, Fibers, 2022, 10, 27 CrossRef CAS.
- K. Yu, Q. Shi, M. L. Dunn, T. Wang and H. J. Qi, Carbon fiber reinforced thermoset composite with near 100% recyclability, Adv. Funct. Mater., 2016, 26, 6098–6106 CAS.
- P. J. Schilling, B. R. Karedla, A. K. Tatiparthi, M. A. Verges and P. D. Herrington, X-ray computed microtomography of internal damage in fiber reinforced polymer matrix composites, Compos. Sci. Technol., 2005, 65, 2071–2078 CAS.
- H. Zrida, P. Fernberg, Z. Ayadi and J. Varna, Microcracking in thermally cycled and aged Carbon fibre/polyimide laminates, Int. J. Fatigue, 2017, 94, 121–130 CrossRef CAS.
- S. Stock, X-ray microtomography of materials, Int. Mater. Rev., 1999, 44, 141–164 CAS.
- B. Yu, R. Bradley, C. Soutis and P. Withers, A comparison of different approaches for imaging cracks in composites by X-ray microtomography, Philos. Trans. R. Soc., A, 2016, 374, 20160037 CrossRef CAS PubMed.
- S. Carmignato, W. Dewulf and R. Leach, Industrial X-ray computed tomography, Springer, 2018, vol. 10 Search PubMed.
- K. Orhan, et al., Micro-computed Tomography (micro-CT) in Medicine and Engineering, Springer technical report, 2020 Search PubMed.
- S. Garcea, Y. Wang and P. Withers, X-ray computed tomography of polymer composites, Compos. Sci. Technol., 2018, 156, 305–319 CAS.
- P. J. Withers and M. Preuss, Fatigue and damage in structural materials studied by X-ray tomography, Annu. Rev. Mater. Res., 2012, 42, 81–103 CrossRef CAS.
- A. Moffat, P. Wright, J.-Y. Buffière, I. Sinclair and S. M. Spearing, Micromechanisms of damage in 0 splits in a [90/0] s composite material using synchrotron radiation computed tomography, Scr. Mater., 2008, 59, 1043–1046 CAS.
- A. Scott, M. Mavrogordato, P. Wright, I. Sinclair and S. Spearing, In situ fibre fracture measurement in carbon–epoxy laminates using high resolution computed tomography, Compos. Sci. Technol., 2011, 71, 1471–1477 CAS.
- Y. Wang, T. L. Burnett, Y. Chai, C. Soutis, P. J. Hogg and P. J. Withers, X-ray computed tomography study of kink bands in unidirectional composites, Compos. Struct., 2017, 160, 917–924 Search PubMed.
- D. Bull, S. Spearing, I. Sinclair and L. Helfen, Three-dimensional assessment of low velocity impact damage in particle toughened composite laminates using micro-focus X-ray computed tomography and synchrotron radiation laminography, Composites, Part A, 2013, 52, 62–69 CrossRef CAS.
- D. D. Symons, Characterisation of indentation damage in 0/90 lay-up T300/914 CFRP, Compos. Sci. Technol., 2000, 60, 391–401 CrossRef CAS.
- B. Yu, R. Bradley, C. Soutis, P. Hogg and P. Withers, 2D and 3D imaging of fatigue failure mechanisms of 3D woven composites, Composites, Part A, 2015, 77, 37–49 CrossRef CAS.
- F. Cosmi and A. Bernasconi, Micro-CT investigation on fatigue damage evolution in short fibre reinforced polymers, Compos. Sci. Technol., 2013, 79, 70–76 CrossRef CAS.
- S. M. Sisodia, S. Garcea, A. George, D. Fullwood, S. Spearing and E. Gamstedt, High-resolution computed tomography in resin infused woven carbon fibre composites with voids, Compos. Sci. Technol., 2016, 131, 12–21 CrossRef CAS.
- J. Vilà, F. Sket, F. Wilde, G. Requena, C. González and J. Llorca, An in situ investigation of microscopic infusion and void transport during vacuum-assisted infiltration by means of X-ray computed tomography, Compos. Sci. Technol., 2015, 119, 12–19 CrossRef.
- A. Jumahat, C. Soutis, F. Jones and A. Hodzic, Fracture mechanisms and failure analysis of carbon fibre/toughened epoxy composites subjected to compressive loading, Compos. Struct., 2010, 92, 295–305 CrossRef.
- G. Requena, G. Fiedler, B. Seiser, P. Degischer, M. Di Michiel and T. Buslaps, 3D-Quantification of the distribution of continuous fibres in unidirectionally reinforced composites, Composites, Part A, 2009, 40, 152–163 CrossRef.
- Y. Wang, S. C. Garcea and P. J. Withers, 7.6 Computed tomography of composites, 2018 Search PubMed.
- L. P. Djukic, G. M. Pearce, I. Herszberg, M. K. Bannister and D. H. Mollenhauer, Contrast enhancement of microct scans to aid 3D modelling of carbon fibre fabric composites, Appl. Compos. Mater., 2013, 20, 1215–1230 CrossRef CAS.
- S. M. Spearing and P. W. Beaumont, Fatigue damage mechanics of composite materials. I: Experimental measurement of damage and post-fatigue properties, Compos. Sci. Technol., 1992, 44, 159–168 CrossRef CAS.
- S. Garcea, I. Sinclair and S. Spearing, In situ synchrotron tomographic evaluation of the effect of toughening strategies on fatigue micromechanisms in carbon fibre reinforced polymers, Compos. Sci. Technol., 2015, 109, 32–39 CrossRef CAS.
- O. C. Zienkiewicz, R. L. Taylor and J. Z. Zhu, The finite element method: its basis and fundamentals, Elsevier, 2005 Search PubMed.
- Y. Sinchuk, P. Kibleur, J. Aelterman, M. N. Boone and W. Van Paepegem, Variational and deep learning segmentation of very-low-contrast X-ray computed tomography images of carbon/epoxy woven composites, Materials, 2020, 13, 936 CrossRef CAS PubMed.
- A. J. Thompson, B. El Said, D. Ivanov, J. P. Belnoue and S. R. Hallett, High fidelity modelling of the compression behaviour of 2D woven fabrics, Int. J. Solids Struct., 2018, 154, 104–113 CrossRef CAS.
- G. Nikishkov, Y. Nikishkov and A. Makeev, Finite element mesh generation for composites with ply waviness based on X-ray computed tomography, Adv. Eng. Softw., 2013, 58, 35–44 CrossRef.
- P. Badel, E. Vidal-Sallé, E. Maire and P. Boisse, Simulation and tomography analysis of textile composite reinforcement deformation at the mesoscopic scale, Compos. Sci. Technol., 2008, 68, 2433–2440 CrossRef CAS.
- Y. Li, B. Sun and B. Gu, Impact shear damage characterizations of 3D braided composite with X-ray micro-computed tomography and numerical methodologies, Compos. Struct., 2017, 176, 43–54 CrossRef.
- J. Ali, J. K. Farooqi, D. Buckthorpe, A. Cheyne and P. Mummery, Comparative study of predictive FE methods for mechanical properties of nuclear composites, J. Nucl. Mater., 2009, 383, 247–253 CrossRef CAS.
- N. Naouar, E. Vidal-Salle, J. Schneider, E. Maire and P. Boisse, 3D composite reinforcement meso FE analyses based on X-ray computed tomography, Compos. Struct., 2015, 132, 1094–1104 CrossRef.
- R. Böhm, J. Stiller, T. Behnisch, M. Zscheyge, R. Protz, S. Radloff, M. Gude and W. Hufenbach, A quantitative comparison of the capabilities of in situ computed tomography and conventional computed tomography for damage analysis of composites, Compos. Sci. Technol., 2015, 110, 62–68 CrossRef.
- L. P. Djukic, I. Herszberg, W. R. Walsh, G. A. Schoeppner, B. G. Prusty and D. W. Kelly, Contrast enhancement in visualisation of woven composite tow architecture using a MicroCT Scanner. Part 1: Fabric coating and resin additives, Composites, Part A, 2009, 40, 553–565 CrossRef.
- J. Hainfeld, D. Slatkin, T. Focella and H. Smilowitz, Gold nanoparticles: a new X-ray contrast agent, Br. J. Radiol., 2006, 79, 248–253 CrossRef CAS PubMed.
- J. Cai, X. Ge, S. Lu, Y. Wang, H. Cui, R. Zhan, S. Su, W. Shan, Z. Cai and C. Wu, et al., Au/FeNiPO4-Based Multiple Spectra Optoacoustic Tomography/CT Dual-Mode Nanoprobe for Systemic Screening of Atherosclerotic Vulnerable Plaque, Adv. Funct. Mater., 2024, 2406192 CrossRef CAS.
- P. J. Bonitatibus Jr., A. S. Torres, G. D. Goddard, P. F. FitzGerald and A. M. Kulkarni, Synthesis, characterization, and computed tomography imaging of a tantalum oxide nanoparticle imaging agent, Chem. Commun., 2010, 46, 8956–8958 RSC.
- M. H. Oh, N. Lee, H. Kim, S. P. Park, Y. Piao, J. Lee, S. W. Jun, W. K. Moon, S. H. Choi and T. Hyeon, Large-scale synthesis of bioinert tantalum oxide nanoparticles for X-ray computed tomography imaging and bimodal image-guided sentinel lymph node mapping, J. Am. Chem. Soc., 2011, 133, 5508–5515 CrossRef CAS PubMed.
- A. Jakhmola, N. Anton, H. Anton, N. Messaddeq, F. Hallouard, A. Klymchenko, Y. Mely and T. F. Vandamme, Poly-ε-caprolactone tungsten oxide nanoparticles as a contrast agent for X-ray computed tomography, Biomaterials, 2014, 35, 2981–2986 CrossRef CAS PubMed.
- L. Wen, L. Chen, S. Zheng, J. Zeng, G. Duan, Y. Wang, G. Wang, Z. Chai, Z. Li and M. Gao, Ultrasmall biocompatible WO3−x nanodots for multi-modality imaging and combined therapy of cancers, Adv. Mater., 2016, 28, 5072–5079 CrossRef CAS PubMed.
- O. Rabin, J. Manuel Perez, J. Grimm, G. Wojtkiewicz and R. Weissleder, An X-ray computed tomography imaging agent based on long-circulating bismuth sulphide nanoparticles, Nat. Mater., 2006, 5, 118–122 CrossRef CAS PubMed.
- K. Ai, Y. Liu, J. Liu, Q. Yuan, Y. He and L. Lu, Large-Scale Synthesis of Bi 2 S 3 Nanodots as a Contrast Agent for In Vivo X-ray Computed Tomography Imaging, Adv. Mater., 2011, 42, 4886–4891 CrossRef PubMed.
- T. L. McGinnity, O. Dominguez, T. E. Curtis, P. D. Nallathamby, A. J. Hoffman and R. K. Roeder, Hafnia (HfO 2) nanoparticles as an X-ray contrast agent and mid-infrared biosensor, Nanoscale, 2016, 8, 13627–13637 RSC.
- F. Ostadhossein, P. Moitra, N. Gunaseelan, M. Nelappana, C. Lowe, M. Moghiseh, A. Butler, N. de Ruiter, H. Mandalika and I. Tripathi, et al., Hitchhiking probiotic vectors to deliver ultra-small hafnia nanoparticles for ‘Color’ gastrointestinal tract photon counting X-ray imaging, Nanoscale Horiz., 2022, 7, 533–542 RSC.
- Y. Li, Y. Qi, H. Zhang, Z. Xia, T. Xie, W. Li, D. Zhong, H. Zhu and M. Zhou, Gram-scale synthesis of highly biocompatible and intravenous injectable hafnium oxide nanocrystal with enhanced radiotherapy efficacy for cancer theranostic, Biomaterials, 2020, 226, 119538 CrossRef CAS PubMed.
- L. Deblock, B. Descamps, I. Goemaere, E. Goossens, G. Vergauwen, J. Debacker, P. Tummers, K. Remaut, I. Van Driessche and K. De Buysser, et al., Dual-Modality Hafnium Oxide Nanocrystals for in Vivo Computed Tomography and Fluorescence Imaging of Sentinel Lymph Nodes, Chem. Mater., 2023, 35, 8883–8896 CAS.
- E. Goossens, L. Deblock, L. Caboor, D. V. d. Eynden, I. Josipovic, P. R. Isaacura, E. Maksimova, M. Van Impe, A. Bonnin and P. Segers, et al., From Corrosion Casting to Virtual Dissection: Contrast-Enhanced Vascular Imaging using Hafnium Oxide Nanocrystals, Small Methods, 2024, 2301499 CrossRef CAS PubMed.
- J. De Roo, F. Van den Broeck, K. De Keukeleere, J. C. Martins, I. Van Driessche and Z. Hens, Unravelling the surface chemistry of metal oxide nanocrystals, the role of acids and bases, J. Am. Chem. Soc., 2014, 136, 9650–9657 CAS.
- D. Van den Eynden, R. Pokratath and J. De Roo, Nonaqueous Chemistry of Group 4 Oxo Clusters and Colloidal Metal Oxide Nanocrystals, Chem. Rev., 2022, 122, 10538–10572 CAS.
- E. Goossens, O. Aalling-Frederiksen, P. Tack, D. Van den Eynden, Z. Walsh-Korb, K. M. Jensen, K. De Buysser and J. De Roo, From Gel to Crystal: Mechanism of HfO2 and ZrO2 Nanocrystal Synthesis in Benzyl Alcohol, J. Am. Chem. Soc., 2024, 146(15), 10723–10734 CAS.
- Z. Hens and J. C. Martins, A solution NMR toolbox for characterizing the surface chemistry of colloidal nanocrystals, Chem. Mater., 2013, 25, 1211–1221 CAS.
- J. De Roo, N. Yazdani, E. Drijvers, A. Lauria, J. Maes, J. S. Owen, I. Van Driessche, M. Niederberger, V. Wood and J. C. Martins, et al., Probing solvent–ligand interactions in colloidal nanocrystals by the NMR line broadening, Chem. Mater., 2018, 30, 5485–5492 CAS.
- Z. Hens, Ligands on Nanocrystal Surfaces, the 1H Nuclear Magnetic Resonance Fingerprint, Acc. Chem. Res., 2023, 4290–4301 Search PubMed.
- B. Masschaele, M. Dierick, D. Van Loo, M. N. Boone, L. Brabant, E. Pauwels, V. Cnudde and L. Van Hoorebeke, J. Phys.: Conf. Ser., 2013, 012012 Search PubMed.
- O. Brunke, E. Neuser and A. Suppes, Internal Symposium on Digital Industrial Radiology and Computed Tomography, Berlin, Germany, 2011, pp. 20–22 Search PubMed.
- J. Bai, Advanced fibre-reinforced polymer (FRP) composites for structural applications, Woodhead Publishing, 2022 Search PubMed.
- M. Nikforooz, O. Verschatse, L. Daelemans, K. De Clerck and W. Van Paepegem, Experimental method for in situ real-time measurement of mixed mode fibre/matrix interface debonding and comparison of fibre sizings in single short fibre composites, Compos. Sci. Technol., 2024, 110488 CrossRef CAS.
- A. T. Akinola, Fatigue performance of glass/epoxy nanocomposites for wind turbine blades, 2010 Search PubMed.
- E. Dümichen, M. Javdanitehran, M. Erdmann, V. Trappe, H. Sturm, U. Braun and G. Ziegmann, Analyzing the network formation and curing kinetics of epoxy resins by in situ near-infrared measurements with variable heating rates, Thermochim. Acta, 2015, 616, 49–60 CrossRef.
- C. Buggisch, D. Gibhardt, N. Felmet, Y. Tetzner and B. Fiedler, Strain sensing in GFRP via fully integrated carbon nanotube epoxy film sensors, Composites, Part C, 2021, 6, 100191 CAS.
- S. van der Heijden, L. Daelemans, B. De Schoenmaker, I. De Baere, H. Rahier, W. Van Paepegem and K. De Clerck, Interlaminar toughening of resin transfer moulded glass fibre epoxy laminates by polycaprolactone electrospun nanofibres, Compos. Sci. Technol., 2014, 104, 66–73 CrossRef CAS.
- M. V. Kovalenko, L. Manna, A. Cabot, Z. Hens, D. V. Talapin, C. R. Kagan, V. I. Klimov, A. L. Rogach, P. Reiss and D. J. Milliron, et al., Prospects of nanoscience with nanocrystals, ACS Nano, 2015, 9, 1012–1057 CrossRef CAS PubMed.
- S. Yamashita, T. Sudo, H. Kamiya and Y. Okada, Colloidal Stability of TiO2 Nanoparticles: the roles of phosphonate ligand length and solution temperature, Chem. – Eur. J., 2022, 28, e202201560 CrossRef CAS PubMed.
- S. Kango, S. Kalia, A. Celli, J. Njuguna, Y. Habibi and R. Kumar, Surface modification of inorganic nanoparticles for development of organic–inorganic nanocomposites—A review, Prog. Polym. Sci., 2013, 38, 1232–1261 CrossRef CAS.
- S. Fu, Z. Sun, P. Huang, Y. Li and N. Hu, Some basic aspects of polymer nanocomposites: A critical review, Nano Mater. Sci., 2019, 1, 2–30 CrossRef.
- E. Reynaud, T. Jouen, C. Gauthier, G. Vigier and J. Varlet, Nanofillers in polymeric matrix: a study on silica reinforced PA6, Polymer, 2001, 42, 8759–8768 CrossRef CAS.
- S. Lee, H.-J. Shin, S.-M. Yoon, D. K. Yi, J.-Y. Choi and U. Paik, Refractive index engineering of transparent ZrO2-polydimethylsiloxane nanocomposites, J. Mater. Chem., 2008, 18, 1751–1755 RSC.
- M. Sabzi, S. Mirabedini, J. Zohuriaan-Mehr and M. Atai, Surface modification of TiO2 nano-particles with silane coupling agent and investigation of its effect on the properties of polyurethane composite coating, Prog. Org. Coat., 2009, 65, 222–228 CrossRef CAS.
- B. Fritzinger, R. K. Capek, K. Lambert, J. C. Martins and Z. Hens, Utilizing self-exchange to address the binding of carboxylic acid ligands to CdSe quantum dots, J. Am. Chem. Soc., 2010, 132, 10195–10201 CrossRef CAS PubMed.
- J. De Roo, Y. Justo, K. De Keukeleere, F. Van den Broeck, J. C. Martins, I. Van Driessche and Z. Hens, Carboxylic-Acid-Passivated Metal Oxide Nanocrystals: Ligand Exchange Characteristics of a New Binding Motif, Angew. Chem., Int. Ed., 2015, 54, 6488–6491 CrossRef CAS PubMed.
- G. Garnweitner, L. M. Goldenberg, O. V. Sakhno, M. Antonietti, M. Niederberger and J. Stumpe, Large-Scale Synthesis of Organophilic Zirconia Nanoparticles and their Application in Organic–Inorganic Nanocomposites for Efficient Volume Holography, Small, 2007, 3, 1626–1632 CrossRef CAS PubMed.
- L. Deblock, E. Goossens, R. Pokratath, K. De Buysser and J. De Roo, Mapping out the aqueous surface chemistry of metal oxide nanocrystals: Carboxylate, phosphonate, and catecholate ligands, JACS Au, 2022, 2, 711–722 CAS.
- R. Gomes, A. Hassinen, A. Szczygiel, Q. Zhao, A. Vantomme, J. C. Martins and Z. Hens, Binding of phosphonic acids to CdSe quantum dots: A solution NMR study, J. Phys. Chem. Lett., 2011, 2, 145–152 CAS.
- J. De Roo, Z. Zhou, J. Wang, L. Deblock, A. J. Crosby, J. S. Owen and S. S. Nonnenmann, Synthesis of phosphonic acid ligands for nanocrystal surface functionalization and solution processed memristors, Chem. Mater., 2018, 30, 8034–8039 CrossRef CAS.
- E. Dhaene, S. Coppenolle, L. Deblock, K. De Buysser and J. De Roo, Binding Affinity of Monoalkyl Phosphinic Acid Ligands toward Nanocrystal Surfaces, Chem. Mater., 2023, 35, 558–569 CrossRef CAS.
- Y. Okada, H. Asama, N. Koike, S. Yamashita, N. Maeta, A. Uesaka and H. Kamiya, Direct Ordering of Anchoring Events at the Surface of Iron Oxide Nanoparticles Enabled by A Stepwise Phase-Transfer Strategy, ChemistrySelect, 2018, 3, 8458–8461 CrossRef CAS.
- E. Guénin, Y. Lalatonne, J. Bolley, I. Milosevic, C. Platas-Iglesias and L. Motte, Catechol versus bisphosphonate ligand exchange at the surface of iron oxide nanoparticles: towards multi-functionalization, J. Nanopart. Res., 2014, 16, 1–13 Search PubMed.
- K. Hossain, L. Florean, A. Del Tedesco, E. Cattaruzza, M. Geppi, S. Borsacchi, P. Canton, A. Benedetti, P. Riello and A. Scarso, Modification of Amorphous Mesoporous Zirconia Nanoparticles with Bisphosphonic Acids: A Straightforward Approach for Tailoring the Surface Properties of the Nanoparticles, Chem. – Eur. J., 2021, 27, 17941–17951 CAS.
- H. Rijckaert, J. De Roo, M. Van Zele, S. Banerjee, H. Huhtinen, P. Paturi, J. Bennewitz, S. J. Billinge, M. Bäcker and K. De Buysser, et al., Pair distribution function analysis of ZrO2 nanocrystals and insights in the formation of ZrO2-YBa2Cu3O7 nanocomposites, Materials, 2018, 11, 1066 Search PubMed.
- M. Egorov, S. Aoun, M. Padrines, F. Redini, D. Heymann, J. Lebreton and M. Mathé-Allainmat, A One-Pot Synthesis of 1-Hydroxy-1, 1-bis (phosphonic acid) s Starting from the Corresponding Carboxylic Acids, 2011 Search PubMed.
- J. De Roo, K. De Keukeleere, Z. Hens and I. Van Driessche, From ligands to binding motifs and beyond; the enhanced versatility of nanocrystal surfaces, Dalton Trans., 2016, 45, 13277–13283 CAS.
- F. Ondreas, P. Lepcio, M. Zboncak, K. Zarybnicka, L. E. Govaert and J. Jancar, Effect of nanoparticle organization on molecular mobility and mechanical properties of polymer nanocomposites, Macromolecules, 2019, 52, 6250–6259 CAS.
- B. J. Ash, D. F. Rogers, C. J. Wiegand, L. S. Schadler, R. W. Siegel, B. C. Benicewicz and T. Apple, Mechanical properties of Al2O3/polymethylmethacrylate nanocomposites, Polym. Compos., 2002, 23, 1014–1025 CAS.
- N. Jouault, F. Dalmas, F. Boué and J. Jestin, Multiscale characterization of filler dispersion and origins of mechanical reinforcement in model nanocomposites, Polymer, 2012, 53, 761–775 CrossRef CAS.
- Y. Lv, B. Lu, S. Zhang, J. Li, G. Lu, H. Sun, S. Liang and Z. Liu, Mechanical enhancement of amine-functionalized TiO2 reinforced polyimine composites, J. Appl. Polym. Sci., 2018, 135, 46446 CrossRef.
- B. Zewde, P. Pitliya and D. Raghavan, The role of surface modified TiO 2 nanoparticles on the mechanical and thermal properties of CTBN toughened epoxy nanocomposite, J. Mater. Sci., 2016, 51, 9314–9329 CrossRef CAS.
- P. Pötschke, A. R. Bhattacharyya, A. Janke and H. Goering, Melt mixing of polycarbonate/multi-wall carbon nanotube composites, Compos. Interfaces, 2003, 10, 389–404 CrossRef.
- J. N. Coleman, U. Khan, W. J. Blau and Y. K. Gun'ko, Small but strong: a review of the mechanical properties of carbon nanotube–polymer composites, Carbon, 2006, 44, 1624–1652 CrossRef CAS.
- Z. Wang, Z. Liang, B. Wang, C. Zhang and L. Kramer, Processing and property investigation of single-walled carbon nanotube (SWNT) buckypaper/epoxy resin matrix nanocomposites, Composites, Part A, 2004, 35, 1225–1232 CrossRef.
- O. Bianchi, J. A. Cruz, L. Paim, A. Lavoratti, Z. Al-Maqdasi, S. C. Amico, P. Fernberg and R. Joffe, Rheology, curing and time-dependent behavior of epoxy/carbon nanoparticles systems, J. Appl. Polym. Sci., 2024, 141, e54821 CAS.
- S. Halder, S. Ahemad, S. Das and J. Wang, Epoxy/glass fiber laminated composites integrated with amino functionalized ZrO2 for advanced structural applications, ACS Appl. Mater. Interfaces, 2016, 8, 1695–1706 CrossRef CAS PubMed.
- M. Shokrieh, S. Ghoreishi and M. Esmkhani, Toughening mechanisms in composite materials, Elsevier, 2015, pp. 295–320 Search PubMed.
- S. K. Kumar and R. Krishnamoorti, Nanocomposites: structure, phase behavior, and properties, Annu. Rev. Chem. Biomol. Eng., 2010, 1, 37–58 CAS.
- B. De'Nève and M. Shanahan, Water absorption by an epoxy resin and its effect on the mechanical properties and infra-red spectra, Polymer, 1993, 34, 5099–5105 Search PubMed.
- H. Zhao and R. K. Li, Effect of water absorption on the mechanical and dielectric properties of nano-alumina filled epoxy nanocomposites, Composites, Part A, 2008, 39, 602–611 CrossRef.
- O. Becker, R. J. Varley and G. P. Simon, Thermal stability and water uptake of high performance epoxy layered silicate nanocomposites, Eur. Polym. J., 2004, 40, 187–195 CrossRef CAS.
- P. Nogueira, C. Ramirez, A. Torres, M. Abad, J. Cano, J. Lopez, I. López-Bueno and L. Barral, Effect of water sorption on the structure and mechanical properties of an epoxy resin system, J. Appl. Polym. Sci., 2001, 80, 71–80 CrossRef CAS.
- C. Ng, B. Ash, L. Schadler and R. Siegel, A study of the mechanical and permeability properties of nano-and micron-TiO2 filled epoxy composites, Adv. Compos. Lett., 2001, 10, 096369350101000301 CrossRef.
- H. Choi, K. Ahn, J.-D. Nam and H. Chun, Hygroscopic aspects of epoxy/carbon fiber composite laminates in aircraft environments, Composites, Part A, 2001, 32, 709–720 CrossRef.
- D.-W. Suh, M.-K. Ku, J.-D. Nam, B.-S. Kim and S.-C. Yoon, Equilibrium water uptake of epoxy/carbon fiber composites in hygrothermal environmental conditions, J. Compos. Mater., 2001, 35, 264–278 CrossRef CAS.
- H. Alamri and I. M. Low, Effect of water absorption on the mechanical properties of nano-filler reinforced epoxy nanocomposites, Mater. Des., 2012, 42, 214–222 CrossRef CAS.
- W. Liu, S. V. Hoa and M. Pugh, Fracture toughness and water uptake of high-performance epoxy/nanoclay nanocomposites, Compos. Sci. Technol., 2005, 65, 2364–2373 CrossRef CAS.
- G. E. Guloglu and M. C. Altan, Moisture absorption of carbon/epoxy nanocomposites, J. Compos. Sci., 2020, 4, 21 CrossRef CAS.
- C. Zou, J. C. Fothergill and S. W. Rowe, The effect of water absorption on the dielectric properties of epoxy nanocomposites, IEEE Trans. Dielectr. Electr. Insul., 2008, 15, 106–117 CAS.
- R. Adams and L. Bischof, Seeded region growing, IEEE Trans. Pattern Anal. Mach. Intell., 1994, 16, 641–647 CrossRef.
- H. Toda, S. Masuda, R. Batres, M. Kobayashi, S. Aoyama, M. Onodera, R. Furusawa, K. Uesugi, A. Takeuchi and Y. Suzuki, Statistical assessment of fatigue crack initiation from sub-surface hydrogen micropores in high-quality die-cast aluminum, Acta Mater., 2011, 59, 4990–4998 CrossRef CAS.
- V. Le Saux, Y. Marco, S. Calloch and P. Charrier, Evaluation of the fatigue defect population in an elastomer using X-ray computed micro-tomography, Polym. Eng. Sci., 2011, 51, 1253–1263 CrossRef CAS.
- D. B. G. Williams and M. Lawton, Drying of organic solvents: quantitative evaluation of the efficiency of several desiccants, J. Org. Chem., 2010, 75, 8351–8354 CrossRef CAS PubMed.
- L. Manxzer, J. Deaton, P. Sharp and R. Schrock, 31. Tetragtdrfuran Complexes of Selected Early Transition Metals, Inorg. Synth., 1982, 21, 135–140 CrossRef.
- S. Akoka, L. Barantin and M. Trierweiler, Concentration measurement by proton NMR using the ERETIC method, Anal. Chem., 1999, 71, 2554–2557 CrossRef CAS PubMed.
- J. Schindelin, I. Arganda-Carreras, E. Frise, V. Kaynig, M. Longair, T. Pietzsch, S. Preibisch, C. Rueden, S. Saalfeld and B. Schmid, et al., Fiji: an open-source platform for biological-image analysis, Nat. Methods, 2012, 9, 676–682 CrossRef CAS PubMed.
- O. Ronneberger, P. Fischer and T. Brox, in Medical image computing and computer-assisted intervention–MICCAI 2015: 18th international conference, Munich, Germany, October 5–9, 2015, proceedings, part III 18, 2015, pp. 234–241.
Footnote |
| † Electronic supplementary information (ESI) available: Additional NMR, DLS, TEM, SEM, TGA and micro-CT data, characterization of synthesized bisPA-mPEG[2] and bisPA-mPEG[6] ligands, deep learning segmentation process, precursor characterization, schematics, tables and calculations. See DOI: https://doi.org/10.1039/d4nr04561k |
| This journal is © The Royal Society of Chemistry 2025 |