DOI:
10.1039/D5MA00327J
(Paper)
Mater. Adv., 2025,
6, 4881-4892
Photo-crosslinked persistent micelle templates with near universal solvent compatibility†
Received
4th April 2025
, Accepted 9th June 2025
First published on 18th June 2025
Abstract
The controlled preparation of porous nanomaterials from block polymer templates is important for diverse applications from energy devices to optical coatings and sorbents. The architectural dimensions determine the overall performance for many of these applications, however the independent control of these dimensions has remained limited to a narrow range of solvent conditions for kinetically trapped (“persistent”) micelle templates. Polymers with a photo-reactive chemistry are shown to enable cross-linked micelle templates that universally remain persistent under diverse solvent conditions. Specifically, poly(poly(ethylene glycol methacrylate))-b-poly(butyl methacrylate-co-coumarin methacrylate) (OBC) was prepared by RAFT polymerization and the resulting micelles were crosslinked with UV light (X-OBC). TEM images revealed that crosslinking in MeOH-only led to a mixture of micelles and unimers whereas crosslinking in MeOH–H2O led to pure micelles. Absorbance measurements indicated 50% coumarin dimerization (crosslinking) occurred after 180 min of UV exposure. The near universal solvent compatibility of X-OBC persistent micelles is demonstrated with DLS measurements in toluene, DCM, DMF, THF, EtOH, and MeOH, whereas OBC is shown to release unimers in DCM, DMF, THF, and toluene, or precipitate from EtOH due to a morphology change. Spectroscopic testing of micelle crosslinking occurred at each templating step. Micelle entrapment was also validated with SAXS and SEM measurements after nanomaterial templating where X-OBC enabled sample series with constant pore size whereas OBC did not. Furthermore, the results uniquely show X-OBC micelle persistence when processed from non-selective solvents. These results highlight how molecular engineering and tailored processing can enable the generalized synthesis of controlled porous nanomaterials.
Introduction
Block polymer micelles are vital for the controlled synthesis of materials with nanoscale porosity,1–6 which can be applied to optical coatings,7–9 sorbents,10,11 and the next generation of energy devices.12–19 Generally amphiphilic block polymers are used where material precursors selectively assemble with a hydrophilic block while a hydrophobic block phase separates. The self-assembly of unimers into micelles is driven by the interfacial enthalpy of the solvophobic block contacting the solvent which can be minimized by aggregation.20,21 This core–solvent interaction can be quantified by χN, where χ is a largely enthalpic term corresponding to the energy of a two-body interface and N scales with the core-block degree of polymerization.20–22 A typical challenge for micelle templates is that sequential additions of material precursors change the solution thermodynamics and thus induce changes to the micelle dimensions.23 The resulting architecture series from such dynamic micelles tend to be a complex convolution of dimension changes without independent control of any particular feature size such as the pore dimension or wall thickness.24,25
Controlling the rate of chain exchange can thus enable improved control over nanomaterial synthesis. Micelle chain exchange can occur by several processes,26–31 where the single chain exchange (SCE) mechanism has well described kinetics.31–37 The rate of SCE depends on a double exponential of χN which has been described as hypersensitive to chain length.31 Notably, the same formula likewise implies a hypersensitivity of the chain exchange rate to χ.31 Persistent micelle templates (PMT) can take advantage of such rate laws by slowing chain exchange with high-χN conditions to realize kinetically trapped micelles with constant aggregation number.25 When PMTs are combined with material precursors they uniquely enable sample series with constant pore size (constant micelle size) and independent control of the wall thickness.25,38 The most demonstrated PMT modality relied on high-χN conditions to suppress chain exchange rates via this energetic barrier.24,38–41 This is typically achieved by making the solution relatively hydrophilic with the addition of water to organic solvents. In one example, processing PMTs from tetrahydrofuran required ∼8 wt% water whereas analogous examples using alcohols required just 2 wt% water.24,39,41 This water-dependence for χN control of PMTs has limited the advancement of anhydrous chemistries for diverse materials. A second PMT modality relies upon immobilization of core chains by vitrification (glassy core).42,43 A challenge with this modality is that it also limits solvent selection to non-plasticizers for the core block which excludes common solvents such as tetrahydrofuran (THF), methylene chloride (DCM), toluene, and others for many polymer systems. This collection of constraints on processing solvents has limited the diversity of material chemistries used with PMTs and motivates the creation of a universal class of PMTs.
Covalently crosslinking a micelle core can stabilize micelles under changing solution conditions and has primarily been developed towards drug delivery and bio-imaging.44–47 To the best of our knowledge, core-crosslinked micelles have not been demonstrated as soft templates for producing controlled porous materials. Diverse synthetic methods such as, photo-induced cycloadditions,48–51 azobenzenes52 click chemistries,44,53–56 and disulfide linkages57–59 can produce core-crosslinked micelles. A challenge with reagent-based crosslinking routes is that the thermodynamics of the solution conditions used for the reaction can affect the micelle aggregation state. In contrast, photo-dimerization does not require chemical reagents, produces no byproducts, and can be conducted in arbitrary solvents. Coumarin moieties are particularly convenient where they undergo olefin photo-dimerization under long wavelength UV light (>320 nm), which induces a [2+2] cycloaddition yielding a cylcobutane dimer that can also undergo cycloreversion under shorter wavelength irradiation (<260 nm).60 The implementation of coumarins in polymer assemblies has caught widespread attention due to the ease of synthesis, reversibility, and UV-vis signature for easily tracked kinetics. Coumarin containing polymers have been implemented in additive manufacturing,61,62 and self-healing materials,63,64 while coumarin containing polymer micelles have been applied to antifogging coatings, emulsion stabilizers65,66 and again, targeted drug delivery and bio-imaging.67–70
In this work we employ photo-active coumarin dimers to produce core-crosslinked polymer micelles to serve as robust soft templates that are persistent, i.e. incapable of chain exchange (Scheme 1). Coumarin monomer units are directly copolymerized into the hydrophobic block (at a molar fraction of 24%) of the amphiphilic polymer via RAFT to produce poly(poly(ethylene glycol methacrylate)-b-butyl methacrylate-co-coumarin methacrylate) (OBC). The micelle solution behavior is first investigated with a combination of TEM and UV-Vis to understand the required processing conditions for persistence before crosslinking can properly occur within the micelle core, producing X-OBC. We note that the result of crosslinking a micelle to yield a single molecule is debatable whether or not to continue calling it a “micelle” since the aggregation number becomes 1 and there is no longer a critical micelle concentration. Literature precedents towards this are varied with many reports44,46,54,56 maintaining the “micelle” descriptor despite these limitations, perhaps to designate the formation pathway. In the same spirit as glassy-core micelles,42,43 single-chain micelles,71,72 and prior reports of crosslinked micelles, we prefer the “micelle” descriptor. The stability of the micelle was then probed via DLS to ensure colloidal stability in various media in all realms of solubility space before use as a template. A unique feature of the coumarin containing micelles is that cross-linking is verifiable at every step of the templating process, all the way from solution phase to within a material matrix. Lastly, a series of nanomaterials are quantitatively compared to show that X-OBC persistent micelles can uniquely be processed in challenging solvent conditions including pure-THF.
 |
| | Scheme 1 Overview showing the preparation and use of X-OBC templates. (a) OBC micelles are irradiated with UV light to induce crosslinking where subsequent addition of nanoparticles selectively associate with the hydrophilic corona. (b) With persistent micelle templates, the resulting pore size is constant with such material additions which enable tailored wall thickness. (c) Subsequent calcination removes the polymer and results in pores. | |
Experimental
Materials
Sodium iodide (NaI) and titanium tetraisopropoxide (TTiP, 99%, Acros) were stored in an argon glovebox prior to use. 1,4 Dioxane (BeanTown Chemical, 99.0%), anisole (99%, BeanTown Chemical), methanol (MeOH, 99.8%, Fisher), methylene chloride (DCM, 99%, Fisher) and N′N-dimethylformamide (DMF, ACS Grade, VWR) were dried at room temperature over 50% w/w of molecular sieves (3 Å, 8–12 mesh, Acros Organics).73 Acetone (ACS Grade, Fisher), DI water (ACS Grade, Ward's Science), anhydrous diethyl ether (Certified ACS, Fischer), ethanol (EtOH, 200 proof, Deacon Laboratories), hexanes (>98.5%, Macron Fine Chemicals), concentrated hydrochloric acid (HCl, 37 wt%, ACS Grade, VWR), 7-hydroxy-4-methylcoumarin (97%, Fisher), 4-dimethaminopryidine (DMAP, 99%, BeanTown Chemical), potassium carbonate (K2CO3, 99%, Fisher), tetraethoxysilane (TEOS, 98%, Alfa Aesar), were all used as received. 2,2′-Azobis(2-methylpropnitrile) (AIBN, 98%, Sigma) was recrystallized from methanol and stored in a freezer (−20 °C). 4-Cyano-4(phenylcarbonothioylthio)pentanoic acid (4CPDB, AmBeed, 98.16%), and 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC, 98.03%, Ambeed) were stored in a freezer. Poly(ethylene glycol)methacrylate (PEGMA, 500 Mn, Sigma) and n-butyl methacrylate (nBMA, >99.0%, TCI) were stored in a refrigerator and passed over a basic alumina column prior to use. Methacrylic acid (MAA, 99%, BeanTown Chemical), 2-bromoethanol (95%, TCI), and 1% uranyl acetate (UrAc, Electron Microscope Sciences) were stored in a refrigerator. Dialysis tubing with 3500 MWCO was purchased from ThermoFisher Scientific. The carbon-coated 300 mesh copper grids for the TEM measurement were purchased from Electron Microscopy Sciences.
Synthesis of PPEGMA
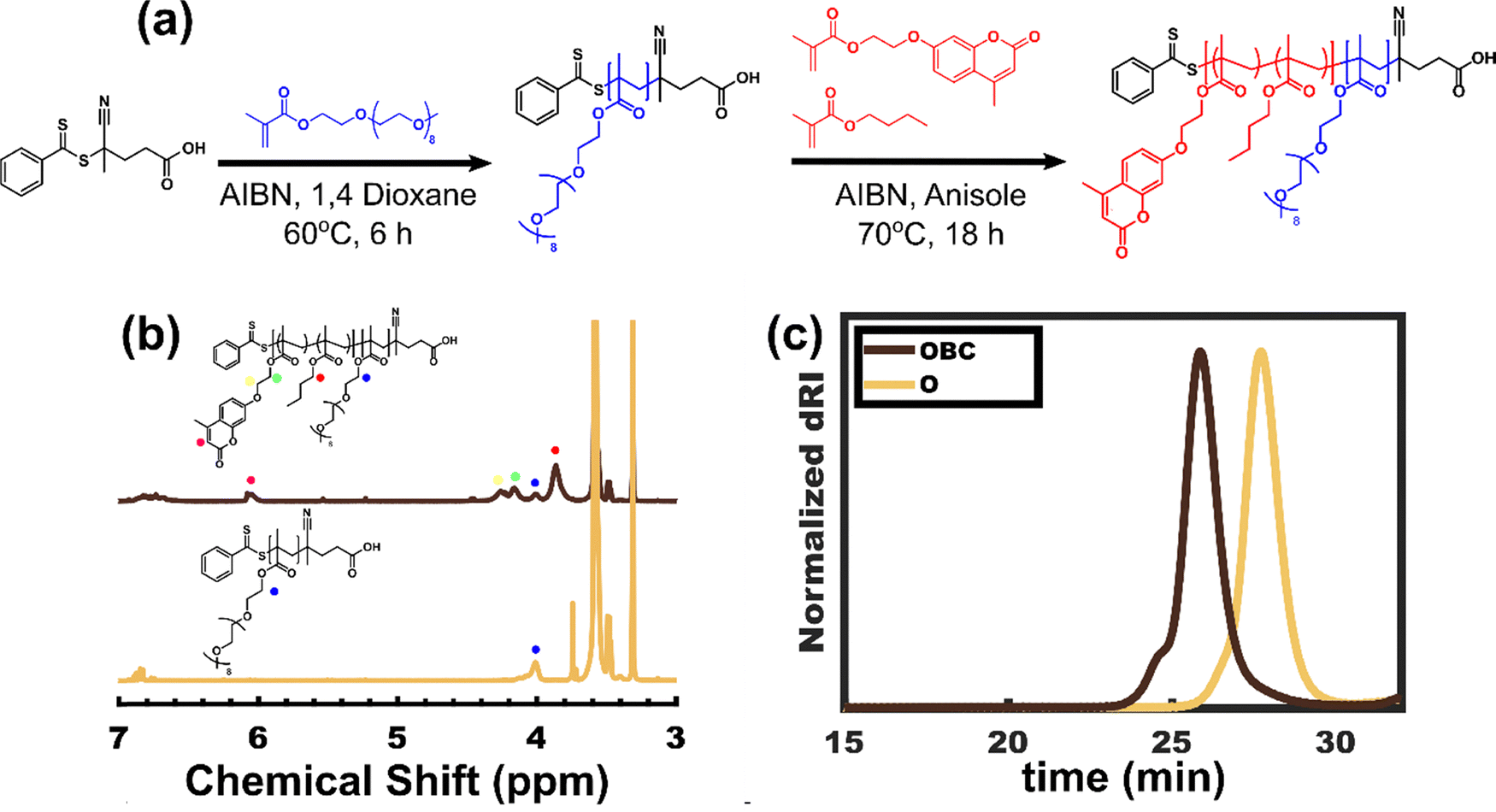
4CPDB (200 mg, 0.72 mmol, 1 equiv.), PEGMA (5.90 mL, 13.0 mmol, 18.0 equiv.), AIBN (120 mg, 0.72 mmol, 0.10 equiv.) in a 10 mg mL−1 1,4 dioxane solution, and 4.00 mL of 1,4 dioxane were combined in a Schlenk flask and underwent three cycles of freeze–pump–thaw to remove dissolved gases. The reaction flask was then brought into an argon-filled glovebox to backfill with inert gas. The polymerization was then carried out in a preheated oil bath at 60 °C for 6 hours. Once the polymerization was complete, the reaction was cooled in a freezer for two hours before venting. An aliquot of crude product was collected for conversion analysis, showing a 97% conversion and was verified by end group analysis leaving a final Mn of 8.50 kg mol−1. The product was purified by washing 3 times with an 80/20 mixture of hexanes and diethyl ether to remove unreacted monomer with polymer separation at each step using centrifugation (7000 rpm, 2 min) with decanting of the supernatant. The product was then dried under vacuum at room temperature overnight. The molar mass and conversion were determined by proton nuclear magnetic resonance (1H-NMR) in CDCl3. The molar-mass dispersity (Đ) was verified by gel permeation chromatography (GPC).
Synthesis of PPEGMA-b-(PnBMA-co-PCouMA), (OBC)
The synthesis of Coumarin Methacrylate (CouMA) is described in the ESI† (S1 and S2). PPEGMA (2.00 g, 8.50 kg mol−1, 0.235 mmol, 1 equiv.) and CouMA (2.51 g, 8.7 mmol, 37 equiv.) were first dissolved in 25 mL of Anisole while gently heating in a Schlenk flask. After allowing the homogeneous solution to cool, nBMA (4.75 mL, 30.1 mmol, 128 equiv.) and AIBN (5.8 mg, 0.035 mmol, 0.15 equiv.) were added to the solution. The mixture then underwent three cycles of freeze–pump–thaw. The reaction flask was then brought into an argon-filled glovebox to backfill with inert gas. The polymerization was then carried out in a preheated oil bath at 70 °C for 18 hours. Aliquots were periodically collected to check the progress of the monomer conversion (S3, ESI†). Once the polymerization was complete, the reaction was cooled in a freezer for two hours before venting. An aliquot of crude product was collected for conversion analysis to estimate monomer consumption, showing a 73.0% conversion of the CouMA monomer and a 68.8% conversion of the nBMA monomer (S3, ESI†). The crude product then underwent dialysis against acetone, changing the dialysis solvent every 12 hours, three times. The purified product was then dried under reduced pressure.
Polymer characterization
The molar mass of the OBC diblock was determined using 1H-NMR spectroscopy (S4 and S5, ESI†) while GPC was used to evaluate the molar mass dispersities (Fig. 1c) of the homopolymer macroinitiator and subsequent diblock. All 1H-NMR spectra were collected using a Bruker Avance III-HD 400. The GPC was composed of a Waters 515 HPLC pump, a Waters 2410 refractive index detector, and three Styragel columns (HR2, HR4, and HR5) in the effective molecular weight range of 0.5–20, 5–600, 50–4000 kg mol−1, respectively. The eluent was THF at a temperature of 25 °C and a flow rate of 1 mL min−1. The data was analyzed using custom MATLAB scripts.74 The instrument was calibrated with polystyrene standards (800, 560, 320, 180, 100, 43, 20, 10, 4.5, 2.1, 1.0, 0.27 kg mol−1) received from Polymer Standards Service. The GPC samples were prepared by dissolution in THF at a concentration of ∼10 mg mL−1 and were filtered through a syringe filter with a pore diameter of 0.2 μm before injection.
 |
| | Fig. 1 (a) Synthesis scheme for O homopolymer (PPEGMA) followed by chain extensions of BC using RAFT polymerization. Both O and OBC were characterized by (b) 1H-NMR spectra and (c) GPC analysis. | |
Micellization of PPEGMA-b-(PCouMA-co-PnBMA)
Micelle solutions were prepared by dissolving 100 mg OBC polymer in 9 mL of anhydrous MeOH with minor agitation until completely dissolved. To decrease the critical micelle concentration, 1 mL of DI water (10 vol%) was added to the solution with sonication for 5 min.30,75,76 Micelles were crosslinked irradiated with a UVP Blak-Ray Long Wave Ultraviolet Lamp Model B 100AP (365 nm), 100 Watts, 9.65 × 6.00′′ lamp head (248 × 140 mm). The micelle solutions are then exposed to UV light for 16 hours and crosslinking was tracked with a Shimadzu UV-2450 spectrophotometer. After the micelles were crosslinked, the subsequent solutions were transferred into pure solvents by either dialysis (MeOH, EtOH, and THF) or rotary evaporation (Toluene and DMF) to reach a final concentration of 5 mg mL−1. Micelles in DCM were produced by drying the micelle solution via rotary evaporation and directly redispersing the micelles in DCM.
DLS measurements
Dynamic light scattering (DLS) measurements were performed on (X-)OBC in various solvents. The DLS measurements to determine hydrodynamic diameter were performed using a Zetasizer Nanoseries Zen3690 instrument. Solutions for DLS were prepared at a concentration of ∼5.00 mg mL−1 and were filtered through a 0.2 μm syringe filter prior to measurement. All measurements were performed in triplicate to ensure reproducibility at 25 °C. All DLS measurements were performed in pure solvents.
Use of X-OBC as micelle templates
Various combinations of OBC and X-OBC micelles with different loadings of TiO2 precursors were examined. An ex situ TiO2 nanoparticle solution was prepared by rapidly adding 5 mL of TTIP to 1.2 mL of conc. HCl and 2 mL of anhydrous MeOH inside of a 20 mL scintillation vial with rapid stirring.41 The solution was then allowed to stir for 20 minutes before use. Five 0.20 mL, aliquots of the previously described micelle stock at 5.00 mg mL−1 were placed in different ½ dram vials and mixed with a predetermined volume of the TiO2 stock solutions to realize the desired material-to-template (M![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :T) mass ratio, assuming complete conversion of TTIP to TiO2. After mixing the two, a 10 μL aliquot of the solution was then spin coated on a 9 × 9 mm cover glass slide for 30 s at 1500 rpm with a 15% relative humidity,38 after which the slide was transferred to a pre-heated hotplate at 200 °C and allowed to age for two hours. Samples spun on silicon substrates were prepared in similar fashion and were later calcined in a furnace at 350 °C for one hour with a 5 °C min−1 ramp rate.
:T) mass ratio, assuming complete conversion of TTIP to TiO2. After mixing the two, a 10 μL aliquot of the solution was then spin coated on a 9 × 9 mm cover glass slide for 30 s at 1500 rpm with a 15% relative humidity,38 after which the slide was transferred to a pre-heated hotplate at 200 °C and allowed to age for two hours. Samples spun on silicon substrates were prepared in similar fashion and were later calcined in a furnace at 350 °C for one hour with a 5 °C min−1 ramp rate.
Ex situ TEOS hydrolysis and solid-state UV-vis
The spectroscopic features of X-OBC were tracked throughout a material templating procedure. Solid-state UV-vis absorption was measured with X-OBC micelles embedded within SiO2 matrix. Following previously described methods,38 briefly, 0.270 mL of TEOS was mixed with 0.120 mL of 0.5 M HCl and 0.850 mL of MeOH. The solution was allowed to rapidly stir for 1 hours on a hotplate at 60 °C. Next, 1 mL of OBC/X-OBC at a concentration of 10 mg mL−1 was mixed with 0.170 mL of SiO2 nanoparticles. The subsequent polymer/material solution was drop cast onto a fused quartz slide that was on a hot plate set to 60 °C for 3 hours.
X-ray measurements
X-ray measurements were performed at the South Carolina SAXS Collaborative (SCSC) using a SAXSLab Ganesha (Xenocs Inc.) instrument. A Xenocs GeniX 3D microfocus source was used with a copper target to produce a monochromatic beam with a λ = 0.154 nm. The instrument was calibrated prior to use with a National Institute of Standards and Technology (NIST) reference material 640d silicon powder with a peak position at 28.44° 2θ, where 2θ is the total scattering angle. A Pilatus 300k detector (Dectris) was used to collect two-dimensional (2D) scattering pattern with a nominal pixel dimension of 172 × 172 μm2. The SAXS data was acquired with an X-ray flux of ∼4.1 M photons per second incident upon the sample with a sample to detector distance of 1040 mm. The 2D images were azimuthally integrated to yield the scattering vector and intensity. Peak positions were fitted using custom MATLAB scripts. The measured SAXS derived-values were reported as the average ± standard error-of-the-mean.
TEM
Transmission electron microscopy (TEM) images were collected in bright-field imaging mode using a JEOL 1400 Plus Transmission Electron Microscope with an accelerating voltage of 120 keV. TEM grids were prepared by placing a 20 μL droplet of the desired X-OBC micelle solution at 1 mg mL−1 with the solution being gently wicked through the other side of the TEM grid. Staining was performed by adding a 20 μL drop of 1 wt% aqueous UrAc solution that was wicked in the same fashion.
SEM
Top view images of calcined films were obtained using a Zeiss Ultraplus thermal field emission scanning electron microscopy (SEM) system using an accelerating voltage of 3 keV and an in-lens secondary electron detector. The working distance was maintained ∼4.1 mm as well as constant magnification for all compared measurements.24 Hundreds of measurements were acquired on each sample to yield statistically significant metrics on pore size and wall thickness. The wall thickness was measured as the shortest distance between neighboring pores. The derived values are presented as averaged values with the standard-error-of-the-mean.
Results
Design considerations and OBC synthesis
There are several considerations when designing a chemistry platform to realize micelles with “universal” kinetic entrapment via crosslinking. (1) The micellization process should be decoupled from the crosslinking process. Here, the introduction of chemical reagents to induce crosslinking modify the thermodynamics of the micelle solution and cause a shift in the equilibrium size. When the time scale for the corresponding chain exchange process is commensurate with the time for crosslinking, then the size dispersity is expected to increase. Thus, using an external stimuli such as the photo-crosslinking of coumarin in PCouMA satisfies this objective (Fig. 1a). (2) The location of crosslinks matter. Micelles have been crosslinked via either the core or the corona regions.44 The templating process, however, requires the corona blocks to wrap around nanoparticle precursors so it is most logical to place the crosslinks amongst the core blocks as done in the PnBMA-co-PCouMA core used here.25,77 (3) The cross-linkable block must have sufficient mobility to enable coupling. PCouMA has a high glass transition temperature (Tg ∼ 160 °C) where the polymer chains are completely immobilized at room temperature.78 This can be countered, however, with the addition of a “filler” block with a low Tg which must also have a sufficiently high upper critical solution temperature (UCST) to favor micellization.79,80 In this case, PnBMA has suitable Tg ∼ 20 °C and exhibits partial solubility in room temperature MeOH and complete solubility at elevated temperatures.80 This partial solubility still allows for the formation of micelles via direct dispersion in MeOH while also plasticizing the overall core region to enable coumarin dimerization. (4) Solvent compatibility of the overall micelle is largely determined by the corona block, including its volume fraction, chemical identity, and architecture.20,81–83 PEO and related PEGMA polymers have some of the broadest ranges of good solvents where the Wiley Database of Polymer Properties84 lists only a few non-solvents for PEO including aliphatic hydrocarbons, ether, and hot water. Towards these ends a PEO-like PEGMA was selected for the corona block. (5) The corona block must be able to interact with material precursors (negative χ) via e.g. hydrogen bonding.77,85,86 PEO and PEGMA are the prototypical hydrophilic blocks towards this end. (6) Synthetic ease is the last noted consideration where the design of OBC is entirely based on methacrylates that are facile to polymerize via RAFT with high conversion extents and narrow dispersities.
The synthesis of OBC was carried out with sequential RAFT polymerizations. RAFT was selected due to its convenient recipe development owing to its use of a minimal number of reagents (Fig. 1a).87 PPEGMA was grown first, followed by a block of PnBMA-co-PCouMA. The hydrophobic block was chosen to have 25 mol% coumarin, similar to a prior report using 29 mol% to achieve crosslinked bulk polymer morphologies. This fraction was intended to ensure multiple crosslinks per block.88 The products were characterized by 1H-NMR to track conversion and determine Mn while GPC was used to validate controlled growth as evidenced with a narrow molar mass dispersity (Đ < 1.2) at each step. The data for OBC synthesis are shown in Fig. 1b and c and are summarized in Table 1.
Table 1 Summary of measured characteristics for the utilized polymers
| Sample name |
Total Mna (kDa) |
M
n
(kDa) |
Molar mass dispersityb (Đ) |
DPPEGMa |
DPnBMAa |
DPCouMAa |
f
hydrophobic
(vol) |
%CouMAa,c,d (mol%) |
|
Determined by 1H-NMR spectroscopy.
Determined by GPC.
Volume fraction of (PnBMA-co-PCouMA) block using literature densities values for PPEGMA (ρ = 1.08 g cm−3),89 PCouMA (ρ = 1.23 g cm−3),78 and PnBMA (ρ = 1.07 g cm−3).37
% CouMA is relative to the hydrophobic block.
|
| PPEGMA |
8.50 |
9.78 |
1.13 |
17.0 |
— |
— |
— |
— |
| OBC |
30.10 |
27.51 |
1.19 |
17.0 |
93.6 |
28.8 |
71 |
0.24 |
Micellization of OBC
The micellization of OBC was first examined. Due to the partial solubility of the core block,80 OBC was directly dispersible in MeOH. The dispersion was photo-crosslinked with UV light so that the solution phase was preserved for TEM imaging at room temperature. The resulting X-OBC–MeOH was deposited on a TEM grid and was stained with UrAc to enhance contrast with the PPEGMA appearing dark with brightfield imaging. The corresponding electron micrographs (Fig. 2a) revealed spherical micelles amongst a “fingerprint background.” This fingerprint feature was interpreted as the result of free unimers in solution, i.e. chains not aggregated within micelles at the point of photo-crosslinking. Thus, in the interest of isolating micelles-alone, ∼10 vol% water was added to increase χN and reduce the critical micelle concentration (Fig. 2b). Crosslinking this sample resulted in X-OBC–MeOH–H2O where the corresponding electron micrographs (Fig. 2c) revealed micelles-alone without apparent unimers. TEM measurements of numerous micelles were made to determine the average core size of X-OBC–MeOH–H2O to be 31.2 ± 0.3 nm (standard-error-of-the-mean). Please note that TEM is carried out under high vacuum so this average core diameter corresponds to micelles without solvent swelling. Also note that the overall micelle is larger when including the corona. Regardless, the aggregation number can be calculated based on the non-swollen core volume (15902 nm3) as compared to the volume of a single core block. The volume of a single core block (31.8 nm3 per block) was calculated from the average core molecular mass (BC-only 21600 g mol−1) and the average core density (volume-weighted average 1.13 g cm−3). Thus the aggregation number was calculated as Nagg = 500 (15902 nm3/31.8 nm3 per block), similar to other reports of similar micelles34,40,43
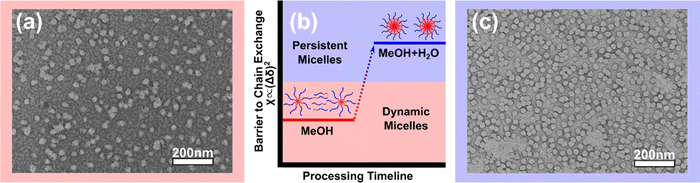 |
| | Fig. 2 (a) TEM image of X-OBC–MeOH showing a mixture of micelles and unimers (“fingerprint” background). (b) Water was added to the processing solution to reduce the critical micelle concentration and suppress unimers (increasing χN). (c) TEM of X-OBC–MeOH–H2O showing micelles alone. TEM samples were stained with UrAc so that the PPEGMA appears dark. | |
X-OBC crosslinking kinetics and “universal” solvent compatibility
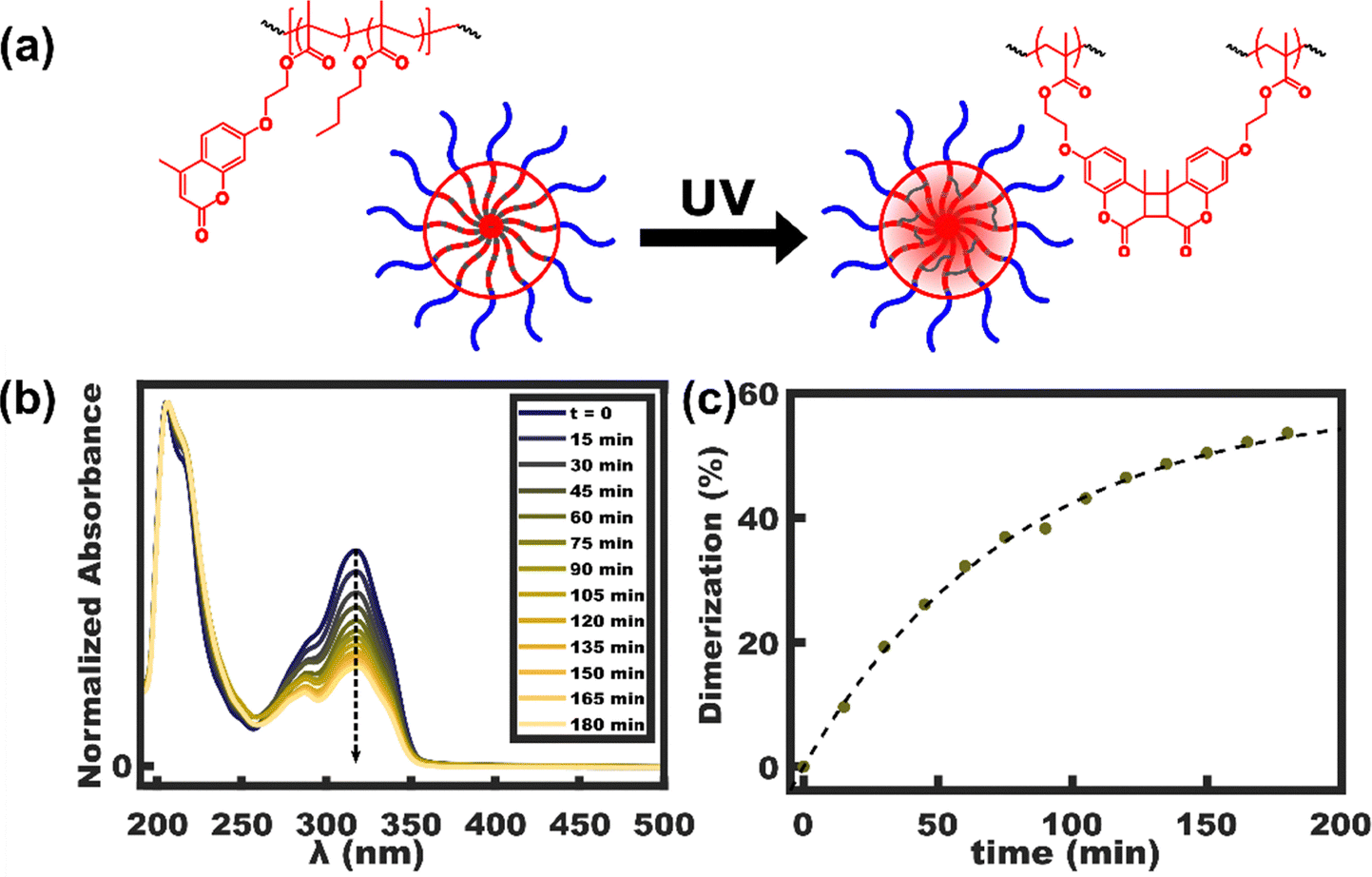
The kinetics of photo-crosslinking were investigated first. The crosslinking reaction extent may be tracked spectroscopically since the change in conjugation substantially modifies the absorbance spectrum. Fig. 3(a) shows the corresponding reaction scheme for coumarin dimerization and the corresponding absorbance spectra are shown in Fig. 3b. Here the UV absorbance decreases markedly during photo-crosslinking, most notable the peak at 320 nm. The rate of absorbance decrease slows markedly after 90 min of UV exposure. The dimer conversion was calculated from the reduction of the peak intensity at 320 nm with the data normalized using the invariant peak at 206 nm. With the initial condition setting the reference point for 100% coumarin (0% dimerized), the data allow quantification of the reaction extent whereafter 180 min there are 46.4% coumarin (53.6% dimer). Here it is interesting to consider the effect of even partial crosslinking upon chain exchange kinetics. The first dimerization of two chains results in a single molecule with 2× the nominal molar mass and thus a 2× increase in N.25,31 Recalling the hypersensitive double exponential dependence of the chain exchange rate on χN, such molar mass increases substantially halt the rate of chain exchange by several orders of magnitude.31 In the limit of high crosslinking density, the result is that each micelle becomes a single molecule where the notion of chain exchange ceases to be relevant. Both inter and intramolecular coumarin reactions are expected to have identical spectroscopic signatures. The extent of intramolecular crosslinking, however, can be crudely estimated based on the number of coumarins per chain and per micelle. Based on DPCouMA = 28.8 and Nagg = 500, the average micelle has 14k coumarins. From this perspective, a minor 0.2% of intramolecular crosslinking is anticipated with random mixing. Based on this underestimate and the 53.6% coumarin crosslinking measured, one may estimate that ∼13 coumarins per chain are dimerized intermolecularly, thus these micelles are sufficiently crosslinked to be considered single molecules.
 |
| | Fig. 3 (a) Schematic representation of the coumarin dimerization within the micelles core. (c) Photo-crosslinking was tracked spectroscopically in OBC micelles. (b) The percent of coumarin dimerized was tracked based on the absorbance at 320 nm. Dashed line provided to guide the eye. | |
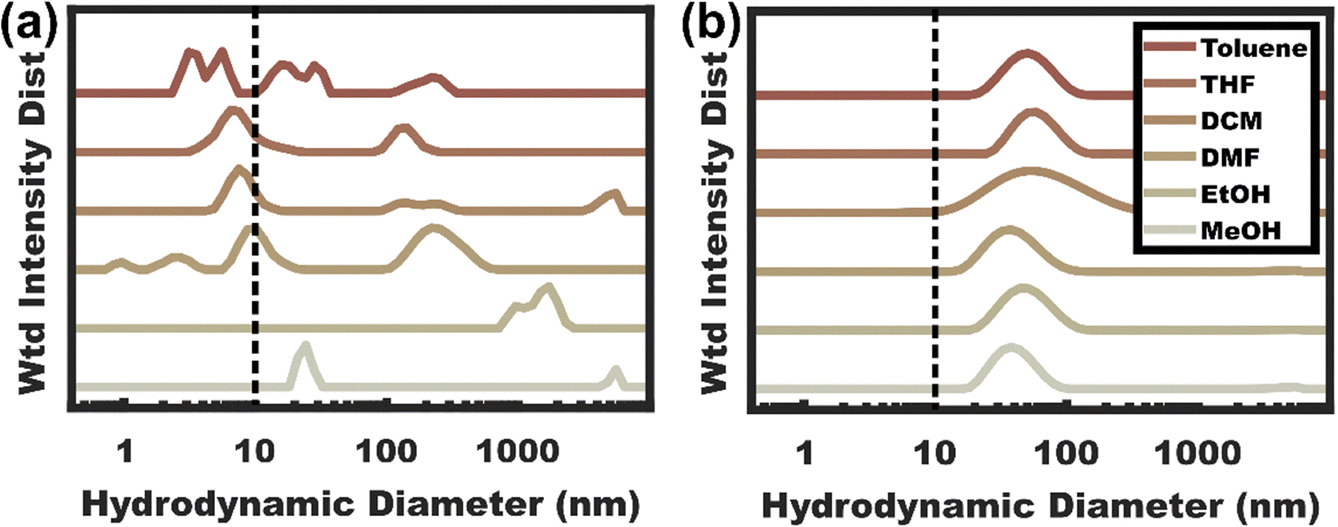
The capability of the resulting X-OBC micelles to exhibit “universal” micelle entrapment in diverse solvents was examined with DLS. Here OBC was dispersed in numerous solvents that span a wide range of Hildebrand parameters and are later compared to X-OBC that was transferred to the same solvent conditions. The solvents included lower alcohols, highly polar DMF, a cyclic ether THF, a water immiscible organochlorine DCM, and an aromatic hydrocarbon toluene with corresponding Hildebrand values90 ranging from 18.3–29.7 MPa0.5. Fig. 4a shows the hydrodynamic diameter of non-crosslinked OBC dispersed in this range of solvents. The resulting aggregate sizes were highly varied with e.g. ∼25 nm micelles in MeOH and ∼1 μm features in EtOH, presumably associated with micelle aggregation due to the low PPEGMA fraction leading to semi-bald/crewcut micelles.91,92 The remaining solvents examined were substantially less-selective, i.e. capable of dissolving all the used polymer chemistries (PPEMGA, PnBMA, and PCouMA). Here THF, DMF, DCM, and toluene all exhibited a mixture of micelles (>10 nm) and unimers (<10 nm) by DLS, indicating a lack of chain entrapment. It is important to note that the measured DLS intensities increase significantly with object size (intensity ∝ size6). It follows that OBC is mostly present as unimers in THF, DMF, DCM, and toluene. In contrast, Fig. 4b shows X-OBC micelles that were crosslinked in MeOH and then transferred to the same range of solvent conditions. Notably X-OBC is dispersible in this complete range of solvent conditions without observable unimers. The main difference between the DLS data for different X-OBC conditions is the extent of micelle swelling which varies with solvent quality. Thus, these data demonstrate the “universal” entrapment of X-OBC micelles in wide ranging solvent conditions.
 |
| | Fig. 4 (a) DLS plots of OBC directly dispersed in various solvents with many conditions releasing substantial unimer fractions (<10 nm, dashed line) as compared to (b) DLS of X-OBC micelles which persisted as micelles under all conditions tested without detectable unimer release. | |
Facile persistence verification
The detection of kinetically trapped persistent micelles has remained historically challenging. Solution small-angle X-ray scattering (SAXS), for example, is unable to detect the steady-state exchange of chains under equilibrium but can detect size changes.93,94 Analogously, SAXS or SEM measurements on micelle templated samples can be used to determine if the results were consistent with persistent micelles as evidenced by comparison with the PMT model where series of samples have constant pore size and variable wall thickness depending on the material-to-template ratio.24,25 In contrast, absorbance measurements are facile and widely available where the spectroscopic signature of dimerized coumarin makes the detection of crosslinked micelles much simpler. Fig. 5a shows solution absorbance spectra for OBC micelles as compared to X-OBC micelles which are clearly distinguishable. Likewise Fig. 5b shows OBC as compared to X-OBC when cast as a polymer film (solvent-free). From this perspective, the fingerprint vs. pure-micelle conditions shown in Fig. 2 are easily distinguishable spectroscopically. Finally, Fig. 5c shows that crosslinked X-OBC is also detectable after use as a template for SiO2. As will be shown later, non-crosslinked OBC micelles have variable size when used as templates due to dynamic reorganization. Thus, absorbance measurements are convenient and facile method to confirm the micelle crosslinking associated with persistent micelles. To date, this is the simplest method to differentiate between micelle templated samples having persistent vs. dynamic micelles.
 |
| | Fig. 5 Absorbance spectra for coumarin containing micelles used to validate the crosslinking associated with micelle persistence at each processing stage from (a) solution, (b) to polymer films, and (c) after use as templates for nanomaterials. The spectra for non-crosslinked and crosslinked samples are easily distinguished in each case. Data is normalized at 210 nm. | |
Material templating with OBC vs. X-OBC
Persistent micelles are also detectable when used as templates with a material titration series. Here pre-formed micelles are combined with various amounts of material precursors in the form of nanoparticles that later crosslink and fuse during thermal processing. Solvent evaporation drives self-assembly of the material around the micelle coronas where persistent micelles uniquely preserve constant pore size through the series whereas dynamic micelles exhibit variable pore size corresponding to a convolution of shifting kinetic rates and shifting equilibrium size. First a representative sample is described before elaborating trends across a titration series. Sample X-OBC-THF-1.20 is first presented based on X-OBC micelles transferred to THF and then combined with TiO2 nanoparticles to reach a material-to-template ratio of 1.20. Fig. 6 shows the corresponding SAXS and scanning electron microscopy (SEM) data. The 2D diffraction pattern inset in Fig. 6a is isotropic, indicating no preferred orientation. The radial integration of this SAXS data exhibits a prominent scattering peak at q* = 0.18 nm−1, corresponding to a d-spacing (d = 2π/qpeak) of 34.1 nm. The SAXS pattern has a second apparent shoulder at ∼2q* which is suggestive of limited long-range ordering, as typically observed for PMT samples which tend to be most consistent with randomly packed spheres.95Fig. 6b presents the SEM micrograph after thermal removal of the polymer template where the continuous porous network (dark) is apparent amongst the matrix of TiO2 (light). The short-range ordering of randomly packed spherical templates is also apparent in this SEM image. The corresponding spatial distribution function was calculated using the CORDERLY software package,96 which also confirmed the isotropic distribution of randomly packed spheres. The pore-to-pore distance of 37.5 nm (S6, ESI†) is similar to the SAXS d-spacing of 34.1 nm. Additional statistical descriptors were derived from direct measurements on SEM images to derive an average pore size of 28.4 ± 0.2 nm and an average wall-thickness of 9.1 ± 0.6 nm (mean and standard-error-of-the-mean). Notably the average pore size here closely matches the core diameter of the X-OBC micelles measured by TEM (31.2 nm).
 |
| | Fig. 6 Representative dataset for sample X-OBC–THF including (a) SAXS and (b) SEM. The integrated SAXS pattern is shown with the 2D pattern inset. The color scale corresponds to the log-scale of the X-ray intensity. The SEM image has light areas that correspond to the TiO2 whereas the dark areas correspond to pores produced by removing the X-OBC template. The inset in (b) is the 2D spatial distribution function derived from the SEM image. Scale bar is 200 nm. | |
A material titration series was first carried out with non-crosslinked OBC using THF as the processing solvent. Prior PMT works with THF all required careful water content control to maintain kinetic micelle control via a χN barrier to chain exchange. This is in part why many PMT reports use alcohols (MeOH, EtOH, etc.) for processing to maintain χN control or avoid plasticizers in the case of glassy core persistent micelles.42,43 The resulting SAXS data (Fig. 7a) show sporadic peak shifts when increasing the TiO2 loading. The corresponding d-spacing trend was also sporadic and is suggestive of micelle size changes that make these values inconsistent with the established PMT model (Fig. 7b). The presence of dynamic OBC micelles was further evidenced by direct measurements (Fig. S6, ESI†) of the resulting pore sizes which ranged from 12.4–15.4 nm across the series (Fig. 7c). The data for this titration series thus indicate that OBC micelles are dynamic under these solvent conditions.
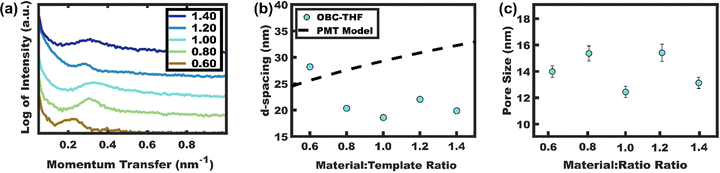 |
| | Fig. 7 (a) SAXS data for TiO2 combined with uncrosslinked OBC with variable material:template loading. (b) The corresponding d-spacing and (c) average pore sizes were similarly sporadic, suggesting dynamic micelles that were inconsistent with the expectations for persistent micelles. Samples were processed from THF and the SAXS data were shifted vertically for clarity. | |
A similar material titration was carried out with crosslinked X-OBC for comparison. In this case, the SAXS data (Fig. 8a) exhibited a monotonic leftward shift with increasing material:template ratio. The corresponding d-spacing trend indicates the expected lattice expansion as additional material is placed between persistent micelles. The d-spacing trend was quantitatively consistent with the PMT model (R2 = 0.97, Tables S1 and S2, ESI†). Furthermore, direct SEM measurements of the resulting samples confirmed constant pore size of 28.4 ± 0.2 nm (Fig. 8d and 9a–c) with the average wall thickness increasing progressively from 5.7 to 11.5 nm with increasing material:template ratio (Fig. 8b and c). The trend of increasing wall thickness was also well-fitted by the PMT model (R2 = 0.92). Thus, these data show that X-OBC is consistent with persistent micelle behavior. Remarkably, X-OBC persistent micelle templates were processed starting from a nearly pure THF solution (<1% water) which previously led to dynamic micelle templates or transitions to bulk phases.25 The “universal” solvent compatibility demonstrated with X-OBC is promising for the future development of persistent micelles that are compatible with ligating solvents (e.g. THF), non-hydrolytic chemistries, and/or water-sensitive materials.
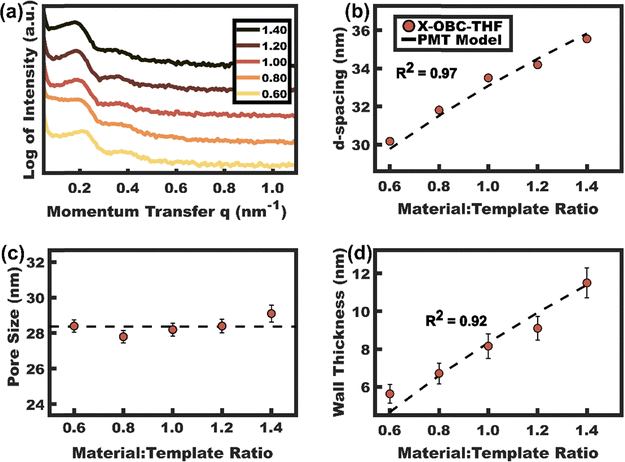 |
| | Fig. 8 (a) SAXS data for TiO2 combined with crosslinked X-OBC with variable loading extents. (b) The corresponding d-spacings were consistent with the PMT model. Direct SEM measurements of (c) pore size and (d) wall thickness was also both consistent with the PMT model. Samples were processed from THF and the SAXS data were shifted vertically for clarity. | |
 |
| | Fig. 9 SEM images of X-OBC–THF series with material:template ratio: (a) 0.6, (b) 1.0, (c) 1.4. Scale bars are 200 nm. | |
Conclusion
This work establishes molecular design criteria that enables persistent micelles via photo-crosslinking. This design included a PEO-like corona block for wide sweeping solvent compatibility. The micellization conditions were found to determine the fraction of OBC present in micelles and unimers where low-CMC conditions favored micelle-only dispersions. The crosslinking kinetics of the resulting micelles were studied spectroscopically to measure the conversion of coumarin to dimers where the reaction plateaued after about 1 hour. The absorbance signature of coumarin dimers also enables facile verification of micelles crosslinking as an indicator of persistent templates at every processing step from solution to within a matrix of material. DLS measurements confirmed that X-OBC micelles remained persistent and without unimers for all solvents tested, spanning a wide range of Hildebrand parameters. Finally, the kinetic control of OBC and X-OBC micelles were tested by analyzing a titration series with nanoparticles. While OBC micelles were dynamic with correspondingly sporadic pore size changes, X-OBC micelles exhibited d-spacing, pore size, and wall thickness trends that were consistent with persistent micelles. A new modality of persistent micelles was thus developed that enables nearly “universal” persistence with wide sweeping solvent compatibility. This solvent generalizability expands persistent micelle capabilities to enable diverse materials chemistries and the use of pure coordinating solvents.
Author contributions
Coby S. Collins: conceptualization, methodology, data curation, formal analysis, writing, editing. Mengxue Zhang: methodology, data curation, proofreading. CJ Sturgill: methodology, data curation, proofreading. Christian X. Ruff: methodology, proofreading. Bryce Melton: methodology, proofreading. Morgan Stefik: conceptualization, methodology, writing, editing, funding acquisition.
Conflicts of interest
There are no conflicts to declare.
Data availability
The data supporting this article have been included as part of the ESI.† Custom Matlab scripts used for GPC analysis are available at: https://stefikgroup.com//GPC/. Raw data are available upon request.
Acknowledgements
C. S. C., M. Z., and M. S. acknowledge support by the NSF CAREER program, NSF Award No. DMR-1752615. C. X. R. and B. M. acknowledge USC Honors College Research Grant support. This work made use of the South Carolina SAXS Collaborative (SCSC).
References
- S. A. Bagshaw, E. Prouzet and T. J. Pinnavaia, Science, 1995, 269, 1242–1244 CrossRef PubMed.
- Z. Qiang, J. Xue, G. E. Stein, K. A. Cavicchi and B. D. Vogt, Langmuir, 2013, 29, 8703–8712 CrossRef CAS PubMed.
- B. D. Vogt, V. L. Chavez, M. Dai, M. R. C. Arreola, L. Song, D. Feng, D. Zhao, G. M. Perera and G. E. Stein, Langmuir, 2011, 27, 5607–5615 CrossRef CAS PubMed.
- A. Labiano, M. Dai, W.-S. Young, G. E. Stein, K. A. Cavicchi, T. H. Epps and B. D. Vogt, J. Phys. Chem. C, 2012, 116, 6038–6046 CrossRef CAS.
- P. Yang, D. Zhao, D. I. Margolese, B. F. Chmelka and G. D. Stucky, Nature, 1998, 396, 152–155 CrossRef CAS.
- T. H. Epps III and R. K. O’Reilly, Chem. Sci., 2016, 7, 1674–1689 RSC.
- B. J. Scott, G. Wirnsberger and G. D. Stucky, Chem. Mater., 2001, 13, 3140–3150 CrossRef CAS.
- S. Guldin, P. Kohn, M. Stefik, J. Song, G. Divitini, F. Ecarla, C. Ducati, U. Wiesner and U. Steiner, Nano Lett., 2013, 13, 5329–5335 CrossRef CAS PubMed.
- M. Stefik, S. Guldin, S. Vignolini, U. Wiesner and U. Steiner, Chem. Soc. Rev., 2015, 44, 5076–5091 RSC.
- M. R. Benzigar, S. N. Talapaneni, S. Joseph, K. Ramadass, G. Singh, J. Scaranto, U. Ravon, K. Al-Bahily and A. Vinu, Chem. Soc. Rev., 2018, 47, 2680–2721 RSC.
- Y. Hu, J. Florek, D. Larivière, F. Fontaine and F. Kleitz, Chem. Rec., 2018, 18, 1261–1276 CrossRef PubMed.
- W. Van Den Bergh and M. Stefik, Adv. Funct. Mater., 2022, 32, 2204126 Search PubMed.
- L. Zu, W. Zhang, L. Qu, L. Liu, W. Li, A. Yu and D. Zhao, Adv. Energy Mater., 2020, 10, 2002152 CrossRef CAS.
- A. Walcarius, Chem. Soc. Rev., 2013, 42, 4098 RSC.
- P. Docampo, S. Guldin, M. Stefik, P. Tiwana, M. C. Orilall, S. Hüttner, H. Sai, U. Wiesner, U. Steiner and H. J. Snaith, Adv. Funct. Mater., 2010, 20, 1787–1796 Search PubMed.
- M. G. Fischer, X. Hua, B. D. Wilts, I. Gunkel, T. M. Bennett and U. Steiner, ACS Appl. Mater. Interfaces, 2017, 9, 22388–22397 CrossRef CAS PubMed.
- T. C. Lin, Y. Yan, S. C. King, C.-H. Lai and S. H. Tolbert, ACS Appl. Mater. Interfaces, 2020, 12, 33775–33784 CrossRef CAS PubMed.
- I. E. Rauda, V. Augustyn, B. Dunn and S. H. Tolbert, Acc. Chem. Res., 2013, 46, 1113–1124 CrossRef CAS PubMed.
- T. Brezesinski, J. Wang, J. Polleux, B. Dunn and S. H. Tolbert, J. Am. Chem. Soc., 2009, 131, 1802–1809 Search PubMed.
- Y. Mai and A. Eisenberg, Chem. Soc. Rev., 2012, 41, 5969 Search PubMed.
- S. E. Mastroianni and T. H. Epps, Langmuir, 2013, 29, 3864–3878 CrossRef CAS PubMed.
- L. Leibler, Macromolecules, 1980, 13, 1602–1617 CrossRef CAS.
- E. G. Kelley, T. P. Smart, A. J. Jackson, M. O. Sullivan and T. H. Epps, Soft Matter, 2011, 7, 7094 RSC.
- A. Sarkar and M. Stefik, J. Mater. Chem. A, 2017, 5, 11840–11853 RSC.
- M. Stefik, J. Mater. Res., 2022, 37, 25–42 CrossRef CAS.
- T. P. Lodge, C. L. Seitzinger, S. C. Seeger, S. Yang, S. Gupta and K. D. Dorfman, ACS Polym. Au, 2022, 2, 397–416 CrossRef CAS PubMed.
- L. R. Parent, E. Bakalis, A. Ramírez-Hernández, J. K. Kammeyer, C. Park, J. De Pablo, F. Zerbetto, J. P. Patterson and N. C. Gianneschi, J. Am. Chem. Soc., 2017, 139, 17140–17151 CrossRef CAS PubMed.
- E. G. Kelley, R. P. Murphy, J. E. Seppala, T. P. Smart, S. D. Hann, M. O. Sullivan and T. H. Epps, Nat. Commun., 2014, 5, 3599 CrossRef PubMed.
- R. P. Murphy, E. G. Kelley, S. A. Rogers, M. O. Sullivan and T. H. Epps, ACS Macro Lett., 2014, 3, 1106–1111 CrossRef CAS PubMed.
- K. A. Lantz, A. Sarkar, K. C. Littrell, T. Li, K. Hong and M. Stefik, Macromolecules, 2018, 51, 6967–6975 CrossRef CAS.
- S.-H. Choi, T. P. Lodge and F. S. Bates, Phys. Rev. Lett., 2010, 104, 047802 Search PubMed.
- S.-H. Choi, F. S. Bates and T. P. Lodge, Macromolecules, 2011, 44, 3594–3604 CrossRef CAS.
- D. Zhao, Y. Ma and T. P. Lodge, Macromolecules, 2018, 51, 2312–2320 CrossRef CAS.
- E. Wang, J. Lu, F. S. Bates and T. P. Lodge, Macromolecules, 2018, 51, 3563–3571 Search PubMed.
- J. Lu, S. Choi, F. S. Bates and T. P. Lodge, ACS Macro Lett., 2012, 1, 982–985 CrossRef CAS PubMed.
- J. Lu, F. S. Bates and T. P. Lodge, ACS Macro Lett., 2013, 2, 451–455 Search PubMed.
- Y. Ma and T. P. Lodge, Macromolecules, 2016, 49, 9542–9552 CrossRef CAS.
- H. N. Lokupitiya, A. Jones, B. Reid, S. Guldin and M. Stefik, Chem. Mater., 2016, 28, 1653–1667 Search PubMed.
- A. Sarkar, L. Evans and M. Stefik, Langmuir, 2018, 34, 5738–5749 CrossRef CAS PubMed.
- E. R. Williams, W. Van Den Bergh and M. Stefik, Soft Matter, 2022, 18, 7917–7930 RSC.
- K. A. Lantz, N. B. Clamp, W. Van Den Bergh, A. Sarkar and M. Stefik, Small, 2019, 15, 1900393 CrossRef PubMed.
- E. R. Williams, P. L. McMahon, J. E. Reynolds, J. L. Snider, V. Stavila, M. D. Allendorf and M. Stefik, Mater. Adv., 2021, 2, 5381–5395 RSC.
- E. R. Williams, C. X. Ruff and M. Stefik, Soft Matter, 2024, 20, 2288–2300 RSC.
- R. K. O’Reilly, C. J. Hawker and K. L. Wooley, Chem. Soc. Rev., 2006, 35, 1068 RSC.
- R. K. O’Reilly, M. J. Joralemon, C. J. Hawker and K. L. Wooley, New J. Chem., 2007, 31, 718–724 RSC.
- M. Talelli, M. Barz, C. J. F. Rijcken, F. Kiessling, W. E. Hennink and T. Lammers, Nano Today, 2015, 10, 93–117 CrossRef CAS PubMed.
- X. Liu, G. Yang, L. Zhang, Z. Liu, Z. Cheng and X. Zhu, Nanoscale, 2016, 8, 15323–15339 Search PubMed.
- J. Jiang, B. Qi, M. Lepage and Y. Zhao, Macromolecules, 2007, 40, 790–792 Search PubMed.
- Y. Shi, R. M. Cardoso, C. F. Van Nostrum and W. E. Hennink, Polym. Chem., 2015, 6, 2048–2053 RSC.
- J. Ding and G. Liu, Chem. Mater., 1998, 10, 537–542 CrossRef CAS.
- J. K. Elter, J. Eichhorn and F. H. Schacher, Macromol. Rapid Commun., 2021, 42, 2100485 CrossRef CAS PubMed.
- X. Tong, G. Wang, A. Soldera and Y. Zhao, J. Phys. Chem. B, 2005, 109, 20281–20287 Search PubMed.
- X. Zhang, H. Dong, S. Fu, Z. Zhong and R. Zhuo, Macromol. Rapid Commun., 2016, 37, 993–997 CrossRef CAS PubMed.
- X. Jiang, J. Zhang, Y. Zhou, J. Xu and S. Liu, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 860–871 CrossRef CAS.
- R. K. O’Reilly, M. J. Joralemon, K. L. Wooley and C. J. Hawker, Chem. Mater., 2005, 17, 5976–5988 CrossRef.
- X.-Q. Yi, Q. Zhang, D. Zhao, J.-Q. Xu, Z.-L. Zhong, R.-X. Zhuo and F. Li, Polym. Chem., 2016, 7, 1719–1729 RSC.
- L. Zhang, W. Liu, L. Lin, D. Chen and M. H. Stenzel, Biomacromolecules, 2008, 9, 3321–3331 CrossRef CAS PubMed.
- S. Matsumoto, R. J. Christie, N. Nishiyama, K. Miyata, A. Ishii, M. Oba, H. Koyama, Y. Yamasaki and K. Kataoka, Biomacromolecules, 2009, 10, 119–127 CrossRef CAS PubMed.
- K. Miyata, Y. Kakizawa, N. Nishiyama, Y. Yamasaki, T. Watanabe, M. Kohara and K. Kataoka, J. Controlled Release, 2005, 109, 15–23 CrossRef CAS PubMed.
- S. R. Trenor, A. R. Shultz, B. J. Love and T. E. Long, Chem. Rev., 2004, 104, 3059–3078 CrossRef CAS PubMed.
- C. P. Kabb, C. S. O’Bryan, C. C. Deng, T. E. Angelini and B. S. Sumerlin, ACS Appl. Mater. Interfaces, 2018, 10, 16793–16801 Search PubMed.
- B. Tarek Benkhaled, K. Belkhir, T. Brossier, C. Chatard, A. Graillot, B. Lonetti, A.-F. Mingotaud, S. Catrouillet, S. Blanquer and V. Lapinte, Eur. Polym. J., 2022, 179, 111570 Search PubMed.
- J. Ling, M. Z. Rong and M. Q. Zhang, J. Mater. Chem., 2011, 21, 18373 RSC.
- C. S. Wong, N. I. Hassan, M. S. Su’ait, M. A. Pelach Serra, J. A. Mendez Gonzalez, L. A. Granda and K. H. Badri, Ind. Crops Prod., 2019, 140, 111613 CrossRef CAS.
- F. Wang and H. Liu, J. Phys. Chem. C, 2018, 122, 3434–3442 CrossRef CAS.
- F. Wang, X. Yang, L. Zhang, W. Xu and H. Liu, Macromol. Mater. Eng., 2017, 302, 1700173 Search PubMed.
- Y.-B. Long, W.-X. Gu, C. Pang, J. Ma and H. Gao, J. Mater. Chem. B, 2016, 4, 1480–1488 Search PubMed.
- N. Kumar, Udayabhanu, A. A. Alghamdi, K. M. Mahadevan and G. Nagaraju, J. Mol. Struct., 2021, 1223, 129208 Search PubMed.
- P. Samanta, K. Kapat, S. Maiti, G. Biswas, S. Dhara and D. Dhara, J. Colloid Interface Sci., 2019, 555, 132–144 CrossRef CAS PubMed.
- N. Jiang, Y. Cheng and J. Wei, Colloids Surf., A, 2017, 522, 28–37 CrossRef CAS.
- W. Cao and L. Zhu, Macromolecules, 2011, 44, 1500–1512 Search PubMed.
- X. Fan, Z. Li and X. J. Loh, Polym. Chem., 2016, 7, 5898–5919 Search PubMed.
- D. B. G. Williams and M. Lawton, J. Org. Chem., 2010, 75, 8351–8354 Search PubMed.
- DIY Gel Permeation Chromatograph – Stefik Group, https://stefikgroup.com/gpc/, (accessed 4 April 2025).
- T. Larison, E. Williams, C. S. Collins, S. V. Pingali and M. Stefik, Macromolecules, 2023, 56, 7818–7826 Search PubMed.
- H. N. Lokupitiya and M. Stefik, Nanoscale, 2017, 9, 1393–1397 Search PubMed.
- B. C. Garcia, M. Kamperman, R. Ulrich, A. Jain, S. M. Gruner and U. Wiesner, Chem. Mater., 2009, 21, 5397–5405 Search PubMed.
- M. B. Sims, B. Zhang, Z. M. Gdowski, T. P. Lodge and F. S. Bates, Macromolecules, 2022, 55, 3317–3324 Search PubMed.
- J. He, X. Tong and Y. Zhao, Macromolecules, 2009, 42, 4845–4852 CrossRef CAS.
- A. B. Dwyer, P. Chambon, A. Town, T. He, A. Owen and S. P. Rannard, Polym. Chem., 2014, 5, 3608–3616 Search PubMed.
- R. Lund, L. Willner, P. Lindner and D. Richter, Macromolecules, 2009, 42, 2686–2695 Search PubMed.
- E. B. Zhulina, M. Adam, I. LaRue, S. S. Sheiko and M. Rubinstein, Macromolecules, 2005, 38, 5330–5351 Search PubMed.
- G. Chen and E. Dormidontova, Macromolecules, 2022, 55, 5222–5232 Search PubMed.
-
D. R. Bloch, The Wiley Database of Polymer Properties, John Wiley & Sons, Ltd, 2003 Search PubMed.
- T. Larison, E. R. Williams, M. Wright, M. Zhang, J. Tengco, M. G. Boebinger, C. Tang and M. Stefik, ACS Nano, 2024, 18, 20133–20141 Search PubMed.
- K. Chiad, S. H. Stelzig, R. Gropeanu, T. Weil, M. Klapper and K. Müllen, Macromolecules, 2009, 42, 7545–7552 CrossRef CAS.
- N. P. Truong, G. R. Jones, K. G. E. Bradford, D. Konkolewicz and A. Anastasaki, Nat. Rev. Chem., 2021, 5, 859–869 Search PubMed.
- M. B. Sims, B. Zhang, Z. M. Gdowski, T. P. Lodge and F. S. Bates, Macromolecules, 2022, 55, 3317–3324 Search PubMed.
- C. Zheng, B. Zhang, F. S. Bates and T. P. Lodge, Soft Matter, 2023, 19, 4519–4525 RSC.
-
A. F. M. Barton, CRC Handbook of Solubility Parameters and Other Cohesion Parameters, Routledge, New York, 2nd edn, 2017 Search PubMed.
- U. Sander and B. A. Wolf, Angew. Makromol. Chem., 1986, 139, 149–156 Search PubMed.
- A. Halperin and S. Alexander, Macromolecules, 1989, 22, 2403–2412 Search PubMed.
- S. Gupta and T. P. Lodge, Macromolecules, 2023, 56, 2137–2148 CrossRef CAS.
- S. Gupta, L. Liberman and T. P. Lodge, Macromolecules, 2023, 56, 4874–4883 Search PubMed.
- J. S. Pedersen, Adv. Colloid Interface Sci., 1997, 70, 171–210 Search PubMed.
- N. Mac Fhionnlaoich, R. Qi and S. Guldin, Langmuir, 2019, 35, 16605–16611 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ,
Mengxue
Zhang
,
Mengxue
Zhang