
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Effect of monomer composition on the formation of hybrid polymer-liquid electrolytes for lithium-ion batteries†
Samuel
Emilsson‡
 ,
Gabriele
Maffeis‡
,
Martina
Cattaruzza
and
Mats
Johansson
*
,
Gabriele
Maffeis‡
,
Martina
Cattaruzza
and
Mats
Johansson
*
Department of Fibre & Polymer Technology, Division of Coating Technology, KTH Royal Institute of Technology, SE-100 44 Stockholm, Sweden. E-mail: matskg@kth.se
First published on 14th April 2025
Abstract
The electrolyte plays a key role in the performance of novel lithium-ion battery concepts. Hybrid polymer-liquid electrolytes (HEs) are suitable candidates for novel concepts of lithium-ion batteries (LIBs) and lithium-metal batteries (LMBs), where high ionic conductivity coupled with mechanical integrity are required at the same time. HEs are produced through polymerization-induced phase separation (PIPS) of a monomer/electrolyte mixture which allows for the formation of a two-phase system where the domains create a bicontinuous structure. Electrochemical performance and thermomechanical behavior can be tailored through several variables e.g., monomer and solvent chemistries, solvent concentration, and curing conditions. The present study is focused on the chemical structure of the monomer where methacrylate and acrylate monomers are compared as homopolymers or copolymers in HEs. The number of ethylene oxide (EO) units in the backbone of the monomers are furthermore analyzed as a structural parameter. The results show that the monomer structure not only affects the electrochemical and thermomechanical properties, but also defines the morphology of the HEs obtained, which can be in the form of a bicontinuous structure, a gel, or a mixture of the two, according to the kinetic and thermodynamic variables affecting the phase separation and the ultimate Tg of the polymer.
1. Introduction
Today, lithium-ion batteries (LIBs) are the most competitive energy storage systems thanks to their high energy density,1 which makes them the dominating battery technology on the market.1–3 However, as the demand for batteries continues to increase, LIBs are starting to reach their ceiling in terms of attainable energy density. This has led to a surge of interest in next-generation batteries. This includes batteries with higher energy densities, such as lithium metal batteries (LMBs), but also new concepts like structural batteries4 in which the batteries are integrated into the load-bearing structure, or flexible batteries for flexible electronics.5 New electrolyte materials need to enable safe, efficient and durable cycling of batteries. First, the electrolyte must be compatible with the electrodes,6 where the solid electrolyte interface (SEI) that is created affects the cycling stability.7 Other important factors are safety and durability issues, e.g. flammability and lithium dendrite growth.8,9 Electrolyte separators with high mechanical integrity are also desired to improve these features.6Next-generation batteries also put additional demands on the electrolyte. Structural LIBs, for example, require a multifunctional electrolyte with at least 100 MPa storage modulus and 10−4 S cm−1 ionic conductivity.10 Liquid electrolytes (LEs) and gel polymer electrolytes (GPEs) are not suitable for this purpose because they do not ensure the stiffness required, while solid polymer electrolytes (SPEs) generally have too low ionic conductivities at ambient temperature.10–12
One novel concept is represented by hybrid polymer-liquid electrolytes (HEs), which feature a bicontinuous structure with two phases, a solid polymer network and a liquid that percolates the porous polymer phase.13 Unlike GPEs, HEs exhibit a higher storage modulus while retaining a sufficient ionic conductivity, at least within the variety of HEs that have been reported so far in the literature.10,12,13 Furthermore, this bicontinuous structure can be formed in situ, infused into the battery assembly.14
HEs are obtained through polymerization-induced phase separation (PIPS), which exploits the decreasing solubility of the forming polymer compared to the monomers in the solvent12,15,16 The obtained morphology of the porous system depends on the chemical composition and polymerization mechanism/conditions.17,18
PIPS was initially developed for chromatography media, which led to fundamental investigations of the PIPS process. Multiple important works17,19–21 showed the effect of solvent choice, monomer choice, and polymerization method. These factors have also extensively been reviewed.15,21–23
For battery applications, several systems have been explored. Epoxy resins combined with ionic liquids for structural supercapacitor applications.18,24–26 The effect of monomer structure, solvent and salt concentration have also been investigated for these systems. HEs based on methacrylate monomers and carbonate solvent electrolytes have also been extensively explored showing the effect of monomer structure14,27 and electrolyte content.27 The effect of solvent molecular weight28 and composition29 has also been studied.
To summarize, a complex interplay of these factors affects the PIPS process, which in turn have a large effect on the properties of the obtained HEs. An ideal HE has pores of sufficient size that allow adequate ionic conductivity, while too large pores lead to a brittle material with insufficient stability.29,30 The choice of the monomer affects the mechanical properties of the polymer phase, but also how pores are formed during PIPS. The solvent and salt choice affect the electrochemical performance of the liquid phase, while also governing pore formation. All these factors need to be considered when developing new HEs based on PIPS.
The present study aims to relate how the chemical structure of the monomers and curing mechanism affect the final morphology and HE properties. Three variables are investigated, i.e., methacrylate versus acrylate monomers, ethoxylate unit content, and homo-versus copolymers (see Fig. 1). The properties investigated are conversion, reaction rate, pore size, ionic conductivity, storage modulus, and glass transition temperature (Tg).
 | ||
| Fig. 1 Chemical structures of all the monomers used for the synthesis of HEs. | ||
2. Experimental
2.1. Materials
Bisphenol A ethoxylate (EO = 2) dimethacrylate (BPAMA) (Mn = 540 g mol−1) was donated by Alkema, while bisphenol A ethoxylate (EO = 1) diacrylate (BPAA1) (Mn = 424 g mol−1, stabilized with 750 ppm 4-methoxyphenol (MeHQ)), bisphenol A ethoxylate (EO = 2) diacrylate (BPAA2) (Mn = 510 g mol−1) and bisphenol A ethoxylate (EO = 4) diacrylate (BPAA4) (Mn = 688 g mol−1, stabilized with 250 ppm MeHQ) were provided by Sigma Aldrich. The initiator 2,2′-azobisisobutyronitrile (AIBN) was purchased from Sigma Aldrich, with purity >98%. Ethylene carbonate (EC), propylene carbonate (PC) and lithium trifluoromethanesulphonate imide (LiTFSI) were purchased from Sigma Aldrich. All the compounds were used as received, without any further purification.2.2. Synthesis
A total of seven different HEs were synthesized with the denotation and compositions presented in Table S1 in the ESI† and illustrated in Fig. 1. Pure polymer samples were also synthesized (i.e. without the presence of EC, PC and LiTFSI) in order to study the bulk properties of the respective polymers and are denoted with a “p” prior to the sample formulation name.The synthesis of the different HE formulations was conducted in a glovebox under inert argon atmosphere (<5 ppm O2) in dry conditions (<5 ppm H2O). The formulation was prepared by mixing EC and PC in equal volume fractions, with EC being heated at 60 °C upon complete melting. LiTFSI was dissolved into the mixture at 1 M concentration. The formulation consisted of 55 wt% monomer, 45 wt% solvent, and 1 wt% AIBN with respect to the monomer weight. The electrolyte was injected into aluminum molds 30 mm long, 6 mm wide and 0.5 mm thick, covered with a glass plate and clamped at the edges. The structure was sealed into a pouch bag and transferred from the glovebox to the preheated oven, where the curing reaction was performed for 45 minutes at 90 °C. The pure polymers were instead polymerized outside the glovebox at the same curing conditions.
2.3. Electrochemical impedance spectroscopy (EIS)
The evaluation of the ionic conductivity was performed by a Gamry Series G 750 potentiostat/galvanostat/ZRA interface connected to a four-point resistance setup, with golden electrodes (two reference electrodes 5 mm apart and two working electrodes 20 mm apart). The frequency range chosen was between 300 kHz and 1 Hz. The ionic conductivity was derived from the resistance and the geometrical parameters through Ohm's first law, given by eqn (1), where l is the distance between the reference electrodes, A the cross-sectional area and Rb the resistance measured by the software as the low-frequency intercept on the real axis in the resulting Nyquist plot.| σ = l/(A·Rb) | (1) |
2.4. Fourier-transformation infrared spectroscopy (FTIR)
FTIR analysis was executed to follow the curing performance using a PerkinElmer spectrum 100 FTIR spectrometer equipped with a deuterated triglycine sulfate detector and the golden gate diamond (Specac Ltd) attenuated total reflectance (ATR) accessory and acquired data processed using PerkinElmer Spectrum software. Two samples of each formulation were analyzed both before and after curing. Eight scans per sample were integrated in one spectrum with a resolution of 4 cm−1. Curing performance was evaluated by comparing the area underneath the vinyl peak in the range between 1627–1647 cm−1.2.5. Real time FTIR
The polymerization rate of all formulations was studied using real time FTIR at atmospheric conditions, i.e., outside the glovebox. Real time FTIR analysis was performed by the PerkinElmer spectrum 100 FTIR Spectrometer equipped with a deuterated triglycine sulfate detector and the golden gate diamond (Specac Ltd) attenuated total reflectance (ATR) accessory and evaluated using PerkinElmer Spectrum software. The plate was pre-heated at 90 °C before the resin was spread on top of the crystal and covered with a microscope slide. The peak area underneath the FTIR spectra between 1627 and 1647 cm−1 was measured and the depletion of the signal with time was evaluated. The conversion was calculated through eqn (2), where C% is the conversion percent and A0 and A is the area underneath the peak in the range 1627–1647 cm−1, before and after the measurement, respectively.| C% = 100 (1 − A/A0) | (2) |
2.6. Dynamic mechanical analysis (DMA)
DMA analysis was performed to determine the thermomechanical properties of the HEs using a DMA Q800 V21.3 Build 96 instrument equipped with an ACS-3 cooling module in tensile film mode. A preload force of 0.125 N was applied and a 1 Hz amplitude strain of 0.1% was used. The clamps were regulated between 10 mm and 15 mm distance from each other before the film was clamped. The temperature was scanned between −50 °C and 200 °C, with a stabilization step for 5 min at −50 °C before the scanning started with a heating rate of 3 °C min−1. The experimental values of Tg were extrapolated from the maximum of the tan![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) δ peak. Wet films (after curing), dried films and pure polymers were tested.
δ peak. Wet films (after curing), dried films and pure polymers were tested.
2.7. Scanning electronic microscopy (SEM)
SEM was used to study the morphology of the cross section of dried HEs Before the analysis, the samples underwent 24 h leaching in water and 24 h vacuum drying in an oven at 50 °C in order to extract the liquid electrolyte. Subsequently, the specimens were cryo-fractured through immersion in liquid nitrogen. The cross sections were analyzed by a Hitachi S-4800 microscope equipped with a cold field-emission electron source. The specimens were mounted on an SEM sample holder with conductive carbon tape, and they were subsequently coated with Pt/Pd using a Cressington 208HR sputter coater for 15 s (corresponding to ca. 2 nm of the conductive coating layer) at a current of 40 mA. The micrographs were captured with 1 kV accelerating voltage, 10 μA current and 8–9 mm working distance.2.8. Solvent exclusion
Solvent exclusion was performed to evaluate the percolating structure formation on samples BPAMA-co-BPAA1 and BPAMA-co-BPAA2. The specimens were soaked into abundant deionized water (>20 mL) and put on a shaking table for 24 h in order to leach the liquid electrolyte out. Subsequently, the samples were dried in a vacuum oven at 50 °C for 24 h and weighed to compare the mass before and after solvent extraction. Two samples of each formulation were analyzed.3. Results and discussion
BPAMA was chosen as a benchmark since this monomer has been extensively studied previously in the context of polymer-based electrolyte materials. It has been demonstrated to have high thermal and thermomechanical stability, without thermal transitions.29 In addition, the ionic transport properties like ionic conductivity and transport number of HEs based on BPAMA have been studied.31 Studies have also shown that this monomer can be applied as an HE32 or separator29 in half cell configurations as well as structural battery applications,33,34 showing stable cycling performance.3.1. The effect of monomer functionality
The termination via coupling leads to a faster rise in molecular weight and an earlier gelation point for the acrylates compared to the methacrylates. Moreover, polyacrylates have a significantly lower Tg compared to polymethacrylates.35,36 This needs to be considered as vitrification effects are possible i.e., if the ultimate Tg is above the curing temperature. The polarity difference between the methacrylates and the acrylates is minor considering the overall size of the monomers.The curing performance of sample BPAA2 is shown in Fig. 2a, while the others are reported in Fig. S1a–f (ESI†). The shift of the carbonyl peak from 1718 to 1728 is caused by the disappearance of the neighbouring C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C vinyl peak (at 1637 cm−1) during polymerization. The disappearance of this peak was also followed in real-time, to monitor the reaction kinetics. HEs demonstrated to polymerize faster than pure polymers, as shown in Fig. 2b for BPAMA and BPAA2 as examples. The conversion rates for the other polymers are reported in Fig. S2a–e (ESI†). Only minor polymerization rate differences are found among the different HEs since the viscosity of the system and the diffusion limitations are lowered in the presence of the solvent. Moreover, HEs experience a plasticizing effect due to the solvents, which leads to a higher conversion compared to the pure polymer samples. Larger differences are found comparing pBPAMA and pBPAA2 where the reaction depends on the intrinsic properties of the polymer (such as the reaction kinetics and the ultimate Tg), as shown in Fig. 2b. The methacrylate, as expected, polymerized slower and presents a lower conversion due to the intrinsic lower propagation rate and a higher ultimate Tg. The polymerization rate differences are small for all HEs and they all exhibit final double bond conversions above 90%, as shown in Table S2 (ESI†).
C vinyl peak (at 1637 cm−1) during polymerization. The disappearance of this peak was also followed in real-time, to monitor the reaction kinetics. HEs demonstrated to polymerize faster than pure polymers, as shown in Fig. 2b for BPAMA and BPAA2 as examples. The conversion rates for the other polymers are reported in Fig. S2a–e (ESI†). Only minor polymerization rate differences are found among the different HEs since the viscosity of the system and the diffusion limitations are lowered in the presence of the solvent. Moreover, HEs experience a plasticizing effect due to the solvents, which leads to a higher conversion compared to the pure polymer samples. Larger differences are found comparing pBPAMA and pBPAA2 where the reaction depends on the intrinsic properties of the polymer (such as the reaction kinetics and the ultimate Tg), as shown in Fig. 2b. The methacrylate, as expected, polymerized slower and presents a lower conversion due to the intrinsic lower propagation rate and a higher ultimate Tg. The polymerization rate differences are small for all HEs and they all exhibit final double bond conversions above 90%, as shown in Table S2 (ESI†).
 | ||
| Fig. 2 FTIR spectra before and after curing for sample BPAA2 (a) and curing performance of BPAMA and BPAA2 as HEs and bulk polymers (b). | ||
Fig. 3a compares the storage modulus of BPAMA and BPAA2. Although they exhibit similar stiffness in the glassy state (at −50 °C), the modulus of BPAA2 decreases more rapidly, and it reaches its rubbery plateau at 50 °C. On the other hand, the modulus of BPAMA does not decrease as sharply as BPAA2, reaching a plateau at 100–150 °C, likely due to the stiffening effect given by solvent evaporation at elevated temperatures. Increasing the temperature further eventually leads to a storage modulus decrease. The storage moduli at 25 °C are listed in Table S3 (ESI†). It should be noted that the resulting storage modulus for BPAA2 is significantly lower than for BPAMA. Moreover, Fig. 3b shows a broad tanδ curve for BPAMA, where a peak cannot be recognized, while BPAA2 has a narrow curve with a clear peak at 14 °C. The differences in Fig. 3 are also corroborated by the differences seen when comparing the pure polymers pBPAMA and pBPAA2 in Fig. S3 (ESI†), which shows that pBPAA2 has a significantly lower Tg compared to pBPAMA.
 | ||
| Fig. 3 Storage modulus versus temperature (a) and tanδ curve (b) for BPAMA and BPAA2. | ||
BPAMA and BPAA2 have ionic conductivities in the order of 10−4 S cm−1. The results obtained are coherent with those present in the literature.13 Some minor difference exists between the two HEs (Table 1) which could be ascribed to the morphology as discussed below.
| Sample | Ionic conductivity (× 10−4 S cm−1) |
|---|---|
| BPAMA | 1.4 ± 0.1 |
| BPAA1 | 1.3 ± 0.1 |
| BPAA2 | 1.0 ± 0.1 |
| BPAA4 | 1.4 ± 0.1 |
It is expected that the polarity of acrylate compared to the corresponding methacrylate is minor because the methyl group is a small part compared to the bisphenol and EO moieties. Therefore, it can be assumed that the solubility in the polar solvent is comparable between the two species and no dramatic differences should be observed during phase separation and in the final morphology. However, the SEM micrographs of the cross section show a larger porous structure for BPAMA compared to BPAA2 which features smaller pores (Fig. 4). This only gives a qualitative understanding of the porosity, but previous studies have shown a correlation between the size of these structures and a lower tortuosity.30,31 This is likely the source of the slightly higher ionic conductivity in BPAMA compared to BPAA2. One explanation to the different porosity is provided by the kinetics of reaction, where acrylates polymerize faster than methacrylates, and have a different termination mechanism.37 Acrylates also undergo autoacceleration to a larger extent.38,39 Two other mechanisms can promote or hamper growth after nucleation and depend on the solvent quality17 and the state of molecular tension of the crosslinked polymer.21 The ultimate Tg may also play a role, as it may affect the capability of the growing polymer phase to interact with the monomers or solvents. However, a deeper investigation is needed to clarify this aspect.
 | ||
| Fig. 4 SEM micrographs of the surface porosity of cross sections for BPAMA (a) and BPAA2 (b). | ||
3.2. The effect of polarity when having the same polymerizable group
As a second parameter, the length of the ethoxy chain was varied (see structures in Fig. 1). The introduction of EO groups makes the monomer more soluble because EO units are slightly polar and interact more with the solvents and salt. Moreover, the length of the spacers is increased, and the aliphatic contribution prevails over the aromatic character. Therefore, the resulting material is expected to be softer. The tanδ curves for homoacrylates are presented in Fig. 5b. Increasing the polarity causes a delay in phase separation, because the oligomer is more soluble in a polar solvent than the oligomer made of the same number of structural units but with less polar functional groups. Hence, the molecular weight of the polymer when phase separation occurs is governed by the solubility.17 Also, the crosslinking density decreases when the monomer length is extended by adding functional groups. In the present case, the gradual increase in polarity is accompanied with a change in the thermomechanical properties. The Tg and the storage modulus at room temperature decrease because part of the solvent coexists with the crosslinked network in a one-phase system similar to a gel. A gradual decrease in Tg is observed from BPAA1 to BPAA2 and BPAA4, where the Tgs are 50 °C, 14 °C and −38 °C, respectively. In Fig. 5a, it can be observed that the storage modulus in the glassy state is comparable for BPAA1 and BPAA2, while it is one order of magnitude lower for BPAA4, because the formed network is still soluble in the solvent to some extent and phase separation has not occurred completely.
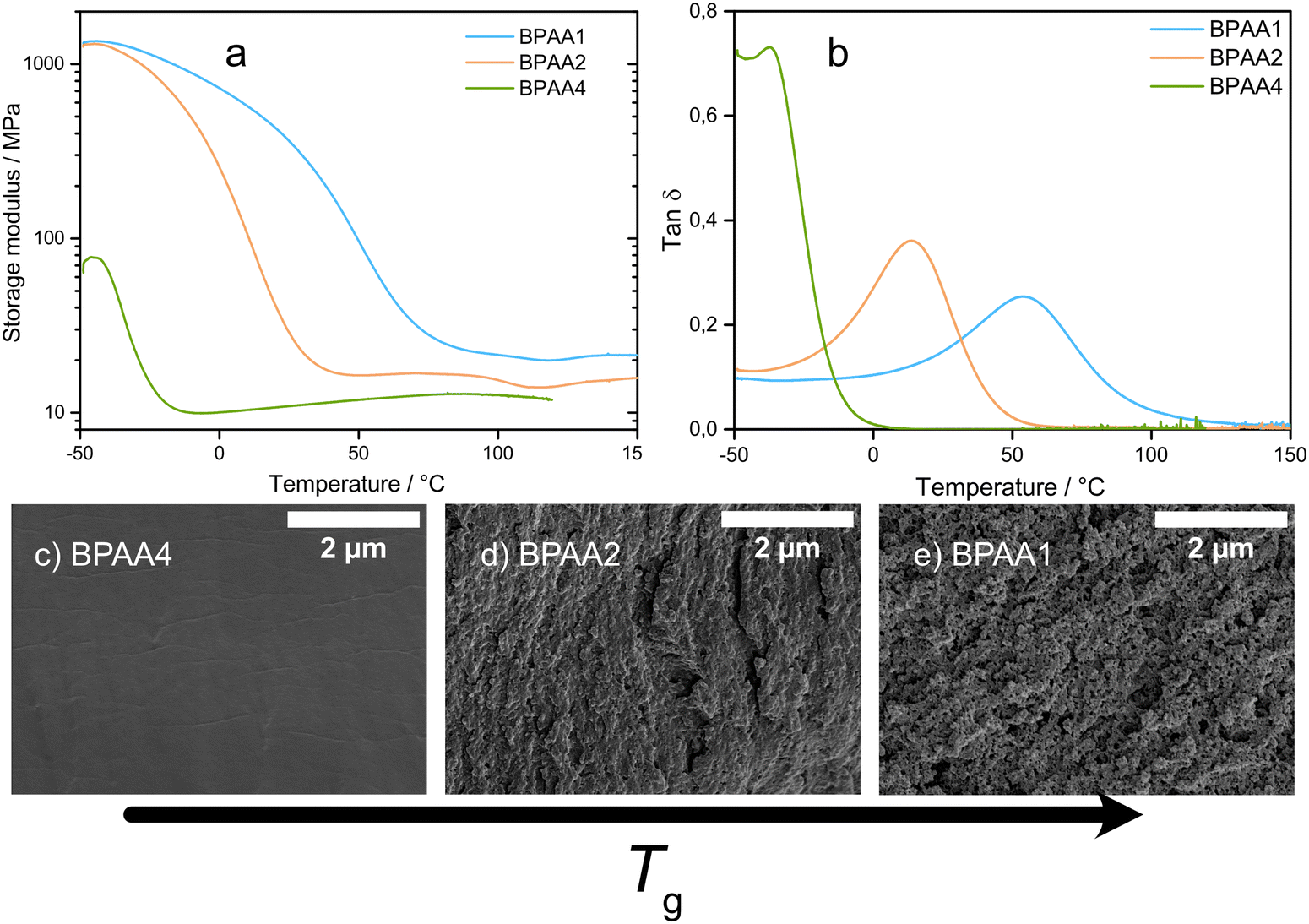 | ||
| Fig. 5 Storage modulus versus temperature (a) and tanδ curve (b) and SEM micrographs of cross sections of (c) BPAA4, (d) BPAA2 and (e) BPAA4. | ||
Solvent exclusion indicates the existence of a percolating liquid phase as most of the solvent was extracted after leaching and drying (Table S4, ESI†). A slight difference was observed in BPAA2, where 10–15% of the solvent was trapped in the system. Solvent exclusion does not confirm the presence of a bicontinuous structure, unless it is corroborated by SEM micrographs. Indeed, it is illustrated in Fig. 5b that the pore structure size decreases from BPAA1 to BPAA2, and it is absent in BPAA4, at least up to ×30.0 k magnitude (Fig. S4, ESI†), assuming that the phase separation partially occurs in BPAA2 and does not occur in BPAA4. All the solvent can be leached out of BPAA4 because it is one order of magnitude softer than the other HEs at room temperature and therefore facilitates the diffusion of solvent, similar to a gel (Table S4, ESI†). Instead, BPAA2 is stiffer and retains a fraction of solvent. However, it cannot be concluded whether the morphology of BPAA2 exhibits a closed porosity or a mixture of bicontinuous and a gel-like structure.
All the samples evaluated in this section belong to the class of acrylates, so they exhibit similar polymerization and termination rates. However, the polarity is highly affected by the introduction or removal of EO units as spacers, that are slightly polar and promote the solubilization into the solvent. The comparison between the acrylates in Table 1 shows that BPAA1 and BPAA4 have similar ionic conductivities, while BPAA2 is around 30% less conductive. DMA and solvent exclusion results, combined with SEM micrographs, prove that the morphology is different for the homoacrylates analysed, and it can be observed that a gradual transition from a bicontinuous system to a gel occurs when the length and solubility of the monomer are increased. Ion transport in HEs is driven by the percolating liquid phase, while in GPEs (with low solvent content) it is dominated by the flexibility of the polymer macromolecules swollen by the solvent.40 For this combination of solvent and acrylate monomers, it is observed that the ionic conductivity increases when there is a clear prevalence of either the bicontinuous structure or gel, but these mechanisms compete when they coexist. It should be mentioned that a previous study used the BPAAn monomers to develop in situ generated GPEs, where the EO units were varied from BPAA4 to BPAA15, all exhibiting the characteristics of a gel, further validating that a transition from a HE to a GPE occurs upon extending the EO chain.41
3.3. The effect of copolymerization
By synthesizing copolymers, the PIPS process can be tailored even further to enhance the appreciable electrochemical performance of BPAMA. The reactivity ratios of the monomer and the respective polymer give information about the type of copolymer that can be obtained. It has been shown in the literature that methacrylates prefer propagating as homopolymers, contrary to acrylates, that rather copolymerize.37 This gives that in the first stage the copolymer is richer in methacrylate than expected from the concentration in the solvent, while this deviation is balanced towards higher conversions.In general, a slow decrease in the storage modulus is combined with a broad tanδ curve. The broadness indicates the presence of a more heterogeneous network structure.12 Due to the reactivity of the radicals and possible diffusion limitations, intramolecular reactions can take place first, until nanogels are formed,42 then the reaction proceeds and loose connections are formed between nanogels.43 The reactivity ratios available for methacrylates and acrylates37 suggest the intermediate behavior of copolymers. Indeed, DMA results in Fig. 6b underline broad curves for all the copolymers. However, unlike the homoacrylates, a clear Tg cannot be detected due to the broad distribution. The storage moduli represented in Fig. 6a and listed in Table S3 (ESI†) are intermediate between BPAMA and the homoacrylates, and they follow the same order as the homoacrylates, where BPAMA-co-BPAA1 is stiffer than BPAMA-co-BPAA2, while BPAMA-co-BPAA4 is the softest in the whole temperature interval. BPAMA-co-BPAA2 was further analysed to compare the wet/dried HE and pure polymer in Fig. S5 (ESI†). The pure polymer exhibits higher storage modulus than wet/dried HE since the pores create defects in the material. On the other hand, the HE dry films maintain a higher modulus at high temperature. This is likely due to a higher concentration of unreacted monomer in the pure polymer sample acting as a plasticizer.
 | ||
| Fig. 6 Storage modulus versus temperature (a) and tanδ curve (b) for BPAMA-co-BPAA1, BPAMA-co-BPAA2 and BPAMA-co-BPAA4. | ||
The ionic conductivities of the HEs based on homopolymers and copolymers are summarized in Fig. 7. Copolymers exhibit improved conductivities compared to the individual components, as proven by BPAMA-co-BPAA2. By combining the enhanced capability of BPAMA to undergo phase separation (Fig. S6a, ESI†), the copolymers benefit from PIPS, as confirmed by the difference in the microstructure between BPAA4 and BPAMA-co-BPAA4 (Fig. 7c and d), BPAA2 and BPAMA-co-BPAA2 (Fig. S6b and c, ESI†). BPAA1 and BPAMA-co-BPAA1 (Fig. 7a and b), instead, show limited differences in the microstructure because BPAA1 already exhibits pores similar to BPAMA. A minimum in ionic conductivity is seen for pure acrylates when EO = 2, while this minimum is not observed in copolymers. For even longer EO chains, also the copolymer could eventually transition into a gel. Although BPAA4 does not exhibit any porosity (Fig. 7d), it is as conductive as BPAA1 (Fig. 7b), which clearly contains pores. This confirms that the conduction mechanism for BPAA4 is based on polymer flexibility typical of GPEs. BPAMA-co-BPAA1 is at least 40% more conductive than the other HEs, meaning that ion transport is promoted by the high porosity (Fig. 7a). A direct relationship between pore size and opaqueness is underlined when considering the specimens in Fig. S7 (ESI†), because light is scattered in the presence of defects in the material.
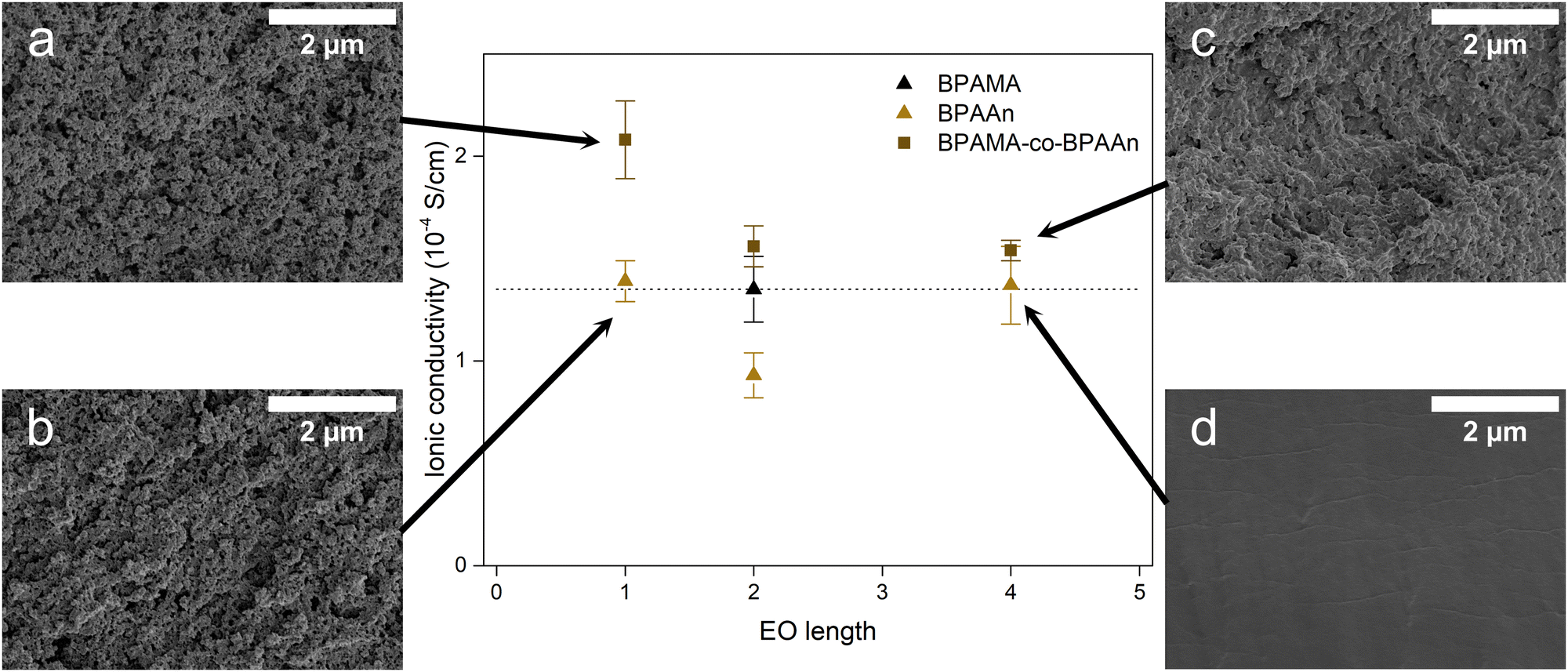 | ||
| Fig. 7 Ionic conductivity for all the samples considered and micrographs referred to (a) BPAMA-co-BPAA1, (b) BPAA1, (c) BPAMA-co-BPAA4 and (d) BPAA4. | ||
4. Conclusions
A number of HEs based on different monomer compositions were synthesized and characterized with respect to their electrochemical and thermomechanical properties, and related to their morphology. It was confirmed that the ionic conductivity is dependent on the average pore size and plasticization of the polymer phase. Homoacrylates have low stiffness at 25 °C, but the respective copolymers, obtained by BPAMA and BPAAn 50/50 vol%, show higher thermomechanical stability, and they could in principle be used to replace BPAMA. All the HEs exhibited ionic conductivities in the order of 10−4 S cm−1, with BPAMA-co-BPAA1 being more conductive than BPAMA. A trend can be identified with respect to the monomer structure and composition, where methacrylates promote the formation of pores and subsequent increase in ionic conductivity. The introduction of EO groups reduces the pore formation because solubility is increased. This leads to a transition from HEs to GPEs, having different conductivity mechanisms. Lastly, mixing methacrylate and acrylate monomers is shown to be a route to tailor (optimize) the obtained morphology and properties of the electrolyte systems.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
KTH Royal Institute of Technology, Polytechnic University of Turin, the Swedish Energy Agency (grant #48488) and Batteries Sweden (BASE) (Vinnova grant: 2019–00064) are gratefully acknowledged for their support.References
- T. Placke, R. Kloepsch, S. Dühnen and M. Winter, J. Solid State Electrochem., 2017, 21, 1939–1964 CrossRef CAS.
- M. Armand, P. Axmann, D. Bresser, M. Copley, K. Edström, C. Ekberg, D. Guyomard, B. Lestriez, P. Novák, M. Petranikova, W. Porcher, S. Trabesinger, M. Wohlfahrt-Mehrens and H. Zhang, J. Power Sources, 2020, 479, 228708 CrossRef CAS.
- Y. Nishi, Chem. Rec., 2001, 1, 406–413 CrossRef CAS PubMed.
- L. E. Asp, M. Johansson, G. Lindbergh, J. Xu and D. Zenkert, Funct. Compos. Struct., 2019, 1, 042001 CrossRef CAS.
- Y. Zhao and J. Guo, InfoMat, 2020, 2, 866–878 CrossRef CAS.
- J. Kalhoff, G. G. Eshetu, D. Bresser and S. Passerini, ChemSusChem, 2015, 8, 2154–2175 CrossRef CAS PubMed.
- K. Xu, Chem. Rev., 2014, 114, 11503–11618 CrossRef CAS PubMed.
- K. E. Okedu, Management and Applications of Energy Storage Devices, BoD–Books on Demand, 2022 Search PubMed.
- X. Q. Xu, X. B. Cheng, F. N. Jiang, S. J. Yang, D. Ren, P. Shi, H. Hsu, H. Yuan, J. Q. Huang, M. Ouyang and Q. Zhang, SusMat, 2022, 2, 435–444 CrossRef CAS.
- N. Ihrner, W. Johannisson, F. Sieland, D. Zenkert and M. Johansson, J. Mater. Chem. A, 2017, 5, 25652–25659 RSC.
- H. Zhang, S. Kulkarni and S. L. Wunder, J. Phys. Chem. B, 2007, 111, 3583–3590 CrossRef CAS PubMed.
- M. Willgert, M. H. Kjell, E. Jacques, M. Behm, G. Lindbergh and M. Johansson, Eur. Polym. J., 2011, 47, 2372–2378 CrossRef CAS.
- M. Cattaruzza, Y. Fang, I. Furó, G. Lindbergh, F. Liu and M. Johansson, J. Mater. Chem. A, 2023, 11, 76–715 RSC.
- N. Ihrner, W. Johannisson, F. Sieland, D. Zenkert and M. Johansson, J. Mater. Chem. A, 2017, 5, 25652–25659 RSC.
- F. Svec, J. Chromatogr. A, 2010, 1217, 902–924 CrossRef CAS PubMed.
- W. Johannisson, N. Ihrner, D. Zenkert, M. Johansson, D. Carlstedt, L. E. Asp and F. Sieland, Compos. Sci. Technol., 2018, 168, 81–87 CrossRef CAS.
- C. Viklund, F. Svec, J. M. J. Fréchet and K. Irgum, Chem. Mater., 1996, 8, 744–750 CrossRef CAS.
- N. Shirshova, A. Bismarck, S. Carreyette, Q. P. V. Fontana, E. S. Greenhalgh, P. Jacobsson, P. Johansson, M. J. Marczewski, G. Kalinka, A. R. J. Kucernak, J. Scheers, M. S. P. Shaffer, J. H. G. Steinke and M. Wienrich, J. Mater. Chem. A, 2013, 1, 15300 RSC.
- F. Svec and J. M. J. Frechet, Anal. Chem., 1992, 64, 820–822 CrossRef CAS.
- H. Minakuchi, K. Nakanishi, N. Soga, N. Ishizuka and N. Tanaka, Anal. Chem., 1996, 68, 3498–3501 CrossRef CAS PubMed.
- H. M. J. Boots, J. G. Kloosterboer, C. Serbutoviez and F. J. Touwslager, Macromolecules, 1996, 29, 7683–7689 CrossRef CAS.
- F. Svec, E. C. Peters, D. Sýkora and J. M. J. Fréchet, J. Chromatogr. A, 2000, 887, 3–29 CrossRef CAS PubMed.
- F. Svec and J. M. J. Fréchet, Science, 1996, 273, 205–211 CrossRef CAS PubMed.
- N. Shirshova, A. Bismarck, E. S. Greenhalgh, P. Johansson, G. Kalinka, M. J. Marczewski, M. S. P. Shaffer and M. Wienrich, J. Phys. Chem. C, 2014, 118, 28377–28387 CrossRef CAS.
- Y. H. Song, T. Kim and U. H. Choi, Chem. Mater., 2020, 32, 3879–3892 CrossRef CAS.
- S. J. Kwon, T. Kim, B. M. Jung, S. B. Lee and U. H. Choi, ACS Appl. Mater. Interfaces, 2018, 10, 35108–35117 CrossRef CAS PubMed.
- H. Krebs, L. Yang, N. Shirshova and J. H. G. Steinke, React. Funct. Polym., 2012, 72, 931–938 CrossRef CAS.
- S. Emilsson, V. Vijayakumar, J. Mindemark and M. Johansson, Electrochim. Acta, 2023, 449, 142176 CrossRef CAS.
- S. Emilsson, G. Lindbergh and M. Johansson, J. Mater. Chem. A, 2024, 44, 30442–30453 RSC.
- S. Duan, M. Cattaruzza, V. Tu, R. M. Auenhammer, R. Jänicke, M. K. G. Johansson, F. Liu and L. E. Asp, Commun. Mater., 2023, 4, 49 CrossRef CAS.
- M. Cattaruzza, Y. Fang, I. Furó, G. Lindbergh, F. Liu and M. Johansson, J. Mater. Chem. A, 2023, 11, 7006–7015 RSC.
- M. Cattaruzza, M. Johansson, G. Lindbergh and F. Liu, ChemElectroChem, 2025, 12, e202400561 CrossRef CAS.
- R. Chaudhary, J. Xu, Z. Xia and L. E. Asp, Adv. Mater., 2024, 36, 2409725 CrossRef CAS.
- L. E. Asp, K. Bouton, D. Carlstedt, S. Duan, R. Harnden, W. Johannisson, M. Johansen, M. K. G. Johansson, G. Lindbergh, F. Liu, K. Peuvot, L. M. Schneider, J. Xu and D. Zenkert, Adv. Energy Sustainable Res., 2021, 2, 2000093 CrossRef.
- L. J. Hughes and G. L. Brown, J. Appl. Polym. Sci., 1961, 5, 580–588 CrossRef CAS.
- S. Krause, J. J. Gormley, N. Roman, J. A. Shetter and W. H. Watanabe, J. Polym. Sci., Part A: Gen. Pap., 1965, 3, 3573–3586 CrossRef CAS.
- S. Ren, L. Hinojosa-Castellanos, L. Zhang and M. A. Dubé, Macromol. React. Eng., 2017, 11, 1600050 CrossRef.
- K. Dusek, Angew. Chem., 1996, 240, 1–15 CAS.
- G. Odian, Principles of Polymerization, John Wiley & Sons, Incorporated, United States, 2004, pp. 464–543 DOI:10.1002/047147875x.ch6.
- S. Sen and F. H. Richter, Adv. Sci., 2023, 10, 2303985 CrossRef CAS.
- Y. Kang, K. Cheong, K.-A. Noh, C. Lee and D.-Y. Seung, J. Power Sources, 2003, 119–121, 432–437 CrossRef CAS.
- B. H. Jones, T. M. Alam, S. Lee, M. C. Celina, J. P. Allers, S. Park, L. Chen, E. J. Martinez and J. L. Unangst, Polymer, 2020, 205, 122783 Search PubMed.
- S. Seiffert, Polym. Chem., 2017, 8, 4472–4487 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ma00125k |
| ‡ These authors contributed equally and should be regarded as co-first authors. |
| This journal is © The Royal Society of Chemistry 2025 |