
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCharge redistribution induced by well-dispersed cobalt oxide nanoparticles on Co3(PO4)2 surfaces enhances OER catalytic activity†
Abdelhadi
El Jaouhari‡
a,
Jamal
Bencaid‡
a,
Anouar
Belhboub
b,
Mustapha
Matrouf
a,
Ikram
Cheras
a,
Jinhua
Zhu
 c,
Bouchaib
Manoun
ad and
Fouad
Ghamouss
*a
c,
Bouchaib
Manoun
ad and
Fouad
Ghamouss
*a
aDepartment of Materials Science, Energy, and Nano-Engineering (MSN) Mohamed VI Polytechnic University, Ben Guerir, 43150, Morocco. E-mail: fouad.ghamouss@um6p.ma
bCentrale Casablanca, Complex Systems and Interactions Research Center, Bouskoura, Ville verte, Casablanca, Morocco
cHenan International Joint Laboratory of Medicinal Plants Utilization, College of Chemistry and Chemical Engineering, Henan University, Kaifeng, China
dLaboratory of Radiation-Matter and Instrumentation S3M, FST Settat, Hassan First University, Settat, 6000, Morocco
First published on 8th April 2025
Abstract
Developing electrocatalysts for the oxygen evolution reaction (OER) with high efficiency and durability to simulate industrial application conditions is essential for addressing environmental issues and the energy crisis. Decorating or anchoring nanoparticles onto catalyst surfaces shows promise in improving catalytic performance. However, the intrinsic mechanism behind this approach is not yet fully understood. Herein, varying amounts of cobalt oxide nanoparticles (1, 2.5, 5, 10 and 20% mass ratios) were in situ synthesized on the surface of amorphous cobalt orthophosphate (Co3(PO4)2) to deeply investigate the behavior of the decorated catalysts. Interestingly, the results indicate that the cobalt orthophosphate decorated with a low amount of cobalt oxide nanoparticles (Co3(PO4)2@1%Co3O4) exhibits the highest catalytic activity (low overpotential of 313.01 mV at 20 mA cm−2 and high stability for 100 hours) compared to samples with higher amounts of these nanoparticles. The electrochemical results reflect that the well-distributed low concentration of Co3O4 induced an inductive effect on the surface of Co3(PO4)2 leading to the redistribution of electron configuration on the surface. These findings can be confirmed by DFT calculations, which reveal a stronger electronic coupling between neighboring cobalt oxide nanoparticles. This stronger interaction minimizes their interaction with cobalt orthophosphate resulting in a decrease in catalytic activity.
Introduction
Transitioning to sustainable energy sources is essential for addressing the urgent concerns of fossil fuel scarcity and environmental issues. Hydrogen, known for being a clean and efficient energy carrier, has become a key element in this major shift.1–3 Among the various methods of hydrogen production, electrochemical water splitting (electrolysis) presents a promising route due to its simplicity and potential for integration with renewable energy sources.4–6 However, the efficiency of electrolysis is significantly hindered by the high overpotential required for the anodic oxygen evolution reaction (OER), which necessitates the development of efficient and durable electrocatalysts. Various materials have been tested as catalysts for the oxygen evolution reaction (OER), including transition metal oxides, hydroxides, oxyhydroxides, alloys, chalcogenides, and phosphides.7–12 However, many of them suffer from numerous issues such as low intrinsic activity, limited active sites, and poor stability, which leads to insufficient performance for widespread commercialization and industrial applications. Therefore, significant research efforts are still ongoing to improve the catalytic activity and durability of electrocatalysts for the OER. Advanced approaches have been adapted to improve the OER catalytic activity of the materials, such as creating vacancies, doping, morphological and size tuning, decorating, and varying structural composition.13–17 Some recent advancements in this context have focused on decorating or anchoring nanoparticles onto catalyst surfaces, showing promise in improving catalytic activity and stability through various mechanisms.16,18–22 For instance, Jingyan Zhang et al.16 proposed the in situ growth of Co3O4 nanoparticles on the surface of NiO nanosheets to enhance catalytic activity toward the OER. The enhanced catalytic activity was attributed to the increased oxygen vacancy ratio of NiO/Co3O4 compared to those of both pristine oxides. This increase reduces the coordination numbers of neighboring metal atoms and optimizes the energetics of the catalytic reaction. Additionally, the authors observed that the NiO/Co3O4 heterostructure exhibited an increase in Ni3+ and a decrease in Co3+, resulting in intermediate spin configurations that are advantageous for OER catalysis. In another study, Weiwei Han et al.23 evaluated the evolution of the OER catalytic activity of ultrasmall IrO2 nanoparticles anchored on hollow Co–Mo multi-oxide heterostructure nanocages. The IrO2@Co3O4–CoMoO4 heterostructure exhibits enhanced catalytic activity and stability for the oxygen evolution reaction compared to IrO2 alone. The investigation showed that the uniform dispersion of ultrasmall iridium oxide nanoparticles on the hollow Co3O4–CoMoO4 provides abundant active sites and the heterogeneous interfaces accelerate the charge transport during the OER process. Furthermore, density functional theory (DFT) calculations demonstrated that the incorporation of Mo into the heterostructure further enhances electron transfer at the interface, thereby improving electron mobility. Yulin Xing et al.24 conducted a study on atomic iridium (Ir)-incorporated nickel hydroxide (Ni(OH)2) nanosheets as enhanced electrocatalysts for the oxygen evolution reaction (OER). The study demonstrated that the catalytic performance of these nanosheets is enhanced through an inductive effect-based mechanism. Due to the electronegativity difference between Ir and Ni, the inductive effect facilitates the formation of high-valent Ni species, increasing the electrophilicity of the catalyst surface. This enhanced electrophilicity improves the adsorption of nucleophilic key intermediates, such as O* and OOH*, thereby accelerating OER kinetics. Huihuang Chen et al.25 investigated the effect of single-atom iridium (Ir) anchored on the surface of cobalt oxide (Co3O4). The anchoring of Ir single atoms promotes the formation of oxygen vacancies on the Co3O4 surface, which shifts the d-band center closer to the Fermi level. This synergistic effect optimizes the adsorption configuration, enhances electron transfer, and strengthens the adsorption of reaction intermediates, thereby improving catalytic activity toward the oxygen evolution reaction (OER).In the present work, amorphous cobalt orthophosphate was synthesized via a simple ultrasound assisted precipitation at room-temperature, followed by thermal treatment under an inert gas to enhance the catalytic activity. The surface of the prepared cobalt orthophosphate underwent in situ growth of varying amounts of nanosized cobalt oxide via a hydrothermal approach. The results obtained indicate that different amounts of anchored Co3O4 affect the catalytic activity differently, depending on their distribution on the surface of Co3(PO4)2. Well-dispersed and non-agglomerated distribution of Co3O4 leads to enhanced OER catalytic activity compared to condensed anchoring. Specifically, well dispersed Co3O4 exhibits an inductive effect, capable of modifying the electronic structure of the Co atoms in the amorphous Co3(PO4)2, resulting in improved catalytic activity. DFT calculations indicate that the interaction between neighboring Co3O4 nanoparticles (NPs) is more favorable compared to the adsorption of NPs on the surface of cobalt orthophosphate. This stronger interaction highlights the tendency of Co3O4 nanoparticles (NPs) to cluster together rather than remain individually dispersed on the surface.
Results and discussion
Cobalt orthophosphate was synthesized using an ultrasonic-assisted co-precipitation method at room temperature to facilitate the dispersion of particles, leading to more catalytically active sites.26 Subsequently, the hydrated cobalt orthophosphate Co3(PO4)2·8H2O underwent thermal treatment under argon gas to create an amorphous phase of Co3(PO4)2 thereby enhancing its catalytic activity. In the last step, in situ growth of cobalt oxide (Co3O4) nanoparticles on the surface of cobalt orthophosphate was achieved using a hydrothermal method in the presence of ammonia. This approach was employed to control the size and morphology of cobalt oxide nanoparticles. Various characterization analyses were performed to thoroughly investigate the structural composition and morphological properties of the synthesized electrocatalysts. X-ray diffraction was exploited to investigate the crystal structure. Fig. 1a displays the XRD diffractograms of all prepared samples. The diffractogram corresponding to Co3(PO4)2·8H2O reflects the presence of all its characteristic peaks of monoclinic cobalt orthophosphate hydrate, including (110) at 11.28°, (020) at 13.27°, (200) at 18.30°, (20−1) at 23.20°, (13−1) at 27.99°, and (221) at 33.219, as indicated by JCPDS No. 41-0375.27 It can be observed from the diffractogram of amorphous Co3(PO4)2 that the phase transformed into a completely amorphous structure after thermal treatment (Fig. S1a, ESI†). This transformation could be explained by the loss of the water molecules from the hydrated structure of orthophosphate during the thermal treatment as can be confirmed from the TGA curve of Fig. S1b (ESI†). The diffractogram of Co3O4 illustrates the characteristic peaks of the face-centered cubic structure of spinel Co3O4 (JCPDS No. 74-2120) such as (111) at 19.05°, (220) at 31.29°, (311) at 36.83°, (511) at 59.34° and (440) at 65.40°.28 The XRD patterns of the samples corresponding to the amorphous Co3(PO4)2 decorated with 1% of Co3O4 do not exhibit characteristic peaks of Co3O4 (Fig. S1a, ESI†), indicating that the small amount of Co3O4 present is below the detection limit of XRD. However, the bands (corresponding to Co3O4) are detectable in the Raman spectra of the decorated samples. In full detail, Fig. 1b shows the Raman spectra of all samples. The Co3(PO4)2·8H2O spectrum is characterized by the main peak located at 953.35 cm−1 which is ascribed to the Raman active PO-stretching vibration.29–31 Another peak can be detected at 1041.02 cm−1 corresponding to the phosphate PO-antisymmetric stretching vibrations.29 The same main peak corresponding to PO-stretching vibration appears in the Raman spectra of amorphous Co3(PO4)2 with low intensity. A small positive shift is shown, and this shift is due to the disorder in the amorphous structure resulting in a broader distribution of vibrational modes, leading to a reduction in peak intensity.32,33 The Raman spectrum of Co3O4 reveals its four characteristic peaks located at 189.48, 462.91, 505.70 and 662.60 cm−1, respectively, attributed to F2g, Eg, F2g and A1g (Fig. 1b) vibration modes of this spinel oxide which are in excellent agreement with the previously published results.34 Furthermore, the growth of Co3O4 with varying amounts on the surface of amorphous Co3(PO4)2 can be confirmed through the Raman spectra of the decorated amorphous Co3(PO4)2@1%Co3O4, Co3(PO4)2@5%Co3O4 and Co3(PO4)2@10%Co3O4. It can be observed that there is a gradual growth (as a function of the varying amount of Co3O4) of peaks corresponding to Co3O4, such as F2g, Eg, and A1g, in the spectra of amorphous Co3(PO4)2 (Fig. 1b). Besides, there is a decrease by the same order in the intensity of the main peak corresponding to the PO-stretching vibration.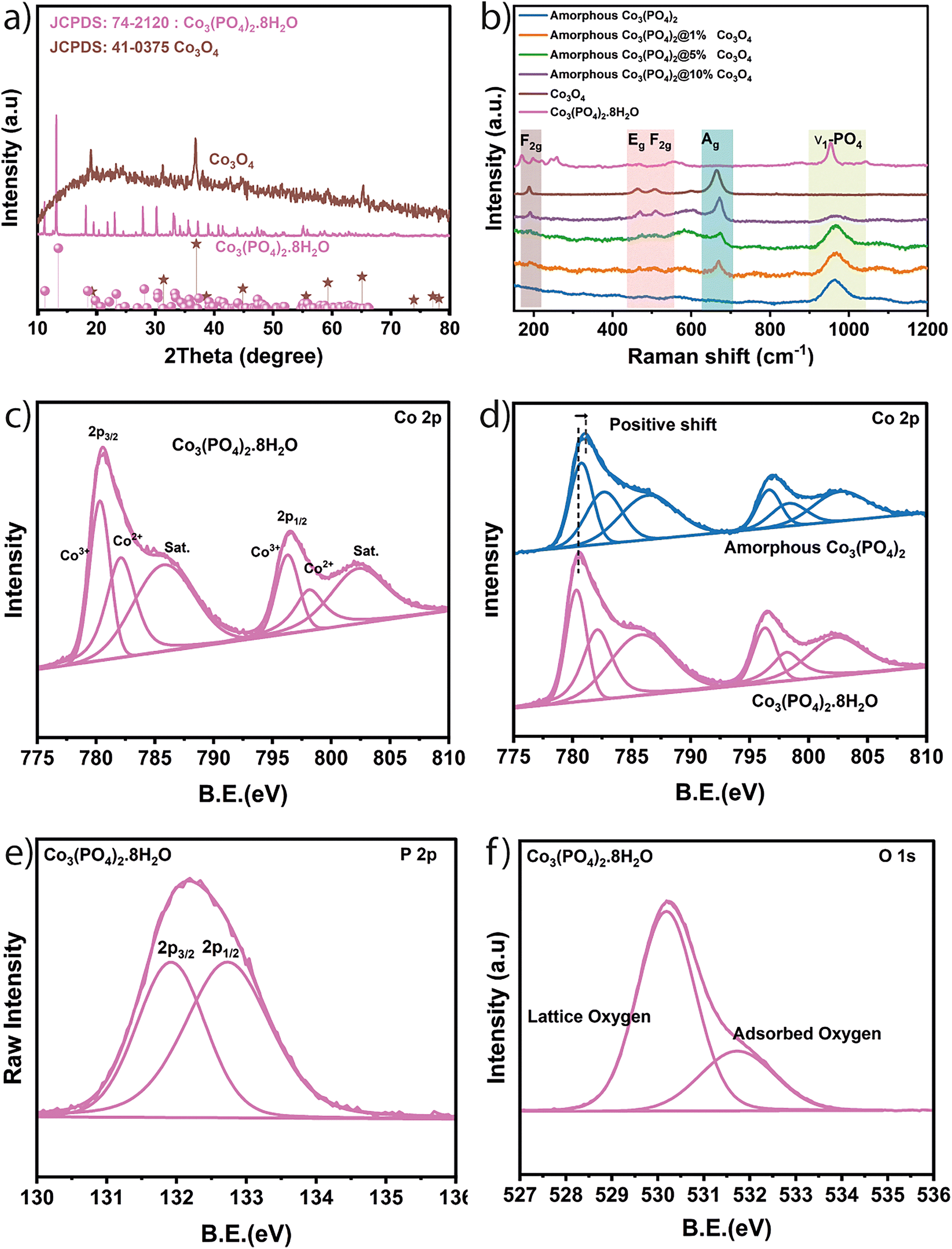 | ||
| Fig. 1 (a) XRD pattern, (b) Raman, (c) XPS Co 2p, (d) XPS comparison of the prepared crystalline Co3(PO4)2·8H2O and amorphous Co3(PO4)2. (e) XPS P 2p and (f) XPS O 1s. | ||
X-ray photoelectron spectroscopy (XPS) has been used to investigate the chemical composition on the surface of samples. The survey spectrum of Co3(PO4)2·8H2O (Fig. S2, ESI†) reflects the presence of the main peaks corresponding to Co 2p, O 1s and P 2p with different electronic states. The Co 2p3/2 peak (Fig. 1c) can be deconvoluted to cover Co3+ at 780.34 eV, Co2+ at 782.11 eV and a satellite peak at 785.81 eV. Similarly, Co 2p1/2 reflects three peaks at 796.31, 798.19 and 802.44 eV corresponding to Co3+, Co2+, and satellite, respectively.35,36 The O 1s peak (Fig. 1f) is present in the form of lattice oxygen of the structure at 530.17 eV and surface adsorbed oxygen at 531.74 eV. Meanwhile P 2p (Fig. 1e) can be decomposed to P 2p3/2 located at 131.92 and P 2p1/2 at 132.72 eV.35,36 SEM has been used to assess the morphological characteristics of the various prepared catalysts, and micrographs depicting different zones of the catalysts are presented in Fig. 2. A homogeneous distribution of 2D lamellar sheets with varying sizes (few micrometers) with nanometric thickness is evident in the micrograph corresponding to Co3(PO4)2·8H2O (Fig. 2a), and its surface appears relatively smooth. After thermal treatment (amorphous Co3(PO4)2), the samples generally maintain the same 2D sheets form but exhibit a rougher surface (Fig. 2b), which is suspected to be beneficial for OER activity as discussed in previous reports.37,38 The corresponding elemental mapping images (Fig. 2c) illustrate a homogenous distribution of cobalt, oxygen and phosphorus throughout the amorphous Co3(PO4)2. Besides, Fig. S3 (ESI†) shows the energy-dispersive X-ray curve with the atomic ratio (%) of Co, P and O in the amorphous Co3(PO4)2. In the activated electrocatalysts Co3(PO4)2@Co3O4, all samples are decorated with varying amounts and densities of dispersed Co3O4 on the surface of cobalt orthophosphate. The surface of Co3(PO4)2 is entirely coated with cauliflower-like nanoparticles of Co3O4 in the Co3(PO4)2@10%Co3O4 case (Fig. 2d). Conversely, for Co3(PO4)2@5%Co3O4, the surface is partially covered with cobalt oxide (Fig. 2e), and only few areas exhibit cobalt oxide in the case of Co3(PO4)2@1%Co3O4 (Fig. 2f).
 | ||
| Fig. 2 SEM images of (a) Co3(PO4)2, (b) amorphous Co3(PO4)2, (d) Co3(PO4)2@10% Co3O4, (e) Co3(PO4)2@5% Co3O4 and (f) Co3(PO4)2@1% Co3O4. (c) EDX Mapping of Co3(PO4)2 and (g) and (h) HR-TEM of Co3(PO4)2@1% Co3O4. | ||
The well-distributed Co3O4 on amorphous Co3(PO4)2 in Co3(PO4)2@1%Co3O4 can be confirmed by high resolution transmission electron microscopy (HR-TEM), as shown in Fig. 2g and h. The micrograph in Fig. 2g displays the distribution of spherical nanoparticles of Co3O4 on the surface of amorphous Co3(PO4)2. Additionally, Fig. 2h shows a lattice fringe with a spacing of 0.24 nm corresponding to the (311) plane of the crystalline Co3O4.39
The electrochemical tests such as linear sweep voltammetry (LSV), cyclic voltammetry (CV), electrochemical impedance spectroscopy (EIS) and chronopotentiometry, were used to gain insights into the electrocatalytic activity and durability of the proposed catalysts. The LSV were conducted employing a conventional three-electrode cell in a 1 M KOH alkaline solution within a potential range of 1.00 to 2.00 V vs. RHE at a scan rate of 2 mV s−1 (Fig. 3a). The overpotentials were assessed at current densities of 10, 20, 100 and 200 mA cm−2 (Fig. 3c). During the initial analysis of the findings, it becomes evident that the amorphous Co3(PO4)2 exhibits enhanced OER catalytic activity compared to its crystalline counterpart (Co3(PO4)2)·8H2O (Fig. S4a, ESI†). This conclusion can be drawn from the lower overpotentials required to achieve all the chosen current densities in amorphous Co3(PO4)2 compared to the pristine Co3(PO4)2·8H2O (Fig. S4a, ESI†), as well as better kinetics indicated by the smallest Tafel slope (Fig. S4b, ESI†). The enhanced catalytic activity of the amorphous catalyst has been investigated in previous reports for a various range of materials.40 In this work, it can be concluded from the results of double layer capacitance (Fig. S4c, ESI†) that there is an increase in the electrochemical active surface area of amorphous Co3(PO4)2 compared to the crystalline catalyst leading to more active sites for catalytic activity. The enhanced catalytic activity and increased number of the active sites for the OER in the case of amorphous Co3(PO4)2 can be explained by the altered electrochemical behavior of amorphous Co3(PO4)2 compared to its crystalline counterpart in 1 M KOH. The cyclic voltammetry of crystalline Co3(PO4)2·8H2O set from 0.6 to 1.6 V vs. RHE at 10 mV s−1, exhibits a distinct oxidation wave (at 1.16 V vs. RHE) corresponding to the oxidation of Co(II) into Co(III) (Fig. S4d, ESI†).40 However, in the voltammogram of amorphous Co3(PO4)2, a significant increase in the current density is observed in the Co(II) to Co(III) oxidation wave, indicating the formation of additional cobalt species during thermal treatment. The increase in redox activity leads to higher catalytic activity, as confirmed by previous investigations.40–42 The transformation of the cobalt surface to a more oxidized state during the amorphization process can also be inferred from the XPS results (Fig. 1d). A notable observation is the positive shift in the Co 2p peak of amorphous Co3(PO4)2 compared to its crystalline counterpart. This shift suggests a higher valence state of cobalt following thermal treatment.
 | ||
| Fig. 3 (a) LSV polarization curves. (b) TOF curves. (c) Tafel and overpotential values. (c) Double layer and electrochemical surface area values. (e) Stability test for 100 h in 1 M KOH. (f) Raman spectroscopy comparison of the prepared pristine 1% decorated amorphous Co3(PO4)2 with PTFE before and after the stability test. | ||
As we progress, our focus shifts to the examination of the results derived from the electrocatalysts featuring Co3O4 decorating amorphous Co3(PO4)2. The LSV curves (Fig. 3a) and Fig. S5 (ESI†) illustrate that decorated catalysts exhibit significantly lower overpotentials to achieve all investigated current densities compared to the pristine amorphous Co3(PO4)2 and pristine Co3O4. As illustrated in Fig. 3c, amorphous Co3(PO4)2@1%Co3O4 required only 313.01 mV to achieve a current density of 20 mA cm−2, exhibiting a small Tafel slope of 67.03 mV dec−1. This outperforms both Co3(PO4)2@2.5%Co3O4 (316.09 mV at 20 mA cm−2; 69.72 mV dec−1) and Co3(PO4)2@5%Co3O4 (333.89 mV at 20 mA cm−2; 71.91 mV dec−1), all of which surpass the values observed for pristine amorphous Co3(PO4)2 (367.16 mV at 20 mA cm−2; 81.09 mV dec−1) and pristine Co3O4 (327 mV at 20 mA cm−2; 76.66 mV dec−1). The comparison was conducted at 20 mA cm−2 instead of 10 mA cm−2 due to potential confusion at the latter current density, where it coincides with the oxidation peak corresponding to the transition from Co(III) to Co(IV). These results indicate that all decorated electrocatalysts demonstrate enhanced catalytic activity with a faster kinetic response, since TOF calculations further confirm the previous contrast in electrochemical behavior upon decoration (Fig. 3b). The increased number of electrochemically active sites following the anchoring of well-dispersed Co3O4 on amorphous cobalt orthophosphate is supported by the enhanced double-layer capacitance and electrochemical surface area (ECSA) values, as shown in Fig. 3d.
Confirmation of the enhanced kinetic behavior can be derived from the results of the electrochemical impedance spectroscopy (EIS), where decorated samples exhibit a diminished charge transfer value compared to both amorphous Co3(PO4)2 and Co3O4 (refer to the ESI† Fig. S6 for more details). The stability of Co3(PO4)2@1%Co3O4 has been examined using the chronoamperometry mode at 30 mA cm−2 for 100 hours and compared with RuO2. It can be seen from Fig. 3e that Co3(PO4)2@1%Co3O4 exhibits good stability as evidenced by the low level of decay in the measured potential over the 100 hours. The Raman comparison after the stability test (Fig. 3f) reflects that the surface of the catalyst transformed into cobalt oxyhydroxide as active sites for the OER process, which is in full agreement with the previous investigation.43,44
To provide further insight into these findings, the electrochemical behavior was investigated using cyclic voltammetry (CV) in the range of 0.6 to 1.6 V vs. RHE at a scan rate of 10 mV s−1. Initially, the voltammogram corresponding to Co3O4 displays a singular redox wave at 1.42 V vs. RHE, attributed to the Co(III)/Co(IV) transition (Fig. 4a). In contrast, amorphous Co3(PO4)2 exhibits an additional wave around 1.1 V vs. RHE, corresponding to Co(II)/Co(III), albeit with a relatively small intensity, alongside the Co(III)/Co(IV) wave. Notably, the voltammogram of Co3(PO4)2@1%Co3O4 demonstrates a significant enhancement of the Co(II)/Co(III) wave with an anodic shift of approximately 40 mV. Co3(PO4)2@2.5%Co3O4 and Co3(PO4)2@5%Co3O4 exhibit a peak similar to that of Co3(PO4)2@1%Co3O4 with a cathodic shifts compared to Co3(PO4)2@1%Co3O4 (Fig. 4b).
 | ||
| Fig. 4 Cyclic voltammetry (CV) in the range of 0.6 to 1.6 V vs. RHE at a scan rate of 10 mV s−1 for (a) comparison between amorphous and 1% decorated amorphous Co3(PO4)2 with Co3O4 against Co3O4 only, and (b) comparison the decorated amorphous Co3(PO4)2 with Co3O4 at percentages of 1%, 2.5%, and 5%. (c) XPS comparison of amorphous and decorated Co3(PO4)2 with 1% of Co3O4 and native Co3O4. | ||
The shift value found in Co3(PO4)2@2.5%Co3O4 is about 30 mV and 50 mV for Co3(PO4)2@5%Co3O4. The anodic shift for Co3(PO4)2@1%Co3O4 observed in the oxidation peak of cobalt Co(II)/Co(III) has been previously reported in cases involving doping oxides, hydroxides and oxyhydroxides of cobalt/nickel by introducing a foreign element in their structure, such as iron, nickel or manganese leading to enhanced catalytic activity toward the oxygen evolution reaction.17,45,46 The observed behavior is typically attributed to the inductive effect generated by the dopant atom, which leads to the redistribution of electrons within the structure of cobalt or nickel.47,48 This inductive effect can be confirmed through the results of high-resolution Co 2p XPS of amorphous cobalt orthophosphate, Co3(PO4)2@1%Co3O4 and Co3O4. It can be detected that this peak in the case of Co3(PO4)2@1%Co3O4 shifted positively compared to pristine amorphous cobalt orthophosphate and Co3O4 indicating a redistribution in the electronic state (higher valence state) of cobalt element (Fig. 4c). The presence of such high-valence cobalt would contribute to the observed enhancement in OER performance as reported previously.49 These high-valent metal species exhibit a stronger electrophilic characteristic, which promotes the adsorption and activation of nucleophilic intermediates, including OH, O, and OOH*, through nucleophilic attack.50–53
To summarize, the well-dispersed and non-agglomerated distribution of low-coordination Co3O4 leads to an enhancement of the catalytic activity toward the oxygen evolution reaction. This is likely due to the capacity of Co3O4 to modify the electronic structure of the Co atoms in the amorphous Co3(PO4)2 conducive to a better catalytic activity. This is more noticeable when the cobalt oxide is well dispersed with a low concentration on the surface of Co3(PO4)2.
To correlate the observed electrochemical behavior with changes in the electronic structure and interactions between different elements of the proposed catalysts, we utilize computational modeling through density functional theory.
Fig. 5 illustrates the optimized structures for the interacting systems studied, specifically Co3O4 nanoparticles adsorbed on the surface of Co3(PO4)2 and the interaction between two Co3O4 nanoparticles. In the case of a single Co3O4 NP adsorbed on the Co3(PO4)2 surface (Fig. 5a and b), the equilibrium distance between the NP and the surface is determined to be around 5.8 Å, indicating a stable adsorption configuration. For the system involving two Co3O4 NPs in interaction (Fig. 5c), the equilibrium inter-NP distance is slightly larger at around 6.4 Å, which could suggest, at first glance, a weaker interaction compared to the NP/surface system.
 | ||
| Fig. 5 (a) Side and (b) inclined views of the optimized structure of adsorbed Co3O4 nanoparticles on the Co3(PO4)2 surface. (c) Optimized structure of the two Co3O4 nanoparticles. Co, P and O atoms are represented by blue, purple and red balls, respectively. | ||
The binding energy calculations further clarify the interaction strengths observed in the optimized structures. The binding energy for the heterostructure involving one Co3O4 NP adsorbed on the Co3(PO4)2 surface, calculated as:
| Eb = ENP/surface − (ENP + Esurface) |
| Eb = ENP–NP − (ENP1 + ENP2) |
 | ||
| Fig. 6 (a) and (b) Partial density of states of Co3O4 nanoparticles in isolated and 2 nanoparticles configurations over O p orbitals and Co d orbitals. (c) Partial density of states of isolated and Co3(PO4)2 surface adsorbed Co3O4 nanoparticles over O p orbitals and Co d orbitals. (d) Charge density difference profile in the Co3O4/Co3(PO4)2 heterostructure. (e) Charge density difference profile in the Co3O4–Co3O4 structure. Turquoise and green areas show charge accumulation and depletion, respectively. The density isosurfaces are plotted considering an isovalue of 0.01 e Bohr−3. | ||
This behavior aligns with the binding energy results, where the NP exhibits stronger interaction with a neighboring NP than with the surface, as reduced overlap with the surface bands (fewer NP bands at the Fermi level) would lead to weaker interactions. Fig. 6d and e show the charge density differences for the NP when adsorbed on the surface or interacting with another NP. The charge transfer density is defined in the case of surface adsorption as follows:
| ρtransfer = ρNP/surface − (ρNP + ρsurface) |
| ρtransfer = ρNP–NP − (ρNP1 + ρNP2) |
For surface adsorption, the charge redistribution is primarily localized on the Co atoms of the NPs, which interact more strongly with the surface, and to a lesser extent on some of the O atoms. Additionally, the charge density change profile on the surface shows that the range of interactions with NPs include all atomic layers of the surface as the charge perturbation due to this interplay is visible on regions in the top atomic layers as well as in the bottom ones. When two NPs are in interaction, the charge density perturbation becomes more pronounced, involving a larger number of NP Co atoms.
The more extensive charge redistribution for this interaction further supports the conclusion that neighboring NPs exhibit stronger electronic hybridization. Bader charge analysis provides further insights into the electronic coupling mechanisms for the Co3O4 NPs when adsorbed on the surface or interacting with another NP. The total charge transfer
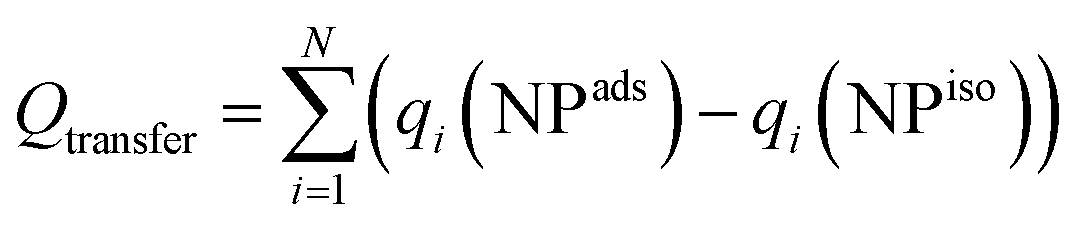
| System | Q transfer (Co) | Q transfer (O) |
|---|---|---|
| Surface/NP | 0.301 | −0.079 |
| 2NPs–NP1 | 0.165 | −0.160 |
| 2NPs–NP2 | 0.104 | −0.109 |
Experimental
Chemicals
All chemicals used were of analytic grade and used without any purification or treatment. Cobalt(II) chloride hexahydrate 99% was obtained from VWR Chemicals, ammonium phosphate dibasic 98%, ammonia solution (NH3·H2O, 28 wt %), potassium hydroxide 98%, iodine 99.7%, acetone (99%) and absolute ethanol were purchased from Sigma Aldrich.Preparation of catalysts
Electrochemical measurements and material characterization
All electrochemical measurements were conducted using a three-electrode conventional cell connected to a Gamry-3000 electrochemical workstation. A mercury/mercury oxide (Hg/HgO) electrode served as the reference electrode, while a platinum wire was utilized as the counter electrode. A carbon cloth electrode (1 cm2), coated with the catalyst, was employed as the working electrode. The catalysts were deposited onto the working electrode surface using electrophoretic deposition in an optimized solution containing 30 mg of the catalyst and 3 mg of I2 dispersed in 30 mL of acetone. The mass loading of the catalyst was estimated to be 0.6 mg cm−2. All potential measurements were referenced to the reversible hydrogen electrode (RHE) using the following equation:| ERHE = EHg/HgO + 0.098 + 0.059 × pH |
The linear sweep voltammetry curves were acquired with 90% iR-compensation in a potential window of 1.00–2.00 V vs. RHE at a scan rate of 2 mV s−1 in 1.0 M KOH electrolyte. Cyclic voltammetry was performed within a potential window of 0.6 to 1.6 V vs. RHE with a scan rate of 10 mV s−1. Electrochemical impedance spectroscopy was conducted in a frequency range of 100 kHz to 0.01 Hz.
For the stability test, a paste of amorphous Co3(PO4)2@1%Co3O4 was prepared by mixing 4 ml of ethanol with 90 wt% of the catalyst with 10 wt% of PTFE dispersed in 60 wt% of water. The paste was then pressed on the surface of a stainless-steel mesh cleaned previously in 0.1 M H2SO4 and ethanol and dried in a vacuum oven.
High-resolution transmission electron microscopy (HR-TEM) measurements were conducted using a JEM 2100F instrument. X-ray photoelectron spectroscopy (XPS) measurements were performed using an Escalab 250xi spectrometer equipped with an Al Ka monochromator X-ray source. High-resolution Raman microscopy at room temperature utilized a laser source with a wavelength of 514 nm (Lab RAM HR Evolution Raman microscopes). X-ray diffractometry was carried out using a Bruker D8 Advance powder instrument, employing an incident X-ray beam emitted from a copper anti-cathode with wavelengths of λ1 = 1.54060 Å (Kα1) and λ2 = 1.54439 Å (Kα2).
Computational details
Periodic first-principles calculations were performed within the density functional theory (DFT) and the generalized gradient approximation (GGA) using the Perdew–Burke–Ernzerhof (PBE) functional54 as implemented in the VASP code.55 The projected augmented wave (PAW) method was used to treat the core electrons.56 Dispersion effects were included using the so-called DFT-D2 method57 which consists in adding a semi-empirical dispersive term directly to the Kohn–Sham energies. Spin-polarized calculations were considered for all systems. The Co3O4 and Co3(PO4)2 crystal structures (cell shape and atomic positions) were optimized considering a plane-wave kinetic energy cutoff of 500 eV. Atomic position relaxations were performed with an energy and maximum residual forces cutoffs of 10−6 eV and 3 × 10−2 eV Å−1, respectively. From the optimized crystal structures, a Co3O4 spherical cluster (28 O atoms and 17 Co atoms) and a (020) Co3(PO4)2 slab, containing five atomic layers, were constructed. To build the hybrid systems, the cluster was placed at an interacting distance of around 6 Å from an identical second cluster to form the two nanoparticles system. The cluster was also adsorbed on the slab by placing it at around 6 Å on top of the surface to form the nanoparticle/surface heterostructure. The surface had dimensions of 23.14 × 29.69 Å2 in the xy plane so as to provide adequate distances between periodic replicas when the cluster is adsorbed, therefore avoiding adsorbate–adsorbate spurious interactions. Indeed, in the nanoparticle/surface heterostructure, the adsorbate–adsorbate separation was at least 14.5 Å in the x direction and 23.5 Å in the y direction, while the adsorbate–surface separation was higher than 26 Å in the z direction (the surface–surface separation in the z direction was about 36.6 Å). For the two nanoparticles system, the separation with periodic replicas, was 14.8 Å, 11.3 Å and 39.5 Å in the x, y and z directions, respectively. Relaxations of the atomic positions of the constructed systems were performed by expanding the wavefunctions of the valence electrons in plane-waves with a kinetic energy cutoff of 450 eV. To relax the atomic positions of the isolated cluster, thresholds of 10−5 eV and 2 × 10−2 eV Å−1 were considered for energy and maximum residual forces, respectively. For the bare surface and hybrid systems, these thresholds were 10−5 eV and 3 × 10−2 eV Å−1. Due to the large dimensions of the bare surface and hybrid systems, the atomic relaxation of all systems was performed by approximating integration over the Brillouin zone by a sum over a 2 × 2 × 1 k-grid using the Monkhorst–Pack scheme.58 For systems involving the surface, atomic relaxation was performed by fixing the two bottom atomic layers, allowing only the three top layers of the surface to move. The obtained optimized structures were used as starting points to calculate the electronic density of states (DOS) considering a denser 3 × 3 × 1 k-grid. A Gaussian smearing of 0.05 eV was adopted. To improve the description of the energy band gaps of the systems, often underestimated by the PBE functional, we considered the PBE+U method using a value of Ueff = U − J around 3 eV for cobalt d orbitals, a shift used previously for cobalt oxide compounds.59,60Conclusions
In summary, Co3O4 nanoparticles in various mass ratios were in situ synthesized on the surface of amorphous Co3(PO4)2 through facile hydrothermal synthesis. All decorated samples exhibited enhanced catalytic activity towards the oxygen evolution reaction. The highest catalytic activity and stability were achieved in the case of Co3(PO4)2@1%Co3O4, demonstrating a low overpotential of 313.01 mV to achieve 20 mA cm−2 and a small Tafel slope of 67.03 mV dec−1, indicating faster kinetics compared to Co3(PO4)2@2.5%Co3O4 and Co3(PO4)2@5%Co3O4. Electrochemical characterization revealed that the well-dispersed cobalt oxide nanoparticles induced an inductive effect capable of modifying the electronic state of cobalt on the catalyst surface. Additionally, Co3(PO4)2@1%Co3O4 exhibited good stability for at least 100 hours, highlighting its potential for industrial applications as a robust catalyst. First-principles calculations reveal stronger binding between Co3O4 NPs, highlighting their tendency to cluster rather than remain isolated on the surface. The extensive charge redistribution observed in NP–NP interactions further supports the conclusion that neighboring NPs exhibit stronger electronic hybridization, suggesting that electronic coupling is more pronounced in the Co3O4–Co3O4 system compared to Co3O4 adsorbed individually on the surface of Co3(PO4)2. This clustering negatively impacts the catalytic activity of Co3O4 nanoparticles on the surface of cobalt orthophosphate.Data availability
The data that support the findings of this study are available from the authors upon request.Conflicts of interest
There are no conflicts to declare.References
- Q. Hassan, A. Z. Sameen, H. M. Salman, M. Jaszczur and A. K. Al-Jiboory, J. Energy Storage, 2023, 72, 108404 CrossRef.
- T. T. Le, P. Sharma, B. J. Bora, V. D. Tran, T. H. Truong, H. C. Le and P. Q. P. Nguyen, Int. J. Hydrogen Energy, 2024, 54, 791–816 CrossRef CAS.
- F. Khalid, I. Dincer and M. A. Rosen, Int. J. Hydrogen Energy, 2016, 41, 7960–7967 CrossRef CAS.
- Z. Chen, S. Yun, L. Wu, J. Zhang, X. Shi, W. Wei, Y. Liu, R. Zheng, N. Han and B.-J. Ni, Nano-Micro Lett., 2023, 15, 4 CrossRef CAS.
- H. Kojima, K. Nagasawa, N. Todoroki, Y. Ito, T. Matsui and R. Nakajima, Int. J. Hydrogen Energy, 2023, 48, 4572–4593 CrossRef CAS.
- N. S. Hassan, A. A. Jalil, S. Rajendran, N. F. Khusnun, M. B. Bahari, A. Johari, M. J. Kamaruddin and M. Ismail, Int. J. Hydrogen Energy, 2024, 52, 420–441 CrossRef CAS.
- A. El Jaouhari, A. Slassi, B. Zhang, A. Pershin, W. Liu, D. Cornil, X. Liu and J. Zhu, J. Power Sources, 2021, 514, 230596 CrossRef CAS.
- A. El Jaouhari, A. Slassi, B. Zhang, W. Liu, D. Cornil, J. Zhu, X. Wu, D. Zhou and X. Liu, Mater. Today Chem., 2022, 23, 100706 CrossRef CAS.
- Y. Zhai, X. Ren, Y. Sun, D. Li, B. Wang and S. (Frank) Liu, Appl. Catal., B, 2023, 323, 122091 CrossRef CAS.
- L. Reith, J. N. Hausmann, S. Mebs, I. Mondal, H. Dau, M. Driess and P. W. Menezes, Adv. Energy Mater., 2023, 13(12), 2203886 CrossRef CAS.
- S. Huang, J. Lu, X. Wu, H. Zhu, X. Shen, S. Cui and X. Chen, Appl. Catal., A, 2023, 664, 119331 CrossRef CAS.
- W. Cao, R. Zhao, G. Liu, L. Wu and J. Li, Appl. Surf. Sci., 2023, 607, 154905 CrossRef CAS.
- Z. Xiao, Y.-C. Huang, C.-L. Dong, C. Xie, Z. Liu, S. Du, W. Chen, D. Yan, L. Tao, Z. Shu, G. Zhang, H. Duan, Y. Wang, Y. Zou, R. Chen and S. Wang, J. Am. Chem. Soc., 2020, 142, 12087–12095 CrossRef CAS PubMed.
- B.-J. Kim, E. Fabbri, D. F. Abbott, X. Cheng, A. H. Clark, M. Nachtegaal, M. Borlaf, I. E. Castelli, T. Graule and T. J. Schmidt, J. Am. Chem. Soc., 2019, 141, 5231–5240 CrossRef CAS PubMed.
- X. Chang, S. Li, L. Wang, L. Dai, Y. Wu, X. Wu, Y. Tian, S. Zhang and D. Li, Adv. Funct. Mater., 2024, 34(21), 2313974 CrossRef CAS.
- J. Zhang, J. Qian, J. Ran, P. Xi, L. Yang and D. Gao, ACS Catal., 2020, 10, 12376–12384 CrossRef CAS.
- M. S. Burke, M. G. Kast, L. Trotochaud, A. M. Smith and S. W. Boettcher, J. Am. Chem. Soc., 2015, 137, 3638–3648 CrossRef CAS.
- Y. Yang, W. Zhang, Y. Xiao, Z. Shi, X. Cao, Y. Tang and Q. Gao, Appl. Catal., B, 2019, 242, 132–139 CrossRef CAS.
- D.-H. Kim and Y.-K. Lee, Chem. Eng. J., 2024, 490, 151701 CrossRef CAS.
- S. Zuo, Y. Liao, C. Wang, A. B. Naden and J. T. S. Irvine, Small, 2023, 20(11), 2308867 CrossRef.
- X. Guo, H. Zhang, W. Xia, M. Ma, D. Cao and D. Cheng, Adv. Funct. Mater., 2024, 34(32), 2316539 CrossRef CAS.
- Q. Liu, X. Mu, F. Kang, S. Xie, C. Yan and Y. Tang, Small, 2024, 20(35), 2402726 CrossRef CAS PubMed.
- W. Han, Y. Qian, F. Zhang, Y. He, P. Li and X. Zhang, Chem. Eng. J., 2023, 473, 145353 CrossRef CAS.
- Y. Xing, J. Ku, W. Fu, L. Wang and H. Chen, Chem. Eng. J., 2020, 395, 125149 CrossRef CAS.
- H. Chen, S. Chen, Z. Zhang, L. Sheng, J. Zhao, W. Fu, S. Xi, R. Si, L. Wang, M. Fan and B. Yang, ACS Catal., 2022, 12, 13482–13491 CrossRef CAS.
- D. Gupta, A. Kafle and T. C. Nagaiah, Small, 2023, 19(24), 2208272 CrossRef CAS.
- W. Shi, C. Liu, M. Li, X. Lin, F. Guo and J. Shi, J. Hazard. Mater., 2020, 389, 121907 CrossRef CAS.
- H. Zeng, M. Oubla, X. Zhong, N. Alonso-Vante, F. Du, Y. Xie, Y. Huang and J. Ma, Appl. Catal., B, 2021, 281, 119535 CrossRef CAS.
- R. L. Frost, W. Martens, P. A. Williams and J. T. Kloprogge, Mineral. Mag., 2002, 66, 1063–1073 CrossRef CAS.
- J. Qi, Y. Lin, D. Chen, T. Zhou, W. Zhang and R. Cao, Angew. Chem., 2020, 132, 9002–9006 CrossRef.
- C. J. Eom and J. Suntivich, J. Phys. Chem. C, 2019, 123, 29284–29290 CrossRef CAS.
- P. Stoch, A. Stoch, M. Ciecinska, I. Krakowiak and M. Sitarz, J. Non-Cryst. Solids, 2016, 450, 48–60 CrossRef CAS.
- B. M. Al-Hasni, G. Mountjoy and E. Barney, J. Non-Cryst. Solids, 2013, 380, 141–152 CrossRef CAS.
- Y. Wang, X. Wei, X. Hu, W. Zhou and Y. Zhao, Catal. Lett., 2019, 149, 1026–1036 CrossRef CAS.
- M. S. Al-Sharif, P. Arunachalam, T. Abiti, M. S. Amer, M. Al-Shalwi and M. A. Ghanem, Arabian J. Chem., 2020, 13, 2873–2882 CrossRef CAS.
- R. Zhang, G. van Straaten, V. di Palma, G. Zafeiropoulos, M. C. M. van de Sanden, W. M. M. Kessels, M. N. Tsampas and M. Creatore, ACS Catal., 2021, 11, 2774–2785 CrossRef CAS PubMed.
- J. Chang, Y. Xiao, M. Xiao, J. Ge, C. Liu and W. Xing, ACS Catal., 2015, 5, 6874–6878 CrossRef CAS.
- N. L. W. Septiani, Y. V. Kaneti, K. B. Fathoni, K. Kani, A. E. Allah, B. Yuliarto, N. Nugraha, H. K. Dipojono, Z. A. Alothman, D. Golberg and Y. Yamauchi, Chem. Mater., 2020, 32(16), 7005–7018 CrossRef CAS.
- Q. Lai, V. Vediyappan, K.-F. Aguey-Zinsou and H. Matsumoto, Adv. Energy Sustainability Res., 2021, 2(11), 2100086 CrossRef CAS.
- D. González-Flores, I. Sánchez, I. Zaharieva, K. Klingan, J. Heidkamp, P. Chernev, P. W. Menezes, M. Driess, H. Dau and M. L. Montero, Angew. Chem., Int. Ed., 2015, 54, 2472–2476 CrossRef.
- D. A. Kuznetsov, B. Han, Y. Yu, R. R. Rao, J. Hwang, Y. Román-Leshkov and Y. Shao-Horn, Joule, 2018, 2, 225–244 CrossRef CAS.
- L. J. Enman, M. S. Burke, A. S. Batchellor and S. W. Boettcher, ACS Catal., 2016, 6, 2416–2423 CrossRef CAS.
- J. Bencaid, A. El Jaouhari, A. Belhboub, M. Matrouf, I. Cheras, M. Adnane, A. Ghanam, B. Manoun and F. Ghamouss, ACS Appl. Energy Mater., 2025, 8(6), 3676–3687 CrossRef CAS.
- X. Zhang, A. El Jaouhari, C. Li, M. Adnane, W. Liu, A. Mellalou, F. Ghamouss and Y. Lin, Electrocatalysis, 2024, 15, 344–352 CrossRef CAS.
- L. J. Enman, M. S. Burke, A. S. Batchellor and S. W. Boettcher, ACS Catal., 2016, 6, 2416–2423 CrossRef CAS.
- D. A. Kuznetsov, B. Han, Y. Yu, R. R. Rao, J. Hwang, Y. Román-Leshkov and Y. Shao-Horn, Joule, 2018, 2, 225–244 CrossRef CAS.
- Z. Zhang, C. Jia, P. Ma, C. Feng, J. Yang, J. Huang, J. Zheng, M. Zuo, M. Liu, S. Zhou and J. Zeng, Nat. Commun., 2024, 15, 1767 CrossRef CAS.
- Y. Dou, C.-T. He, L. Zhang, M. Al-Mamun, H. Guo, W. Zhang, Q. Xia, J. Xu, L. Jiang, Y. Wang, P. Liu, X.-M. Chen, H. Yin and H. Zhao, Cell Rep. Phys. Sci., 2020, 1, 100077 CrossRef.
- Y. Xing, J. Ku, W. Fu, L. Wang and H. Chen, Chem. Eng. J., 2020, 395, 125149 CrossRef CAS.
- M. Gao, Y. Xu, J. Jiang, Y. Zheng and S. Yu, J. Am. Chem. Soc., 2012, 134, 2930–2933 CrossRef CAS.
- B. S. Yeo and A. T. Bell, J. Am. Chem. Soc., 2011, 133, 5587–5593 CrossRef CAS.
- S. Klaus, Y. Cai, M. W. Louie, L. Trotochaud and A. T. Bell, J. Phys. Chem. C, 2015, 119, 7243–7254 CrossRef CAS.
- H. Chen, S. Chen, Z. Zhang, L. Sheng, J. Zhao, W. Fu, S. Xi, R. Si, L. Wang, M. Fan and B. Yang, ACS Catal., 2022, 12, 13482–13491 CrossRef CAS.
- J. P. Perdew, K. Burke and Y. Wang, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 16533–16539 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B, 1976, 13, 5188–5192 CrossRef.
- H. Keil, M. Hellström, C. Stückl, R. Herbst-Irmer, J. Behler and D. Stalke, Chem. – Eur. J., 2019, 25, 15786–15794 CrossRef CAS PubMed.
- W. Hu, X.-M. Cao and P. Hu, J. Phys. Chem. C, 2018, 122, 19593–19602 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ma00276a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |