
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Novel biobased, flexible blocky copolyesters based on poly(lactic acid) and poly(ethylene azelate)†
Rafail O.
Ioannidis
,
Zoe
Terzopoulou
 *,
Alexandra
Zamboulis
,
Nikolaos D.
Bikiaris
,
Michiel Jan
Noordam
and
Nikolaos
Nikolaidis
*
*,
Alexandra
Zamboulis
,
Nikolaos D.
Bikiaris
,
Michiel Jan
Noordam
and
Nikolaos
Nikolaidis
*
Laboratory of Polymer and Colors, Chemistry and Technology, Aristotle University of Thessaloniki, GR-541 24, Thessaloniki, Greece. E-mail: terzoe@gmail.com; nfnikola@chem.auth.gr
First published on 14th April 2025
Abstract
The synthesis and characterization of a series of novel, high molecular weight poly(lactic acid)-b-poly(ethylene azelate) (PLA-b-PEAz) blocky copolyesters are reported for the first time. The copolyesters were synthesized by the ring-opening polymerization (ROP) of L-lactide, using oligo(ethylene azelate) as a macroinitiator. Four different comonomer mass ratios were used in the feed, namely 97.5-2.5, 95-5, 90-10, and 80-20, the minor comonomer being PEAz. Gel permeation chromatography (GPC) and intrinsic viscosity measurements [η] confirmed the high number average molecular weight  of the materials, ranging from 10 to 80 kg mol−1, while the chemical structure was studied via nuclear magnetic resonance (NMR) and Fourier transform infrared spectroscopy (FTIR). NMR analysis indicated the formation of block copolymers; however, confirming the presence of triblock structures proved challenging. Therefore, a system consisting of PLA-b-PEAz block copolyesters along with PLA segments was proposed and described as blocky copolyesters. According to differential scanning calorimetry (DSC), the melting temperatures of the copolymers exhibited only slight shifts toward lower values, whereas the glass transition and cold crystallization temperatures decreased significantly, indicating enhanced flexibility. Furthermore, isothermal crystallization experiments from the melt suggested that the crystallization ability of the PLA-based copolyesters was improved compared to PLA. The thermal stability of most copolyesters was enhanced. The mechanical performance was assessed via tensile and flexural measurements, revealing high elongation and Young's modulus values, indicating tough and strong materials. Moreover, during the three-point bending tests, none of the copolyesters fractured.
of the materials, ranging from 10 to 80 kg mol−1, while the chemical structure was studied via nuclear magnetic resonance (NMR) and Fourier transform infrared spectroscopy (FTIR). NMR analysis indicated the formation of block copolymers; however, confirming the presence of triblock structures proved challenging. Therefore, a system consisting of PLA-b-PEAz block copolyesters along with PLA segments was proposed and described as blocky copolyesters. According to differential scanning calorimetry (DSC), the melting temperatures of the copolymers exhibited only slight shifts toward lower values, whereas the glass transition and cold crystallization temperatures decreased significantly, indicating enhanced flexibility. Furthermore, isothermal crystallization experiments from the melt suggested that the crystallization ability of the PLA-based copolyesters was improved compared to PLA. The thermal stability of most copolyesters was enhanced. The mechanical performance was assessed via tensile and flexural measurements, revealing high elongation and Young's modulus values, indicating tough and strong materials. Moreover, during the three-point bending tests, none of the copolyesters fractured.
1. Introduction
Due to the rapid production of fossil-based polymers and their extensive accumulation in the environment, new legislations have been implemented regarding their safe-by-design manufacture by the European Union.1 In recent years, the annual growth of plastic production and its contribution to greenhouse gas emissions has exceeded 400 million tons and 1.7 billion tons. Given the depletion of fossil fuels and their contribution to climate change, the development of polymers from renewable resources is crucial as a sustainable alternative to replace fossil-based counterparts.2 These polymers must ensure top quality and performance for each application while being environmentally friendly and posing no threat to all living organisms at the same time.3,4The field of renewable resource-based polymers has advanced rapidly in recent decades, with researchers exploring a wide range of alternative materials to replace fossil-based counterparts, e.g. epoxy resins, polymers with long-chain aliphatic chains or with rigid structures, such as bio-poly(ethylene terephthalate) (bio-PET), bio-polyethylene (bio-PE), poly(2,5-furan dicarboxylate) (PEF), and poly(lactic acid) (PLA). These polymers can be obtained, for example, via the valorization of biomass such as sweet corn, sugar cane, or agricultural waste.5–7 PLA is such a biobased polyester (i.e.L-lactide is primarily derived from corn) and can be used in a plethora of industry sectors as an alternative to PET and poly(propylene terephthalate) (PPT) for the production of plastic and disposable bags, fibers, and textiles.8 It is important to note that the non-renewable energy and greenhouse gas (GHG) emissions produced during the manufacturing of PLA are significantly lower compared to those of PET.9,10 In this light, plant-based resources can be utilized for producing PLA or PEF to substitute PET in the engineering sectors.11–13
High molecular weight (MW) PLA samples can be synthesized by implementing one of the most efficient methods, the ring-opening polymerization (ROP) of L-lactide. All of PLA's properties, including mechanical and thermal, are affected by its MW.14,15 Consequently, increasing the MW of PLA leads to higher thermal transitions, such as a higher melting temperature of approximately 170–180 °C. In addition, additives can further enhance the thermal and crystallization properties of PLA.16,17 Unsurprisingly, PLA has made a significant impact due to its versatility, finding applications in diverse fields such as packaging, screen-printed electronics, pharmaceutical drug delivery, agriculture, and construction.18 End-of-life management is critical for material development (e.g. polyesters), and given PLA's low degradation rate, new recycling methods must be developed. In this context, Siddiqui et al.19 investigated the impact of alkaline hydrolysis assisted by microwave irradiation for PLA recycling and reported a 100% depolymerization of PLA under relatively mild conditions.
Properties such as brittleness, crystallization rate, low biodegradability, and barrier properties of PLA must be improved to expand its range of applications.20,21 Synthesizing copolyesters based on PLA is a facile method to improve the properties of PLA by combining desirable features of two or three comonomers. For instance, PLA can be a strong material as it exhibits high Young's modulus values, and adding a long aliphatic polyester can improve its elongation, resulting in a strong and tough substrate.14,22 The presence of a second comonomer in the macromolecular chains of the predominant polymer can enhance the crystallization rate and mechanical performance of the copolymers. For instance, in PLA-b-poly(ethylene glycol) (PEG) block copolymers, PEG as the second block increased the crystallization rate of PLA under melt isothermal conditions and significantly enhanced its elongation.23,24 Designing copolyesters with tunable properties can be challenging, as achieving high MW—which are critical for optimal material performance—remains difficult (e.g., over 50 kg mol−1). Thus, obtaining high MW copolyesters is still a task at hand and one of the most crucial factors that must be considered for extrusion purposes.25
PEAz is a biobased and biodegradable,26,27 long-chain aliphatic polyester derived from azelaic acid (AzA) and ethylene glycol (EG) that can be used to tune the properties of PLA through copolymerization. Valorization of biomass can lead to the synthesis of both AzA and EG, and the most common starting materials for their production are oleic acid and cellulose, respectively.28 The mechanical performance of PLA, especially its elongation, can be improved by the addition of long aliphatic building blocks.29 Papageorgiou et al.27,30 studied the crystallization behavior of PEAz and found a high crystallization rate, which means that it can further improve the molecular mobility of the macromolecular chains of PLA and, therefore, its mechanical performance.31 This study aims to provide valuable insights into the synthesis of novel biobased and potentially biodegradable PLA-b-PEAz copolyesters with high MW, intended for a broad range of applications including food packaging and printed electronics (PE).22,32,33 To address PLA's inherent brittleness, flexible PLA-based copolyesters incorporating PEAz were designed to enhance its mechanical performance by increasing elongation to values exceeding 50%, approaching 100%, while simultaneously achieving high Young’s modulus values above 1.5 GPa. To be suitable for engineering applications, their excellent mechanical performance must be complemented by high melting temperatures.8
The goal of this study is to synthesize novel biobased flexible copolyesters with high MW, suitable for engineering applications. Thus, five different flexible poly(lactic acid)-b-poly(ethylene azelate) (PLA-b-PEAz) blocky copolyesters were synthesized via the ring-opening polymerization (ROP) of L-lactide. The homopolyesters PLA and PEAz were synthesized through ROP of L-lactide, and a two-stage melt polycondensation reaction, respectively. Gel permeation chromatography (GPC) and intrinsic viscosity [η] measurements revealed the high MW of the materials. Nuclear magnetic resonance (NMR) and Fourier transform infrared spectroscopy (FTIR) were used to study the chemical structure of the materials. In the case of PLA-b-PEAz copolyesters, blocky structures were proposed, that include both block copolymer and PLA homopolymer chains. The thermal and crystallization behavior was further studied via differential scanning calorimetry (DSC) and X-ray diffraction (XRD), where all materials exhibited high melting temperatures. Isothermal melt crystallization experiments were performed using DSC and polarized light microscopy (PLM) to investigate the crystallization kinetics of the copolymers. Almost all copolymers exhibited enhanced thermal stability compared to PLA, which was examined via thermogravimetric analysis (TGA). The presence of the second comonomer significantly improved the mechanical performance of PLA. As a function of the PEAz content, all the synthesized materials outperformed PLA, with each copolyester showing distinct differences in their mechanical response. Finally, the off-white color of the materials was confirmed by color measurements.
2. Experimental section
2.1. Materials
Azelaic acid (AzA) (purity >99.0%) was purchased from Fluka (Steinheim, Germany), 1,2-ethanediol (anhydrous, >99.8%), 1-dodecanol and Tin(ii) 2-ethylhexanoate (Sn(Oct)2) was supplied from Aldrich Co., (London, UK). Titanium butoxide (Ti(OBu)4, purity: >97.0%) was purchased from Sigma Aldrich Chemical Co (Steinheim, Germany), and L-lactide (LA) (99.9%) was purchased by PURAC Biochem BV (Gorinchem, The Netherlands) under the brand name PURASORB®L. All other materials and solvents used were of analytical grade.2.2. Synthesis of aliphatic (co)-polyesters
 | ||
| Fig. 1 Synthesis of (a) PEAz, (b) PLA, and (c) PLA-b-PEAz blocky copolyesters. | ||
Poly(lactic acid) (PLA) was synthesized via the ring opening polymerization (ROP) of L-lactide (Fig. 1b), where 400 ppm of Sn(Oct)2 and 1-dodecanol (dissolved in acetone), with a molar ration of L-lactide to 1-dodecanol 1.08 × 103, were added in a round-bottom flask, at 160 °C for 2 h (350 rpm), under nitrogen flow (N2). The unreacted monomer was removed from the flask by applying slowly high vacuum (5.0 Pa) distillation for 15 min at 180 °C (400 rpm). The polymerization reaction was finished by rapidly cooling the flask at room temperature.
2.3. Polymer characterization
 | (1) |
The semicrystalline polyester samples were heated at 20 °C min−1 up to a temperature T = Tm + 40 °C to evaluate their melting temperature (Tm). Amorphous materials were obtained by heating the samples to 40 °C above the melting temperature and holding it there for 3 minutes to erase their thermal history, followed by a cooling step in the DSC at the highest rate possible.31 Thus, the glass transition temperature (Tg), cold (Tcc) and Tm were measured for the quenched samples, during the heating step at 20 °C min−1. Finally, the materials were cooled from the melt (Tm + 40 °C) at 10 °C min−1 to a temperature below Tg (Tg – 40 °C) for the determination of the crystallization temperature (Tc) and the crystallization enthalpy (ΔHc). The relative degree of crystallinity (Xc) was calculated with equation 2:25
 | (2) |
During isothermal melt crystallization, the samples were heated at 40 °C above their Tm and held there for 3 minutes to erase any thermal history, then a cooling step in the DSC at the highest rate possible was performed to the selected crystallization temperature. The final step was a subsequent heating at 40 °C above their Tm.
3-Point bending tests were performed using a Shimadzu EZ Flexural Tester Model EZ-LX, with a 2 kN load cell, according to ASTM D790-17. Compression molded samples were prepared using a thermopress with appropriate dimensions; 12.7 mm wide and <1.6 mm thickness. The samples were tested flatwise on the support span, resulting in a support span-to-depth ratio of 16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (tolerance ±1). For every sample, at least five measurements were made, and the mean values were calculated by averaging the data of flexural’ s modulus and flexural strength. One-way ANOVA was used to determine the statistical significance.
:1 (tolerance ±1). For every sample, at least five measurements were made, and the mean values were calculated by averaging the data of flexural’ s modulus and flexural strength. One-way ANOVA was used to determine the statistical significance.
3. Results and discussion
3.1. Synthesis and structure of PLA-b-PEAz copolyesters
GPC (Fig. S1, ESI†) and intrinsic viscosity [η] measurements were conducted, investigating the and dispersity Đ, of the homo- and copolyesters (Table 1). The addition of the second comonomer resulted in the reduction of the
and dispersity Đ, of the homo- and copolyesters (Table 1). The addition of the second comonomer resulted in the reduction of the  of the copolyesters. Higher azelate content (macroinitiator) resulted in an increase of the hydroxyl end-groups and, thus, a larger number of initiation sites. So, by maintaining the same quantity of catalyst, the molar ratio of lactide to catalyst was constant, while the catalyst's active sites remained constant, resulting in a decrease of the
of the copolyesters. Higher azelate content (macroinitiator) resulted in an increase of the hydroxyl end-groups and, thus, a larger number of initiation sites. So, by maintaining the same quantity of catalyst, the molar ratio of lactide to catalyst was constant, while the catalyst's active sites remained constant, resulting in a decrease of the  .25
.25
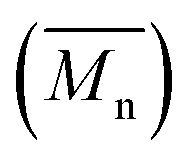 and Đ (GPC), and intrinsic viscosity [η] of PLA, PEAz, and their copolyesters
and Đ (GPC), and intrinsic viscosity [η] of PLA, PEAz, and their copolyesters
| Materials |
|
Đ | [η] (dL g−1) |
|---|---|---|---|
 : number average molecular weight, Đ: dispersity. : number average molecular weight, Đ: dispersity. |
|||
| PLA | 78100 |
3.14 | 1.89 |
| PLA-b-PEAz_97.5-2.5 | 76100 |
2.87 | 1.85 |
| PLA-b-PEAz_95-5 | 68800 |
2.98 | 1.76 |
| PLA-b-PEAz_90-10 | 27500 |
2.80 | 1.24 |
| PLA-b-PEAz_80-20 | 11600 |
3.64 | 0.82 |
| PEAz | 4900 | 4.11 | 0.42 |

Fig. 2a shows the ATR-FTIR spectra of the homopolyesters PLA, PEAz, and the copolyesters. The main characteristic peaks of PLA can be seen at 2997–2923 cm−1 related to C–H stretching, at 1746 cm−1 to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching, at 1453 cm−1 to –CH3 asymmetric bending, and at 1181 cm−1 to the stretching of the –C–O–C– groups. For all copolyesters, the main peaks that appeared in the ATR-FTIR spectra were related to PLA because of the low PEAz content, but some peaks could be detected due to the presence of azelate segments. One of the most essential peaks was in the carbonyl's region. Fig. 2b shows the zoomed area of that region. A peak shoulder appeared only for copolyesters with 10% and 20% of PEAz, thus confirming the presence of the azelate segments. A weak peak at 1269 cm−1 can be observed corresponding to the C–O/C–C stretch and it was also related to the azelate segments.
O stretching, at 1453 cm−1 to –CH3 asymmetric bending, and at 1181 cm−1 to the stretching of the –C–O–C– groups. For all copolyesters, the main peaks that appeared in the ATR-FTIR spectra were related to PLA because of the low PEAz content, but some peaks could be detected due to the presence of azelate segments. One of the most essential peaks was in the carbonyl's region. Fig. 2b shows the zoomed area of that region. A peak shoulder appeared only for copolyesters with 10% and 20% of PEAz, thus confirming the presence of the azelate segments. A weak peak at 1269 cm−1 can be observed corresponding to the C–O/C–C stretch and it was also related to the azelate segments.
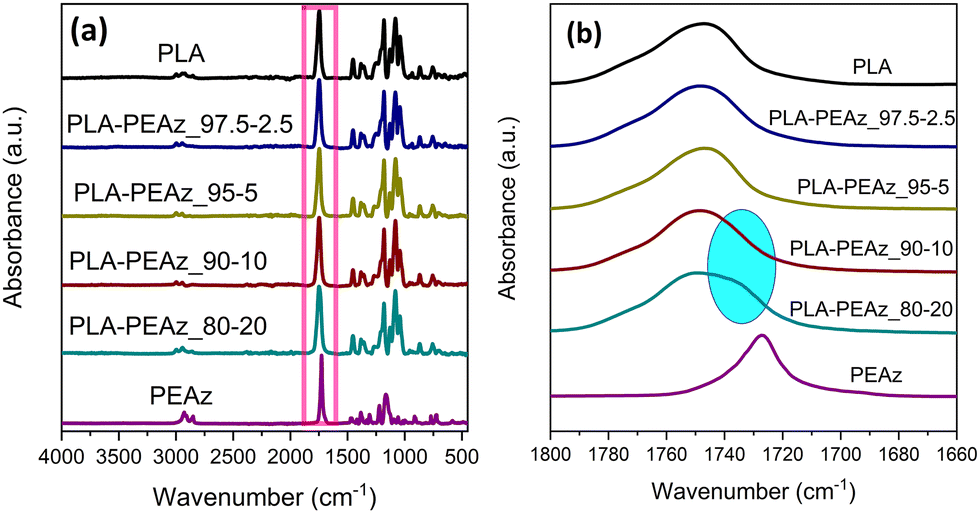 | ||
| Fig. 2 ATR-FTIR spectra of (a) PLA, PEAz, and their copolymers, and (b) zoom at carbonyl's region. | ||
Fig. 3 displays the 1H NMR and 13C NMR spectra of the synthesized polyesters. In the 1H spectra, the appearance of resonance signals attributed to the –OCH– (B) and –OCH2– (1) groups of PLA and PEAz, at 5.16 and 4.26 ppm confirm the successful polymerization. The signal at 2.32 ppm is attributed to the C(O)CH2– (3) methylene group, the signal at 1.58 ppm to the methyl group (C), and the ones at 1.61 and 1.32 ppm to the –CH2– groups (5,6). Traces of residual unreacted lactide (around 5 ± 1%) can be observed for all materials (signal at 5.04 ppm) except for the PLA-b-PEAz 80-20 copolyester, where no traces of lactide were observed. The strong signals of the CO carbon (A & 2), corresponding to the esters of PLA and PEAz segments, are observed at 169.5 and 173.4 ppm in the 13C spectra. Furthermore, typical resonance signals of PLA and PEAz units were observed: 68.9 ppm (–OCH– B), 62.8 ppm (–OCH2– 1), 33.9 ppm (–C(O)CH2– 3), 28.8 & 24.7 (–CH2– 4,5,6) and 16.6 ppm (–CH3 C).
 | ||
| Fig. 3 (a) 1H NMR, (b) 13C NMR spectra of the synthesized homopolyesters and copolyesters, and (c) the corresponding signal assignments. | ||
End-chain –CHOH groups are observed for the PLA segments at 4.4 ppm, while the corresponding –CH2OH groups are not observed for PEAz segments (expected around 3.5–3.8 ppm).34 This observation is strong evidence of the successful copolymerization and suggests the formation of blocky copolymers PLA-PEAz-PLA, confirming that PEAz, via the end-chain OH groups, acted as a macroinitiator for the successful formation of copolyesters. DSC measurements (Fig. 4) can also support the successful synthesis of block copolymers; however, it was difficult to prove the formation of block copolymers via NMR measurements due to the presence of the aliphatic parts of PLA and PEAz. Thus, PLA-b-PEAz block copolyesters in the presence of PLA homopolymer moieties were suggested and referred to as blocky copolyesters.35,36 Due to the high  of the copolymers and the relative structural similarity of PLA and PEAz (both are aliphatic), it is not possible to detect the units between PLA and PEAz segments to further calculate the micro-structure of the copolymers. Nevertheless, there is strong indirect evidence, vide infra, of the successful copolymerization. The composition of the copolymers is very close to the feed ratio (Table S1, ESI†). The lower PLA content can be attributed to the unreacted lactide and some loss due to sublimation.
of the copolymers and the relative structural similarity of PLA and PEAz (both are aliphatic), it is not possible to detect the units between PLA and PEAz segments to further calculate the micro-structure of the copolymers. Nevertheless, there is strong indirect evidence, vide infra, of the successful copolymerization. The composition of the copolymers is very close to the feed ratio (Table S1, ESI†). The lower PLA content can be attributed to the unreacted lactide and some loss due to sublimation.
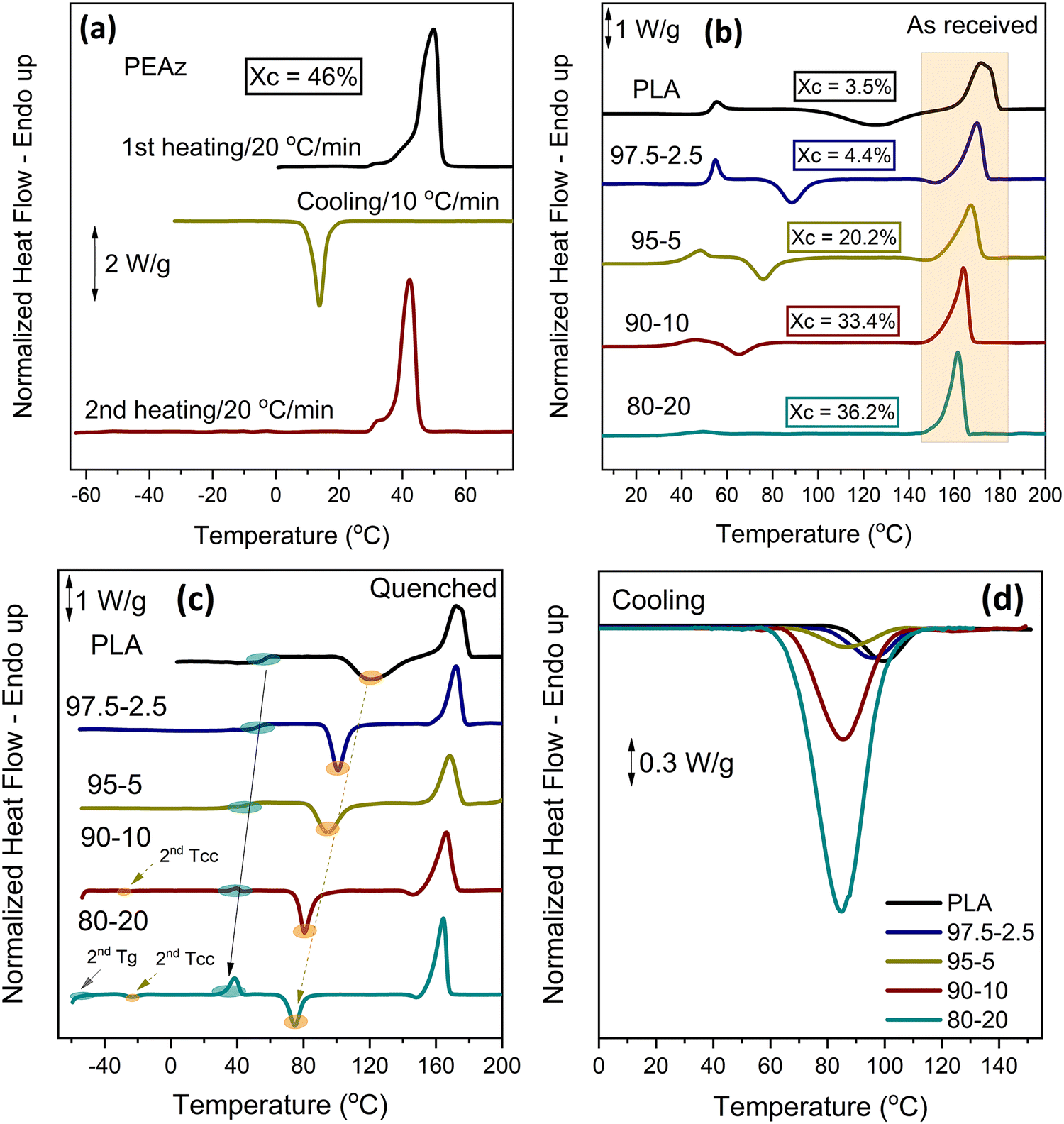 | ||
| Fig. 4 (a) DSC traces of PEAz, and (b) PLA and PLA-b-PEAz copolyesters during heating at 20 °C min−1 of as-received compression molded samples, (c) during heating at 20 °C min−1 of quenched samples, and (d), during cooling at 10 °C min−1. | ||
3.2. Thermal properties and crystalline behavior
Fig. 4 shows the DSC scans of compression molded samples of neat PLA, PEAz, and PLA-b-PEAz copolyesters with heating rates of 20 °C min−1 for the first and the second step (after quenching) and cooling rates of 10 °C min−1 during cooling from the melt.Fig. 4a shows that PEAz was a rapidly crystallizable polymer that crystallized during quenching. On the other hand, the crystallization of PLA from the melt and the glassy state was limited. In the case of the copolyesters, the first DSC scan (Fig. 4b) revealed that the relative degree of crystallinity (Xc) increased systematically with the presence of the long aliphatic segments of PEAz. The melting temperature (Tm) reduction of the copolyesters was minimal due to the formation of stable crystals. As validated by XRD (Fig. 7), the PLA crystal phase dominated. Tg and Tcc (Fig. 4c) were strongly affected by the second long aliphatic comonomer and shifted toward lower temperatures, making the copolyesters flexible materials. In addition, all materials exhibited limited crystallization ability during cooling from the melt (Fig. 4d) with low ΔHc values (Table 2). The copolyester with 10 and 20 wt% of PEAz segments revealed additional thermal transitions. Overall, these results could suggest a partial phase separation, as it will be further discussed.31,37
| Sample | As received | Quenched | Cooling | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| T m (°C) | ΔHm − ΔHcc (J g−1) | X c (%) | T g (°C) | T cc (°C) | ΔHcc (J g−1) | ΔHm (J g−1) | T m (°C) | T c (°C) | ΔHc (J g−1) | |
| PLA | 171.8 | 3.2 | 3.5 | 56.2 | 120.3 | 39.8 | 40.3 | 173.3 | 101.0 | 3.0 |
| 97.5-2.5 | 170.1 | 4.1 | 4.4 | 52.7 | 101.0 | 32.0 | 33.1 | 172.1 | 95.6 | 1.3 |
| 95-5 | 167.3 | 18.8 | 20.2 | 47.0 | 94.6 | 29.4 | 31.6 | 168.3 | 86.4 | 1.2 |
| 90-10 | 163.8 | 31.1 | 33.4 | 36.8 | 80.7/−24.7 | 25.8/0.4 | 37.7 | 166.4 | 85.5/−17.9 | 13.2/0.22 |
| 80-20 | 161.3 | 33.6 | 36.2 | 32.3/−52.6 | 72.9/−23.8 | 19.4/1.63 | 38.8 | 164.4 | 84.7/−15.0 | 25.3/0.37 |
| PEAz | 49.8 | 73.6 | 46 | −53.2 | — | — | 70.5 | 42.3 | 13.7 | 59.6 |
Applying a heating rate of 5 °C min−1 to the quenched samples (Fig. 5), it was possible to observe two distinct Tg for the copolyesters with the highest PEAz content (10 and 20 wt%), once again. Partial phase separation probably occurred as the azelate blocky units trapped within the macromolecular chains of the PLA block segments and acted as plasticizers. Also, a weak Tm corresponding to PEAz moieties was revealed only for the copolyester PLA-b-PEAz_80-20. In this case, during heating at slow rates, crystals with enhanced thermodynamic stability were formed. Subsequently, the melting temperatures of the materials shifted slightly towards higher temperatures.
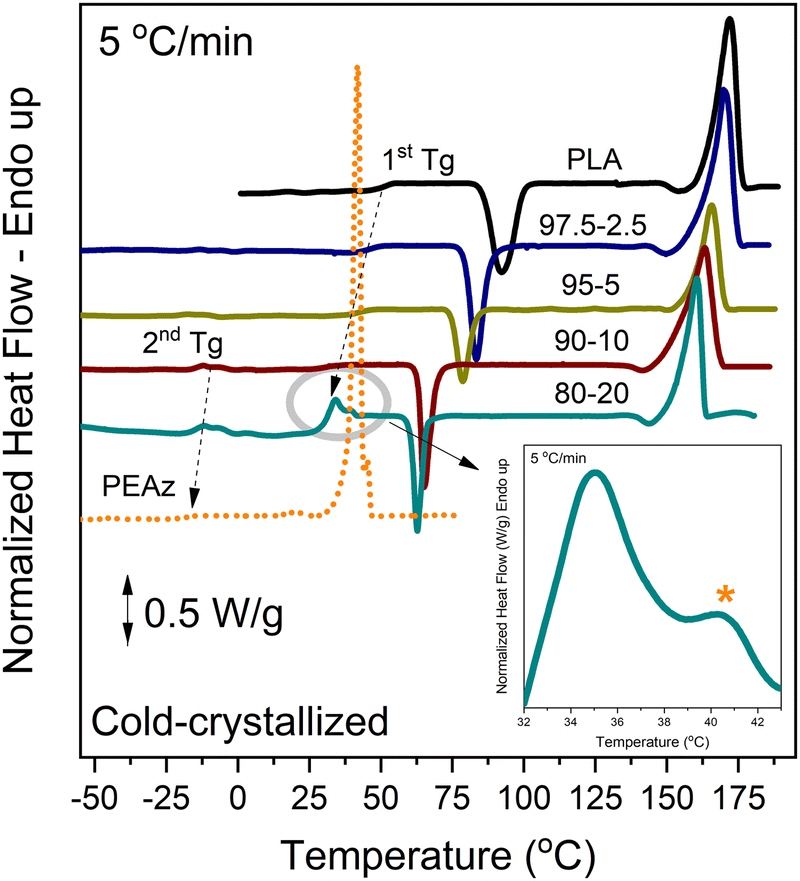 | ||
| Fig. 5 DSC traces during heating of quenched samples at 5 °C min−1. | ||
In order to increase the degree of crystallinity of the PLA-based copolyesters, annealing experiments were performed. After annealing the compression molded samples of PLA and copolyesters, sharp endotherm peaks appeared in the DSC scans of all samples with high Xc (%) values, which were calculated using eqn (1). In this way, for all materials, the melting temperatures were enhanced towards higher temperatures than those of the as-received samples, making them once again suitable substrates for engineering-like applications (Fig. 6).
 | ||
| Fig. 6 DSC traces of the materials after annealing at Tcc (onset) – 10 °C for 1 h for the compression molded samples. The DSC traces were recorded during the heating rate of 20 °C min−1. | ||
Fig. 7a shows the XRD patterns of the compression molded samples of the synthesized homopolyesters and copolyesters. Only PEAz exhibited strong diffraction peaks, whereas for the PLA and PLA-based copolyesters, the intensity of their peaks was relatively small because of their high MW, PLA's short aliphatic segment, and the presence of a side methyl group.38 The copolyester with 20% azelate content exhibited stronger diffraction peaks than the other copolymers. PLA exhibited two main diffraction peaks, at 15.6 and 17.0°, corresponding to the crystal planes (010) and (200/110), respectively (Fig. 7a). After annealing (Fig. 7b), only a small fraction of the PEAz segments could crystallize within the main crystal lattice of the copolyesters. As a matter of fact, only the main diffraction peak of the PEAz at 22.0° emerged (Fig. 7b). This indicated that the long aliphatic blocky segments of PEAz were excluded to the amorphous regions. Meaning that the long blocky aliphatic parts did not act as defects within the predominant crystal of PLA, thus the Tm of the copolyesters remained almost unchanged (Fig. 4a). This is in agreement with previous studies in the literature.25,31,37,39 In all cases, the intensity of the diffraction peaks was increased.
 | ||
| Fig. 7 XRD patterns of PLA, PEAz and their copolymers (a) using as-received compression molded samples and (b) after annealing of the compression molded samples at Tcc (onset) – 10 °C for 1 h. | ||
3.3. Preliminary study of isothermal melt crystallization via DSC and PLM
Polymer crystallization constitutes a major and important field among the variety of characterization techniques that can be implemented for the investigation of the relationship between the structure and end-of-life properties. Furthermore, the in-depth study of isothermal crystallization kinetics of copolymers could lead to important outcomes regarding the comonomer composition independence to the overall performance of the material.40–42Isothermal melt crystallization kinetics43 of PLA, PEAz, and PLA-b-PEAz copolyesters were examined through DSC at various crystallization temperatures. Crystallization exothermic peaks are presented in Fig. S2 (ESI†). In every case, at small supercoolings (ΔT = Tm − Tc), exothermic peaks became broader because at temperatures close to Tm, crystallization is hindered, but crystals with enhanced thermodynamic stability were formed. The relative degree of crystallinity (X(t)) was calculated based on the equation below to estimate the crystallization rate of the materials. X(t) as a function of crystallization time at different temperatures was obtained (Fig. S3, ESI†) based on the fact the evolution of crystallinity was linearly proportional to the evolution of heat which was released during the crystallization.
 | (3) |
 | ||
| Fig. 8 Inverse τ1/2 as a function of supercooling during isothermal melt crystallization of PLA and PLA-b-PEAz copolyesters at various temperatures. | ||
During isothermal experiments from the melt, PLA exhibited the well-known unusual behavior with a maximum 1/τ1/2 value at 100 °C.16 The presence of the second blocky azelate segment influenced the crystallization behavior of the copolyesters, and more specifically, the copolyesters exhibited higher crystallization rates than PLA at large supercoolings. This happened because, close to the melting temperature, the presence of crystals that can induce crystallization was limited. Also, recrystallization and melting could be two competitive phenomena that might suppress the isothermal melt crystallization process at temperatures close to Tm in the case of the PLA-based copolyesters. Thus, at low supercoolings the crystallization ability of the copolyesters was hindered. The materials exhibited high 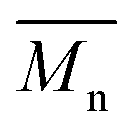 , comparable to PLA, but by adding 10 and 20 wt% PEAz segments resulted in a rapid decrease of the
, comparable to PLA, but by adding 10 and 20 wt% PEAz segments resulted in a rapid decrease of the  . Thus, only the materials with high
. Thus, only the materials with high  were taken into consideration for the comparison of the crystallization rates (Fig. 8) since
were taken into consideration for the comparison of the crystallization rates (Fig. 8) since  strongly affects crystallization kinetics. The PEAz blocky segments enhanced the molecular mobility of the copolymers chains, based on the reduction of the Tcs and Tgs that was observed after quenching for all samples, resulting in higher crystallization rates for the isothermal melt crystallization experiments at large supercoolings. Klonos et al. studied in detail the molecular dynamics of aliphatic PLA-based copolyesters via broadband dielectric spectroscopy (BDS), supporting the improvement of the molecular mobility of PLA-copolyesters based on poly(propylene adipate) (PPAd) and poly(butylene succinate) (PBS).31,37,44
strongly affects crystallization kinetics. The PEAz blocky segments enhanced the molecular mobility of the copolymers chains, based on the reduction of the Tcs and Tgs that was observed after quenching for all samples, resulting in higher crystallization rates for the isothermal melt crystallization experiments at large supercoolings. Klonos et al. studied in detail the molecular dynamics of aliphatic PLA-based copolyesters via broadband dielectric spectroscopy (BDS), supporting the improvement of the molecular mobility of PLA-copolyesters based on poly(propylene adipate) (PPAd) and poly(butylene succinate) (PBS).31,37,44
The melting behavior of the homopolyesters and PLA-b-PEAz copolyesters was examined after isothermal melt crystallization through DSC. All materials exhibited multiple melting behavior, as observed in the DSC scans presented in Fig. 9. The copolymers revealed three endothermic peaks during heating at 20 °C min−1, after the isothermal crystallization from the melt, and especially in the case of the copolyester with 2.5% of PEAz, distinct melting peaks emerged, making the impact of the second comonomer on the main crystal lattice noticeable. It is important to note that the crystallization temperature plays a crucial role in the melting behavior of the materials and that at different supercoolings the melting peaks can correspond to crystals with different thermal stability.
 | ||
| Fig. 9 Subsequent heating at 20 °C min−1 after isothermal melt crystallization at the corresponding temperatures of (a) PLA, PLA-b-PEAz copolyesters, (b), 97.5-2.5, (c) 95-5, (d) 90-10, (e) 80-20, and (f) PEAz. | ||
The melting behavior PLA has been thoroughly studied,45,46 and our results are in agreement with the detailed reports of Yasuniwa et al. on the melting behavior of PLA.47 More specifically, PLA (Fig. 9) exhibited three melting peaks at high supercoolings, and a weak exothermic re-crystallization peak, which appeared after the first melting peak (I). Peak I was associated with the melting of defective crystals, and re-crystallization was observed due to the re-organization of the defective crystal lattice. Thus, the third (III) peak appeared because of the melting of recrystallized crystals, while the second peak (II) resulted from the melting of the primary crystals. Peaks I and II co-existed at Tcs (crystallization temperatures) over 115 °C. On the other hand, at small supercoolings (close to melting temperature), crystals with higher thermodynamic stability were formed, increasing the Tm. At 125 °C, all peaks were merged to one primary melting peak, and at higher crystallization temperatures, an additional peak appeared before the main melting peak.
PLA-b-PEAz copolyesters and PEAz exhibited a similar thermal behavior after isothermal crystallization from the melt (Fig. 9), with the presence of three main melting peaks, except for the copolyester PLA-b-PEAz 97.5-2.5, which revealed an additional one. In the case of the copolyester with 2.5% PEAz, the weak endothermic first peak (I), which developed after the crystallization temperature, was associated with the melting of secondary crystals. The impact of the second comonomer was observed in the multiple thermal behavior, forming a wide endothermic peak with three different distinct melting peaks, implicating the development of crystals with different thermodynamic stability at high supercoolings. Compared with PLA, at low crystallization temperatures (high ΔT), no exothermic re-crystallization shoulder was observed in any part of the heating scan, indicating that either the azelate segments helped the formation of more stable crystals during the crystallization experiments from the melt or the melting area dominated over re-crystallization. Further studies will be implemented with modulated temperature DSC (MDSC) to investigate the mechanism of the multiple melting behavior of the PLA-b-PEAz copolyesters.48
Fig. 10 shows the morphology of the spherulites that developed under isothermal conditions from the melt, as observed via polarized light microscopy (PLM). The pictures were taken at two different time intervals, the first at the early stage of the spherulite development and the other after the complete crystallization of the samples, at 115 °C. For PLA and its copolyesters, large and well-defined spherulites were formed in all compositions, including the Maltese cross morphology in their structure. This may indicate less nucleation and thus, the reduction of the crystal density of the copolyesters.31 PEAz exhibited high nucleation density at 30 °C, forming spherulites with a very small radius. Given the development of large size spherulites of the copolyesters, an in-depth study regarding their growth rate at different intervals of temperature and time will be carried out in the future.38
 | ||
| Fig. 10 PLM observations under isothermal melt crystallization of (a) and (b) PLA, (c) and (d) PLA-b-PEAz_97.5-2.5 copolyester, (e) and (f) PLA-b-PEAz_95-5 copolyester, (g) and (h) PLA-b-PEAz_90-10 copolyester, (i) and (j) PLA-b-PEAz_80-20 copolyester at 115 °C and (k) PEAz at 30 °C. The scale bar was set at 500 μm. | ||
3.4. Thermogravimetric analysis (TGA)
The mass loss of the samples as a function of temperature during heating at 20 °C min−1 under inert atmosphere is presented in Fig. 11a. In Fig. 11b, the derivative TG curves revealed that the maximum rate degradation of PEAz occurred at higher temperatures than the degradation of PLA. More specifically, the degradation of PLA started at 270 °C, and at 410 °C, the process was completed. The degradation of PEAz, on the other hand, started from 400 to 520 °C (Table S2, ESI†). For both homopolyesters, the degradation occurred in one step, whereas for the PLA-b-PEAz copolyesters, two well-defined steps appeared (Fig. 11b). Significant mass loss was observed over 300 °C for all samples, and the maximum thermal degradation rate occurred from 371 to 380 °C for PLA and the copolyesters (Table S2, ESI†). All copolyesters, except the one with 20% PEAz, exhibited slightly higher thermal stability than PLA, based on the peaks of the derivative of TGA curves (Fig. 11b and Table S2, ESI†) compared to PLA. This can probably be attributed to the long aliphatic ethylene azelate segments, which made the degradation process of the copolyesters more difficult.25 This was correlated to the residuals of the materials (Table S2, ESI†), which were higher for the copolyesters with lower PLA segments. | ||
| Fig. 11 (a) TGA curves of PLA, PEAz and their copolymers at 20 °C min−1 in inert atmosphere, and (b) derivative TG curves of PLA, PEAz and their copolymers. | ||
3.5. Investigation of mechanical performance via tensile and 3-point bending measurements
The mechanical performance of the PLA-based copolyesters was investigated through stress–strain measurements of the compression molded samples. Representative curves of the materials are depicted in Fig. 12 and the tensile data are presented in Fig. 13. Fig. S4 and Table S3 (ESI†) show the stress–strain curves and data of the PLA-b-PEAz copolyesters, respectively. The PLA homopolymer exhibited adequate high tensile features for engineering-like applications, i.e., tensile stress at break over 50 MPa and Young's modulus around 5000 MPa, but low elongation.14 Moreover, the PLA samples did not exhibit yielding. The presence of the second comonomer had a substantial impact on the mechanical behavior of the PLA-b-PEAz copolyesters compared to PLA (***p < 0.001). The elongation significantly improved, over 70% for all materials, but they also appeared tough and robust due to the high values of the tensile parameters. For the evaluation of the mechanical performance of the PLA-based copolyesters, the Elongation and Young's modulus were considered the most important parameters. Furthermore, the PLA-b-PEAz_80-20 copolyester significantly differed from the other copolyesters (Fig. 13). However, adding just 2.5 wt% of PEAz segments, the mechanical behavior of PLA improved significantly, maintaining favorable tensile properties and making PLA-based copolyesters promising materials for engineering applications. | ||
| Fig. 12 (a) Tensile stress–strain curves of PLA and PLA-b-PEAz copolyesters and (b) zoom area at low strain values. | ||
 | ||
| Fig. 13 Tensile data of the materials. One-way ANOVA showed a significant difference between PLA and PLA-based copolyesters for all the tensile parameters. Among the copolyesters, a significant difference was also observed, but for the E′. *p < 0.05, **p < 0.01 and ***p < 0.001. | ||
At the same time, all materials had high elongation, even for the copolyesters with lower azelate content, suggesting that the inherent brittleness of PLA can be solved by adding only 2.5 wt% of PEAz.49–52 Interestingly, as the content of the second comonomer increased, particularly for the PLA-b-PEAz_90-10 and 80-20 copolyesters, the stress at break, stress at yield and Young's modulus increased as well, despite their low  , while at the same time they exhibited high elongation (Fig. 13). This is attributed to strain-induced crystallization that occurred during the tensile measurements. During the experiments, the materials were subjected to strain and were being deformed, so the polymer chains arranged to form additional crystalline structures.53 Thus, the copolyesters exhibited high values of Young's modulus, tensile stress at yield, at break, and elongation simultaneously.
, while at the same time they exhibited high elongation (Fig. 13). This is attributed to strain-induced crystallization that occurred during the tensile measurements. During the experiments, the materials were subjected to strain and were being deformed, so the polymer chains arranged to form additional crystalline structures.53 Thus, the copolyesters exhibited high values of Young's modulus, tensile stress at yield, at break, and elongation simultaneously.
Besides the MW, thermal properties and crystallinity are considered important factors to explain the mechanical performance of the copolyesters.54,55 The relative degree of crystallinity, Xc, played a significant role in the mechanical behavior of the materials. During heating of the as-received compression molded samples of PLA and the copolyester with 2.5% of long aliphatic segments were amorphous, but for the copolyesters with higher PEAz content, the crystallinity was higher (Table 2). Based on the above findings, the tensile stress at break, at yield, and Young's modulus increased gradually and followed the trend of crystallinity. The crystalline regions of the polymer matrices of copolyesters caused additional resistance during the stress–strain measurements, which improved their mechanical performance.25 For this reason, the copolyester containing 20% PEAz exhibited probably the best tensile parameter values. Considering also the high  values of PLA-b-PEAz 97.5-2.5 and 95-5 samples, these materials could be promising flexible substrates for engineering-like applications.
values of PLA-b-PEAz 97.5-2.5 and 95-5 samples, these materials could be promising flexible substrates for engineering-like applications.
Aliphatic segments proved to be a reliable tool for tuning the mechanical properties of PLA via copolymerization,56 preparing tough or/and flexible substrates, for engineering applications (i.e. flexible printed electronics).57,58 Based on previous studies, high MW PLA copolyesters with PBS25 showed remarkable mechanical performance, with hardness and elastic modulus over 150 and 5500 MPa, respectively. In a series of different PLA-based copolyesters using poly(propylene adipate) (PPAd), elongation over 100% was reported.39
Apart from the tensile properties, further mechanical performance analysis was necessary to address various possible applications. For instance, for PE applications, it is crucial to examine the substrate's resistance to bending and the maximum strength that can be withstood via the flexural modulus and strength parameters, respectively.58Fig. 14 shows the flexural data of PLA and PLA-b-PEAz copolyesters, where the copolyesters significantly differed from PLA (**p < 0.01 and ***p < 0.001). PLA broke during the tests and exhibited the highest flexural strength and modulus among the materials (Fig. S5, ESI†).59 Flexural strength is the maximum stress the materials can withstand during the experiments. On the other hand, the PLA-based copolyesters did not break during the 3-point bending tests, in line with the high elongation values exhibited during the tensile measurements. The flexural strength and modulus of PLA decreased with the presence of the PEAz chains, and at the same time, the materials exhibited high values of flexural features (Table S4, ESI†). Moreover, the flexural data had a similar trend to the tensile ones as a function of the PEAz content.
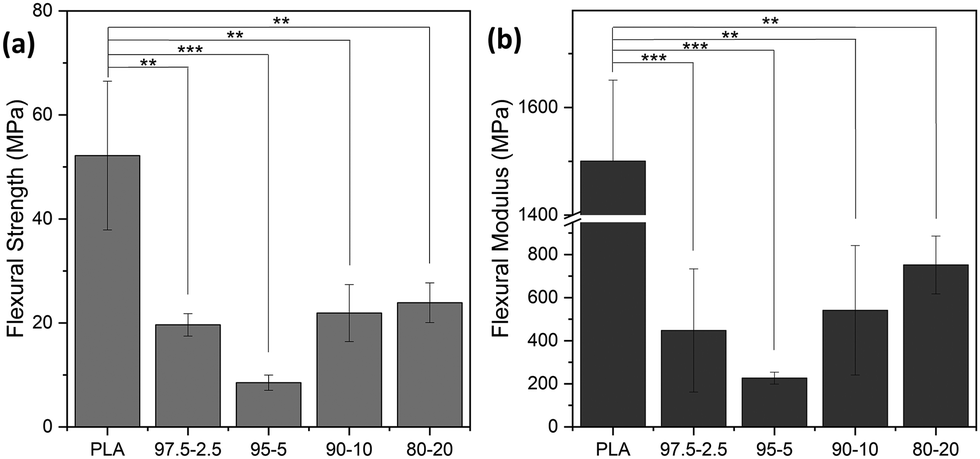 | ||
| Fig. 14 Flexural properties of PLA and PLA-b-PEAz copolyesters. One way ANOVA showed significant difference between PLA and PLA-based copolyesters for the flexural parameters. **p < 0.01 and ***p < 0.001. | ||
3.6. Color measurements
Along with the thermal and mechanical examination of the materials, the coloration of the substrate constitutes another crucial feature for PE and packaging applications. The next figure presents the color measurements of the PLA-based copolymers in terms of CIE coordinates. PLA with L* value close to 85, a* and b* to 0.8 and 6.8, respectively, were considered white color material. PEAz oligomers did not influence the CIE parameters of PLA, meaning that the copolyesters exhibited the white-like color of PLA and not, for example, a yellow one, which could be a common issue of the PLA-based copolyesters (Fig. 15). | ||
| Fig. 15 CIE L*a*b* coordinates of PLA, PEAz, and PLA-b-PEAz copolymers, L*: perceptual lightness, a* and b*: red–green and blue–yell. | ||
4. Conclusions
Innovative, flexible poly(lactic acid)-b-poly(ethylene azelate) (PLA)-b-(PEAz) blocky copolyesters with high molecular weight were successfully synthesized. All the synthesized copolyesters (PLA-b-PEAz 97.5-2.5, 95-5, 90-10, and 80-20) had noteworthy properties. The materials exhibited high viscosity values and from 10 to 80 kg mol−1, making the copolyesters promising materials, for instance, for packaging, printed electronics, and generally for engineering applications. Due to their high
from 10 to 80 kg mol−1, making the copolyesters promising materials, for instance, for packaging, printed electronics, and generally for engineering applications. Due to their high  , all copolyesters exhibited high Tm, while the Tg and Tcc decreased due to the addition of the flexible macromolecular chains of the long aliphatic PEAz blocky segments. XRD patterns revealed that the azelate segments were excluded to the amorphous regions. Isothermal melt crystallization experiments showed that all copolyesters exhibited higher crystallization rates than PLA (depending on supercooling), and through PLM observations, the Maltese cross morphology of the copolyesters was observed. Moreover, the high
, all copolyesters exhibited high Tm, while the Tg and Tcc decreased due to the addition of the flexible macromolecular chains of the long aliphatic PEAz blocky segments. XRD patterns revealed that the azelate segments were excluded to the amorphous regions. Isothermal melt crystallization experiments showed that all copolyesters exhibited higher crystallization rates than PLA (depending on supercooling), and through PLM observations, the Maltese cross morphology of the copolyesters was observed. Moreover, the high 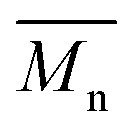 and the presence of the azelate segments facilitated the high thermal stability of the copolyesters. Their mechanical performance was significantly improved, making the copolyesters strong and tough materials, where in all cases, the elongation surpassed 70% and exhibited high values of Young's modulus >1.7 GPa, reaching almost 3 GPa. Furthermore, during the 3-point bending tests, the copolyesters did not break. Last but not least, the color measurements revealed their off-white coloration. Additional studies will be conducted regarding biodegradability, compostability, UV degradation, an in-depth study of the molecular mobility of PLA-b-PEAz blocky copolyesters and scale-up production via cast film extrusion line for engineering applications, specifically for printed electronics.
and the presence of the azelate segments facilitated the high thermal stability of the copolyesters. Their mechanical performance was significantly improved, making the copolyesters strong and tough materials, where in all cases, the elongation surpassed 70% and exhibited high values of Young's modulus >1.7 GPa, reaching almost 3 GPa. Furthermore, during the 3-point bending tests, the copolyesters did not break. Last but not least, the color measurements revealed their off-white coloration. Additional studies will be conducted regarding biodegradability, compostability, UV degradation, an in-depth study of the molecular mobility of PLA-b-PEAz blocky copolyesters and scale-up production via cast film extrusion line for engineering applications, specifically for printed electronics.
Author contributions
Writing – original draft, validation, formal analysis, data curation, conceptualization, P. O. I., Z. T.; writing – review and editing, data curation, methodology, validation, investigation, conceptualization, software, data curation, A. Z., M. J. N.; writing – review, validation, investigation, N. D. B.; writing – review and editing, supervision, N. N. All authors have read and agreed to the published version of the manuscript.Data availability
The DSC, FTIR, NMR, Tensile, TGA and XRD data are available on Zenodo (doi: 10.5281/zenodo.15235199). The rest of the data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The research was funded by the European Union under the GA no 101070556 (Sustain-A-Print, https://www.sustainaprint.eu/). Views and opinions expressed are however those of the author(s) only and do not necessarily reflect those of the European Union or RIA. Neither the European Union nor the granting authority can be held responsible. The publication of the article in OA mode was financially supported by HEAL-Link. The authors also would like to express their gratitude to Dr Antigoni Margellou, and Prof. Konstantinos Triantafyllidis from the Department of Chemistry, AUTH for the TGA measurements. Sincerely, Rafail O. Ioannidis would respectfully acknowledge Dr Panagiotis Klonos for his insightful comments concerning the results on the thermal analysis and isothermal melt crystallization.References
- https://environment.ec.europa.eu/topics/plastics/biobased-biodegradable-and-compostable-plastics_en .
- https://www.european-bioplastics.org/market/# .
- W. Ali, H. Ali, S. Souissi and P. Zinck, Are bioplastics an ecofriendly alternative to fossil fuel plastics?, Environ. Chem. Lett., 2023, 21, 1991–2002 CrossRef CAS.
- S. S. Ali, E. A. Abdelkarim, T. Elsamahy, R. Altohamy, F. Li, M. Kornaros, A. Zuorro, D. Zuoa and J. Sun, Bioplastic production in terms of life cycle assessment: a state-of-the-art review, Environ. Sci. Ecotechnology, 2023, 19, 100254 CrossRef PubMed.
- D. K. Schneiderman and M. A. Hillmyer, 50th Anniversary Perspective: There Is a Great Future in Sustainable Polymers, Macromolecules, 2017, 50, 3733–3749 CrossRef CAS.
- Y. Zhu, C. Romaina and C. K. Williams, Sustainable polymers from renewable resources, Nature, 2016, 540, 354–362 CrossRef CAS PubMed.
- E. E. Mastalygina and K. V. Aleksanyan, Recent Approaches to the Plasticization of Poly(lactic Acid) (PLA) (A Review), Polymers, 2023, 16, 87 CrossRef PubMed.
- K. Babaremu, O. P. Oladijo and E. Akinlabi, Biopolymers: a suitable replacement for plastics in product packaging, Adv. Ind. Eng. Polym. Res., 2023, 6, 333–340 CAS.
- K. Kadac-Czapska, E. Knez and M. Grembecka, Food and human safety: the impact of microplastics, Crit. Rev. Food Sci. Nutr., 2022, 64, 3502–3521 CrossRef PubMed.
- W. Ali, H. Ali, S. Gillani, P. Zinck and S. Souissi, Polylactic acid synthesis, biodegradability, conversion to microplastics and toxicity: a review, Environ. Chem. Lett., 2023, 21, 1761–1786 CrossRef CAS.
- K. Loos, R. Zhang, I. Pereira, B. Agostinho, H. Hu, D. Maniar, A. J. D. Silvestre, N. Guigo and A. F. Sousa, A Perspective on PEF Synthesis, Properties, and End-Life, Front. Chem., 2020, 8, 585 CrossRef CAS PubMed.
- X. Fei, J. Wang, X. Zhang, Z. Jia, Y. Jiang and X. Liu, Recent Progress on Bio-Based Polyesters Derived from 2,5-Furandicarbonxylic Acid (FDCA), Polymers, 2022, 14, 625 CrossRef CAS PubMed.
- L. Shen, E. Worrell and M. K. Patel, Comparing life cycle energy and GHG emissions of bio-based PET, recycled PET, PLA, and man-made cellulosics, Biofuels, Bioprod. Biorefin., 2012, 6, 625–639 CrossRef CAS.
- E. Balla, V. Daniilidis, G. Karlioti, T. Kalamas, M. Stefanidou, N. Bikiaris, A. Vlachopoulos, I. Koumentakou and D. N. Bikiaris, Poly(lactic acid): a versatile biobased polymer for the future with multifunctional properties-from monomer synthesis, polymerization techniques and molecular weight increase to PLA applications, Polymers, 2021, 13(11), 1822 CrossRef CAS PubMed.
- N. D. Bikiaris, I. Koumentakou, C. Samiotaki, D. Meimaroglou, D. Varytimidou, A. Karatza, Z. Kalantzis, M. Roussou, R. Bikiaris and G. Z. Papageorgiou, Recent Advances in the Investigation of Poly(lactic acid) (PLA) Nanocomposites: Incorporation of Various Nanofillers and their Properties and Applications, Polymers, 2023, 15(5), 1196 CrossRef CAS PubMed.
- S. Saeidlou, M. A. Huneault, H. Li and C. B. Park, Poly(lactic acid) crystallization, Prog. Polym. Sci., 2012, 37, 1657–1677 CrossRef CAS.
- S. Liu, S. Qin, M. He, D. Zhou, Q. Qin and H. Wang, Current applications of poly(lactic acid) composites in tissue engineering and drug delivery, Composites, Part B, 2020, 199, 108238 CrossRef CAS.
- V. DeStefano, S. Khan and A. Tabada, Applications of PLA in modern medicine, Eng. Regener., 2020, 1, 76–87 Search PubMed.
- M. N. Siddiqui, L. Kolokotsiou, E. Vouvoudi, H. H. Redhwi, A. A. Al-Arfaj and D. S. Achilias, Depolymerization of PLA by Phase Transfer Catalysed Alkaline Hydrolysis in a Microwave Reactor, J. Polym. Environ., 2020, 28, 1664–1672 CrossRef CAS.
- R. Supthanyakul, N. Kaabbuathong and S. Chirachanchai, Poly(L-lactide-b-butylene succinate-b-L-lactide) triblock copolymer: a multi-functional additive for PLA/PBS blend with a key performance on film clarity, Polym. Degrad. Stab., 2017, 142, 160–168 CrossRef CAS.
- H. Zebiri, H. Van Den Berghe, S. Sayegh, P. E. Chammas, C. Pompée, M. Chammas and X. Garric, Synthesis of PLA-poly(ether urethane)-PLA copolymers and design of biodegradable anti-adhesive membranes for orthopaedic applications, J. Mater. Chem. B, 2021, 9, 832–845 RSC.
- Z. Terzopoulou and D. N. Bikiaris, Biobased plastics for the transition to a circular economy, Mater. Lett., 2024, 362 Search PubMed.
- N. D. Bikiaris, P. A. Klonos, A. Kyritsis and P. Barmpalexis, Structural and thermodynamical investigation in triblock copolymers of polylactide and poly(ethylene glycol), PLA-b-PEG-b-PLA, envisaged for medical applications, Mater. Tod. Commun., 2024, 38 Search PubMed.
- B. H. Rodríguez and A. Lieske, Widening the Application Range of PLA-Based Thermoplastic Materials through the Synthesis of PLA-Polyether Block Copolymers: Thermal, Tensile, and Rheological Properties, Macromol. Mater. Eng., 2024, 309(3), 2300309 CrossRef.
- Z. Terzopoulou, A. Zamboulis, N. D. Bikiaris, A. Margellou, M. A. Valera, A. Mangas, S. Koltsakidis, K. Tsongas, D. Tzetzis and K. S. Triantafyllidis, Properties of PLA-co-PBSu Copolymers Rapidly Synthesized by Reactive Processing, J. Polym. Environ., 2023, 32, 1–15 Search PubMed.
- A. Todea, C. Deganutti, M. Spennato, F. Asaro, G. Zingone, T. Milizia and L. Gardossi, Azelaic acid: a bio-based building block for biodegradable polymers, Polymers, 2021, 13(23), 4091 CrossRef CAS PubMed.
- G. Z. Papageorgiou, D. N. Bikiaris, D. S. Achilias and N. Karagiannidis, Synthesis, crystallization, and enzymatic degradation of the biodegradable polyester poly(ethylene azelate), Macromol. Chem. Phys., 2010, 211, 2585–2595 CrossRef CAS.
- M. K. Wong, S. S. M. Lock, Y. H. Chan, S. J. Yeoh and I. S. Tan, Towards sustainable production of bio-based ethylene glycol: progress, perspective and challenges in catalytic conversion and purification, Chem. Eng. J., 2023, 468, 143699 CrossRef CAS.
- B. Zhang, X. Bian, S. Xiang, G. Li and X. Chen, Synthesis of PLLA-based block copolymers for improving melt strength and toughness of PLLA by in situ reactive blending, Polym. Degrad. Stab., 2017, 136, 58–70 CrossRef CAS.
- G. Z. Papageorgiou, D. N. Bikiaris, D. S. Achilias, E. Papastergiadis and A. Docoslis, Crystallization and biodegradation of poly(butylene azelate): comparison with poly(ethylene azelate) and poly(propylene azelate), Thermochim. Acta, 2011, 515, 13–23 CrossRef CAS.
- P. A. Klonos, Z. Terzopoulou, A. Zamboulis, M. A. Valera, A. Mangas, A. Kyritsis, P. Pissis and D. N. Bikiaris, Direct and indirect effects on molecular mobility in renewable polylactide-poly(propylene adipate) block copolymers as studied via dielectric spectroscopy and calorimetry, Soft Mater., 2022, 18, 3725–3737 RSC.
- A. M. Gorman, A. Clayton, T. O’Connell and D. Johnson, A recyclable screen ink with state-of-the-art performance developed using a bottom-up, safety and sustainability-driven approach, MRS Adv., 2023, 8, 311–316 CrossRef CAS.
- Z. Terzopoulou, L. Papadopoulos, A. Zamboulis, D. G. Papageorgiou, G. Z. Papageorgiou and D. N. Bikiaris, Tuning the properties of furandicarboxylic acid-based polyesters with copolymerization: a review, Polymers, 2020, 12(6), 1209 CrossRef CAS PubMed.
- A. Sonseca and M. El Fray, Enzymatic synthesis of an electrospinnable poly(butylene succinate-co-dilinoleic succinate) thermoplastic elastomer, RSC Adv., 2017, 7, 21258 RSC.
- L. Genovese, M. Soccio, N. Lotti, M. Gazzano, V. Siracusa, E. Salatelli, F. Balestra and A. Munari, Design of biobased PLLA triblock copolymers for sustainable food packaging: thermo-mechanical properties, gas barrier ability and compostability, Eur. Polym. J., 2017, 95, 289–303 CrossRef CAS.
- C. Ba, J. Yang, Q. Hao, X. Liu and A. Cao, Syntheses and physical characterization of new aliphatic triblock poly(L-lactide-b-butylene succinate-b-L-lactide)s bearing soft and hard biodegradable building blocks, Biomacromolecules, 2003, 4, 1827–1834 CrossRef CAS PubMed.
- P. A. Klonos, N. D. Bikiaris, A. Zamboulis, M. A. Valera, A. Mangas, A. Kyritsis and Z. Terzopoulou, Segmental mobility in sustainable copolymers based on poly(lactic acid) blocks built onto poly(butylene succinate) in situ, Soft Mater., 2023, 19, 7846–7858 RSC.
- M. L. Di Lorenzo, The crystallization and melting processes of poly(L-lactic acid), Macromol. Symp., 2006, 234, 176–183 CrossRef CAS.
- Z. Terzopoulou, A. Zamboulis, D. N. Bikiaris, M. A. Valera and A. Mangas, Synthesis, properties, and enzymatic hydrolysis of poly(Lactic acid)-co-poly(propylene adipate) block copolymers prepared by reactive extrusion, Polymers, 2021, 13(23), 4121 CrossRef CAS PubMed.
- Z. Refaa, M. Boutaous and D. A. Siginer, PLA crystallization kinetics and morphology development, Int. Polym. Process., 2018, 33, 336–344 CrossRef CAS.
- S. Vyazovkin, A. K. Burnham, J. M. Criado, L. A. Pérez-Maqueda, C. Popescu and N. Sbirrazzuoli, ICTAC Kinetics Committee recommendations for performing kinetic computations on thermal analysis data, Thermochim. Acta, 2011, 520, 1–19 CrossRef CAS.
- R. M. Van Horn, M. R. Steffen and D. O’Connor, Recent progress in block copolymer crystallization, Polym. Cryst., 2018, 1(4), e10039 Search PubMed.
- A. J. Müller, R. M. Michell and A. T. Lorenzo, Isothermal Crystallization Kinetics of Polymers, in Q. Guo, Polymer Morphology, 2016 Search PubMed.
- R. M. D’Ambrosio, R. M. Michell, R. Mincheva, R. Hernandez, C. Mijangos, P. Dubois and A. J. Müller, Crystallization and stereocomplexation of PLA-mb-PBS multi-block copolymers, Polymers, 2018, 10 Search PubMed.
- M. Yasuniwa, S. Tsubakihara, Y. Sugimoto and C. Nakafuku, Thermal analysis of the double-melting behavior of poly(L-lactic acid), J. Polym. Sci., Part B: Polym. Phys., 2004, 42, 25–32 CrossRef CAS.
- M. L. Di Lorenzo, The crystallization and melting processes of poly(L-lactic acid), Macromol. Symp., 2006, 234, 176–183 CrossRef CAS.
- M. Yasuniwa, K. Iura and Y. Dan, Melting behavior of poly(L-lactic acid): effects of crystallization temperature and time, Polymer, 2007, 48, 5398–5407 CrossRef CAS.
- S. Solarski, M. Ferreira and E. Devaux, Characterization of the thermal properties of PLA fibers by modulated differential scanning calorimetry, Polymer, 2005, 46, 11187–11192 CrossRef CAS.
- E. Luoma, M. Välimäki, T. Rokkonen, H. Sääskilahti, J. Ollila, J. Rekilä and K. Immonen, Oriented and annealed poly(lactic acid) films and their performance in flexible printed and hybrid electronics, J. Plast. Film Sheeting, 2021, 37, 429–462 CrossRef CAS.
- C. Siracusa, F. Quartinello, M. Soccio, M. Manfroni, N. Lotti, A. Dorigato, G. M. Guebitz and A. Pellis, On the Selective Enzymatic Recycling of Poly(pentamethylene 2,5-furanoate)/Poly(lactic acid) Blends and Multiblock Copolymers, ACS Sustain, Chem. Eng., 2023, 11, 9751–9760 CAS.
- K. Samadi, M. Francisco, S. Hegde, C. A. Diaz, T. A. Trabold, E. M. Dell and C. L. Lewis, Mechanical, rheological and anaerobic biodegradation behavior of a Poly(lactic acid) blend containing a Poly(lactic acid)-co-poly(glycolic acid) copolymer, Polym. Degrad. Stab., 2019, 170, 109018 CrossRef.
- G. Fredi and A. Dorigato, Compatibilization of biopolymer blends: a review, Adv. Ind. Eng. Polym. Res., 2023, 7(4), 373–404 Search PubMed.
- C. Zhou, S. Dong, P. Zhu, J. Liu, D. Wang and X. Dong, Strain-induced form transition and crystallization behavior of the transparent polyamide, Polymers, 2021, 13(7), 1028 CrossRef CAS PubMed.
- G. Perego, D. Glan and C. Cella, Effect of Molecular Weight and Crystallinity on Poly(lactic acid) Mechanical Properties, J. Appl. Polym. Sci., 1996, 59, 37–43 CrossRef CAS.
- C. C. Tsai, R. J. Wu, H. Y. Cheng, S. C. Li, Y. Y. Siao and D. C. Kong, Crystallinity and dimensional stability of biaxial oriented poly(lactic acid) films, Polym. Degrad. Stab., 2010, 95, 1292–1298 CrossRef CAS.
- X. Hu, H. Kang, Y. Li, M. Li, R. Wang, R. Xu, H. Qiao and L. Zhang, Direct copolycondensation of biobased elastomers based on lactic acid with tunable and versatile properties†, Polym. Chem., 2015, 6, 8112–8123 RSC.
- Y. Khan, A. Thielens, S. Muin, J. Ting, C. Baumbauer and A. C. Arias, A New Frontier of Printed Electronics: Flexible Hybrid Electronics, Adv. Mater., 2019, 32(15), 1905279 CrossRef PubMed.
- J. Machiels, A. Verma, R. Appeltans, M. Buntinx, E. Ferraris and W. Deferme, Printed Electronics (PE) As An enabling Technology to Realize Flexible Mass Customized Smart Applications, Proc. CIRP., 2020, 96, 115–120 CrossRef.
- D. Li, Y. Jiang, S. Lv, X. Liu, J. Gu, Q. Chen and Y. Zhang, Preparation of plasticized poly(lactic acid) and its influence on the properties of composite materials, PLoS One, 2018, 13(3), e0193520 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ma00014a |
| This journal is © The Royal Society of Chemistry 2025 |