DOI:
10.1039/D4MA01252F
(Paper)
Mater. Adv., 2025,
6, 3220-3236
Effectiveness comparison of octyltrimethoxysilane and hexadecyltrimethoxysilane functionalized on natural silica-coated magnetic materials for ciprofloxacin and chloramphenicol adsorption†
Received
16th December 2024
, Accepted 28th March 2025
First published on 31st March 2025
Abstract
This article reports the synthesis of natural magnetic silica-coated material (NMM@SiO2) functionalized with hydrophobic properties of octyltrimethoxysilane (OTMS) and hexadecyltrimethoxysilane (HDTMS). Characterization to confirm the success of OTMS and HDTMS functionalization on NMM@SiO2 was performed, and the adsorption properties of ciprofloxacin (CIP) and chloramphenicol (CAP) on the materials were compared. The optimal conditions for CIP and CAP adsorption on both adsorbents were at pH 6, a contact time of 60 min for CIP and 90 min for CAP, and an initial concentration of 250 mg L−1. The OTMS-functionalized adsorbent demonstrated a higher adsorption capacity than HDTMS-one, with capacities of 87.83 mg g−1 for CIP and 56.44 mg g−1 for CAP. The adsorption kinetics followed a pseudo-second-order model, and the adsorption isotherms were consistent with the Freundlich model. Thermodynamic parameters indicated a negative enthalpy change (ΔH), suggesting an exothermic reaction, a negative Gibbs free energy (ΔG), indicating spontaneity, and a positive entropy change (ΔS), suggesting increased randomness. The successful adsorption of CIP and CAP, achieved through introducing hydrophobic groups, promises alternative materials for CIP and CAP removal from water waste.
1. Introduction
In recent years, antibiotics have been growing concerns due to their widespread use in human therapy, agriculture, aquaculture, and water, as they have been identified as potent pollutants.1 The pollution from excessive antibiotic use is a potential threat to human health as it promotes the proliferation of antibiotic-resistant bacteria (ARB).2 Due to this threat, the World Health Organization (WHO) has classified the spread of ARB as one of the three most serious public health threats.2,3 Furthermore, antibiotics are frequently released into the environment through household wastewater discharge and improper disposal of expired medications, posing significant risks to humans.4 Unregulated and hazardous antibiotics include ciprofloxacin and chloramphenicol.
Ciprofloxacin (CIP) is a fluoroquinolone antibiotic,5 while chloramphenicol (CAP) is an amphenicol antibiotic. Both antibiotics are widely used and active against a broad spectrum of Gram-negative and Gram-positive bacteria.6,7 However, uncontrolled use of these antibiotics is often detected in the environment and is genotoxic. Of the administered dose of CIP in humans, 45–62% is excreted unmetabolized in the urine and 15–25% in feces.7 CIP affects mammalian tissues and cells through binding to bacterial DNA gyrase, topoisomerase IV, and type II topoisomerase enzymes.8 Chloramphenicol's hazards include anemia, hematologic disorders, allergies, and liver toxicity. Ciprofloxacin can cause tendon damage, neurological effects, heart complications, nausea, and diarrhea. Due to the dangers posed by these antibiotics, it is necessary to remove them from the environment as early as possible.
Several methods have been reported for the removal of CIP and CAP from the environment, including the photo-Fenton oxidation reaction,6 photocatalytic processes,9 hydrolysis,10 biodegradation,11 electrochemical oxidation,12 ozonation,13 pulsed discharge plasma,14 and adsorption.15–17 However, most methods besides adsorption have drawbacks, such as high costs, maintenance expenses, complex treatment procedures, and the generation of toxic sludge.18 The advantages of the adsorption method over others include high efficiency, simplicity, low cost, environmental friendliness, and the absence of harmful by-products.18 Based on the reported studies, a comparison of octyl trimethoxysilane (OTMS) and hexadecyltrimethoxysilane (HDTMS) for the removal of CIP and CAP has not yet been explored.
It is essential to consider the characteristics of the adsorbate to improve the efficiency of adsorbate adsorption on adsorbents.19 Based on their structure, CIP and CAP contain hydrophobic groups, so hydrophobic adsorbents are required to enhance adsorption performance. Several studies have reported on hydrophobic interactions in adsorption mechanisms. Peng et al. (2015)20 used mesoporous and bamboo-based carbon, demonstrating that adsorption predominantly occurs through hydrophobic interactions. Similarly, Zhou et al. (2019)21 employed a Fe3O4/graphene oxide/citrus peel-derived biochar-based nanocomposite for CIP adsorption, where hydrophobic groups primarily drove the interaction mechanism. Yu et al. (2019)22 utilized porous graphene hydrogel for CIP adsorption, which occurred through both hydrophobic and π–π interactions. Additionally, Zhao et al. (2016)23 investigated using multi-walled carbon nanotubes for CAP adsorption, revealing that the mechanism involved EDA and hydrophobic interactions. In this article, we compare the functional groups of OTMS and HDTMS, which are expected to possess hydrophobic properties to adsorb CIP and CAP. However, using these functional groups without a ESI† presents limitations such as low physical stability, limited active surface area, difficulty in regeneration, poor resistance to extreme conditions, and aggregation.24 Various ESI† have been used for CIP and CAP adsorption, including Fe3O4@SiO2/SiTBA-ALG,25 Fe3O4@SiO2-APTMS-HBA,26 magnetite–imprinted chitosan nanocomposites,27 and Fe3O4/carbon for CIP adsorption.28 Other materials, like NaOH-modified bamboo charcoal,29 multi-walled carbon nanotubes,23 and magnetic molecularly imprinted polymers (MMIPs) have been used for CAP adsorption.30
One limitation of the adsorption process is the difficulty in separating and recovering adsorbents after use, making the utilization of magnetic materials an effective solution to facilitate separation using an external magnetic field and enhancing process efficiency.31 They are advantageous due to their simplicity, efficiency, sensitivity, and ease of separation from liquid suspensions using an external magnetic field.32 Consequently, magnetic materials have been considered viable alternatives for removing water pollutants due to their high surface area and magnetic properties.33 Magnetic materials are typically derived from metal oxide substances.34 Some oxides used for CIP and CAP adsorption include CoFe2O435 and Fe3O4.26 However, few reports exist on using magnetic materials sourced from iron sand. The advantages of using natural magnetic materials include low cost, abundant availability, environmental friendliness, adequate adsorption capacity, and non-toxicity.18,36,37
The use of magnetic materials has a drawback, as they tend to aggregate due to their nanoscale particle size and the influence of magnetic gravity,38 necessitating a silica coating. Silica is chosen because it protects the magnetic material from oxidation, prevents agglomeration, remains stable over a wide pH range, and enhances chemical stability.39 In this study, natural magnetic material will be combined with SiO2 extracted from natural sand to form natural magnetic material coated with silica (NMM@SiO2), which exhibits superior properties.
This article compares alkylsilane chain lengths, specifically OTMS (C8) and HDTMS (C16), embedded in natural magnetic silica-coated materials. The OTMS-functionalized material is designated as NMM@SiO2/OTMS, while the HDTMS-functionalized material is referred to as NMM@SiO2/HDTMS, which exhibits hydrophobic properties. The natural magnetic material originates from iron sand in Flores Island, NTT, and the silica was extracted from natural sand in Takari River, NTT. Each functional group chain was varied in quantities of 3, 7.5, and 15 mmol on natural magnetic silica-coated material. The synthesized materials were then used to adsorb CIP and CAP. This article also reports the characterization of the adsorbent materials, including functional groups, crystallinity, morphology and surface composition, magnetic properties, and hydrophobic properties. The synthesized adsorbents were used to adsorb CIP and CAP, optimizing parameters such as pH, contact time and kinetics, initial concentration, adsorption isotherms, thermodynamics, desorption, reusability, and adsorption mechanism.
2. Materials and methods
2.1. Materials
The natural magnetic material (NMM) was separated from iron sand collected from Ena Gera-Nagekeo beach, Nusa Tenggara Timur (NTT), Indonesia (coordinates: 8°53′28.64′′S, 121°12′19.43′′E). Silica was extracted from a sand sample collected from Takari River, Timor Island- NTT (coordinates 9°58′21.75 ′′S, 124°5′40.61′′E). The alkylsilanes used in this study were octyltrimethoxysilane (OTMS) and hexadecyltrimethoxysilane (HDTMS) purchased from Sigma Aldrich (95%). The antibiotics studied were ciprofloxacin (purity >98.0%) and chloramphenicol (purity >98%), purchased from Sangon Biotech Co., Ltd (Shanghai, China). Additional chemicals included glacial acetic acid (Merck, 99.5%), crystalline NaOH (pro analysis), HCl (pro analysis, 37%), methanol (Merck, 99.9%), and ethanol (JT Baker, 98%).
2.2. Methods
2.2.1. Preparation of NMM and silica.
The natural magnetic material (NMM) preparation and silica extraction followed procedures previously reported by Naat et al. (2024)15 and Nuryono et al. (2020).34 NMM was obtained by separating the collected iron sand using an external magnetic field. Subsequently, the material attracted to the external magnet was considered raw NMM. A total of 150 g of raw NMM was then dried in an oven at 60 °C for 24 hours, followed by grinding to a particle size of 200 mesh. Afterward, 150 g of the raw material was washed using 250 mL of double-distilled water. The washing was repeated five times to remove non-magnetic components. The sieved product (>200 mesh) was dried in an oven at 95–100 °C for 18 h. The product (1 g) was activated by refluxing it in a 10% HCl solution (1 mL) for 2 h at 75–80 °C. The activated material was then washed with distilled water until a pH of 7.0 was reached and then dried in an oven at 95–100 °C for 18 h. The product was then designated as NMM.
In preparing silica, the sand sample (50 g) was crushed, sieved using a 200-mesh sieve, and stirred in a 2 M HCl solution (200 mL) for 12 h. The material was washed with distilled water until reaching a pH of 7.0 and dried. This material was refluxed using 20 mL of 7 M NaOH solution at 105 °C for 6 h to produce a Na2SiO3 solution. The solution was diluted with 240 mL of distilled water and left to settle for 24 h before being separated through filtration. The resulting filtrate was treated with 2 M HCl solution to reach a pH of 7, and a white precipitate was formed. This precipitate was allowed to settle at room temperature (25 °C) for 24 h and washed five times with 50 mL of hot distilled water.
2.2.2. Synthesis of NMM@SiO2/alkylsilane (alkylsilane = OTMS and HDTMS).
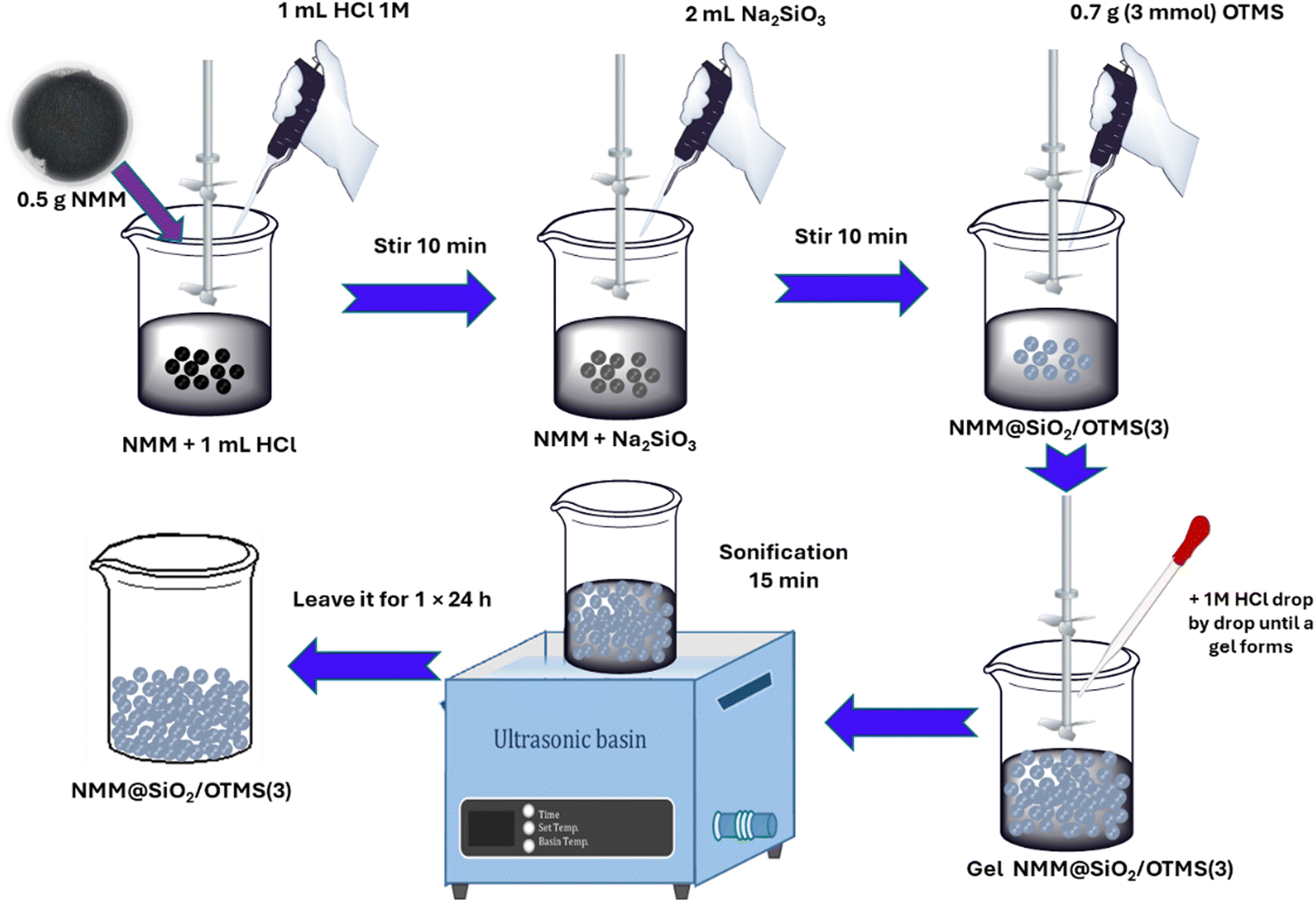
NMM (0.5 g) in a 25 mL glass beaker was added with 1 mL of a 1 M HCl solution and stirred for 30 min with a mechanical stirrer. Separately, 1 g of SiO2 extracted from the sand sample was mixed with 10 mL of 1 M NaOH solution under a continuous stirring for 1 h at 70 °C until a sodium silicate solution was formed. Then, 0.5 g of activated NMM was mixed with 2 mL (3 mmol) of sodium silicate solution (pH ≈ 12) and stirred again for 30 min.40 Then, 0.7 g OTMS (3 mmol) was added to the NMM and sodium silicate mixture. The mixture was stirred mechanically for 1 h while 1 M HCl was added dropwise until a gel formed at pH 7. The gel was measured using a universal pH meter on the formed gel. The gel was sonicated for 15 min, left in a closed container overnight (24 h), washed with double-distilled water, and dried at 60 °C for 24 h. The product obtained was washed to neutrality, separated with an external magnetic field, and dried at 60 °C for 6 h to obtain NMM@SiO2/alkylsilane. The obtained magnetic material was called NMM@SiO2/OTMS(3). The variation in the mmol amounts of OTMS and HDTMS was carried out following the same procedure as described above, with the only difference being the amounts of OTMS and HDTMS added. The specific amounts of OTMS and HDTMS are detailed in Table 1, and the synthesis pathway for NMM@SiO2/OTMS(3) is illustrated in Fig. 1.
Table 1 Material code for various moles of OTMS and HDTMS
| Material codes |
OTMS |
HDTMS |
| Mass, g (mmol) |
Mass, g (mmol) |
| NMM: 0.5 g, and sodium silicate solution: 2 mL. |
| NMM@SiO2/OTMS(3) |
0.70 (3.00) |
0.00 |
| NMM@SiO2/OTMS(7.5) |
1.75 (7.50) |
0.00 |
| NMM@SiO2/OTMS(15) |
3.25 (15.0) |
0.00 |
| NMM@SiO2/HDTMS(3) |
0.00 |
1.04 (3.00) |
| NMM@SiO2/HDTMS(7.5) |
0.00 |
2.60 (7.50) |
| NMM@SiO2/HDTMS(15) |
0.00 |
5.20 (15.0) |
 |
| | Fig. 1 NMM@SiO2/OTMS(3) synthesis pathway. | |
2.2.3. Measurement of adsorbent hydrophobicity.
The hydrophobic properties of the synthesized NMM@SiO2/alkylsilane were determined by measuring the water contact angle (WCA). NMM@SiO2/alkylsilane was sieved, weighed, and evenly distributed on a 1 × 1 cm plate with a thickness of 4 mm. A drop of water was placed on the NMM@SiO2/alkylsilane surface using a Pasteur pipette. As the water contacted the NMM@SiO2/alkylsilane surface, a photograph was taken with a Canon EOS 10 DSLR camera. Contact angles were measured using ImageJ software.41
2.2.4. Determination of pHPZC of NMM@SiO2/alkylsilane.
As much as 10 mL of 0.01 M NaCl solution, adjusted to pH 1–10 with 0.1 M HCl or 0.1 M NaOH, was added to 10 mg of NMM@SiO2/alkylsilane (the volume to weight ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). The mixture was shaken for 60 min, allowed to stand for 3 d. The final pH of each solution was recorded.
:1). The mixture was shaken for 60 min, allowed to stand for 3 d. The final pH of each solution was recorded.
2.2.5. Adsorption experiment.
CIP and CAP antibiotic adsorption on adsorbents in Table 1 were conducted in a batch system by varying pH, contact time, and initial concentration. A stock antibiotic solution of 1000 mg L−1 was prepared by dissolving 1 g of antibiotic in double-distilled water and adjusting the volume to 1 L. Subsequently, various concentrations were prepared by diluting the stock solution. Calibration curves for each antibiotic were prepared to determine concentrations before and after adsorption. The solution was adjusted at pH 2–10, and contact time was varied 10, 20, 30, 40, 50, 60, 90, 120, and 150 min, and initial concentrations of 10, 20, 40, 60, 80, 100, 150, 200, 250, and 300 mg L−1. Each adsorption was stirred mechanically at 250 rpm at room temperature (25 °C). After adsorption, the sample was separated with an external magnet and filtered, and the antibiotic content was analyzed using UV-Vis at CIP = 270 nm and CAP = 278 nm.15 Each adsorption was performed in duplicate, and the adsorbed antibiotics were calculated from the difference between antibiotic concentration before and after adsorption using eqn (1):42–44| |  | (1) |
where qe (mg g−1) is the adsorption capacity, V (L) is the solution volume, C0 is the initial concentration, Ce is the final concentration, and m (g) is the mass of the adsorbent used.
2.2.6. Characterization of adsorbents.
The adsorbents were characterized using multiple techniques: Brunauer–Emmett–Teller (BET) surface area (Micromeritics Instrument Corp, Gemini VII Version 5.03), FTIR spectrometer (Nicolet Avatar 360 IR), X-ray diffractometer (XRD) (Rigaku Multiplex), scanning electron microscope (SEM)/energy-dispersive X-ray (EDX) spectroscopy (JEOL JSM-6360LA), and vibrating sample magnetometer (VSM) (Physical Properties Measurement System Quantum Design PPMS® VersaLab™ Cryogen-free 3 Tesla).
2.2.7. Determination of kinetics, isotherms, and thermodynamics adsorption.
The kinetic models used to study CIP and CAP adsorption on NMM@SiO2/alkylsilane included pseudo-first-order, pseudo-second-order, and Elovich models. Isotherm models applied were Langmuir, Freundlich, and Redlich–Peterson (R–P) models. The kinetics and isotherm models are presented in Tables S1 and S2 (ESI†), respectively. Thermodynamic data were obtained from temperature variations at 303, 313, and 323 K, each with 50, 100, 150, 200, and 250 mg L−1 concentrations. K Values were plotted for each temperature as lnK vs. 1/T, allowing calculations of ΔH°, ΔS° dan ΔG° from the resulting equations. The equations used to calculate thermodynamics are presented in eqn (S1)–(S3) (ESI†).
2.2.8. Desorption and reusability of NMM@SiO2/alkylsilane.
Desorption tests for CIP and CAP were performed using three eluents: double-distilled water, 0.1 M NaOH, and 40% ethanol. The antibiotic desorption capacity (qedes, mg g−1) was calculated with eqn (2), and the desorption percentage using eqn (3):15| |  | (2) |
| |  | (3) |
where V (L) is the eluent volume, m (g) is the mass of NMM@SiO2/alkylsilane, Cf (mg L−1) is the CAP concentration in the eluent solution, and D represents the desorption efficiency (%). The reusability of NMM@SiO2/alkylsilane was tested over five cycles of adsorption–desorption. After each desorption under optimal conditions, the adsorption capacity for reusability was calculated using eqn (1).
3. Results and discussion
3.1. FTIR characterization
Fig. 2 shows the FTIR spectra of NMM, NMM@SiO2, NMM@SiO2/OTMS(3, 7.5, and 15), and NMM@SiO2/HDTMS(3, 7.5, and 15). Fig. 2 confirms the successful functionalization of OTMS and HDTMS on silica-coated NMM. The asymmetric and symmetric C–H stretching peaks of CH2 appear at wavenumbers 2926 and 2856 cm−1, respectively, with a bending peak at 1466 cm−1.45,46 These peaks are present in the spectra of NMM@SiO2/OTMS(3), NMM@SiO2/OTMS(7.5), and NMM@SiO2/OTMS(15), showing varying intensities without any shift. Similarly, the asymmetric and symmetric C–H stretching of –CH2 is observed at 2922 and 2852 cm−1, with a bending peak at 1468 cm−1 in the spectra of NMM@SiO2/HDTMS(3), NMM@SiO2/HDTMS(7.5), and NMM@SiO2/HDTMS(15), also with different intensities and no shift.47,48 The appearance of these absorption peaks confirms the silanization process and the presence of octyl and hexadecyl groups on the synthesized adsorbent material.45 The difference between OTMS and HDTMS lies in the higher and narrower intensity of HDTMS, indicating a longer –CH2 chain length than OTMS. The increase in functional group intensity correlates with the amount of OTMS and HDTMS (in mmol) added, where a greater amount results in a higher observed intensity. A broad absorption band at 3443–3452 cm−1 and a sharp band at 1636–1640 cm−1 correspond to the stretching and bending of hydrogen bonds (O–H), respectively.47,49 The peak with the highest intensity and absorption is associated with siloxane (Si–O–Si). The antisymmetric and symmetric vibrations appear in the wavenumber ranges 1075–1087 cm−1 and 795–804 cm−1, respectively,47 with siloxane bending occurring at 460–466 cm−1.45,46
 |
| | Fig. 2 FTIR spectra of NMM@SiO2/alkylsilane materials. | |
These peaks indicate a silylation reaction in which silane groups replace the single bonds of polar (–OH) groups on silica, leading to the polymerization of silane molecules.45 This silylation reaction provides evidence of chemical modification between the silica surface and OTMS and HDTMS. This chemical bonding imparts hydrophobicity to the synthesized NMM@SiO2/alkylsilane material. As reported by Ramezani et al. (2014),46 functionalization of isooctyltrimethoxysilane on silica shows characteristic asymmetric and symmetric –CH2 stretching, as well as siloxane peaks, and results in increased hydrophobicity of the synthesized material. Similarly, Xu & Zhang (2021)47 and Parera et al. (2019)48 successfully modified HDTMS on nano-silica surfaces, achieving hydrophobic properties.
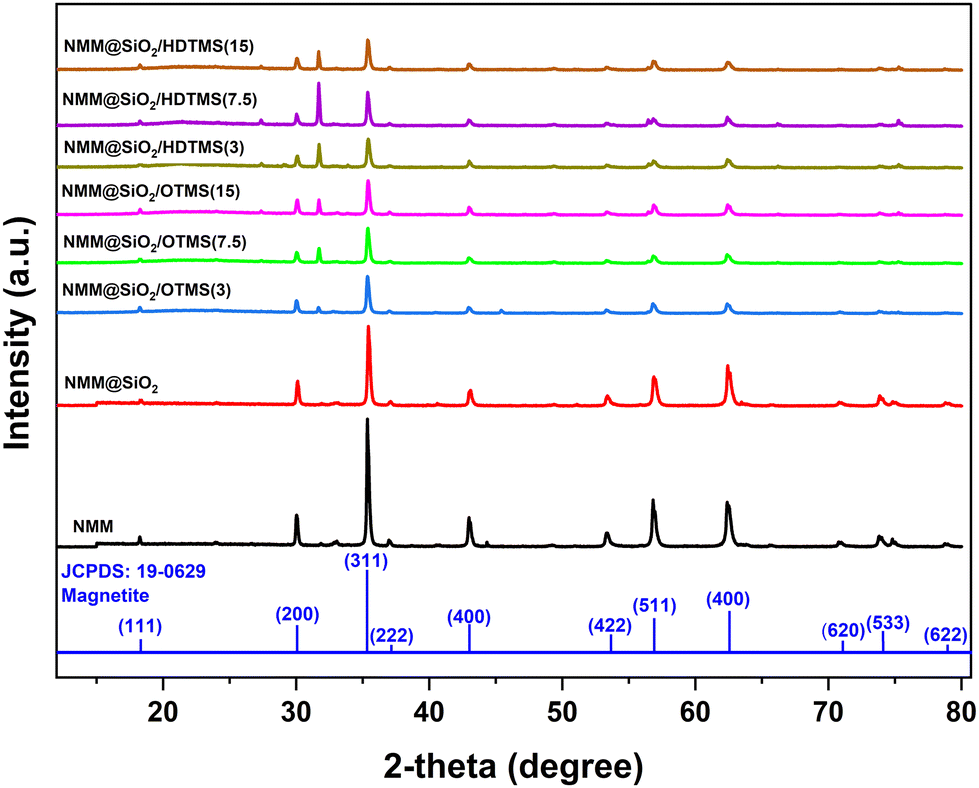
3.2. XRD characterization
Fig. 3 shows the XRD characterization results of the adsorbent materials NMM, NMM@SiO2, NMM@SiO2/OTMS, and NMM@SiO2/HDTMS. The XRD results, consistent with JCPDS standard number 19-0629, indicate that the crystal phase of magnetite (Fe3O4) exhibits the highest correspondence.15 The XRD analysis shows peaks matching the crystal plane orientations (111), (220), (311), (222), (400), (422), (511), (440), (620), (533), and (622), appearing at 2θ values of 18.26°, 30.03°, 35.37°, 36.99°, 42.96°, 53.36°, 56.86°, 62.45°, 70.86°, 73.86°, and 75.25°, respectively. Fig. 3 also indicates that coating with OTMS and HDTMS does not alter the core crystal structure of the natural magnetic material, although changes in intensity and crystallinity are observed. The intensity of the crystal plane orientations decreases with increasing mmol quantities of OTMS and HDTMS. The crystallinity of NMM@SiO2/OTMS decreases with higher amounts of OTMS, while NMM@SiO2/HDTMS shows an increase. The variation in crystallinity due to the quantity of added mmol remains insignificant, with a change of ≤ 2%. Crystallinity values for NMM@SiO2/OTMS (3, 7.5, and 15) are 52.6%, 51.4%, and 50.8%, respectively, while for NMM@SiO2/HDTMS (3, 7.5, and 15), they are 55.7%, 57.5%, and 57.7%. The increase in crystallinity with more HDTMS added is likely due to a denser atomic arrangement, resulting in a more structured material. These findings are also related to the hydrophobicity values (Fig. 4) and the surface morphology observed in SEM images (Fig. 6a–c). The increased HDTMS amount corresponds to higher hydrophobicity due to a denser layer formation. The surface morphology of NMM@SiO2/HDTMS appears dense, rigid, non-granular, and non-porous. In similar studies, such as those of Wei et al. (2016),50 coating starch nanocrystals with HDTMS resulted in dense, hydrophobic, multilayered materials.
 |
| | Fig. 3 XRD patterns of NMM@SiO2/alkylsilane materials. | |
 |
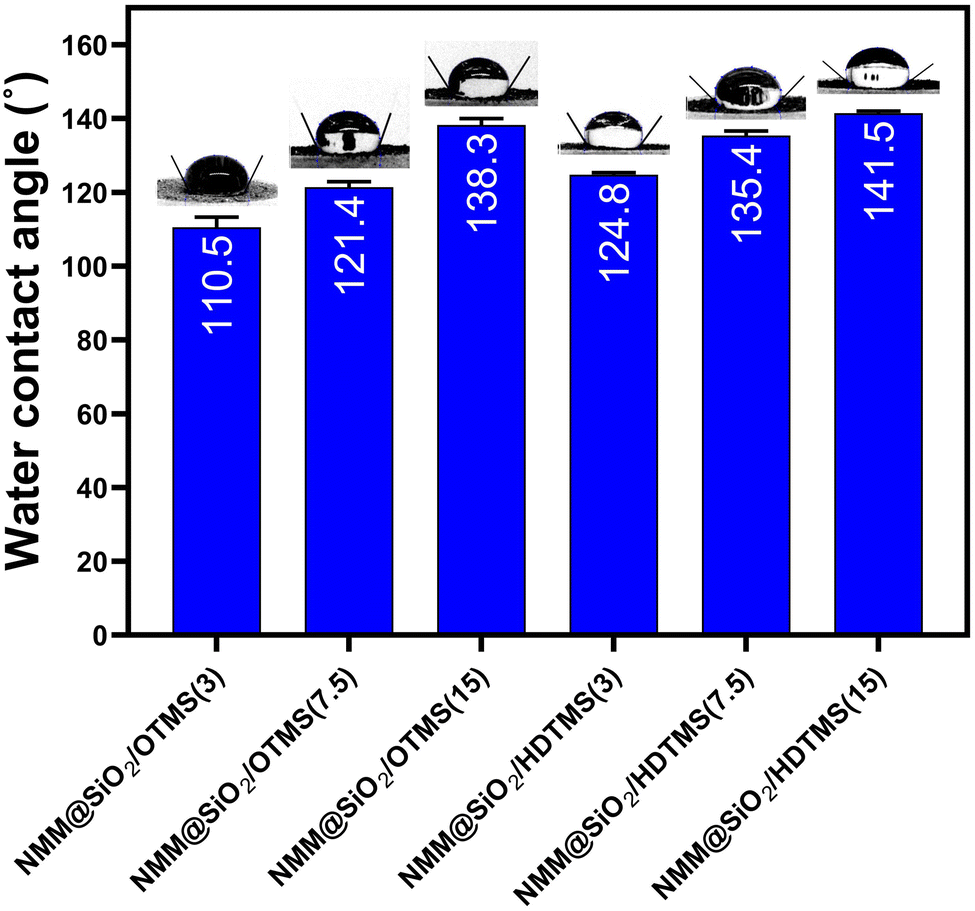
| | Fig. 4 Water contact angle of materials adsorbent. | |
3.3. Hydrophobicity of materials
The hydrophobicity values of NMM@SiO2/OTMS and NMM@SiO2/HDTMS are shown in Fig. 4, measured through water contact angle (WCA). The hydrophobicity values for NMM@SiO2/OTMS(3), NMM@SiO2/OTMS(7.5), and NMM@SiO2/OTMS(15) are 110.5 ± 1.5°, 121.4 ± 1.3°, and 138.3 ± 1.6°, respectively. Meanwhile, the values for NMM@SiO2/HDTMS(3), NMM@SiO2/HDTMS(7.5), and NMM@SiO2/HDTMS(15) are 124.8 ± 0.5°, 135.4 ± 1.2°, and 141.5 ± 0.6°, respectively. Hydrophobicity increases with higher mmol amounts of OTMS or HDTMS due to the stability of the hydrophobic layer formed through stronger hydrophobic interactions between alkyl groups, creating a more stable surface. NMM@SiO2/HDTMS exhibits the highest hydrophobicity among these functional groups due to its longer alkyl chain (16 carbon atoms), while OTMS has only 8. The longer alkyl chains create a denser and more structured layer on the material surface as a physical barrier to water absorption. The correlation between hydrophobicity and surface area of HDTMS-modified materials is also evident. Similarly, Pyo & Chang (2021)51 reported that increasing the alkyl chain length on mesoporous silica surfaces also increases WCA values, as observed by Novak et al. (2020)52 with octyltriethoxysilane-modified MFI zeolites.
3.4. Adsorbent variation
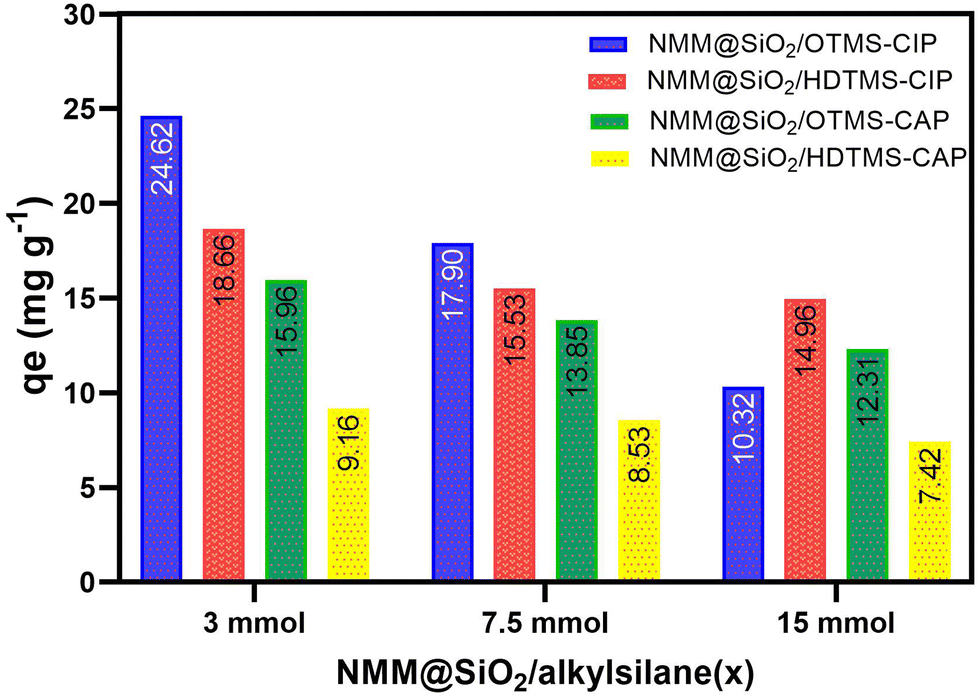
Fig. 5 compares CIP and CAP adsorption for different adsorbent variations, with 12 variations in Table 1, specifically 3, 7.5, and 15 mmol for each OTMS and HDTMS variation. Notably, The 3 mmol OTMS and 3 mmol HDTMS showed the highest adsorption capacities among the others (7.5 and 15 mmol). The adsorption capacities for CIP with NMM@SiO2/OTMS(3) and NMM@SiO2/HDTMS(3) were 24.62 and 18.66 mg g−1, respectively, while for CAP, they were 15.96 and 9.16 mg g−1. Due to the highest adsorption capacities, these NMM@SiO2/OTMS(3) and NMM@SiO2/HDTMS(3) adsorbents were further studied for pH, contact time, initial concentration, kinetic model, isotherm, thermodynamics, desorption, and reusability in subsequent sections.
 |
| | Fig. 5 Comparison of modified NMM@SiO2/OTMS and NMM@SiO2/HDTMS [C0 = 100 mg L−1, m = 20 mg, V = 20 mL, time = 60 min (CIP), 90 min (CAP), pH = 6 (CIP and CAP), rotary speed = 250 rpm, T = 25 °C]. | |
3.5. SEM-EDX mapping characterization
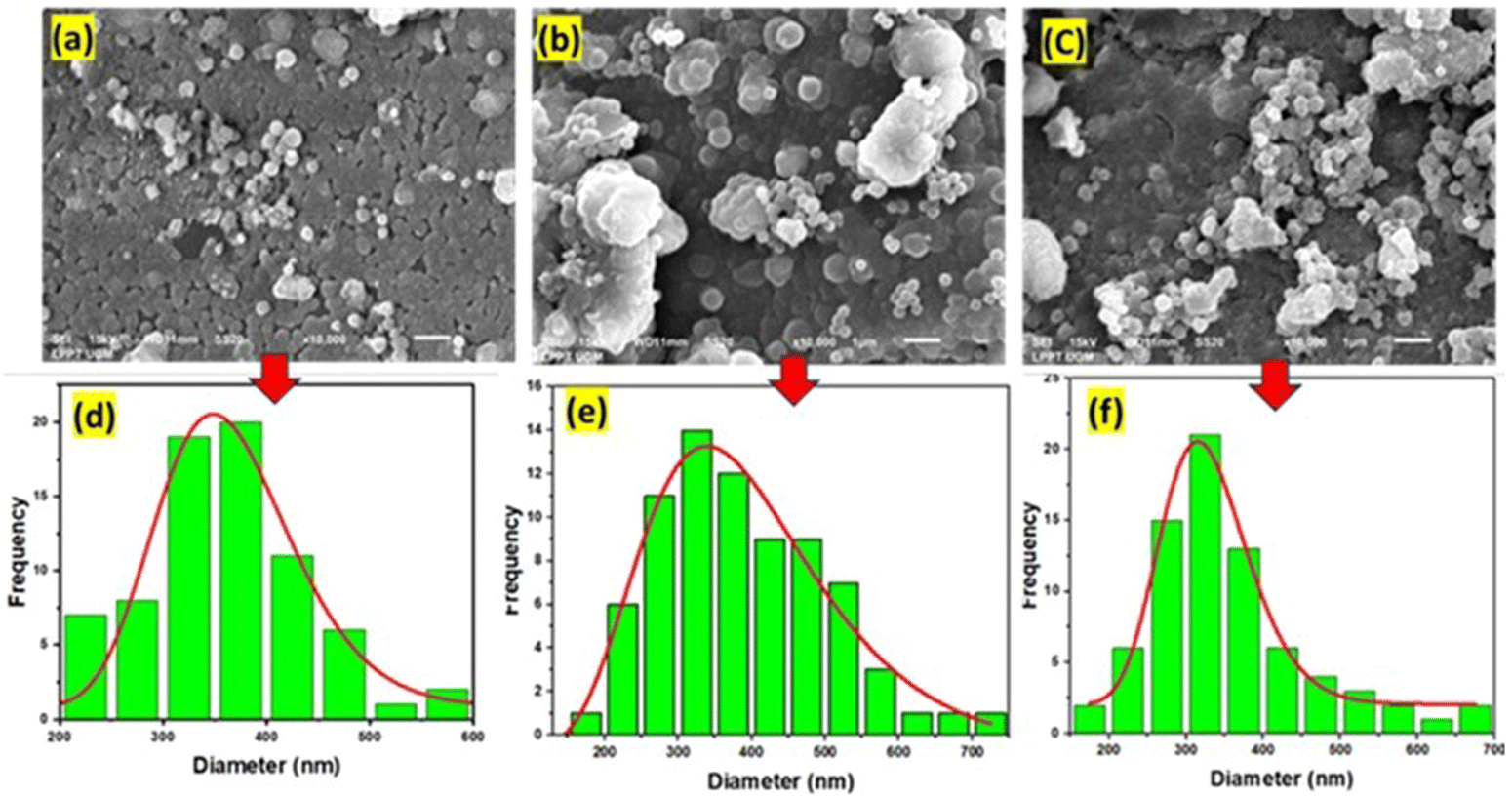
Fig. 6 shows the surface morphology and particle size of NMM@SiO2/OTMS (3, 7.5, and 15). The surface morphology of NMM@SiO2/HDTMS (3, 7.5, and 15) can be seen in Fig. S1 (ESI†). The surface morphology of NMM@SiO2/OTMS (3, 7.5, and 15) shows particles in the form of grains with clearly defined grain boundaries. These spherical particles are nearly uniformly distributed on the adsorbent surface. The particles also form larger clusters through agglomeration. The SEM images reveal that increasing the OTMS quantity results in greater agglomeration on the material surface. Similarly, Novak et al. (2020)52 have modified MFI zeolites with 0, 5, 10, and 15% OTMS, producing varying surface morphologies. At 10 and 15% OTMS, irregularly aggregated particles with different sizes and shapes were observed. Particle size analysis of NMM@SiO2/OTMS(3, 7.5, and 15) revealed sizes between 100–700 nm, with the highest frequency between 300–400 nm. In contrast to OTMS, the surface morphology of NMM@SiO2/HDTMS (Fig. S1, ESI†) appears dense, rigid, non-granular, and non-porous. This phenomenon is due to the longer HDTMS chains covering the NMM@SiO2 surface. Similar findings were reported by Li et al. (2023),53 who coated SiO2-HDTMS on AZ91 alloy, resulting in a dense surface with HDTMS uniformly covering the AZ91 alloy. Similarly, Sinha Ray et al. (2021)54 demonstrated that HDTMS coating on silica nanoparticles-coated carbon nanofibers produced a dense, non-hollow, non-porous surface. Coating HDTMS on starch nanocrystals (SNC) by Wei et al. (2016)50 created a dense, hydrophobic, and multilayered surface due to the longer carbon chain of HDTMS.
 |
| | Fig. 6 SEM images of (a) NMM@SiO2/OTMS(3), (b) NMM@SiO2/OTMS(7.5), and (c) NMM@SiO2/OTMS(15), and particle size of (d) NMM@SiO2/OTMS(3), (e) NMM@SiO2/OTMS(7.5), and (f) NMM@SiO2/OTMS(15). | |
The EDX results, showing the elemental composition of NMM@SiO2/OTMS(3) and NMM@SiO2/HDTMS(3), are shown in Fig. 7(c and d). The elements present in the material include carbon, oxygen, silicon, and iron, with respective percentages of 5.14%, 53.38%, 12.01%, and 29.46% for NMM@SiO2/OTMS(3) and 5.30%, 61.22%, 7.87%, and 25.60% for NMM@SiO2/HDTMS(3). Carbon in both materials confirms the successful modification with OTMS and HDTMS, as evidenced by the mapping results. Based on the EDX results, the percentages of OTMS and HDTMS bound to the material are 62.66% (1.88 mmol) and 77.27% (2.32 mmol), respectively. Fig. 7(a and b) shows the mapping results for NMM@SiO2/OTMS(3) and NMM@SiO2/HDTMS(3). The mapping of NMM@SiO2/OTMS and NMM@SiO2/HDTMS demonstrates that elements are uniformly distributed across the material surface. The mapped elements include dark blue for carbon, red for oxygen, light blue for silicon, and green for iron (Fig. 7a and b). SEM-EDX mapping shows the successful modification with OTMS and HDTMS on the silica-coated natural magnetic material. This finding is consistent with the VSM curve (Fig. 8a), which shows a decrease in the magnetic saturation (Ms) value of the natural magnetic material from 58.43 emu g−1 to 36.49 emu g−1 and 31.69 emu g−1 after being coated with silica and functionalized with OTMS and HDTMS, respectively.15 The FTIR curve (Fig. 2) also shows characteristic OTMS and HDTMS functional groups at specific wavelengths. The WCA measurement results (Fig. 4) indicate hydrophobic properties, confirming the successful synthesis of the material.
 |
| | Fig. 7 SEM mapping of (a) NMM@SiO2/OTMS(3), (b) NMM@SiO2/HDTMS(3), and EDX spectra of (c) NMM@SiO2/OTMS(3), (d) NMM@SiO2/HDTMS(3). | |
 |
| | Fig. 8 (a) VSM hysteresis VSM hysteresis and adsorbent separation using an external magnetic field, and (b) BET analysis curves. | |
3.6. VSM characterization
Fig. 8(a) shows the magnetic properties of the synthesized materials NMM@SiO2/OTMS(3) and NMM@SiO2/HDTMS(3). The saturation magnetization (Ms), remanent magnetization (Mr), and coercivity field (Hc) values for NMM@SiO2/OTMS(3) are 31.69, 0.38 emu g−1, and 1.62 emu g−1, respectively, while for NMM@SiO2/HDTMS(3), they are 36.49, 0.29 emu g−1, and 4.63 emu g−1. Both materials, with Ms values of 31.69 and 36.49 emu g−1, are still separable using an external magnetic field (Fig. 8a). The Ms and Mr values show that both materials can respond effectively to an external magnetic field and are well-dispersed after the magnetic field is removed.55 As shown in Fig. 8(a), both materials can be easily separated from suspension systems after use. The complete separation of both adsorbents after the adsorption process occurs within one minute. The S-like curves in the magnetic hysteresis loops, with low remanence and coercivity values, indicate soft ferromagnetic properties at room temperature. Similar studies have been reported by Mosayebi et al. (2020),56 which functionalized OTMS on GO-Fe3O4 for transesterification reactions, yielding an Ms of 29.1 emu g−1. Chang et al. (2011)57 coated hexadecyltrimethoxysilane on C16/SiO2–Fe3O4 NPs for Rhodamine 6G removal, achieving an Ms of 35 emu g−1, effective for magnetic material separation. Peng et al. (2016)58 coated HDTMS on a superhydrophobic magnetic cellulose sponge (SMCS), obtaining a Ms of 12.84 emu g−1, indicating strong magnetization.
3.7. BET analysis
Fig. 8(b) shows the adsorption isotherms for NMM@SiO2/OTMS(3) and NMM@SiO2/HDTMS(3), which follow a Type IV isotherm, as shown by the hysteresis loops based on IUPAC classification.59 The specific surface areas of NMM@SiO2/OTMS(3) and NMM@SiO2/HDTMS(3) are 15.89 m2 g−1 and 33.50 m2 g−1, respectively, with NMM@SiO2/HDTMS(3) exhibiting a higher surface area due to its longer HDTMS chains covering the entire NMM@SiO2 surface, resulting in a more orderly structure. The HDTMS chain has a longer alkyl chain than OTMS, which leads to stronger intermolecular interactions and the formation of a denser hydrophobic layer on the surface of NMM@SiO2. This finding is consistent with Novak et al. (2020),52 who reported that 5% of OTMS-functionalized MFI zeolites exhibited hysteresis loops at p/p0 > 0.4. Pyo & Chang (2021)51 modified mesoporous silica with alkylsilanes (C3, C8, C12, and C18), resulting in Type IV hysteresis loops and increased surface area with longer alkyl chain lengths.
3.8. Optimal pH
Fig. 9(a) shows the of NMM@SiO2/OTMS(3) and NMM@SiO2/HDTMS(3). The pHPZC values for both adsorbents are 6.01 and 6.02, respectively. A pHPZC of 6.01 indicates that the adsorbent charge is neutral, with positive charges below 6.01 and negative charges above 6.01. Fig. 9(b) also shows the optimal pH for CIP and CAP adsorption using NMM@SiO2/OTMS(3) and NMM@SiO2/HDTMS(3). The pH range reported in this study is from pH 2–10. The optimal pH for CIP adsorption occurs at pH 6, with 27.84 and 21.50 mg g−1 adsorption capacities for NMM@SiO2/OTMS(3) and NMM@SiO2/HDTMS(3). The highest adsorption capacity at pH 6 is due to hydrophobic interactions between the adsorbent and CIP. CIP also has pKa values of 5.90 and 8.89.60 Below pH 5.90, CIP+ is formed due to protonation of the piperazine amine group, whereas above pH 8.89, CIP− is formed due to deprotonation of the carboxyl group (Fig. 9c).61 At pH 6, the adsorbent (pHPZC = 6.01) and CIP are neutral, forming hydrophobic bonds between the adsorbent and CIP. Similar findings were also reported by Movasaghi et al. (2019)62 and Li et al. (2014),63 who found that hydrophobic bonds could form at pH 6–7 because, at this pH, CIP is in the CIP0 form. Aydin et al. (2019)64 adsorbed CIP using Fe3O4/red mud nanoparticles, finding an optimal pH of 6. CIP exists in cationic form due to the protonation of the secondary amine on the piperazine group when the solution pH is below 5.90. When the solution pH is between 5.90 and 8.90, CIP exists as a zwitterion. If the pH is above 8.89, CIP is in an anionic form (Fig. 9c). Movasaghi et al. (2019) CIP adsorption over a pH range of 3–10 was also reported. The optimal pH for CAP adsorption also occurs at pH 6 for NMM@SiO2/OTMS(3) and NMM@SiO2/HDTMS(3), with capacities of 16.25 and 14.71 mg g−1, respectively. The optimal CAP adsorption at pH 6 is due to the CAP neutral form (Fig. 9d), making it more nonpolar and allowing interaction with the adsorbent's hydrophobic surface. The dominant interaction between the adsorbents, CIP, and CAP is hydrophobic interaction with van der Waals forces.
 |
| | Fig. 9 Curve for pHPZC determination (a) effect of pH on adsorption (b) species at pH variations: CIP (c), and CAP (d). | |
Overall, the adsorption capacity of NMM@SiO2/OTMS(3) is higher than that of NMM@SiO2/HDTMS(3) for both CIP and CAP. This high adsorption capacity is attributed to the alkyl chain length and hydrophobic properties. The longer alkylsilane chains in HDMS-modified adsorbents create a more significant steric barrier, reducing the number of active sites available for interaction with CIP and CAP molecules on the adsorbent surface. The long alkyl chains of NMM@SiO2/HDTMS can form a dense hydrophobic layer, thereby limiting the interaction of CAP and CIP molecules with the adsorbent surface. The longer alkyl chains may overlap, creating a steric barrier that further restricts the interaction of CIP and CAP molecules. In contrast, NMM@SiO2/OTMS, with its shorter alkyl chains, provides more open space between functional groups, allowing easier interaction of CIP and CAP molecules despite having a smaller specific surface area than NMM@SiO2/HDTMS. The hydrophobic properties are due to the adsorbents functionalized with octyltrimethoxysilane and hexadecyltrimethoxysilane, exhibiting hydrophobicity levels of 110.5° (OTMS) and 124.8° (HDTMS). The shorter alkyl chains in OTMS allow for more effective adsorption of CIP and CAP molecules.
3.9. Optimum contact time and adsorption kinetics models for CIP and CAP
The optimal contact time was studied to determine the most effective duration for CIP and CAP adsorption by the adsorbent.65Fig. 10(a) shows the optimization of contact time for CIP and CAP adsorption using NMM@SiO2/OTMS(3) and NMM@SiO2/HDTMS(3) adsorbents. The optimal contact time for CIP adsorption by NMM@SiO2/OTMS(3) and NMM@SiO2/HDTMS(3) was 60 min, with adsorption capacities of 28.03 and 24.15 mg g−1, respectively. The optimal contact time for CAP adsorption by NMM@SiO2/OTMS(3) was 90 min, with an adsorption capacity of 17.02 mg g−1, and for NMM@SiO2/HDTMS(3), it was 50 min with an adsorption capacity of 14.04 mg g−1. The increase in adsorption over time indicates that more empty sites were available for adsorption until reaching the optimum condition. No empty sites were available after the optimal contact time, resulting in an adsorbent-adsorbate equilibrium.
 |
| | Fig. 10 Curves of correlation between the amount of adsorbed antibiotics versus contact time (a), adsorption kinetic models (b)–(d). | |
Fig. 10(b–d) shows the kinetic model, and Table 2 presents the adsorption parameters for CIP and CAP using both NMM@SiO2/OTMS(3) and NMM@SiO2/HDTMS(3) adsorbents. This article reports three kinetic models used to study CIP and CAP adsorption: PFO, PSO, and Elovich. Based on the modelling results in Fig. 10(b–d), the adsorption isotherm of CIP and CAP using NMM@SiO2/OTMS(3) and NMM@SiO2/HDTMS(3) adsorbents follows a pseudo-second-order (PSO) kinetic model, as the R values are closest to 1.66 The PSO kinetic model suggests that the adsorption rate is proportional to the number of active sites on the adsorbent surface. The adsorption capacity and rate for CIP using NMM@SiO2/OTMS(3) and NMM@SiO2/HDTMS(3) were 27.624 mg g−1 (k2 = 1.416 g mg−1 min−1) and 24.630 mg g−1 (k2 = 0.795 g mg−1 min−1), respectively. For CAP, the adsorption capacity and rate using NMM@SiO2/OTMS(3) and NMM@SiO2/HDTMS(3) were 16.313 mg g−1 (k2 = 0.484 g mg−1 min−1) and 15.863 mg g−1 (k2 = 0.122 g mg−1 min−1), respectively. The results are similar and comparable to those previously reported. Aydin et al. (2019)64 studied CIP adsorption using Fe3O4/red mud nanoparticles and found that the PSO kinetic model was followed with R2 = 0.9971, qe = 111.11 mg g−1, and a rate of 8.79 × 10−4 min−1. Danalıoğlu et al. (2017)67 studied CIP adsorption using levulinic acid-coated Fe3O4 and found that the PSO model was followed with R2 = 0.99. Wu et al. (2018)68 and Idris et al. (2020)69 used nZVI/AC and silica-grafted MIPs for CAP adsorption, and both studies found that the PSO kinetic model was followed, with R2 = 0.999.
Table 2 Kinetic adsorption parameters of CIP and CAP
| Adsorbent |
Antibiotics |
PFO |
PSO |
Elovich |
|
q
e (mg g−1) |
k
1 (min−1) |
R
2
|
q
e (mg−1) |
k
2 (g mg−1 min−1) |
R
2
|
β
|
α mg L−1 min−1 |
R
2
|
| NMM@SiO2/OTMS(3) |
CIP |
4.22 |
0.028 |
0.955 |
27.624 |
1.416 |
0.999 |
0.77 |
3.00 × 106 |
0.941 |
| NMM@SiO2/HDTMS(3) |
6.24 |
0.126 |
0.968 |
24.630 |
0.795 |
0.998 |
0.54 |
1.33 × 104 |
0.912 |
| NMM@SiO2/OTMS(3) |
CAP |
5.93 |
0.024 |
0.896 |
16.313 |
0.484 |
0.993 |
0.64 |
464.01 |
0.794 |
| NMM@SiO2/HDTMS(3) |
8.08 |
0.182 |
0.679 |
15.863 |
0.122 |
0.925 |
0.26 |
5.40 |
0.847 |
3.10. Optimum initial concentration and isotherm models for CIP and CAP adsorption
Fig. 11(a) reports the optimal CIP and CAP adsorption concentration curve using NMM@SiO2/OTMS(3) and NMM@SiO2/HDTMS(3). The concentration range studied was from 10 to 300 mg L−1. The optimal adsorption concentration for CIP and CAP occurred at 250 mg L−1 for both adsorbents. The adsorption capacities of CIP at this optimal concentration for NMM@SiO2/OTMS(3)and NMM@SiO2/HDTMS(3) were 87.83 and 59.54 mg g−1, respectively, while for CAP, they were 56.44 and 39.13 mg g−1. As shown in Fig. 11(a), at low concentrations below 250 mg L−1, most active sites on the adsorbent surface are unoccupied, allowing CIP and CAP molecules to adsorb. However, as the concentration increases, more CIP and CAP molecules occupy the active sites or form hydrophobic interactions on the surfaces of NMM@SiO2/OTMS(3) and NMM@SiO2/HDTMS(3) adsorbents. At a concentration of 250 mg L−1, all active sites are engaged in hydrophobic interactions, forming optimally filled layers. Above 250 mg L−1, the active sites begin to saturate, resulting in stability and a decline in adsorption on the adsorbent surface.
 |
| | Fig. 11 (a) Correlation curves between the amount of adsorbed antibiotics versus initial concentration, and (b)–(d) adsorption isotherm models. | |
Fig. 11(b–d) shows the isotherm model plot for CIP and CAP adsorption using both adsorbents, while Table 3 presents the adsorption isotherm parameters for CIP and CAP. Three isotherm models were used to study CIP and CAP adsorption: Langmuir, Freundlich, and Redlich–Peterson. Fig. 11(b–d) and Table 3 shows that the adsorption isotherm follows the Freundlich model, as the R2 value is closest to 1. The Freundlich model indicates that CIP and CAP adsorption by NMM@SiO2/OTMS(3) and NMM@SiO2/HDTMS(3) occurs through multilayer adsorption on heterogeneous binding sites via hydrophobic interactions. If the value of n < 1, then the adsorption process is a chemical process; if the value of n > 1, adsorption is a physical process.70 The n value provides valuable information about the adsorption process. In this study (Table 3), the value n > 1 was obtained, indicating that adsorption is a physical process. Several studies related to CIP and CAP adsorption following the Freundlich isotherm model include Zhao et al. (2020),71 who adsorbed CAP using media-molecularly imprinted polymers (CAP-RAM-MIPs), and Idris et al. (2020),69 who used molecularly imprinted polymers (MIPs). Zhou et al. (2019)21 adsorbed CIP using Fe3O4/graphene oxide/citrus peel-derived materials and obtained the Freundlich model. Aydin et al. (2019)64 adsorbed CIP using Fe3O4/red mud nanoparticles, Movasaghi et al. (2019)62 used pretreated oat hulls, and Yin et al. (2018) used schorl as an adsorbent; all followed the Freundlich isotherm model.
Table 3 Isotherm Adsorption Parameters of CIP and CAP
| Adsorbent |
Antibiotics |
Langmuir |
Freundlich |
Redlich-Piterson |
|
q
m (mg g−1) |
K
L (L mg−1) |
R
2
|
K
F (L g−1) |
n
F
|
q
e (mg g−1) |
R
2
|
β (mg L−1)−β |
K
RP (L g−1) |
R
2
|
| NMM@SiO2/OTMS(3) |
CIP |
312.50 |
0.001 |
0.025 |
0.566 |
1.135 |
87.83 |
0.931 |
0.1184 |
0.5689 |
0.197 |
| NMM@SiO2/HDTMS(3) |
217.39 |
0.001 |
0.020 |
0.340 |
1.109 |
59.54 |
0.929 |
0.0985 |
1.0797 |
0.136 |
| NMM@SiO2/OTMS(3) |
CAP |
63.694 |
0.003 |
0.423 |
0.253 |
1.101 |
56.44 |
0.949 |
0.0917 |
1.373 |
0.162 |
| NMM@SiO2/HDTMS(3) |
99.010 |
0.002 |
0.101 |
5.737 |
1.036 |
39.13 |
0.935 |
0.0349 |
1.746 |
0.019 |
3.11. Thermodynamics of CIP and CAP adsorption
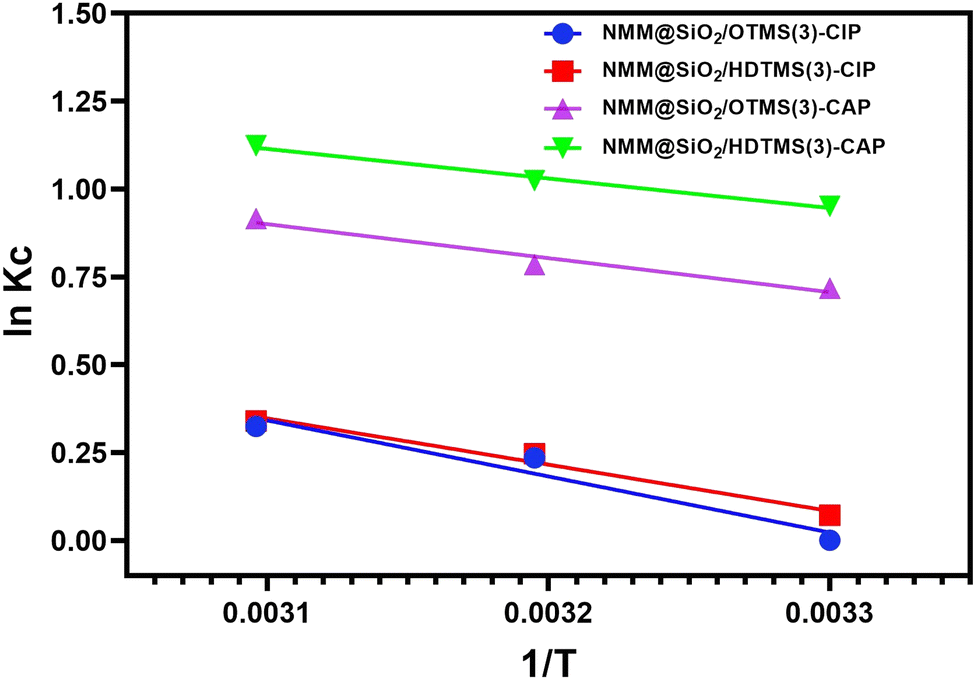
Fig. 12 and Table 4 report the thermodynamic analysis results for CIP and CAP adsorption using the adsorbents NMM@SiO2/OTMS(3) and NMM@SiO2/HDTMS(3). The enthalpy change (ΔH) values for CAP on NMM@SiO2/OTMS(3) and NMM@SiO2/HDTMS(3) are −8.017 and −6.993 kJ mol−1, respectively, while for CIP, they are −13.208 and −10.924 kJ mol−1. The negative signs indicate that the adsorption model for CIP and CAP follows an exothermic reaction.15 Based on the ΔH (kJ mol−1) values, the interaction between CIP and CAP can be classified as physical adsorption or physisorption since the ΔH values (kJ mol−1) fall within the 5–20 kJ mol−1 range. These results align with the Freundlich adsorption isotherm model obtained for CIP and CAP, indicating the presence of hydrophobic and van der Waals interactions. Studies on hydrophobic interactions in CIP and CAP adsorption have been reported by Peng et al. (2015),20 Zhou et al. (2019),21 and Peng et al. (2018).72 The negative ΔG (kJ mol−1) values indicate spontaneous adsorption. Increasingly negative ΔG values suggest a system's tendency to reach equilibrium, and these antibiotics’ adsorption is more favorable at higher temperatures. The positive ΔS (kJ mol−1 K−1) values indicate increased randomness as CIP and CAP molecules bind to the adsorbent surface, increasing system entropy. The high R2 values, close to 1, suggest that the adsorption of these two antibiotics reliably depends on the values of ΔH°, ΔS°, and ΔG°. The signs and magnitudes of the adsorption parameters for CIP and CAP are consistent with studies by Hu & Wang (2016),73 Huang et al. (2014),74 Yin et al. (2018),70 Yu et al. (2018),75 Zheng et al. (2018),76 and Zhu et al. (2015).77
 |
| | Fig. 12 The thermodynamic curve between lnKcversus 1/T. | |
Table 4 Thermodynamic parameters of CAP and CIP adsorption
| Adsorbent |
Adsorbate |
T (K) |
ΔG (kJ mol−1) |
ΔH (kJ mol−1) |
ΔS (kJ mol−1 K−1) |
R
2
|
| NMM@SiO2/OTMS(3) |
CIP |
303 |
−26.474 |
−13.208 |
13.766 |
0.945 |
| 313 |
−26.912 |
| 323 |
−27.350 |
| NMM@SiO2/HDTMS(3) |
CIP |
303 |
−22.086 |
−10.924 |
13.766 |
0.975 |
| 313 |
−22.454 |
| 323 |
−22.823 |
| NMM@SiO2/OTMS(3) |
CAP |
303 |
−17.815 |
−8.017 |
0.0323 |
0.960 |
| 313 |
−18.138 |
| 323 |
−18.461 |
| NMM@SiO2/HDTMS(3) |
CAP |
303 |
−16.368 |
−6.993 |
0.0309 |
0.989 |
| 313 |
−16.677 |
| 323 |
−16.987 |
3.12 Desorption and reusability
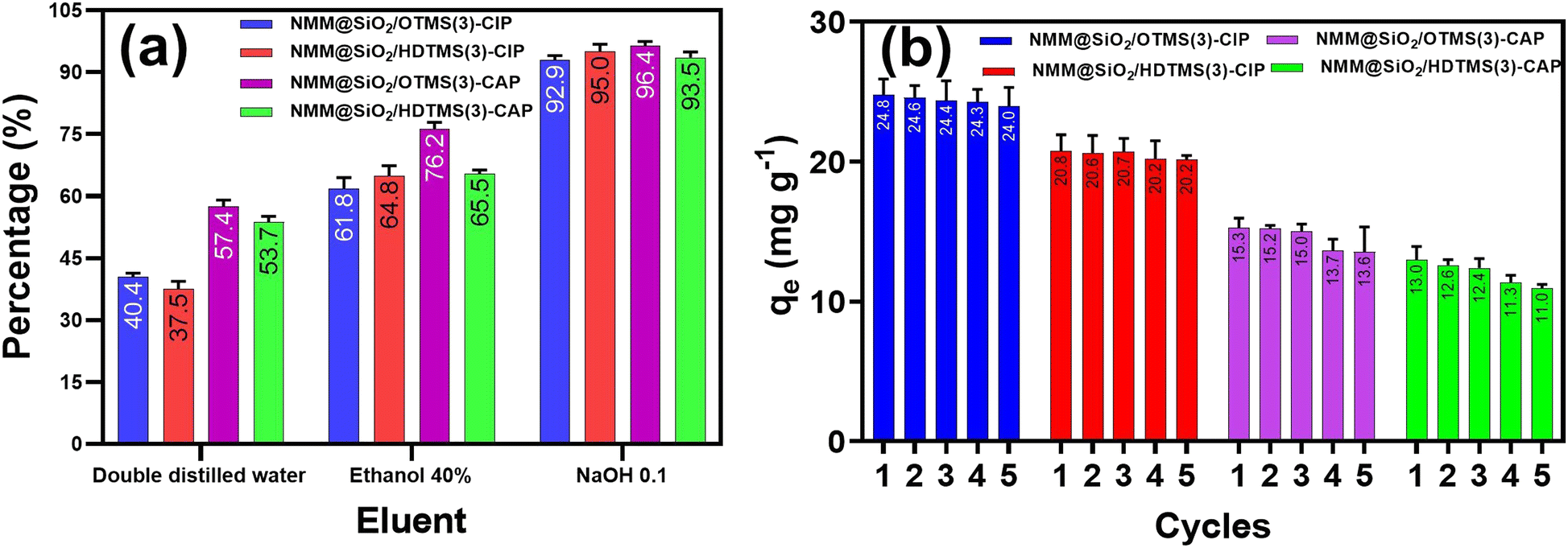
Desorption and reusability studies ensure the potential for reusing the adsorbent and preventing environmental issues if the spent adsorbent is discarded without regeneration.78Fig. 13 shows the desorption and reusability of CIP and CAP adsorption using the adsorbents NMM@SiO2/OTMS(3) and NMM@SiO2/HDTMS(3). Three eluents were used for CIP and CAP desorption: double-distilled water, 40% ethanol, and 0.1 M NaOH. Results indicated that 0.1 M NaOH was the most effective eluent for desorbing CIP and CAP, achieving over 90% desorption. The desorption percentages for CIP were 92.9% and 95% for NMM@SiO2/OTMS(3) and NMM@SiO2/HDTMS(3), respectively; for CAP, they were 96.4% and 93.5%. Ethanol (40%) showed around 61.8–76.2% desorption rates, while double-distilled water achieved between 37.5–57.4%. In similar studies, Ahammad et al. (2021)79 used NaOH as an eluent for CAP, and Chandrasekaran et al. (2020)80 and Duan et al. (2018)81 used NaOH for CIP, all achieving high desorption rates.
 |
| | Fig. 13 (a) Desorbed antibiotic percentage from adsorbent with different eluents and (b) reusability of CIP and CAP adsorption in five cycles. [Conditions: adsorbent = NMM@SiO2/OTMS(3) and NMM@SiO2/HDTMS(3), time = 90 min (CAP), 60 min (CIP), C0 = 100 mg L−1, pH = 6, m = 20 mg, V = 20 mL, rotary speed = 250 rpm, T = 25 °C]. | |
Reusability was tested over five adsorption–desorption cycles using 0.1 M NaOH, which provided the highest desorption efficiency. Results indicated that NMM@SiO2/OTMS(3) and NMM@SiO2/HDTMS(3) adsorbents remained stable for up to five cycles, showing potential as reusable materials for CIP and CAP adsorption–desorption.
3.13. Adsorption mechanisms
The adsorption mechanism can be explained based on isotherm models, thermodynamic parameters, and the properties of the adsorbent material. The Freundlich isotherm model suggests that CIP and CAP adsorption occurs through multilayer adsorption on heterogeneous binding sites. Thermodynamic parameters show that the enthalpy change (ΔH) for CIP and CAP falls within 5–20 kJ mol−1, indicating physical adsorption (physisorption) interactions between the adsorbent and adsorbate. The synthesized OTMS and HDTMS-functionalized adsorbents exhibit hydrophobic properties. Therefore, CIP and CAP adsorption is primarily driven by hydrophobic, dipole–dipole interactions, van der Waals interactions, and hydrogen bond, as illustrated in Fig. 14. The hydrophobic interactions of NMM@SiO2/OTMS(3) and NMM@SiO2/HDTMS(3) involve hydrophobic alkyl chains (C8 and C16). CAP has an aromatic ring and methyl groups attached to its aromatic ring. At the same time, CIP contains a quinolone and cyclopropyl ring, both of which are hydrophobic, allowing for hydrophobic interactions between NMM@SiO2/OTMS(3)-CAP/CIP and NMM@SiO2/HDTMS(3)-CAP/CIP.
 |
| | Fig. 14 Mechanism model of interaction between NMM@SiO2/OTMS and NMM@SiO2/HDTMS for CIP and CAP. | |
The functionalization of OTMS and HDTMS enhances surface hydrophobicity; however, unreacted silanol groups (–Si–OH) may still be present on the silica layer. These residual groups can interact with polar functional groups in CIP and CAP, such as carboxyl (–COOH), amine (–NH), and carbonyl (–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) in CIP, as well as hydroxyl (–OH) and carbonyl (–CO) in CAP, through dipole–dipole interactions. The dipole–dipole interaction between CIP and the adsorbent occurs through the interaction of carbonyl (–CO) with hydrogen atom of silanol (–Si–OH) groups, carboxyl (–COOH) and hydroxyl (-OH) groups with oxygen atoms of the siloxane (–Si–O–Si–). Similarly, in the case of CAP, dipole–dipole interactions take place between nitro (–NO2) and carbonyl (–CO) groups with hydrogen atoms silanol (–Si–OH) groups. Additionally, interaction through hydrogen bonding may occur in CIP adsorption between –NH and –OH with –Si–OH in the silica layer.
O) in CIP, as well as hydroxyl (–OH) and carbonyl (–CO) in CAP, through dipole–dipole interactions. The dipole–dipole interaction between CIP and the adsorbent occurs through the interaction of carbonyl (–CO) with hydrogen atom of silanol (–Si–OH) groups, carboxyl (–COOH) and hydroxyl (-OH) groups with oxygen atoms of the siloxane (–Si–O–Si–). Similarly, in the case of CAP, dipole–dipole interactions take place between nitro (–NO2) and carbonyl (–CO) groups with hydrogen atoms silanol (–Si–OH) groups. Additionally, interaction through hydrogen bonding may occur in CIP adsorption between –NH and –OH with –Si–OH in the silica layer.
van der Waals interactions occur due to the attraction between adjacent molecules at very close distances. In this study, van der Waals interactions likely happen between the C8 and C16 alkyl chains and the aromatic rings in CIP and CAP molecules. The illustration of interaction models that may occur is presented in Fig. 14. Related studies on CIP and CAP adsorption through hydrophobic interactions are listed in Table 5.
Table 5 Comparison of adsorbents in this study with other studies
| Adsorbents |
Adsorbate |
q
max/qe (mg g−1) |
Ref. |
| ZnO nanoparticles |
CIP |
0.160 |
Dhiman & Sharma et al. (2019)82 |
| Montmorillonite |
CIP |
0.600 |
Avci et al. (2019)83 |
| Humic acid/cellulose |
CIP |
10.87 |
Wang et al. (2020)84 |
| Biosorbent from Enteromorpha prolifera |
CIP |
21.70 |
Wu et al. (2015)85 |
| Levulinic acid coated Fe3O4 |
CIP |
53.76 |
Danalıoğlu et al. (2017)67 |
| NMM@SiO2/C1(3) |
CIP |
106.81 |
Naat et al. (2024)15 |
| magnetite imprinted chitosan polymer nanocomposites (Fe-CS NCs) |
CIP |
142 |
Rasoulzadeh et al. (2019)27 |
| Fe3O4@SiO2/SiTBA-ALG |
CIP |
464.423 |
Soares et al. (2019)25 |
| Fe3O4@SiO2-APTMS-HBA |
CIP |
415.3 |
Amirmahani et al. (2020)26 |
| NMM@SiO2/OTMS(3) |
CIP |
87.83 |
This work |
| NMM@SiO2/HDTMS(3) |
CIP |
59.54 |
This work |
| Bamboo charcoal-based biochar |
CAP |
8.1 |
Liao et al. (2013)86 |
| Bamboo charcoal-based biochar |
CAP |
0.65 |
Fan et al. (2020)29 |
| Core–shell molecularly non-imprinted polymers based on magnetic chitosan |
CAP |
11.18 |
Ma et al. (2015)30 |
| Chitosan-based non-molecularly imprinted polymers |
CAP |
6.0 |
Wang et al. (2014)87 |
| Si@MIPs-CAP |
CAP |
32.26 |
Idris et al. (2020)69 |
| Si@NIPs-CAP |
CAP |
29.6 |
Idris et al. (2020)69 |
| NMM@SiO2/C1(3) |
CAP |
58.46 |
Naat et al. (2024)15 |
| Magnetic carboxyl-modified hyper crosslinked resins (MA-50) |
CAP |
193.88 |
Jin et al. (2017)88 |
| Powdered activated carbon (PAC) |
CAP |
304.878 |
Tian et al. (2017)89 |
| NMM@SiO2/OTMS(3) |
CAP |
56.44 |
This work |
| NMM@SiO2/HDTMS(3) |
CAP |
39.13 |
This work |
The adsorption capacities of CIP and CAP observed in this study, as shown in Table 5, fall within a moderate range: 87.83 and 59.54 mg g−1 for CIP and 56.44 and 39.13 mg g−1 for CAP, respectively. Based on Table 5, the decrease in CIP adsorption with increasing alkyl chain length (HDTMS < OTMS < MTMS) indicates that greater hydrophobicity can restrict the movement of CIP molecules to the active sites of the adsorbent. NMM@SiO2/OTMS exhibits a higher adsorption capacity than NMM@SiO2/HDTMS due to its shorter OTMS alkyl chain, which allows for additional interactions such as dipole–dipole interactions and hydrogen bonding with CIP, in contrast, the longer HDTMS alkyl chain can hinder CIP diffusion. A notable advantage of this research is the comparison of two adsorbents with distinct hydrophobic properties, achieved by modifying alkylsilane chain lengths OTMS (C8) and HDTMS (C16), which led to unique adsorbent characteristics. These adsorbents were synthesized from natural materials, with iron sand as the magnetic material source and natural sand as the silica source. The results indicate that the synthesized materials exhibit strong potential as effective adsorbents for removing CIP and CAP in wastewater treatment applications.
4. Conclusions
In this study, we successfully synthesized silica-coated natural magnetic materials functionalized with octyltrimethoxysilane (NMM@SiO2/C8) and hexadecyltrimethoxysilane (NMM@SiO2/C16), both possessing hydrophobic properties. Using a batch adsorption method, these materials were compared for their ability to adsorb ciprofloxacin (CIP) and chloramphenicol (CAP). Results showed that the adsorbents with the highest adsorption capacities were NMM@SiO2/OTMS(3) and NMM@SiO2/HDTMS(3), used for CIP and CAP adsorption. The optimal pH for CIP and CAP adsorption by both adsorbents was 6, with optimal contact times of 90 min for CIP and 60 min for CAP. The optimal concentration for CIP and CAP adsorption was 250 mg L−1. NMM@SiO2/C8(3) demonstrated the highest adsorption capacity, with 87.83 mg g−1 for CIP and 56.44 mg g−1 for CAP. The adsorption kinetics of CIP and CAP followed a pseudo-second-order model, while the adsorption isotherm followed the Freundlich model. Thermodynamic parameters, including negative enthalpy (ΔH), indicate an exothermic reaction, negative ΔG values show spontaneity, and positive ΔS values suggest increased system randomness. The OTMS-functionalized adsorbent showed higher adsorption capacity than the HDTMS-functionalized material. The successful adsorption of CIP and CAP, achieved through introducing hydrophobic groups, promises alternative materials for CIP and CAP removal from water waste.
Abbreviations
| CIP | Ciprofloxacin |
| CAP | Chloramphenicol |
| OTMS | Octyltrimethoxysilane |
| NMM | Natural magnetic materials |
| NMM@SiO2/OTMS(3) | Natural magnetic materials-silica-octyltrimethoxysilane-3 mmol |
| NMM@SiO2/HDTMS(3) | Natural magnetic materials-silica-hexadecyltrimethoxysilane-3 mmol |
|
q
e,(adsorption)
| Adsorption capacity (mg g−1) or adsorbate concentration at equilibrium time (mg g−1) |
|
q
t
| Adsorbate concentration of antibiotic at time t (min) (mg g−1) |
|
k
| Adsorption rate constant |
|
C
0
| Initial concentration of adsorbate in solution (mg L−1) |
|
C
e
| Final concentration (mg L−1) |
|
C
t
| Concentration of antibiotic in solution at time t (mg L−1) |
|
V
| Volume of solution (mL) |
|
m
| Adsorbent mass (g) |
|
q
max
| Maximum amount of adsorbate sorbed (mg g−1) |
|
K
L
| The Langmuir adsorption constant |
|
K
f
| Freundlich constant |
|
n
| The value indicating the degree of linearity between adsorbate solution and the adsorption process |
|
α
| Initial adsorption rate in the Elovich model (mg g−1 s−1) |
|
β
| Constant related to the extent of surface coverage and activation energy for chemisorption in the Elovich model (g mg−1) |
|
β
| Redlich–Peterson constant (mg L−1)−β |
|
K
RP
| Redlich–Peterson constant (L mol−1). |
Author contributions
Johnson Nune Naat: data curation, investigation, writing the original draft. Suyanta: methodology, validation, supervision, writing, review, and editing. Nuryono: conceptualization, methodology, validation, supervision, writing, review, and editing.
Data availability
The data supporting this article have been included in the ESI.†
Conflicts of interest
The authors declare no conflicts of interest.
Acknowledgements
The authors express their gratitude to the Indonesian Education Scholarship (BPI), Center for Higher Education Funding and Assessment Ministry (PPAPT) of Higher Education, Science, and Technology of Republic Indonesia, and Indonesian Endowment Funds for Education (LPDP) for providing PhD scholarship for Johnson Nune Naat and funding for this research on the decree No. 01595/J5.2.3./BPI.06/9/2022. All charts are created using GraphPad Prism 9.
References
- Y. Gao, Y. Li, L. Zhang, H. Huang, J. Hu, S. Mazhar Shah and X. Su, J. Colloid Interface Sci., 2012, 368, 540–546, DOI:10.1016/j.jcis.2011.11.015.
- A. A. Inyinbor, O. S. Bello, A. E. Fadiji and H. E. Inyinbor, J. Environ. Chem. Eng., 2018, 6, 784–793, DOI:10.1016/j.jece.2017.12.056.
- K. O’Malley, W. McDonald and P. McNamara, Environ. Sci., 2023, 9, 2188–2212, 10.1039/D3EW00356F.
- S. Singh, P. Sharma, N. Pal, D. K. Sarma and M. Kumar, Environ. Monit. Assess., 2024, 196, 325, DOI:10.1007/s10661-024-12425-4.
- Q. T. Tran, T. H. Do, X. L. Ha, H. P. Nguyen, A. T. Nguyen, T. C. Q. Ngo and H. D. Chau, Appl. Sci., 2022, 12, 8770, DOI:10.3390/app12178770.
- F. Cheng, P. Zhou, Y. Liu, X. Huo, J. Zhang, Y. Yuan, H. Zhang, B. Lai and Y. Zhang, Sci. Total Environ, 2021, 797, 149097, DOI:10.1016/j.scitotenv.2021.149097.
- C. Girardi, J. Greve, M. Lamshöft, I. Fetzer, A. Miltner, A. Schäffer and M. Kästner, J. Hazard. Mater., 2011, 198, 22–30, DOI:10.1016/j.jhazmat.2011.10.004.
- J. C. Nwabuife, C. A. Omolo and T. Govender, J. Controlled Release, 2022, 349, 338–353, DOI:10.1016/j.jconrel.2022.07.003.
- S. K. Mondal, A. K. Saha and A. Sinha, J. Cleaner Prod., 2018, 171, 1203–1214, DOI:10.1016/j.jclepro.2017.10.091.
- M. Zhou, C. Li, L. Zhao, J. Ning, X. Pan, G. Cai and G. Zhu, Sci. Total Environ, 2021, 752, 142261, DOI:10.1016/j.scitotenv.2020.142261.
- Z. Tan, X. Yang, L. Chen, Y. Liu, X. Hui-Juan, Y. Li and B. Gong, Bioresour. Technol., 2022, 344, 126280, DOI:10.1016/j.biortech.2021.126280.
- R. Ennouri, R. Lavecchia, A. Zuorro, S. C. Elaoud and E. Petrucci, J. Water Process Eng, 2021, 41, 101995, DOI:10.1016/j.jwpe.2021.101995.
- E. Asgari, A. Sheikhmohammadi, H. Nourmoradi, S. Nazari and M. Aghanaghad, Process Saf. Environ. Prot., 2021, 147, 356–366, DOI:10.1016/j.psep.2020.09.041.
- H. Guo, N. Jiang, H. Wang, H. Wang, N. Lu, K. Shang, J. Li and Y. Wu, J. Hazard. Mater., 2019, 371, 666–676, DOI:10.1016/j.jhazmat.2019.03.051.
- J. N. Naat, S. Suyanta and N. Nuryono, Case Stud. Chem. Environ. Eng., 2024, 10, 100878, DOI:10.1016/j.cscee.2024.100878.
- L. Qalyoubi, A. Al-Othman and S. Al-Asheh, Environ. Res., 2022, 215, 114182, DOI:10.1016/j.envres.2022.114182.
- G. Wang, Y. Zhang, S. Wang, H. Guo, N. Jiang, H. Wang, Y. Zhang, S. Wang, Y. Wang, H. Song, S. Lva and C. Li, Environ. Sci., 2020, 6, 1568–1575, 10.1039/D0EW00117A.
- S. De Gisi, G. Lofrano, M. Grassi and M. Notarnicola, Sustainable Mater. Technol., 2016, 9, 10–40, DOI:10.1016/j.susmat.2016.06.002.
- Y. Liu, X. Zhang and J. A. Wang, Chemosphere, 2022, 291, 132728, DOI:10.1016/j.chemosphere.2021.132728.
- X. Peng, F. Hu, F. L. Lam, Y. Wang, Z. Liu and H. Dai, J. Colloid Interface Sci., 2015, 460, 349–360, DOI:10.1016/j.jcis.2015.08.050.
- Y. Zhou, S. Cao, C. Xi, X. Li, L. Zhang, G. Wang and Z. Chen, Bioresour. Technol., 2019, 292, 121951, DOI:10.1016/j.biortech.2019.121951.
- F. Yu, Y. Sun, M. Yang and J. Ma, J. Hazard. Mater., 2019, 374, 195–202, DOI:10.1016/j.jhazmat.2019.04.021.
- H. Zhao, X. Liu, Z. Cao, Z. Yi, S. Xiaodong, Y. Yi, Z. Junliang and X. Jiang, J. Hazard. Mater., 2016, 310, 235–245, DOI:10.1016/j.jhazmat.2016.02.045.
- D. Hawthorne, A. Pannala, S. Sandeman and A. Lloyd, J. Drug Delivery Sci. Technol., 2022, 78, 103936, DOI:10.1016/j.jddst.2022.103936.
- S. F. Soares, M. J. Rocha, M. Ferro, C. O. Amorim, J. S. Amaral, T. Trindade and A. L. Daniel-da-Silva, Int. J. Biol. Macromol., 2019, 139, 827–841, DOI:10.1016/j.ijbiomac.2019.08.030.
- N. Amirmahani, H. Mahdizadeh, M. Malakootian, A. Pardakhty and N. O. Mahmoodi, J. Inorg. Organomet. Polym. Mater., 2020, 30, 3540–3551, DOI:10.1007/s10904-020-01499-5.
- H. Rasoulzadeh, A. Mohseni-bandpei, M. Hosseini and M. Safari, Int. J. Biol. Macromol., 2019, 133, 712–721, DOI:10.1016/j.ijbiomac.2019.04.139.
- H. Mao, S. Wang, J. Y. Lin, Z. Wang and J. Ren, J. Environ. Sci., 2016, 49, 179–188, DOI:10.1016/j.jes.2016.05.048.
- Y. Fan, B. Wang, S. Yuan, X. Wu, J. Chen and L. Wang, Bioresour. Technol., 2010, 101, 7661–7664, DOI:10.1016/j.biortech.2010.04.046.
- W. Ma, J. Dai, X. Dai, Z. Da and Y. Yan, Monatshefte für Chemie, 2015, 146, 465–474, DOI:10.1007/s00706-014-1351-1.
- G. Zhou, C. Liu, L. Chu, Y. Tang and S. Luo, Bioresour. Technol., 2016, 219, 451–457, DOI:10.1016/j.biortech.2016.07.038.
- J. Hu, S. Huang, X. Huang, Z. Kang and N. Gan, Microporous Mesoporous Mater., 2014, 197, 180–184, DOI:10.1016/j.micromeso.2014.06.014.
- Y. Deng, Y. Cai, Z. Sun and D. Zhao, Chem. Phys. Lett., 2011, 510, 1–13, DOI:10.1016/j.cplett.2011.04.093.
- R. Bhateria and R. Singh, J. Water Process Eng., 2019, 31, 100845, DOI:10.1016/j.jwpe.2019.100845.
- M. Malakootian, A. Nasiri and H. Mahdizadeh, Water Sci., 2018, 78, 2158–2170, DOI:10.2166/wst.2018.494.
- M. H. Dehghani, S. Ahmadi, S. Ghosh, A. Othmani, C. Osagie, M. Meskini, S. S. AlKafaas., A. Malloum, W. A. Khanday, A. O. Jacob, O. Gökku, A. Oroke, O. M. Chineme, R. R. Karri and E. C. Lima, Arab. J. Chem., 2023, 16, 105303, DOI:10.1016/j.arabjc.2023.105303.
- N. Nuryono, D. Miswanda, C. C. W. Sakti, B. Rusdiarso, P. A. Krisbiantoro, N. Utami, R. Otomo and Y. Kamiya, Mater. Chem. Phys., 2020, 255, 123507, DOI:10.1016/j.matchemphys.2020.123507.
- L. Yang, J. Tian, J. Meng, R. Zhao, C. Li and T. Jin, Molecules, 2018, 23(562), 1–10, DOI:10.3390/molecules23030562.
- N. Zhu, H. Ji, P. Yu, J. Niu, M. U. Farooq, M. W. Akram, I. U. Udego, H. Li and X. Niu, Nanomaterials, 2018, 8, 810, DOI:10.3390/nano8100810.
- N. Nuryono, N. M. Rosiati, B. Rusdiarso, S. C. W. Sakti and S. Tanaka, J. Korean Phys. Soc., 2014, 3, 1–12, DOI:10.1186/2193-1801-3-515.
- P. Wahyudi, A. H. Alimuddin and A. Shofiyani, J. Pure Appl. Chem. Res., 2020, 3, 15–21 Search PubMed.
- Y. A. B. Neolaka, G. Supriyanto and H. S. Kusuma, J. Environ. Chem. Eng., 2018, 6, 3436–3443, DOI:10.1016/j.jece.2018.04.053.
- J. N. Naat, Y. A. B. Neolaka, Y. Lawa, C. L. Wolu, D. Lestarani, S. Sugiarti and D. Iswantini, AIMS Mater Sci, 2022, 9, 36–55, DOI:10.3934/matersci.2022003.
- J. N. Naat, Y. A. B. Neolaka, T. Lapailaka, T. Tj, A. Sabarudin, H. Darmokoesoemo and H. S. Kusuma, Rasayan J. Chem., 2021, 14, 550–560 CrossRef CAS.
- A. Hasan and L. M. Pandey, Mater. Sci. Eng., C, 2016, 68, 423–429, DOI:10.1016/j.msec.2016.06.003.
- M. Ramezani, M. R. Vaezi and A. Kazemzadeh, Appl. Surf. Sci., 2014, 317, 147–153, DOI:10.1016/j.apsusc.2014.08.095.
- B. Xu and Q. Zhang, ACS Omega, 2021, 6, 9764–9770, DOI:10.1021/acsomega.1c00381.
- H. J. Perera, R. Latifi and F. D. Blum, J. Phys. Chem. C, 2019, 123, 19005–19012, DOI:10.1021/acs.jpcc.9b04025.
- D. Y. Nadargi, J. L. Gurav, N. E. Hawi, A. V. Rao and M. Koebel, J. Alloys Compd., 2010, 496, 436–441, DOI:10.1016/j.jallcom.2010.01.157.
- B. Wei, B. Sun, B. Zhang, J. Long, L. Chen and Y. Tian, Carbohydr. Polym., 2016, 136, 1203–1208, DOI:10.1016/j.carbpol.2015.10.025.
- C. E. Pyo and J. H. Chang, ACS Omega, 2021, 6, 16100–16109, DOI:10.1021/acsomega.1c01981.
- S. Novak, T. F. Chaves, L. Martins and S. V. Santilli, Colloids Surf., A, 2020, 585, 124109, DOI:10.1016/j.colsurfa.2019.124109.
- T. Li, F. Sun, Y. Zhao and M. Chen, Prog. Org. Coat., 2023, 174, 107232, DOI:10.1016/j.porgcoat.2022.107232.
- S. S. Ray S, H. K. Lee, D. T. T. Huyen, Y. H. Park, S. E. Nam, I. C. Kim and Y. N. Kwon, Prog. Org. Coat., 2021, 153, 106165, DOI:10.1016/j.porgcoat.2021.106165.
- L. Wang, C. Shen and Y. Cao, J. Phys. Chem. Solids, 2019, 133, 28–34, DOI:10.1016/j.jpcs.2019.05.004.
- M. Mosayebi, Z. Salehi, H. Doosthosseini, P. Tishbi and Y. Kawase, Renew Energy, 2020, 154, 569–580, DOI:10.1016/j.renene.2020.03.040.
- Y. P. Chang, C. L. Ren, Q. Yang, Z. Y. Zhang, L. J. Dong, X. G. Chen and D. S. Xue, Appl. Surf. Sci., 2011, 257, 8610–8616, DOI:10.1016/j.apsusc.2011.05.031.
- H. Peng, H. Wang, J. Wu, G. Meng, Y. Wang, Y. Shi, Z. Liu and X. Guo, Ind. Eng. Chem. Res., 2016, 55, 832–838, DOI:10.1021/acs.iecr.5b03862.
- K. S. W. Sing, Pure Appl. Chem., 1985, 57, 603–619, DOI:10.1351/pac198557040603.
- T. J. Al-Musawi, A. H. Mahvi, A. D. Khatibi and D. Balarak, J. Porous Mater., 2021, 28, 835–852, DOI:10.1007/s10934-021-01039-7.
- C. C. Lin and C. Y. Lee, Mater. Chem. Phys., 2020, 240, 122049, DOI:10.1016/j.matchemphys.2019.122049.
- Z. Movasaghi, B. Yan and C. Niu, Ind. Crops Prod., 2019, 127, 237–250, DOI:10.1016/j.indcrop.2018.10.051.
- H. Li, D. Zhang, X. Han and B. Xing, Chemosphere, 2014, 95, 150–155, DOI:10.1016/j.chemosphere.2013.08.053.
- S. Aydin, M. E. Aydin, F. Beduk and A. Ulvi, Sci. Total Environ, 2019, 670, 539–546, DOI:10.1016/j.scitotenv.2019.03.205.
- M. Y. Badi, A. Azari, H. Pasalari, A. Esrafili and M. Farzadkia, J. Mol. Liq., 2018, 261, 146–154, DOI:10.1016/j.molliq.2018.04.019.
- M. E. Mahmoud, S. R. Saad, A. M. El-Ghanam and R. H. A. Mohamed, Mater. Chem. Phys., 2021, 257, 123454, DOI:10.1016/j.matchemphys.2020.123454.
- S. T. Danalıoğlu, S. S. Bayazit, Ö. Kerkez, B. G. Alhogbi and M. A. Abdel, Chem. Eng. Res. Des., 2017, 123, 259–267, DOI:10.1016/j.cherd.2017.05.018.
- Y. Wu, Q. Yue, Z. Ren and B. Gao, J. Mol. Liq., 2018, 262, 19–28, DOI:10.1016/j.molliq.2018.04.032.
- Z. M. Idris, B. H. Hameed, L. Ye, S. Hajizadeh, B. Mattiasson and A. T. Mohd Din, J. Environ. Chem. Eng., 2020, 8, 103981, DOI:10.1016/j.jece.2020.103981.
- D. Yin, Z. Xu, J. Shi, L. Shen and Z. He, J. Water Reuse Desalin., 2018, 8, 350–359, DOI:10.2166/wrd.2017.143.
- N. Zhao, K. Liu, C. He, J. Gao, W. Zhang, T. Zhao, D. C. W. Tsang and R. Qiu, Environ. Int., 2020, 143, 105899, DOI:10.1016/j.envint.2020.105899.
- X. Peng, F. Hu, T. Zhang, F. Qiu and H. Dai, Bioresour. Technol., 2018, 249, 924–934, DOI:10.1016/j.biortech.2017.10.095.
- D. Hu and L. Wang, Desalin. Water Treat., 2016, 57, 28436–28449, DOI:10.1080/19443994.2016.1183232.
- L. Huang, M. Wang, C. Shi, J. Huang and B. Zhang, Desalin. Water Treat., 2014, 52, 2678–2687, DOI:10.1080/19443994.2013.833873.
- F. Yu, D. Chen and J. Ma, New J. Chem., 2018, 42, 2216–2223, 10.1039/C7NJ03770H.
- X. Zheng, S. Xu, Y. Wang, X. Sun, Y. Gao and B. Gao, J. Colloid Interface Sci., 2018, 527, 202–213, DOI:10.1016/j.jcis.2018.05.054.
- X. Zhu, D. C. W. Tsang, F. Chen, S. Li and X. Yang, Environ. Technol., 2015, 36, 3094–3102, DOI:10.1080/09593330.2015.1054316.
- C. A. Igwegbe, S. N. Oba, C. O. Aniagor, A. G. Adeniyi and J. O. Ighalo, J. Ind. Eng. Chem., 2021, 93, 57–77, DOI:10.1016/j.jiec.2020.09.023.
- N. A. Ahammad, M. A. Zulkifli, M. A. Ahmad, B. H. Hameed and A. T. Mohd Din, J. Environ. Chem. Eng., 2021, 9, 105015, DOI:10.1016/j.jece.2020.105015.
- A. Chandrasekaran, C. Patra, S. Narayanasamy and S. Subbiah, Environ. Res., 2020, 188, 109825, DOI:10.1016/j.envres.2020.109825.
- W. Duan, N. Wang, W. Xiao, Y. Zhao and Y. Zheng, J. Mol. Liq., 2018, 269, 874–881, DOI:10.1016/j.molliq.2018.08.051.
- N. Dhiman and N. Sharma, Indian Chem. Eng., 2019, 61, 67–76, DOI:10.1080/00194506.2018.1424044.
- A. Avcı, I. Inci and N. Baylan, Water, Air, Soil Pollut., 2019, 230, 250, DOI:10.1007/s11270-019-4315-6.
- L. Wang, C. Yang, A. Lu, S. Liu, Y. Pei and X. Luo, Bioresour. Technol., 2020, 302, 122812, DOI:10.1016/j.biortech.2020.122812.
- S. Wu, Y. Li, X. Zhao, Q. Du, Z. Wang, Y. Xia and L. Xia, Int. J. Phytorem., 2015, 17, 957–961, DOI:10.1080/15226514.2014.935288.
- P. Liao, Z. Zhan, J. Dai, X. Wu, W. Zhang, K. Wang and S. Yuan, Chem. Eng. J., 2013, 228, 496–505, DOI:10.1016/j.cej.2013.04.118.
- Y. Wang, E. Wang, H. Dong, F. Liu, Z. Wu, H. Li and Y. Wang, Adsorpt. Sci. Technol., 2014, 32, 321–330, DOI:10.1260/0263-6174.32.4.321.
- J. Jin, T. Feng T, Y. Ma, W. Wang, Y. Wang, Q. Zhou and A. Li, Chemosphere, 2017, 185, 563–573, DOI:10.1016/j.chemosphere.2017.07.058.
- S. Tian, J. Dai, Y. Jiang, Z. Chang, A. Xie, J. He, R. Zhang and Y. Yan, J. Colloid Interface Sci., 2017, 505, 858–869, DOI:10.1016/j.jcis.2017.06.062.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a
*a