
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePioneering wound care solutions: triaxial wet-spun fibers with bioactive agents for chronic wounds – part I (production and characterization of the triaxial fibers)†
Catarina S.
Miranda
a,
Elina
Marinho
 a,
Diana
Rocha
b,
Carla
Silva
b,
M. Manuela P.
Silva
c,
Inge
Schlapp-Hackl
d,
Wenwen
Fang
de,
Michael
Hummel
d,
Susana P. G.
Costa
c,
Natália C.
Homem
af and
Helena P.
Felgueiras
*a
a,
Diana
Rocha
b,
Carla
Silva
b,
M. Manuela P.
Silva
c,
Inge
Schlapp-Hackl
d,
Wenwen
Fang
de,
Michael
Hummel
d,
Susana P. G.
Costa
c,
Natália C.
Homem
af and
Helena P.
Felgueiras
*a
aCentre for Textile Science and Technology (2C2T), University of Minho, Campus of Azurém, 4800-058 Guimarães, Portugal. E-mail: catarina.miranda@2c2t.uminho.pt; elinamarinho@2c2t.uminho.pt; helena.felgueiras@2c2t.uminho.pt; Fax: +351-253-510-293; Tel: +351-253-510-283
bCentre of Biological Engineering (CEB), University of Minho, Campus de Gualtar, 4710-057 Braga, Portugal. E-mail: id9610@alunos.uminho.pt; carla.silva@ceb.uminho.pt
cCentre of Chemistry (CQ), University of Minho, Campus of Gualtar, 4710-057 Braga, Portugal. E-mail: nini@quimica.uminho.pt; spc@quimica.uminho.pt
dDepartment of Bioproducts and Biosystems, School of Chemical Engineering, Aalto University, Espoo 02150, Finland. E-mail: inge.schlapp-hackl@aalto.fi; michael.hummel@aalto.fi; wenwen.fang@aalto.fi
eTechnical University Munich, Campus Straubing for Biotechnology and Sustainability, Schulgasse 16, 94315 Straubing, Germany. E-mail: wenwen.fang@tum.de
fSimoldes Plastics S.A., Rua Comendador António da Silva Rodrigues, 165, 3720-193, Oliveira de Azeméis, Portugal. E-mail: natalia.homem@simoldes.com
First published on 16th April 2025
Abstract
Fiber-based constructs have been produced as an alternative to conventional dressings for the treatment of chronic wounds (CWs), showing good tenability, high surface area and regulable porosity. A commonly used technique for processing such dressings is wet-spinning, which involves precipitating a polymer solution into a coagulation bath containing a non-solvent of that polymer. This process produces fibers with varying diameters and morphologies. In this study, we propose to engineer a triaxial wet-spun fibrous system, consisting of three layers, modified with active agents for wound healing applications. The innermost layer (core) was composed of polycaprolactone (PCL), which imparted the fibers with high elasticity and mechanical properties. This layer was blended with cinnamon leaf oil (CLO), enhancing the system with antibacterial and antioxidant capacities. The intermediate layer contained sodium alginate (SA), conferring a moist environment, loaded with the alanine–alanine–proline–valine (AAPV) tetrapeptide, responsible for regulating the local enzymatic activity. The outermost layer, or shell, was composed of cellulose acetate (CA), which conferred high rigidity and porosity to the fibers. This report represents the initial phase of a broader study, concentrating on the evaluation of the morphological, physical, thermal, and mechanical properties of the proposed triaxial system. The fibers demonstrated maximum elongations at break exceeding 300%, also achieving tenacities up to 41.40 ± 0.03 MPa. They were also found to maintain their structural integrity when exposed to physiological-like conditions, in which the triaxial fibers achieved 9.61 ± 4.08% mass loss after 28 days of incubation, and to exhibit high thermal stability. Furthermore, all fibers attained porosity between 10 and 60% and a dressing composed of these triaxial wet-spun fibers was successfully knitted, serving as proof of concept for the potential application of these fibers in dressing fabrication. The engineered fibers not only possess high mechanical, thermal and structural stability, but also allow for a sustained and orderly release of two active agents, AAPV and CLO, simultaneously controlling local enzymatic activity and reactive oxygen species (ROS) levels and fighting bacterial infections. Overall, the results confirmed the feasibility of the designed wet-spun fibers for future wound healing applications.
1. Introduction
Fiber-based wound dressings have been highlighted in chronic wound (CW) care for their good tenacity, high exudate absorption capacity and oxygen permeability.1–3 Spinning techniques are often employed to produce these kinds of dressings.4,5 Wet-spinning has gathered much interest in recent years because of its straightforward processing. This technique involves the injection of a polymeric solution that quickly changes phase when in contact with a coagulation bath constituted by a non-solvent of the polymeric solution. As a result, coagulation of the polymer occurs, forming wet-spun fibers.1–3 Wet-spinning not only prevents thermal degradation of polymers, but also allows the formation of structures with a variety of diameters, organization levels and tunable chemical and physical properties, dictated by the spinneret design, coagulation bath and the polymeric solution properties (solvent, concentration, presence or absence of additives, etc.).2,6 A wide range of polymers, composites and active agents, including those sensitive to temperature, can be processed through this technique.7 For this reason, wet-spinning is often chosen over other spinning techniques, e.g. melt-spinning, for preventing material degradation.3 Additionally, it is considered a process with a faster mass fabrication of individual and collectable fibers, on which detailed analyses and characterization can be performed, something that is not possible with the electrospinning technique, for instance.8–10To the authors’ knowledge, there are very few reports in the literature regarding the production of triaxial wet-spun fibers (three layers). As an example, triaxial wet-spun structures were developed by Mirabedini et al., which consisted of a core of poly(3,4-ethylenedioxythiophene)polystyrene sulfonate (PEDOT:PSS), surrounded by an intermediate layer of chitosan (CS) and an outermost layer (shell) of a carbon nanotube (CNT)-loaded polymer for electronic applications. The presence of three layers led to higher flexibility and structural stability for their intended application.11 However, more examples involving the use of this technique for coaxial and monolayered fibers have been reported in the literature, including a study from our group, in which polycaprolactone (PCL) and sodium alginate (SA) coaxial wet-spun fibers (with two layers) were produced. Such constructs achieved maximum elongations at break up to 220% and showed high structural stability when exposed to physiological media, along with an effective thermal behavior.10 Braccini et al. produced CS and hyaluronic acid (HA) composite wet-spun scaffolds, in which their filaments’ morphologies were successfully controlled by using a non-solvent coagulation bath, also presenting an interconnected porous structure.12 Additionally, Lu et al. applied the wet-spinning technique to engineer hexagonal boron nitride nanosheets, which presented excellent thermal conductivity and, more importantly, a tensile strength of 192 MPa.13
Several biodegradable polymers have been processed by wet-spinning.1 One of the most frequent is PCL, a synthetic polyester, due to its excellent mechanical properties, elasticity, high solubility in organic solvents, biodegradability and tunable surface roughness.14,15 This polymer can effectively blend and form composites with different polymers and, for this reason, PCL has also been applied as a supporting material for the production of wet-spun fibers, loaded with active agents, for wound healing applications.10,16 Another option is cellulose acetate (CA), a biocompatible polymer gifted with high rigidity and mechanical resistance.17,18 These properties, combined with good biodegradability, hydrolytic stability, chemical resistance and water absorption capacity, led to the recognition of CA as an appealing option for the production of wet-spun fibers.1 Sodium alginate (SA) represents another promising alternative. It can be extracted from the cell wall of brown algae and has been employed for tissue engineering and drug delivery systems, due to its biocompatibility, biodegradability, low toxicity, antiseptic properties, fast ionic gelation after contact with divalent cations and excellent hydration properties.19,20 Curiously, SA has been recently used for the production of coaxial wet-spun fibers for wound healing applications, whose swelling capacities were essential for maintaining a moist environment and, as a consequence, accelerate the healing process.9,10
Previous research from the team has focused on the production of coaxial wet-spun fibers of PCL and SA loaded with an antimicrobial peptide (AMP), which not only enabled a controlled release of the AMP, but also showed great mechanical properties as well as structural stability when exposed to physiological-like media, proving their suitability for wound healing applications.10 In addition, PCL and CA coaxial fibers were also produced and loaded with essential oils (EOs), reaching higher maximum elongations at break and thermal stability compared to the peptide-loaded fibers, in response to the EOs’ plasticizing effect and their affinity towards PCL.21 The present research combines information acquired from the two previous wet-spun systems, to generate an innovative triaxial (three layered) wet-spun fiber for use in the treatment of chronic wounds. Here, PCL and CLO (cinnamon leaf oil; antimicrobial potential) were loaded into the fibers’ core, for endowing the fibers with high mechanical properties and elasticity. CLO is mainly composed of eugenol and cinnamaldehyde, endowing it with high antibacterial, antioxidant and anti-inflammatory properties.21,22 Yet, the plasticizing effect and high volatility of CLO, together with the hydrophobic behavior from the two compounds, justified the presence of an intermediate layer with affinity towards water. To accomplish that, SA, a polymer with excellent hydration properties, loaded with the peptide alanine–alanine–proline–valine (AAPV), was employed as the intermediate layer. AAPV presents the capacity to inhibit the activity of human neutrophil elastase (HNE), an enzyme which presents abnormally high levels of activity during inflammatory processes, thus accelerating the wound healing process.23 Still, to address issues associated with the low mechanical performances of SA which could lead to an early degradation of fibers, an external layer (shell) containing CA was proposed. In the end, this combination of polymers and bioactive agents was expected to generate a structurally stable and porous triaxial wet-spun system. To understand the implications of each component for the physical, chemical, thermal and mechanical properties of the fibers, the morphology, chemical composition, thermal responses and mechanical behaviors of the system were evaluated. In addition, the fibers’ hydration capacities and stability in physiological media were also studied. A knitted dressing made from the engineered triaxial fibers was produced as proof-of-concept. This way, a wound dressing composed of the proposed triaxial wet-spun system not only would maintain the properties of the existing fiber-based dressings but would also be able to offer protection of the active agents from the surrounding media. Moreover, such type of dressing would possess both antibacterial and antioxidant properties, as well as enzymatic regulation capacities, which are fundamental to accelerate and facilitate the wound healing process.21
This work marks the first research effort on the production of triaxial fibers loaded with both a peptide and an EO for potential wound healing applications.
2. Materials and methods
2.1. Materials
CA (C14H16N4; Mn 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000; CAS number 67-56-1), PCL (CH3(C6H10O2)nCH3; Mn 80000; CAS number 24980-41-4) and SA (from brown algae, medium viscosity; NaC6H7O6; CAS number 9005-38-3) were obtained from Sigma-Aldrich (St. Louis, Missouri, USA). N,N-Dimethylformamide (DMF; C3H7NO; Mw 73.09 g mol−1; CAS number 68-12-2) was acquired from Merck (Darmstadt, Germany). Calcium chloride (CaCl2; Mw 110.99 g mol−1; CAS number 10043-52-4; anhydrous) was employed as a coagulation/crosslinking agent during wet-spinning and was obtained from Chem-Lab (Zedelgem, Belgium).
000; CAS number 67-56-1), PCL (CH3(C6H10O2)nCH3; Mn 80000; CAS number 24980-41-4) and SA (from brown algae, medium viscosity; NaC6H7O6; CAS number 9005-38-3) were obtained from Sigma-Aldrich (St. Louis, Missouri, USA). N,N-Dimethylformamide (DMF; C3H7NO; Mw 73.09 g mol−1; CAS number 68-12-2) was acquired from Merck (Darmstadt, Germany). Calcium chloride (CaCl2; Mw 110.99 g mol−1; CAS number 10043-52-4; anhydrous) was employed as a coagulation/crosslinking agent during wet-spinning and was obtained from Chem-Lab (Zedelgem, Belgium).
AAPV was synthesized as described in ref. 10. Pure cinnamon leaf oil (CLO, origin Cinnamomum zeylanicum Blume, ρ = 1.049) was obtained from Folha d’Água Company (Santo Tirso, Portugal).24 Sodium phosphate dibasic (Na2HPO4; Mn 141.98; CAS number 7558-79-4; Sigma-Aldrich), monosodium phosphate monohydrate (KH2PO4; Mn 137.99; CAS number 10049-21-5; Sigma Aldrich), potassium chloride (KCl; Mw 74.56 g mol−1; CAS number 7447-40-7; Merck) and sodium chloride (NaCl; Mw 58.44 g mol−1; CAS number 7647-14-5; Merck) were used in the preparation of phosphate buffer saline solution (PBS at 0.01 M: 1.44 g L−1 of Na2HPO4, 0.24 g L−1 of KH2PO4, 0.20 g L−1 of KCl and 8.00 g L−1 of NaCl, adjusted to physiological pH 7.4). All reagents were used without further purification. The solutions were prepared utilizing ultrapure water (Milli-Q Gradient A10 Water Purification System, Millipore Corporation, MA, US), with a resistivity greater than 18 MΩ cm−1.
2.2. Wet-spun fiber production
10% w/v PCL and CA solutions were separately prepared in DMF and allowed to stand for 1 h, whereas a 2% w/v SA solution was prepared in distilled water (dH2O) and allowed to stand for 3 h – concentrations were selected based on previous research from the team.10 The three polymeric solutions were stirred continuously at 50 °C. CLO was combined with the PCL solution at 16.40 mg mL−1, corresponding to 4× its MIC (amount determined in ref. 21) and left to homogenize for 1 h at 50 °C (PCL-CLO solution). AAPV was added to the SA solution at 50 μg mL−1, a concentration defined according to ref. 25 (SA-AAPV solution). Viscosities were measured using a Brookfield DV-II + Pro viscometer (Boston, USA) with spindle 21, with speeds of 50–70 rpm at 17–35 °C. All solutions were ultrasonicated to remove air bubbles prior to fiber production. The wet-spinning setup consisted of three syringe pumps (NE-300, New Era Pump Systems, Norleq, Santo Tirso, Portugal) to control the rate and ejection volume, a triaxial spinneret (sealed needles of three layers with 21, 15 and 11 Gauge, from inner to the outer layer) and a large tray containing 500 mL of a 2% w/v calcium chloride coagulation bath, at room temperature (RT). All fibers were collected automatically using a cylindrical-shaped collector at speeds of 9–13 rpm and dried for 1 h at RT. Syringes connected to the inner port (core), the intermediate port (core 2) and the outer port (shell) of the triaxial spinneret were loaded with PCL or PCL-CLO solutions, SA or SA-AAPV solutions, and CA solution, respectively. PCL and PCL-CLO were ejected at 0.11 mL min−1, SA and SA-AAPV were ejected at 0.13 mL min−1, whereas CA was ejected at 0.15 mL min−1, forming PCL-CLO/SA-AAPV/CA fibers (final composition). All speeds were optimized. According to the literature, higher speeds may facilitate future up-scale production, that is why they were selected in this study.9,25–29 Control fibers were produced without the shell (just PCL or PCL-CLO and SA or SA-AAPV), without the intermediate layer (just PCL or PCL-CLO and CA) and without the core (hollow fibers of SA or SA-AAPV and CA). Control fibers were also produced containing only the core (PCL or PCL-CLO), only the intermediate layer (SA or SA-AAPV) and only the shell (CA). Moreover, fibers were ejected without some of the components of the triaxial system, namely CLO and AAPV (PCL/SA/CA), CLO (PCL/SA-AAPV/CA), and AAPV (PCL-CLO/SA/CA). Processing parameters for control samples were same as those of the complete system. After drying, the fibers were stored in a cabinet desiccator (Sicco, Grünsfeld, Germany) at 19 °C and a relative humidity of 41%, for further testing. Although several reports state that fibers should be dried at higher temperatures, here, drying conditions were selected in order to prevent EO evaporation and any interferences with the AMP activity.28,29 The identification of the fibers used “/” for separating elements belonging to different layers and “—” for components within the same layer (Table 1).| Fiber typology | Core composition | Intermediate layer composition | Shell composition |
|---|---|---|---|
| PCL | PCL | — | — |
| PCL-CLO | PCL blended with CLO | — | — |
| SA | — | SA | — |
| SA-AAPV | — | SA blended with AAPV | — |
| CA | — | — | CA |
| PCL/SA | PCL | SA | — |
| PCL-CLO/SA | PCL blended with CLO | SA | — |
| PCL/SA-AAPV | PCL | SA blended with AAPV | — |
| PCL-CLO/SA-AAPV | PCL blended with CLO | SA blended with AAPV | — |
| SA/CA | — | SA | CA |
| SA-AAPV/CA | — | SA blended with AAPV | CA |
| PCL/CA | PCL | — | CA |
| PCL-CLO/CA | PCL blended with CLO | — | CA |
| PCL/SA/CA | PCL | SA | CA |
| PCL-CLO/SA/CA | PCL blended with CLO | SA | CA |
| PCL/SA-AAPV/CA | PCL | SA blended with AAPV | CA |
| PCL-CLO/SA-AAPV/CA | PCL blended with CLO | SA blended with AAPV | CA |
2.3. Microfibers’ physical, chemical, thermal and mechanical characterization
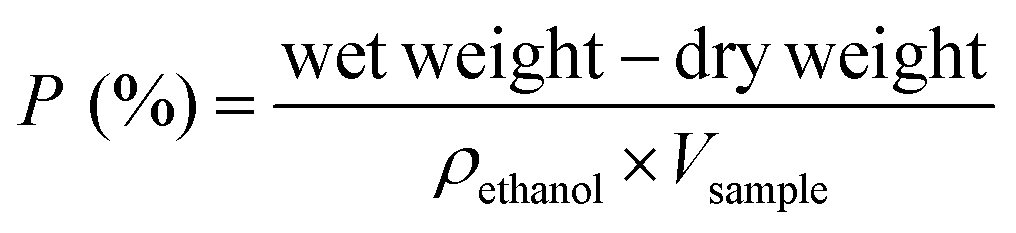 | (1) |
± 2 °C and 65% ± 2% relative humidity [RH]) using a Textechno Herbert Stein Favigraph (Dohrweg, Germany), associated with the TexTechno Favigraph software, following the standard EN ISO 5079. Filaments of 5 cm in length were analyzed at RT with an optimized holding distance (gauge length) starting at 10 mm and increasing until the fibers’ maximum elongation at break was reached. The crosshead speed was established at 10 mm min−1, using a loading cell of 20–100 cN. The area of each sample was calculated and tenacity (MPa) was determined using the following expression: | (2) |
Representative curves, regarding the tenacity and maximum elongation for each fiber typology, were determined, using the GraphPad Prism 8.0 Software (GraphPad Software Inc, USA).
 | (3) |
Water retention (WR) was also determined using the following expression:31
 | (4) |
 | (5) |
2.4. Dressing production and characterization
A dressing made from the engineered triaxial fibers (PCL-CLO/SA-AAPV/CA) was produced using a Vanguard Circular Single Jersey Tricolab (Pennsylvania, USA) knitting machine with 20 needles, connected to an Omron SYSDRIVE 3G3XV Inverter (Kyoto, Japan) to control velocity. The dressing was produced at a speed of 3 ranks per minute in which the opening and closing of the dressing was defined by the distance between the needles, controlled by screws. The obtained knitted dressing was photographed, from a distance of 9 cm and at 10× magnification, using a 108 MP camera of a Redmi Note 12 Pro mobile phone. Contexture was determined by photographing the dressing using a LinenTest™ 6.5 × 20 mm folding magnifier (Carson, Newport, USA) and counting the number of columns and ranks. The thickness was measured with an analog micrometer from Mitutoyo (USA) with 0.01 mm resolution and 18 Pa of applied pressure (10 measurements). Finally, mass per unit of area was also determined by using an analytical balance (3 measurements).2.5. Statistical analysis
All measurements were conducted in triplicate unless otherwise stated in the Experimental sections. Numerical data were reported as mean ± standard deviation (SD). Data were treated using GraphPad Prism 8.0 Software (GraphPad Software Inc, USA). Normality analysis was performed, and the results were analyzed using one-way ANOVA and Tukey tests. Statistically significant differences were considered at p < 0.05.3. Results and discussion
3.1. Fiber morphology
The morphology of the engineered wet-spun fibers was assessed by brightfield microscopy and SEM (Fig. 1 and Fig. S1 (ESI†), respectively). Regarding the monolayered fibers, the combination of CLO with PCL led to an increase in their inner diameter compared to pristine PCL (Fig. 1e). The main component of CLO, eugenol, is known to exhibit a strong affinity for PCL. This is primarily due to the hydroxyl groups in eugenol interacting strongly with the carbonyl groups in PCL.32 This factor, along with the reduction in the viscosity of the polymeric solutions upon the addition of CLO (292.500 cP and 231.00 cP for PCL and PCL-CLO solutions at RT, respectively; Fig. S2 in the ESI†) and the changes in the configuration of the PCL polymeric chains, contributed to the expansion of the fibers’ diameters as there were no restrictions from any intermediate or outer layers.9,25 Contrary to this observation, in the coaxial fibers, the inner diameters were larger than those of the monolayered fibers, despite the presence of an intermediate/outer layer (SA or SA-AAPV or CA) that would restrict the core's dilation. As previously evidenced in work conducted by our team, when SA is in contact with PCL, the latter tends to precipitate very quickly, increasing its volume and partially incorporating the SA layer.10 This theory is also supported by the fact that the intermediate layers’ thicknesses experienced a significant decrease in size for all PCL-containing coaxial fibers (Fig. 1f). Similar outcomes were observed during the production of SA/PCL electrospun composite structures, in which PCL layer's thicknesses were superior to SA layer's thicknesses.33,34 On the other hand, in SA or SA-AAPV hollow fibers along with SA/CA and SA-AAPV/CA coaxial fibers, the thicknesses were superior due to the formation of flattened structures and the absence of PCL layers. The presence of AAPV slightly increased the SA layer's thickness, possibly due to the changes introduced by AAPV in the SA polymeric chains’ configuration, generating a less compact structure, also evidenced by a decrease in the solution's viscosity (5600 cP and 1250 cP for SA and SA-AAPV solutions at RT, respectively; Fig. S2 in the ESI†). There are still no reports in the literature addressing the chemical interactions between AAPV and SA; nevertheless, it is likely that such interactions occurred through the hydroxyl groups from both SA and AAPV. Moreover, the outer layers’ thicknesses were larger in CA fibers, due to the absence of both PCL or PCL-CLO and SA or SA-AAPV inner layers which, once again, allowed more freedom to the CA's polymeric chains to expand (Fig. 1g).9,25 As expected, the presence of a third layer (triaxial fibers) resulted in larger total diameters (Fig. 1h). Here, again, it is likely that the SA or SA-AAPV intermediate layer is partially incorporated into the PCL or PCL-CLO innermost layer since a reduction in the intermediate layer thickness in comparison with PCL and CA layers was observed (Fig. 1f). | ||
| Fig. 1 Micrographs of (a) PCL-CLO, (b) SA/CA and (c) PCL-CLO/SA-AAPV/CA wet-spun fibers’ morphology obtained by brightfield microscopy and (d) PCL-CLO/SA-AAPV/CA fibers’ morphology obtained by SEM. Distribution of (e) core thicknesses, (f) intermediate layer thicknesses, (g) shell thicknesses and (h) complete fiber diameters of all wet-spun fiber typologies. Data are reported as mean ± SD (n = 5). Statistical significance was determined via the Tukey test by applying multiple comparisons between the different fiber typologies (*p < 0.0226, **p < 0.0078, ***p < 0.006, ****p < 0.0001). | ||
3.2. ATR-FTIR
ATR-FTIR spectra were collected from all wet-spun fiber typologies to confirm the presence of all components in the fibers (Fig. 2 and Table S1 in the ESI†). The presence of PCL in all PCL-containing fibers was confirmed by the detection of peaks centered at ≈1728 cm−1 and 2950 cm−1, associated with carbonyl groups and C–H stretching vibrations, respectively. Bands in the range of 1173–1246 cm−1 were also observed, attributed to C–O–C vibrations of PCL.35 On the other hand, in all SA-containing fibers, peaks at 1600 cm−1 and 1420 cm−1 were detected, related to COO− vibrations characteristic of the SA polymer. Moreover, a pronounced band, centered at ≈3300 cm−1, was observed, attributed to the –OH groups characteristic of SA.9 The presence of CA in CA-containing fibers was confirmed by the peaks centered at 1738 cm−1 and 1660 cm−1, corresponding to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibrations. Characteristic vibrations of CH2, acetyl ester groups and C–O stretching vibrations were also detected by peaks centered at 1438, 1222 and 1044 cm−1, respectively.35 Due to the very small concentrations of CLO and AAPV loaded in the fibers, their presence could not be confirmed via this technique.
O stretching vibrations. Characteristic vibrations of CH2, acetyl ester groups and C–O stretching vibrations were also detected by peaks centered at 1438, 1222 and 1044 cm−1, respectively.35 Due to the very small concentrations of CLO and AAPV loaded in the fibers, their presence could not be confirmed via this technique.
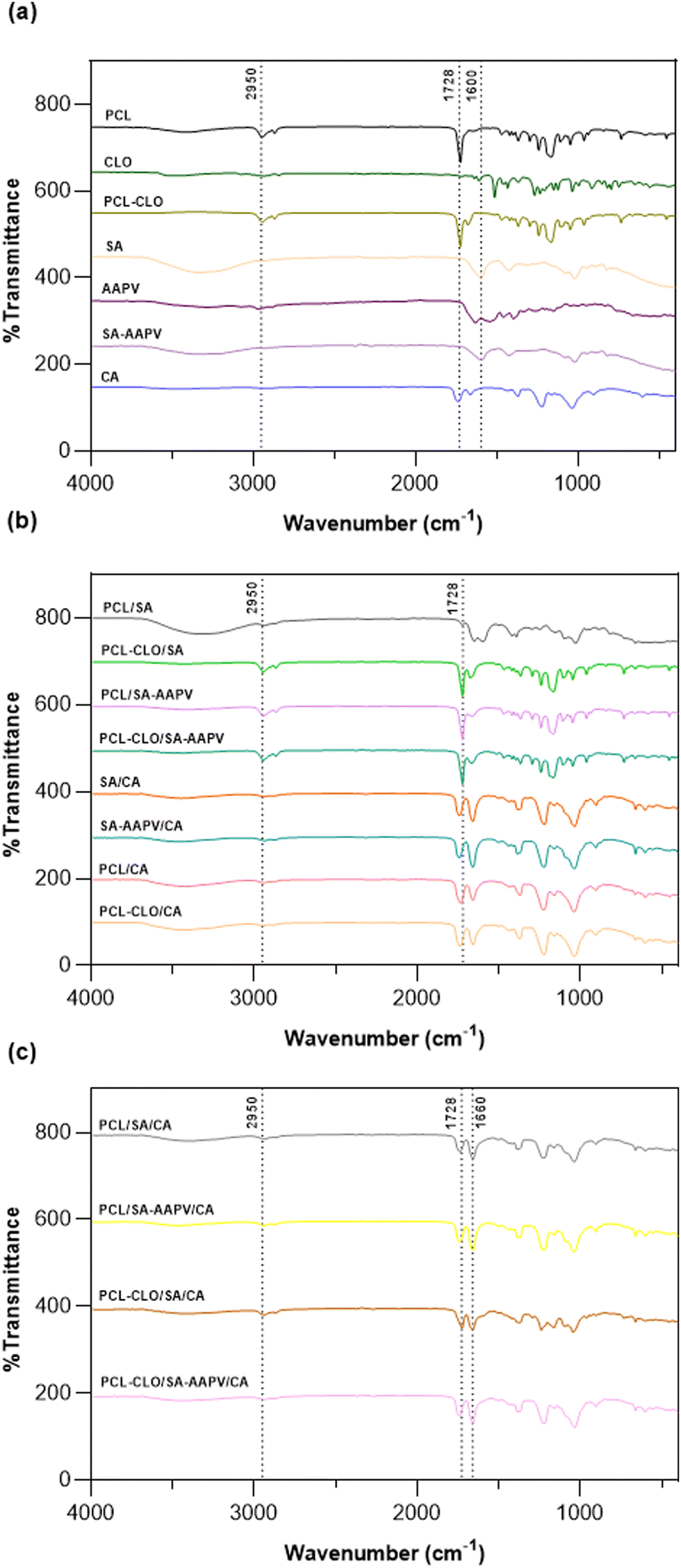 | ||
| Fig. 2 ATR-FTIR spectra of (a) CLO (liquid form), AAPV (powder form) and PCL, PCL-CLO, SA, SA-AAPV and CA, all in monolayered form; (b) PCL/SA, PCL-CLO/SA, PCL/SA-AAPV, PCL-CLO/SA-AAPV, SA/CA, SA-AAPV/CA, PCL/CA and PCL-CLO/CA in coaxial form; and (c) PCL/SA/CA, PCL/SA-AAPV/CA, PCL-CLO/SA/CA and PCL-CLO/SA-AAPV/CA in the form of triaxial fibers. | ||
3.3. Thermal response
The thermal behavior of all wet-spun fiber typologies was assessed by DSC and TGA (Fig. 3 and Table S2 in the ESI†). A PCL characteristic peak centered at ≈58 °C (onset at 49.89 °C) was detected on PCL monolayered fibers, representing the first melting temperature of the polymer, along with the fusion of its short polymer segments (Fig. 3a).36 Apart from this, such sample presented high thermal stability up to ≈350 °C, suffering only 3% mass loss until that temperature (Fig. 3d). Such observation is in accordance with reports that state that PCL's main thermal degradation temperature is, in fact, ≈350 °C.37 The same characteristic PCL peak suffered a small shift upon the addition of CLO (PCL-CLO monolayered fibers; from 58.42 °C to 51.22 °C), which is in line with observations reported in several other studies (Fig. 3a).37,38 In fact, according to Phaiju et al., the present result confirms the effective miscibility between PCL and the EO, due to the low differences of the melting temperatures.37 Additionally, mass losses were detected in the TGA spectrum of CLO-loaded fibers, at earlier temperatures (mass loss of ≈10%) until 100 °C, coherent with the TGA spectra of CLO, which presented a slight degradation at ≈100 °C (thermal degradation associated with the release of occluded water and of the solvents used in the synthesis) and was completely degraded at ≈200 °C. This decrease in the fibers’ thermal stability can be explained by the incorporation of CLO, which may have influenced the polymeric chains’ orientation, also decreasing the crystallinity of the structure and resulting in earlier degradation steps.38 SA hollow fibers presented a peak centered at ≈143 °C (onset at 141.87 °C), frequently attributed to the thermal degradation of the polymer, involving dehydration and depolymerization reactions from its carboxylic groups.9 The AAPV presence in the fibers resulted in slightly earlier degradation, occurring at higher rates (e.g., PCL-CLO/SA-AAPV fibers lost ≈30% of mass until ≈150 °C; Fig. 3e). This observation may result from alterations in the conformation and organization of SA polymeric chains, influenced by AAPV, thus leading to a less thermally stable structure.9,25 On the other hand, two characteristic CA peaks were observed in CA fibers, in which the first one was centered at ≈164.32 °C (onset at 160.69 °C) and the second was centered at ≈271.91 °C (onset at 266.42 °C). It is likely that the former peak is associated with the glass transition temperature of the polymer, whereas the latter represents the crystallization of CA amorphous domains present on its polymeric chains.39,40 From a general perspective, such fibers presented high thermal stability until 300 °C, after which, they degraded at faster rates until ≈380 °C, due to the breakdown of glycosidic bonds.39,40 The presence of PCL, SA and CA characteristic peaks was preserved in all coaxial fibers (Fig. 3b). However, PCL's melting temperature suffered small shifts and reductions in enthalpies (e.g., from 58.42 °C with an enthalpy of 63.73 J g−1 for PCL fibers to 46.20 °C with an enthalpy of 10.24 J g−1 for PCL-CLO/CA fibers).10 Moreover, on PCL/SA coaxial fibers, mass losses were observed early (until 250 °C) because of SA, whose initial degradation step usually occurs at ≈120 °C (Fig. 3e).9 Although the thermal interactions between PCL and SA are not fully disclosed in the literature, it is predicted that both melting and glass transition temperatures of PCL decrease due to the presence of alginate polymeric chains and its water molecules, also affecting the mobility of the two polymers and their thermal stability.41 | ||
| Fig. 3 DSC (left) and TGA thermograms (right) of (a) and (d) CLO (liquid form), AAPV (powder) and PCL, PCL-CLO, SA, SA-AAPV and CA monolayered, (b) and (e) PCL/SA, PCL/SA-AAPV, PCL-CLO/SA, PCL-CLO/SA-AAPV, SA/CA, SA-AAPV/CA, PCL/CA and PCL-CLO/CA coaxial and (c) and (f) PCL/SA/CA, PCL-CLO/SA/CA, PCL/SA-AAPV/CA and PCL-CLO/SA-AAPV/CA triaxial wet-spun fibers, respectively. | ||
On the other hand, PCL/CA fibers presented a shift regarding their first degradation step (from 200 °C to 250 °C), in relation to CA fibers. One explanation for the present outcome relies on the contact between PCL, a semicrystalline polymer, and CA, an amorphous polymer, reducing PCL's melting point and disrupting its crystallization processes.41 Afterwards, at temperatures between 300 and 400 °C, such fibers also presented lower mass losses than CA fibers, confirming that the presence of PCL on both PCL/SA and PCL/CA coaxial fibers translated into higher thermal stability for both coaxial fiber typologies.10 Finally, PCL/SA/CA triaxial fibers showed small mass losses (≈10%) until ≈100 °C and enthalpies related to SA's characteristic peak also decreased (e.g., from 403.49 J g−1 on SA fibers to 254.99 J g−1 on PCL-CLO/SA/CA fibers) (Fig. 3c). Such alterations were likely caused by the presence of the SA component, which was surrounded by a PCL core and a CA shell. Consequently, the SA intermediate layer was endowed with higher protection from the surrounding environment which delayed its thermal degradation. A second event of mass loss was then observed at ≈200 °C, coinciding with the CA degradation temperature, followed by a faster rate of mass loss at ≈300 °C, which refers to the main degradation step of PCL (as evidenced in the TGA spectra of the PCL fibers) (Fig. 3f).37 Such results enabled the detection of the three polymers and CLO, while showing the effective thermal stability of all engineered samples.
3.4. Porosity
The porosity levels were determined by sample immersion in absolute ethanol for 1 h (Fig. 4a). This evaluation is fundamental for the foreseen use in wound dressings, as it influences moisture balance, gas exchanges, and absorption and management of exudates.42 A highly porous structure enhances moisture retention and gas exchange, being fundamental to the maintenance of a moist wound environment and an easier passage of oxygen, supporting tissue repair.43 Moreover, porosity can play an important role in the mechanical properties of fibrous structures, namely in their stiffness and strength.44 Although a structure endowed with high porosity is frequently more flexible, it is also more fragile and attains lower tensile strengths.45 | ||
| Fig. 4 Representation of (a) porosity levels of each wet-spun fiber typology; (b) SEM images of PCL-CLO/SA-AAPV/CA fibers’ surface topology. Data are reported as mean ± SD (n = 3). Statistical significance was determined via Tukey tests by applying multiple comparisons between the different fiber typologies (statistical differences were found between PCL and CA, PCL-CLO/SA, PCL/SA-AAPV, PCL-CLO/SA-AAPV, SA/CA, SA-AAPV/CA, PCL/CA, PCL-CLO/CA and triaxial fibers: ****p < 0.0001). | ||
All wet-spun fibers attained porosity levels lower than 60%. Such values can potentially benefit the overall properties of the constructs since too high porosity rates impact their mechanical performances, as the presence of voids generates more easily breakable sites throughout the structure.42 However, it is still crucial for wound healing-applied fibers to present a balance of sufficient pores so that the surface area can be enhanced for allowing exudate absorption and management to be efficient, simultaneously preventing the infiltration of external pathogens, which would be challenging in highly porous scenarios.46,47 As a result, reaching the correct balance of porosity in wet-spun fibers towards an enhanced healing system, while simultaneously not compromising other relevant properties (e.g., mechanical performances) remains as one of the most difficult requisites to be accomplished. PCL and SA monolayered fibers presented the lowest porosity as expected (13.71% and 11.83%, respectively), as both polymers are not considered porous in their pristine forms.48,49 In this group of fibers, the presence of AAPV and CLO additives did not significantly alter the porosity levels of the samples, as both molecules were loaded at low concentrations. Nevertheless, a slight increase in porosity was evidenced in the PCL/SA coaxial fibers loaded with AAPV and/or CLO. Although PCL and CLO share chemical interactions between hydroxyl groups and carbonyl groups, the presence of a SA external layer may have limited such chemical interactions due to its partial incorporation in the PCL layer, as stated in Section 3.1. To the authors’ knowledge, the chemical interactions and affinities between PCL and SA are not yet well documented in the literature. One explanation for such interactions is associated with the presence of hydroxyl groups in both polymers, allowing for the formation of chemical bonds.26 This way, a less dense network is formed, in which more pores would be opened. Regarding CA hollow fibers, the results showed high porosity levels. It is likely that the paper-like consistency, characteristic of this polymer, might have contributed to the formation of a large number of pores when in contact with the aqueous coagulation bath.50 Similar outcomes have been observed in lignin-cellulose man-made fibers and polyamide-CA membranes.51,52 Both PCL/CA and SA/CA coaxial fibers and the triaxial fibers reached elevated porosity (51.31%, 51.73% and 48.77%, respectively) because of the presence of the external CA layer and potential residual salt entrapment on the surface of the triaxial samples during coagulation (Fig. 4b and Fig. S1d in the ESI†). Data indicated that those groups of fibers present higher porosity, in relation to PCL and SA monolayered samples. Further testing would be required in order to confirm the capacity of the produced triaxial wet-spun system to reach such balance in porosity levels.
3.5. Mechanical analysis
The mechanical behavior of all wet-spun fiber typologies was assessed by determining their maximum elongations at break and tenacities (Table S3, ESI†). In Fig. 5, the mechanical properties are illustrated by representatives, showing the influence of each component (e.g., PCL; SA; CA polymers) on the fibers’ mechanical performances. SA and SA-AAPV hollow fibers attained low maximum elongations (less than 28 ± 6.24%), not only due to the poor mechanical properties inherent to SA, but also due to their flattened structures and heterogeneities in fiber diameters that could be seen macroscopically.10 Contrarily, CA fibers presented the lowest maximum elongations at break (≈29 ± 5.01%). However, such samples showed high tenacity, resultant from the inherent rigidity of the polymer, as discussed in several reports.53 PCL and PCL-CLO monolayered fibers showed an increase in the maximum elongations compared with the previously mentioned groups of fibers (increases of ≈400% and ≈600%, respectively). Such outcome was expected due to the high elastic properties of PCL.54,55 Here, the presence of CLO enhanced both maximum elongations and tenacity of PCL pristine fibers, likely due to the high affinity between the two compounds, which translated into the formation of a stronger and denser structural network. In fact, several reports have addressed the strong affinity between PCL and eugenol (the main component of CLO) through intricate interactions between carbonyl and hydroxyl groups, respectively.32 The elastic properties of PCL also caused high maximum elongations at break in PCL and SA-containing coaxial fibrous samples. In fact, such group of fibers achieved higher elongations compared to PCL monolayered samples (>200% increase in the case of PCL/SA-AAPV samples). One explanation for such occurrence is related to the presence of an external layer (SA or SA-AAPV) that functioned as a barrier and delayed the rupture of the coaxial fibers (Fig. 5g).10 PCL/CA and PCL-CLO/CA samples also presented high maximum elongations at break (<600%) and a pronounced increase in the tenacities compared to pristine PCL fibers. This is explained by the simultaneous presence of PCL and CA polymers, endowed with high mechanical properties, along with the presence of a shell, protecting the sample from its rupture (Fig. 5k).53–56 Moreover, PCL and CA have high affinity to each other, contributing to the formation of mechanically resistant structures.57 However, the absence of PCL (SA/CA and SA-AAPV/CA samples) resulted in lower maximum elongations at break in such fibrous systems.54,55 Still, both fiber typologies attained high tenacities (greater than ≈26%), since CA is characterized by very high rigidity and tensile strength.53,56 Triaxial fibers attained similar maximum elongations at break compared to the coaxial samples. According to previous studies, the PCL matrix can partially incorporate the SA matrix during coaxial fibers’ production, also evidenced by the reduced thicknesses of the intermediate layers of the triaxial fibers (Fig. 1f and Fig. S3, ESI†).10 Here, the separation between the three layers is also difficult to distinguish, resulting from interfacial adhesion. Such event altered the configurations of PCL polymeric chains and their respective mobilities, generating structures with easily breakable sites and reducing the fibers’ mechanical resistance (Fig. 1).9 Also, all PCL-containing fibers achieved maximum elongations exceeding 400%, confirming the elastic nature of the samples and evidencing that the porosity levels of the fibers did not affect their mechanical behaviors (Fig. 4). From a general perspective, such tenacity levels and maximum elongations of the triaxial fibers show their potential to be incorporated in a wound dressing, with high durability and structural integrity.54,55 | ||
| Fig. 5 Representatives of stress–strain curves of all engineered wet-spun fibers: (a–e) individual fiber layers and their additives (controls); (f–m) coaxial forms modified with respective additives (controls); (n–p) triaxial forms modified with bioactive agents (controls); and (q) final triaxial fiber will all additives included. Tensile tests were repeated 10 times for each fiber typology. | ||
3.6. Swelling capacity
The hydration properties of the fibers were assessed by determining their swelling degrees and water retention (WR) after being in contact with PBS for 7 days (Fig. 6). SA hollow fibers degraded at day 7, due to the low mechanical properties of such constructs.9,10 This result was expected since SA is characterized by poor mechanical performance and, consequently, by the low structural integrity of SA-containing constructs.58 Still, SA presented excellent hydration capacity as demonstrated by WR rates greater than 200% up to day 3 of incubation (Table S4, ESI†).59 Apart from that, the presence of residual salts from the CaCl2 coagulation bath may have also contributed to the high swelling ratios of the fibers, as Ca2+ ions could have gone through ion-exchange processes with Na+ ions from PBS, simultaneously binding with the COO− groups of SA. Consequently, the electrostatic repulsion between COO− groups of SA could have resulted in chain relaxation and allowed for higher swelling degrees.60 The addition of AAPV increased the swelling ratios and WR of the fibers. Since both SA and AAPV present a hydrophilic nature, water molecules from PBS are likely to be drawn to fibers more intensely, leading to a higher WR and, subsequently, an enhancement of their swelling ratios.10 Although the chemical interactions between SA and AAPV are not fully understood, it is likely that their affinity was promoted via their hydroxyl and carboxyl groups. Additionally, the changes in SA polymeric chains’ configurations could have led to polymeric chain expansion, as evidenced by the increase of the intermediate layer's thickness upon addition of AAPV (Fig. 1f), resulting in a more porous fiber and enabling more free hydroxyl groups to be exposed for interacting with water molecules from PBS.61 | ||
| Fig. 6 Wet-spun fibers’ swelling ratios (a) and water retention (b) over 7 days of incubation in PBS. | ||
In contrast, the addition of CLO strongly reduced both the degree of swelling and water retention of PCL monolayered fibers, since both CLO and PCL display hydrophobic properties.62 CA monolayered fibers also attained low swelling ratios. Such observation was expected due to the increased hydrophobic profile of CA compared to cellulose.63,64 Similarly, SA/CA and SA-AAPV/CA fibers attained low hydration capacities since the outermost layer was composed of CA, limiting the infiltration and subsequent interactions of both SA and AAPV with PBS.49,63,64 The low degrees of swelling from PCL/CA and PCL-CLO/CA coaxial fibers were predictable due to the absence of any hydrophilic element.49,63,64 Among all PCL- and SA-containing coaxial fibers, PCL-CLO/SA-AAPV samples reached the highest degree of swelling and WR. PCL has the ability to block the interactions between SA and water molecules and, consequently, the polymer's capacity for absorbing water. Because of that, more hydrophobic groups are exposed to PBS, hence reducing fibers’ hydration properties.49 However, in this particular group of fibers, CLO contributed to an increase in swelling. One explanation may be related to the interactions of CLO with PCL, which altered the configurations of its polymeric chains (also evidenced by the increase in diameter with the loading of CLO onto PCL monolayered fibers) and reduced PCL's ability to interfere with the interaction of SA with water molecules. In light of all these polymers and active agents’ mutual interferences, triaxial fibers displayed the lowest swelling ratios. Here, the presence of both PCL and CA, endowed with hydrophobic profiles, resulted in a lower amount of water molecules being absorbed into the fibers, leading to a decrease of WR and interfering with the interactions established between SA polymeric chains/AAPV (hydrophilic components) and water molecules from PBS.10,49,63,64
Data are presented as average percentage of mass loss ± SD (n = 3). Statistical significance was determined via the Tukey test applying multiple comparisons between the different fiber typologies – significance between PCL and SA; SA/AAPV: p < 0.0074 on day 1, significance between PCL and SA: p < 0.0123; PCL and PCL/SA-AAPV: p < 0.0196 on day 3; significance between PCL/SA and SA; SA-AAPV: p < 0.0060 on day 7; significance between PCL/SA and PCL-CLO/SA-AAPV: p < 0.0045 on day 14; significance between PCL and PCL/SA; PCL/SA-AAPV: p < 0.0174 on day 21; significance between PCL/SA and PCL/SA-AAPV; SA-AAPV/CA: p < 0.0476 on day 28.
3.7. Degradation profile
The stability of the wet-spun fibers in physiological media was assessed via their incubation in PBS at 37 °C and pH 7.4. Fibers’ degradation was detected visually and by tracking mass losses throughout 28 days of incubation (Fig. 7). SA and SA-AAPV hollow fibers were completely degraded after the first 14 days of incubation (Table S5, ESI†), likely by polymer dissolution into the media, a result which had also been reported in a previous study conducted by our research team.10 In fact, such occurrence was expected due to the poor mechanical properties of SA, its high affinity towards water (Fig. 5; leading to partial dissolution in PBS), along with the visible flattened structure of the two fiber typologies (Fig. S4, ESI†).9,10,58 | ||
| Fig. 7 Wet-spun fibers’ degradation profiles over 28 days of incubation in PBS. Data are presented as average percentage of mass loss ± SD (n = 3). | ||
On the other hand, all the remaining fibers retained their structural integrity throughout the 28 days of incubation in PBS (Fig. S5, ESI†), as a result of the presence of PCL and/or CA polymers, both characterized by high mechanical and structural properties.53–56 Such conclusion was also supported by the high maximum elongations at break and tensile strengths attained for all PCL-containing fibers (Fig. 5). Other researchers have reported similar outcomes; for instance, Schmitt et al. developed a scaffold composed of PCL wet-spun fibers for cardiac tissue engineering applications, showing that PCL endowed the structures not only with high elongations and Young's modulus but also with elevated structural stability after exposure to physiological media.65 In addition, Yuan et al. confirmed the high rigidity derived from the CA component in dry-jet wet-spun fibers.66
Interestingly, PCL and PCL-CLO monolayered fibers presented negative mass losses, likely due to interactions between PCL and PBS salts. Although the mechanisms involved in such interactions are not yet reported in the literature, according to Zhang et al., the functional groups of both PCL and PBS are capable of being retained effectively upon the formation of PCL/PBS composites.67 One explanation for such occurrence is associated with the formation of a new hydrogen bond system between the carbonyl and methylene group of PCL.67 During the initial 7 days of incubation, CLO led to mass increments, probably because of the changes introduced in the configuration and organization of the PCL polymeric chains upon the interaction.24 However, this effect was less pronounced after the 7-day mark, which is explained by the volatile nature of CLO and its instability in physiological media (Table S5, ESI†).68,69 In contrast, there were no mass increments in CA monolayered fibers. To the authors’ knowledge, there are no reports on the chemical interactions between CA and PBS salts. Still, as CA presents hydrophobic character, it is reasonable to assume a low affinity towards PBS, an aqueous-based buffer.63,64 Because of CA's inherent rigidity, mass losses were negligible.49,63,64 As predicted, the addition of SA and AAPV (SA/CA and SA-AAPV/CA coaxial fibers) resulted in higher mass losses (reduced mechanical performances), potentially affecting the configuration of the CA polymeric chains and, thus, compromising the fibers’ structural integrity.9,10
PCL/SA, PCL/SA-AAPV, PCL-CLO/SA and PCL-CLO/SA-AAPV coaxial fibers have likely suffered from the simultaneous diffusion of buffer particles, the shell degradation effects and SA dissolution. The quick degradation of SA and SA-AAPV layers led to higher exposure of PCL to the media and, therefore, diffusion of the media onto PCL predominated, justifying the mass increments observed for the four fiber typologies.10,49 PCL/CA and PCL-CLO/CA did not appear to suffer from the effect of PCL binding with PBS salts. The presence of CA on the outer layer of the fibers functioned as a barrier, being endowed with high rigidity and structural properties, which limited the interactions of PCL with PBS.10,49,63,64 Hence, the influence of PCL binding with PBS salts on the degradation profiles of the triaxial fibers was also low, once more, due to the presence of the CA outer layer.10,49,63,64 Generally, the triaxial fibers showed low mass losses, translated into high structural stability imposed by the simultaneous influence of PCL's elastic behavior and CA's high rigidity.10,49,63–65 On the other hand, the loading of CLO also displayed some influence on the mass losses of such fibers in the initial 7 days, prior to its evaporation (Table S5, ESI†).24 Still, the influence was lower compared with the monolayered fibers, as the shell components probably hindered the interactions between PCL and CLO.3 All in all, the results proved the fibers’ high stability in physiological media, attesting to their suitability for the desired application.
3.8. Dressing production
A wound dressing formed of PCL-CLO/SA-AAPV/CA triaxial fibers was produced with a circular single jersey knitting machine (Fig. 8). The produced dressing attained 771.70 ± 0.138 μm thickness, corresponding to an increase of 219 μm in comparison with the average total diameter of PCL-CLO/SA-AAPV/CA fibers. Such increment resulted from the combination of several filaments that passed through each needle of the knitting machine in order to attain a more efficient and potentially resilient production of the dressing. The engineered structure possessed an average number of 14 wales and 7 courses per cm2, with 7.360 ± 2.827 mg cm−2 of mass per unit of area. Such task was successfully achieved due to the high elasticity of PCL, as previously discussed in Section 3.5. It is noteworthy that the presence of two external layers (the intermediate layer of SA and the outermost layer of CA) offered protection to a possible fiber's rupture. These preliminary results are a proof-of-concept of the potential of the engineered triaxial fibers for future applications in wound dressings. Nevertheless, further work is required towards optimizing dressing production, including conducting a complete analysis of the produced knitted fabric and study of the processibility of the fibers within varying knitting structures (such as rib, pique and interlock knit, interlaced structure or jacquard weave), and through all that determine the most efficient structure for the foreseen purpose. Additionally, the elasticity of the CA layer could be improved by applying surface modification techniques, such as graft copolymerization with acrylate monomers, coating with elastomeric materials like silicone rubber, or surface functionalization by adding amino groups. These modifications can help mitigate potential rupture events and allow for stronger tensile strengths to be employed during knitting before scaling up to larger production. | ||
| Fig. 8 Photograph of the produced dressing collected at (a) macroscopic scale (photograph was taken at 9 cm distance); (b) brightfield microscopy at 5× magnification. | ||
4. Conclusions
Triaxial wet-spun fibers were successfully produced and processed in the form of a small dressing in this research. Microscopic observations confirmed the effective construction of the triaxial system. The engineered fibers attained good mechanical, physical and thermal properties, along with the capacity to preserve their structural integrity when exposed to physiological medium. The external layer of the fibers showed higher porosity, resulting from its contact with Ca2+ salts from the coagulation bath, which allowed for the opening of pores of pivotal importance for the controlled release of active agents. All data presented, along with the fact that the system is composed of biodegradable polymers and active agents loaded at low concentrations, confirmed the potential and feasibility of this triaxial fibrous system for a future application as a wound dressing for the treatment of CWs. Further improvements in attaining higher homogeneity of the fibers’ diameters, morphologies and lower interfacial layer adhesion will be required, involving the use of different spinnerets and the addition of stretching and washing baths. The enhancement of elasticity of the intermediate and outermost layers will also be necessary, along with the development of different textile dressing structures composed of the proposed triaxial system towards the optimization of the wound dressing production. The porosity of such dressing should also be followed by more accurate porosity assessments, including the use of micro-CT or mercury intrusion porosimety.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was funded by the European Regional Development Fund through the Operational Competitiveness Program and the National Foundation for Science and Technology of Portugal (FCT) under the project UID/CTM/00264/2020 of Centre for Textile Science and Technology (2C2T), on its base (https://doi.org/10.54499/UIDB/00264/2020) and programmatic (https://doi.org/10.54499/UIDP/00264/2020) components, as well as the project UID/QUI/00686/2020 of Centre of Chemistry, on its base (https://doi.org/10.54499/UIDB/00686/2020) and programmatic (https://doi.org/10.54499/UIDP/00686/2020) components. This study was supported by the Portuguese Foundation for Science and Technology (FCT) under the scope of the strategic funding of the UIDB/04469/2020 unit, and by LABBELS – Associate Laboratory in Biotechnology, Bioengineering and Microelectromechanical Systems, LA/P/0029/2020. D. R. acknowledges FCT for the funding via scholarship 2022.13494.BDANA. C. S. M. acknowledges FCT for PhD funding via scholarship 2020.08547.BD, along with extra funding for conducting an international internship in Espoo, Finland. H. P. F. also acknowledges FCT for auxiliary researcher contract 2021.02720.CEEIND.References
- C. S. Miranda, A. R. M. Ribeiro, N. C. Homem and H. P. Felgueiras, Spun Biotextiles in Tissue Engineering and Biomolecules Delivery Systems, Antibiotics, 2020, 9, 174, DOI:10.3390/antibiotics9040174.
- C. S. Miranda, A. F. G. Silva, S. M. M. A. Pereira-Lima, S. P. G. Costa, N. C. Homem and H. P. Felgueiras, Tunable Spun Fiber Constructs in Biomedicine: Influence of Processing Parameters in the Fibers’ Architecture, Pharmaceutics, 2022, 14, 174, DOI:10.3390/pharmaceutics14010164.
- A. Mirabedini, Developing Novel Spinning Methods to Fabricate Continuous Multifunctional Fibres, Doctoral thesis, University of Wollongong, 2017, 1–235.
- R. Nayak and R. Padhye, Nano Fibres by Electro spinning: Properties and Applications, J. Text. Eng. Fashion Technol., 2017, 2, 486–497, DOI:10.15406/jteft.2017.02.00074.
- D. Puppi, C. Mota, M. Gazzarri, D. Dinucci, A. Gloria, M. Myrzabekova, L. Ambrosio and F. Chiellini, Additive manufacturing of wet-spun polymeric scaffolds for bone tissue engineering, Biomed. Microdevices, 2012, 14, 1115–1127, DOI:10.1007/s10544-012-9677-0.
- D. Brüggemann, J. Michel, N. Suter, M. G. de Aguiar and M. Maas, Wet-spinning of magneto-responsive helical chitosan microfibers, Beilstein J. Nanotechnol., 2020, 11, 991–999, DOI:10.3762/bjnano.11.83.
- A. Rohani, A. Nouri and A. Sutti, A perspective on the wet spinning process and its advancements in biomedical sciences, Eur. Polym. J., 2022, 181, 111681, DOI:10.1016/j.eurpolymj.2022.111681.
- J. Xue, T. Wu, Y. Dai and Y. Xia, Electrospinning and electrospun nanofibers: Methods, materials, and applications, Chem. Rev., 2019, 119, 5298–5415, DOI:10.1021/acs.chemrev.8b00593.
- N. C. Homem, T. D. Tavares, C. S. Miranda, J. C. Antunes, M. P. Teresa and H. P. Felgueiras, Functionalization of crosslinked sodium alginate/gelatin wet-spun porous fibers with Nisin Z for the inhibition of Staphylococcus aureus - induced infections, Int. J. Mol., 2021, 22, 1930, DOI:10.3390/ijms22041930.
- C. S. Miranda, A. F. G. Silva, C. L. Seabra, S. Reis, M. M. P. Silva, S. M. M. A. Pereira-Lima, S. P. G. Costa, N. C. Homem and H. P. Felgueiras, Sodium alginate/polycaprolactone co-axial wet-spun microfibers modified with N-carboxymethyl chitosan and the peptide AAPV for Staphylococcus aureus and human neutrophil elastase inhibition in potential chronic wound scenarios, Biomater. Adv., 2023, 151, 213488, DOI:10.1016/j.bioadv.2023.213488.
- A. Mirabedini, Z. Lu, S. Mostafavian and J. Foroughi, Triaxial carbon nanotube/conducting polymer wet-spun fibers supercapacitors for wearable electronics, Nanomaterials, 2021, 11, 1–16, DOI:10.3390/nano11010003.
- G. Chen, S. Braccini, C. Chen, J. Jacqueline, F. Barsotti, C. Ferrario, G. Chen and D. Puppi, Additive manufacturing of wet-spun chitosan/hyaluronic acid scaffolds for biomedical applications Additive manufacturing of wet-spun chitosan/hyaluronic acid scaffolds for biomedical applications, Carbohydr. Polym., 2024, 329, 121788, DOI:10.1016/j.carbpol.2024.121788.
- W. Lu, Q. Deng, M. Liu, B. Ding and Z. Xiong, Coaxial Wet Spinning of Boron Nitride Nanosheet Based Composite Fibers with Enhanced Thermal Conductivity and Mechanical Strength, Nanomicro. Lett., 2024, 1–13, DOI:10.1007/s40820-023-01236-w.
- E. Malikmammadov, T. E. Tanir, A. Kiziltay and V. Hasirci, PCL and PCL-Based Materials in Biomedical Applications, J. Biomater. Sci., 2017, 29, 863, DOI:10.1080/09205063.2017.1394711.
- Y. Hou, W. Wang and P. Bartolo, Materials & Design Investigation of polycaprolactone for bone tissue engineering scaffolds: In vitro degradation and biological studies, Mater. Des., 2022, 216, 110582, DOI:10.1016/j.matdes.2022.110582.
- H. P. Felgueiras, N. C. Homem, M. A. Teixeira, A. R. M. Ribeiro, M. O. Teixeira, J. C. Antunes and M. T. P. Amorim, Biodegradable wet-spun fibers modified with antimicrobial agents for potential applications in biomedical engineering, J. Phys.: Conf. Ser., 2021, 12007, DOI:10.1088/1742-6596/1765/1/012007.
- M. A. Teixeira, M. T. P. Amorim and H. P. Felgueiras, PVA/CA based electrospun nanofibers: Influence of processing parameters in the fiber diameter, IOP Conf. Ser.: Mater. Sci. Eng., 2019, 634, 012040, DOI:10.1088/1757-899X/634/1/012040.
- M. A. Teixeira, M. T. P. Amorim and H. P. Felgueiras, Cellulose Acetate in Wound Dressings Formulations: Potentialities and Electrospinning Capability, Springer International Publishing, 2020, pp. 1515–1525 DOI:10.1007/978-3-030-31635-8.
- D. Yang and K. S. Jones, Effect of alginate on innate immune activation of macrophages, J. Biomed. Mater. Res., Part A, 2009, 90, 411–418, DOI:10.1002/jbm.a.32096.
- J. Sun and H. Tan, Alginate-based biomaterials for regenerative medicine applications, Materials, 2013, 6, 1285–1309, DOI:10.3390/ma6041285.
- C. S. Miranda, E. Marinho, C. L. Seabra, C. Evenou, J. Lamartine, B. Fromy, S. P. G. Costa, N. C. Homem and H. P. Felgueiras, Antimicrobial, antioxidant and cytocompatible coaxial wet-spun fibers made of polycaprolactone and cellulose acetate loaded with essential oils for wound care, Int. J. Biol. Macromol., 2024, 277, DOI:10.1016/j.ijbiomac.2024.134565.
- P. Knauth, Z. L. López, G. J. A. Hernández and M. T. E. Sevilla, Cinnamon essential oil: Chemical composition and biological activities, Biochem. Res. Trends, 2018, 215–243 Search PubMed.
- F. C. Santos, A. M. S. Soares, M. S. T. Gonçalves and S. P. G. Costa, Phototriggered release of tetrapeptide AAPV from coumarinyl and pyrenyl cages, Amino Acids, 2017, 49, 1077–1088, DOI:10.1007/s00726-017-2405-6.
- C. S. Miranda, A. F. G. Silva, C. L. Seabra, S. Reis, M. M. P. Silva, S. M. M. A. Pereira-lima, S. P. G. Costa and H. P. Felgueiras, Sodium alginate/polycaprolactone co-axial wet-spun microfibers modified with N-carboxymethyl chitosan and the peptide AAPV for Staphylococcus aureus and human neutrophil elastase in chronic wound scenarios, Biomater. Adv., 2023, 151, 213488, DOI:10.1016/j.bioadv.2023.213488.
- J. M. Domingues, M. O. Teixeira, M. A. Teixeira, D. Freitas, S. F. da Silva, S. D. Tohidi, R. D. V. Fernandes, J. Padrão, A. Zille, C. Silva, J. C. Antunes and H. P. Felgueiras, Inhibition of Escherichia Virus MS2, Surrogate of SARS-CoV-2, via Essential Oils-Loaded Electrospun Fibrous Mats: Increasing the Multifunctionality of Antivirus Protection Masks, Pharmaceutics, 2022, 14, 303, DOI:10.3390/pharmaceutics14020303.
- C. S. Miranda, A. F. G. Silva, C. L. Seabra, S. Reis, M. M. P. Silva, S. M. M. A. Pereira-Lima, S. P. G. Costa, N. C. Homem and H. P. Felgueiras, Sodium alginate/polycaprolactone co-axial wet-spun microfibers modified with N-carboxymethyl chitosan and the peptide AAPV for Staphylococcus aureus and human neutrophil elastase inhibition in potential chronic wound scenarios, Biomater. Adv., 2023, 151, 213488, DOI:10.1016/j.bioadv.2023.213488.
- H. P. Felgueiras, N. C. Homem, M. A. Teixeira, A. R. M. Ribeiro, J. C. Antunes and M. T. P. Amorim, Physical, thermal, and antibacterial effects of active essential oils with potential for biomedical applications loaded onto cellulose acetate/polycaprolactone wet-spun microfibers, Biomolecules, 2020, 10, 1–20, DOI:10.3390/biom10081129.
- P. R. Schmitt, K. D. Dwyer, A. J. Minor and K. L. K. Coulombe, Wet-Spun Polycaprolactone Scaffolds Provide Customizable Anisotropic Viscoelastic Mechanics for Engineered Cardiac Tissues, Polymers, 2022, 14, 4571, DOI:10.3390/polym14214571.
- J. M. Rocha, R. P. C. L. Sousa, D. Sousa, S. D. Tohidi, A. Ribeiro, R. Fangueiro and D. P. Ferreira, Polycaprolactone-Based Fibrous Scaffolds Reinforced with Cellulose Nanocrystals for Anterior Cruciate Ligament Repair, Appl. Sci., 2025, 15, 2301, DOI:10.3390/app15052301.
- M. Harahap, B. Hararak, I. Khan, S. Pandita and G. Saharman, Wet-spinning of cellulose acetate reinforced with acetylated nano-crystalline cellulose as carbon fibre precursors, IOP Conf. Ser.: Mater. Sci. Eng., 2019, 553, 012038, DOI:10.1088/1757-899X/553/1/012038.
- M. Marques, J. Carneiro, B. Justus, J. T. Espinoza, J. M. Budel, P. V. Farago and J. P. De Paula, Preparation and characterization of a novel antimicrobial film dressing for wound healing application, Braz. J. Pharm. Sci., 2020, 1–11, DOI:10.1590/s2175-97902020000118784.
- Q. Lv, M. Wu and Y. Shen, Enhanced swelling ratio and water retention capacity for novel super-absorbent hydrogel, Colloids Surf., A, 2019, 123972, DOI:10.1016/j.colsurfa.2019.123972.
- H. P. Felgueiras, N. C. Homem, M. A. Teixeira, A. R. M. Ribeiro, J. C. Antunes and M. T. P. Amorim, Physical, thermal, and antibacterial effects of active essential oils with potential for biomedical applications loaded onto cellulose acetate/polycaprolactone wet-spun microfibers, Biomolecules, 2020, 10, 1–20, DOI:10.3390/biom10081129.
- W. W. Hu, Y. C. Wu and Z. C. Hu, The development of an alginate/polycaprolactone composite scaffold for in situ transfection application, Carbohydr. Polym., 2018, 183, 29–36, DOI:10.1016/j.carbpol.2017.11.030.
- A. Dodero, M. Alloisio, M. Castellano and S. Vicini, Multilayer Alginate-Polycaprolactone Electrospun Membranes as Skin Wound Patches with Drug Delivery Abilities, ACS Appl. Mater. Interfaces, 2020, 12, 31162–31171, DOI:10.1021/acsami.0c07352.
- A. Benkaddour, K. Jradi, S. Robert and C. Daneault, Grafting of polycaprolactone on oxidized nanocelluloses by click chemistry, Nanomaterials, 2013, 3, 141–157, DOI:10.3390/nano3010141.
- X. He, Optimization of Deacetylation Process for Regenerated Cellulose Hollow Fiber Membranes, Int. J. Polym. Sci., 2017, 2017(1), 3125413, DOI:10.1155/2017/3125413.
- R. F. P. Pereira, J. P. Donoso, C. J. Magon, I. D. A. Silva, M. A. Cardoso, M. C. Gonçalves, R. C. Sabadini, A. Pawlicka, V. de Zea Bermudez and M. M. Silva, Ion conducting and paramagnetic d-PCL(530)/siloxane-based biohybrids doped with Mn2+ ions, Electrochim. Acta, 2016, 211, 804–813, DOI:10.1016/j.electacta.2016.06.088.
- S. Phaiju, P. Mulmi, D. K. Shahi, T. I. Hwang, A. P. Tiwari, R. Joshi, H. R. Pant and M. K. Joshi, Antibacterial Cinnamon Essential Oil Incorporated Poly(ε-Caprolactone) Nanofibrous Mats: New Platform for Biomedical Application, J. Inst. Sci. Technol., 2020, 25, 9–16, DOI:10.3126/jist.v25i2.33724.
- S. Zhelyazkov, G. Zsivanovits, E. Stamenova and M. Marudova, Physical and barrier properties of clove essential oil loaded potato starch edible films, Biointerface Res. Appl. Chem., 2022, 12, 4603–4612, DOI:10.33263/briac124.46034612.
- S. D. Ribeiro, A. B. Meneguin, H. da, S. Barud, J. M. Silva, R. L. Oliveira, R. M. N. de Asunção, T. F. Tormin, R. A. A. Muñoz, G. R. Filho and C. A. Ribeiro, Synthesis and characterization of cellulose acetate from cellophane industry residues. Application as acetaminophen controlled-release membranes, J. Therm. Anal. Calorim., 2022, 147, 7265–7275, DOI:10.1007/s10973-021-11022-8.
- P. A. Vinodhini, K. Sangeetha, G. Thandapani, P. N. Sudha, V. Jayachandran and A. Sukumaran, FTIR, XRD and DSC studies of nanochitosan, cellulose acetate and polyethylene glycol blend ultrafiltration membranes, Int. J. Biol. Macromol., 2017, 104, 1721–1729, DOI:10.1016/j.ijbiomac.2017.03.122.
- G. Ö. Kayan and A. Kayan, Polycaprolactone Composites/Blends and Their Applications Especially in Water Treatment, Chem. Eng., 2023, 7(6), 104, DOI:10.3390/chemengineering7060104.
- T. Kavitha, S. I. H. Abdi and S. Y. Park, pH-sensitive nanocargo based on smart polymer functionalized graphene oxide for site-specific drug delivery, Phys. Chem. Chem. Phys., 2013, 15(14), 5176–5185 RSC.
- A. Seijo-Rabina, S. Paramés-Estevez, A. Concheiro, A. Pérez-Muñuzuri and C. Alvarez-Lorenzo, Effect of wound dressing porosity and exudate viscosity on the exudate absorption: In vitro and in silico tests with 3D printed hydrogels, Int. J. Pharm.: X, 2024, 8, 100288, DOI:10.1016/j.ijpx.2024.100288.
- Q. Zhang, Y. Jiang, Y. Zhang, Z. Ye, W. Tan and M. Lang, Effect of porosity on long-term degradation of poly (ε -caprolactone) scaffolds and their cellular response, Polym. Degrad. Stab., 2012, 1–10, DOI:10.1016/j.polymdegradstab.2012.10.008.
- P. Norooz Kermanshahi and G. Maria Barbosa Soares, The Iranian Journal of Textile Nano-bio Modification Systematic Review of Wound Dressings: A Movement from the Past to the Present, 2022, https://www.ijtnbm.ir Search PubMed.
- T. Bhattacharjee, D. B. Amchin, J. A. Ott, F. Kratz and S. S. Datta, Article Chemotactic migration of bacteria in porous media, Biophys. J., 2021, 120, 3483–3497, DOI:10.1016/j.bpj.2021.05.012.
- C. D. Weller, V. Team and G. Sussman, First-Line Interactive Wound Dressing Update: A Comprehensive Review of the Evidence, Front. Pharmacol., 2023, 11, 1–13, DOI:10.3389/fphar.2020.00155.
- R. Adrian and K. Szustakiewicz, Structure-property relationships in PCL porous scaffolds obtained by means of the TIPS and TIPS-PL methods, Polym. Test., 2023, 118, 107906, DOI:10.1016/j.polymertesting.2022.107906.
- K. Y. Lee and D. J. Mooney, Alginate: Properties and biomedical applications, Prog. Polym. Sci., 2012, 37, 106–126, DOI:10.1016/j.progpolymsci.2011.06.003.
- S. Wu, X. Qin and M. Li, The structure and properties of cellulose acetate materials: A comparative study on electrospun membranes and casted films, J. Ind. Text., 2014, 44, 85–98, DOI:10.1177/1528083713477443.
- J. Bengtsson, E. Johnsson, H. Ulmefors and T. Köhnke, Revealing pore size distribution in cellulose and lignin cellulose man made fibers – effect of draw ratio and lignin content, Cellulose, 2024, 31, 7003–7013, DOI:10.1007/s10570-024-06007-3.
- I. Ounifi, Y. Guesmi, C. Ursino, H. Agougui, M. Jabli, A. Hafiane, A. Figoli and E. Ferjani, Synthesis of a Thin-Film Polyamide-Cellulose Acetate Membrane: Effect of Monomers and Porosity on Nano-Filtration Performance, J. Nat. Fibers, 2021, 1–18, DOI:10.1080/15440478.2021.2002766.
- R. Erdmann and S. Kabasci, Thermal Properties of Plasticized Cellulose Acetate and Its β -Relaxation Phenomenon Polymers4, Polymers, 2012, 1012–1014, DOI:10.3390/polym4021012.
- G. U. Rani and S. Sharma, Biopolymers, Bioplastics and Biodegradability: An Introduction, Reference Module in Materials Science and Materials Engineering, Elsevier, 2021 DOI:10.1016/B978-0-12-820352-1.00131-0.
- I. Dermatology, M. Christen and F. Vercesi, Polycaprolactone: How a Well-Known and Futuristic Polymer Has Become an Innovative Collagen-Stimulator in Esthetics, Clin., Cosmet. Invest. Dermatol., 2020, 31–48, DOI:10.2147/ccid.S229054.
- X. Dreux, C. Carrot, A. Argoud and C. Vergelati, Viscoelastic behaviour of cellulose acetate/triacetin blends by rheology in the melt state, Carbohydr. Polym., 2019, 1–19, DOI:10.1016/j.carbpol.2019.114973.
- K. Khoshnevisan, H. Maleki, H. Samadian, M. Doostan and M. R. Khorramizadeh, Antibacterial and antioxidant assessment of cellulose acetate/polycaprolactone nanofibrous mats impregnated with propolis, Int. J. Biol. Macromol., 2019, 140, 1260–1268, DOI:10.1016/j.ijbiomac.2019.08.207.
- Y. B. Kim and G. H. Kim, PCL/Alginate Composite Scaffolds for Hard Tissue Engineering: Fabrication, Characterization, and Cellular Activities, ACS Comb. Sci., 2014, 17(2), 87–99, DOI:10.1021/co500033h.
- M. Umar, A. Ullah, H. Nawaz, T. Areeb, M. Hashmi, D. Kharaghani, K. O. Kim and I. S. Kim, Wet-spun bi-component alginate based hydrogel fibers: Development and in-vitro evaluation as a potential moist wound care dressing, Int. J. Biol. Macromol., 2021, 168, 601–610, DOI:10.1016/j.ijbiomac.2020.12.088.
- S. K. Bajpai and S. Sharma, Investigation of swelling/degradation behaviour of alginate beads crosslinked with Ca2+ and Ba2+ ions, React. Funct. Polym., 2004, 59, 129–140, DOI:10.1016/j.reactfunctpolym.2004.01.002.
- L. Fan, Y. Du, B. Zhang, J. Yang, J. Zhou and J. F. Kennedy, Preparation and properties of alginate/carboxymethyl chitosan blend fibers, Carbohydr. Polym., 2006, 65, 447–452, DOI:10.1016/j.carbpol.2006.01.031.
- A. Huang, Y. Jiang, B. Napiwocki, H. Mi, X. Peng and L. S. Turng, Fabrication of poly(ε-caprolactone) tissue engineering scaffolds with fibrillated and interconnected pores utilizing microcellular injection molding and polymer leaching, RSC Adv., 2017, 7, 43432–43444, 10.1039/c7ra06987a.
- A. Kramar and J. González-benito, Materials & Design Preparation of cellulose acetate film with dual hydrophobic-hydrophilic properties using solution blow spinning, Mater. Des., 2023, 227, 111788, DOI:10.1016/j.matdes.2023.111788.
- S. Wu, X. Qin and M. Li, The structure and properties of cellulose acetate materials: A comparative study on electrospun membranes and casted films, J. Ind. Text., 2014, 44, 85–98, DOI:10.1177/1528083713477443.
- P. R. Schmitt, K. D. Dwyer, A. J. Minor and K. L. K. Coulombe, Wet-Spun Polycaprolactone Scaffolds Provide Customizable Anisotropic Viscoelastic Mechanics for Engineered Cardiac Tissues, Polymers, 2022, 14, 4571, DOI:10.3390/polym14214571.
- W. Yuan, K. Wu, N. Liu, Y. Zhang and H. Wang, Cellulose acetate fibers with improved mechanical strength prepared with aqueous NMMO as solvent, Cellulose, 2018, 25, 6395–6404, DOI:10.1007/s10570-018-2032-8.
- J. Huang, C. Cui, G. Yan, J. Huang and M. Zhang, A Study on Degradation of Composite Material PBS/PCL, Polym. Polym. Compos., 2016, 24, 143–148, DOI:10.1177/096739111602400209.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ma01105h |
| This journal is © The Royal Society of Chemistry 2025 |