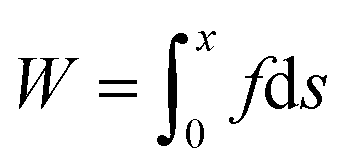DOI:
10.1039/D4MA01102C
(Paper)
Mater. Adv., 2025,
6, 1513-1519
Facile coating of low-molecular-weight stretchable adhesive films leveraging carbodiimide-to-urea conversion and gallic acid for enhanced adhesion†
Received
5th November 2024
, Accepted 13th January 2025
First published on 14th January 2025
Abstract
We present a simple method for bonding an elastomer substrate using a stretchable, low-molecular-weight adhesive. By mixing 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide (EDC) and gallic acid on an aminated surface, EDC is converted into a urea derivative. This derivative features both hydrogen bond donor and acceptor functionalities, combined with bio-inspired adhesive gallol groups, which collectively enhance adhesiveness through extensive hydrogen bonding. Importantly, the adhesive preserves the stretchability of the elastomer.
1. Introduction
Adhesives play a vital role in the robust attachment of biomedical devices to biological tissues or living bodies. Numerous stretchable devices have been reported for biosensing1–8 and therapeutic applications.9–11 In most cases, these devices require an adhesive for long-term use on the target tissue. Potential adhesive materials for stretchable substrates are either polymeric materials (e.g. polydopamine,11,12 graphene oxide-based bioadhesives,7 polyethylene glycol–lactide acid diacrylate hydrogels,8 and gallol-functionalized hydrogels13–15) or low-molecular-weight materials (e.g. ionic liquids16 and supramolecular materials17,18). Although polymeric adhesives have strong adhesive performance due to abundant covalent and cross-linked structures,7,8 these curable adhesives require careful surface treatment and have a risk of impairing the flexibility or stretchability of the elastomer substrates. In contrast, the adhesiveness of low-molecular-weight adhesives (LMWAs) can be tuned based on their various in situ non-covalent interactions, and they have larger absolute adhesion areas owing to a higher degree of freedom for molecular diffusion.19 Although LMWAs are strongly adhesive, most are also highly viscous.16–18 This high viscosity of LMWAs may impair the stretchability of the substrate. Therefore, further optimization of non-covalent bonds of LMWAs is necessary to balance adhesiveness with the stretchability of elastomeric substrates.
To ensure the stretchability of LMWAs, we aimed to increase the formation of additional non-covalent bonds from the precursor solution by applying the conversion of carbodiimide to urea derivatives. Liang et al. created an adhesive that used urea derivatives as hydrogen bond donors for efficient adhesion.20 Urea derivatives are often produced as by-products from carbodiimides such as 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide (EDC) during amide bond formation. EDC has also tertiary amino hydrochloride groups that can be used as hydrogen bond acceptors.20 Therefore, in the present study, we used EDC as an adhesive precursor solution. In addition to exploiting this conversion from carbodiimide to urea derivatives, we introduced bio-inspired adhesive-functional gallol groups as auxiliary hydrogen bond donors.15,21 They were used to supplement the hydrogen bonds from urea and amino groups for stronger adhesiveness.22
Herein, we report a facile method for coating a low-molecular-weight stretchable adhesive on an aminated substrate of polydimethylsiloxane (PDMS), one of the versatile elastomers (Fig. 1a). EDC and gallic acid (GA) are mixed on the surface of PDMS so that the EDC is converted to 1-ethyl-3-(3-dimethylaminopropyl)urea (EDU) for the formation of an adhesive layer with numerous hydrogen bond acceptors and donors from gallol, urea, and amino groups (Fig. 1b and c). The simultaneous modification of the surface of the PDMS substrate by gallol via amino groups produces an adhesive substrate (Fig. 1b). When applied to an aminated PDMS synthesized through hydrophilization by plasma treatment23 followed by a silane coupling reaction24 (Fig. S1, ESI†), the adhesive enhanced the adhesiveness of PDMS fabricated by bar-coating. The adhesive coating does not sacrifice the stretchability of the original PDMS. The simultaneous adhesiveness and stretchability are attributed to the energy dissipation and robust cohesion ensured by hydrogen bonds in the adhesive layer.
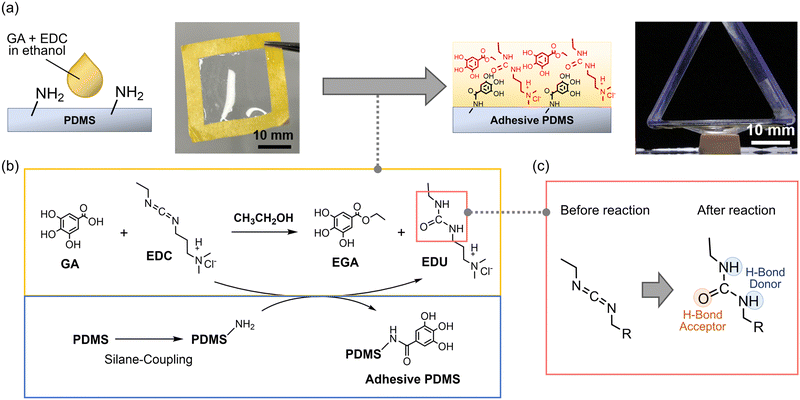 |
| | Fig. 1 (a) Schematic showing the modification of a PDMS film by casting GA and EDC in ethanol to enhance the adhesiveness of the PDMS film, (b) the reaction scheme, and (c) accompanying substitution of carbodiimide to urea which can form hydrogen bonds (PDMS = polydimethylsiloxane; GA = gallic acid; EDC = 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide). | |
2. Materials and methods
2.1. Materials
Polyvinyl alcohol (PVA, MW: 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–23000) was purchased from Kanto Chemical (Tokyo, Japan). Polyethylene terephthalate (PET, 12 cm width, Lumirrors L-25T60) was purchased from Toray Industry Inc. (Tokyo, Japan). Ethyl acetate, hexane, super-dehydrated ethanol (99.5%), dimethyl sulfoxide-d6 (DMSO-d, 99.9%, containing 0.05 vol% tetramethylsilane (TMS)), and 1-ethyl-3-(3-dimethylaminopropyl)urea (EDU) were purchased from FUJIFILM Wako Pure Chemical (Osaka, Japan). Polydimethylsiloxane (PDMS, SILPOT 184) was purchased from Dow-Toray Specialty Materials (Tokyo, Japan). 3-Aminopropyltrimethoxysilane (APTMS), gallic acid (GA), 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDC), and ethyl gallate (EGA) were purchased from Tokyo Chemical Industry Co., Ltd (Tokyo, Japan). Scotch tape (243J Plus) was purchased from 3M Company (MN, USA). The glass substrate (no. 5 thickness, 30 × 30 mm2) was purchased from Matsunami Glass (Osaka, Japan). The nylon mesh (HC-15) was purchased from Sefar Holding AG (Thal, Switzerland). 70% Ethanol (alcohol preparations ET-N) was purchased from UENO FOOD TECHNO INDUSTRY, Ltd (Tokyo, Japan). The bio-skin model was purchased from Beaulax Co., Ltd (Saitama, Japan).
000–23000) was purchased from Kanto Chemical (Tokyo, Japan). Polyethylene terephthalate (PET, 12 cm width, Lumirrors L-25T60) was purchased from Toray Industry Inc. (Tokyo, Japan). Ethyl acetate, hexane, super-dehydrated ethanol (99.5%), dimethyl sulfoxide-d6 (DMSO-d, 99.9%, containing 0.05 vol% tetramethylsilane (TMS)), and 1-ethyl-3-(3-dimethylaminopropyl)urea (EDU) were purchased from FUJIFILM Wako Pure Chemical (Osaka, Japan). Polydimethylsiloxane (PDMS, SILPOT 184) was purchased from Dow-Toray Specialty Materials (Tokyo, Japan). 3-Aminopropyltrimethoxysilane (APTMS), gallic acid (GA), 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDC), and ethyl gallate (EGA) were purchased from Tokyo Chemical Industry Co., Ltd (Tokyo, Japan). Scotch tape (243J Plus) was purchased from 3M Company (MN, USA). The glass substrate (no. 5 thickness, 30 × 30 mm2) was purchased from Matsunami Glass (Osaka, Japan). The nylon mesh (HC-15) was purchased from Sefar Holding AG (Thal, Switzerland). 70% Ethanol (alcohol preparations ET-N) was purchased from UENO FOOD TECHNO INDUSTRY, Ltd (Tokyo, Japan). The bio-skin model was purchased from Beaulax Co., Ltd (Saitama, Japan).
2.2. Fabrication of the PDMS films
A PET film was coated with a sacrificial layer comprising PVA solution (7 wt% in water) using a roll-to-roll gravure printing system (Tabletop Mini-Labo Test Coater, Yasui Seiki Co., Ltd, Tokyo, Japan) under 30 rpm gravure roll rotation and 1.3 m min−1 line speed with in-line thermal treatment at 80 °C. The PVA film was coated with a PDMS solution (the ratio of the base to the curing agent was 10:1 (w/w)) (30 and 60 wt% in a mixed solvent of hexane/ethyl acetate = 4:1 (w/w)) using a desktop coater (TC-1, Mitsui Electric Co., Ltd, Tokyo, Japan) and a wireless bar coater (OSP-100-L60, OSG System Products Co., Ltd, Aichi, Japan) at a moving speed of 10 mm s−1 with a platform at 25 °C to fabricate the 9.4 and 40 μm PDMS films on PVA. The 133 μm thick film was fabricated in a similar way using a 100 wt% PDMS solution and double-layered scotch tape to raise the height of the processing bar. The PDMS with various thicknesses were crosslinked by heating at 100 °C for 1 h. Scotch tape was cut to a square frame with inner dimensions of 20 × 20 mm2 and outer dimensions of 30 × 30 mm2 using a laser processing machine (VLS2.30DT, Universal Laser System, Inc., Kanagawa, Japan). Subsequently, the PDMS/PVA film was peeled off from the PET film using the frame of scotch tape attached around the side of the PDMS film (20 × 20 mm2). Finally, the PVA sacrificial layer in the detached PDMS/PVA film was dissolved in water to obtain the PDMS film.
2.3. Measurement of the thickness of the PDMS films
The PDMS film was dried on a glass substrate and the film was scratched to make the glass surface exposed. The thickness of the film was calculated based on the difference of height between the film surface and glass surface measured using a profilometer (Dektak XT-S, Bruker BioSpin, Kanagawa, Japan, 0.1 nm resolution) with a stylus (12.5 μm radius).
2.4. Adhesive modification of the PDMS films with GA and EDC
Each PDMS film was transferred on a nylon mesh and surface-treated using a plasma cleaner (PDC-001, HARRICK PLASMA, Ithaca, NY, USA) at 7 W for 10 min to introduce hydroxy groups onto the PDMS surface.23 Each film was then immersed in an APTMS solution (10 wt% in methanol) for 30 min. Subsequently, each PDMS film was washed with methanol and dried. GA (0.65 mol L−1) and EDC (1 eq. vs. GA) in super-dehydrated ethanol were cast on the PDMS film and left for 1 h in a vacuum to obtain the adhesive PDMS (adhesive layer: 40 μm). We also prepared adhesive PDMS with a thinner adhesive layer (4.5 or 10 μm) by using 0.065 or 0.325 mol L−1 GA instead of 0.65 mol L−1 GA.
2.5. Fourier-transform infrared (FT-IR) spectroscopy analysis
To investigate the surface structure of the modified PDMS film, the adhesive layer of the adhesive PDMS film was washed with ethanol to obtain PDMS-GA to expose the modified surface. Subsequently, the FT-IR spectra of the freestanding films of pristine PDMS, PDMS-NH2 (after reaction with APTMS), and PDMS-GA were obtained using a Compact FT-IR Spectrometer (ALPHA II-P, C295-W/D, Bruker BioSpin, Kanagawa, Japan).
2.6. Electrospray ionization-mass spectrometry (ESI-MS)
Samples were prepared by diluting pure GA, EDC, and the adhesive layer, which comprised the products from GA and EDC, by 106 times with methanol. The ESI-MS spectra of the samples were obtained using an electrospray ionisation time-of-flight mass spectrometry system (micrOTOF II, Bruker BioSpin, Kanagawa, Japan). Positive mode ESI was used for the measurement of pure GA, EDC and the adhesive layer, and negative mode ESI was used for the measurement of the adhesive layer.
2.7. Proton nuclear magnetic resonance (1H NMR) spectroscopy
The adhesive layer, which comprised the products from GA and EDC, was concentrated using a rotary evaporator (N-1100, Tokyo Rikakikai Co., Ltd, Japan) to remove the ethanol. Ethyl acetate was added to the products from GA and EDC followed by water. After stirring thoroughly, the aqueous layer was separated from the organic layer. The products in the aqueous and organic layers were concentrated under vacuum, and DMSO-d6 was added to each product to dissolve it. The 1H NMR spectrum of each product was obtained using a nuclear magnetic response apparatus (Agilent 400-MR spectrometer at 400 MHz, Agilent Technologies International Japan, Ltd, Japan).
2.8. Measurement of the thickness of the adhesive layer
Adhesive PDMS (PDMS: 40 μm) was prepared on a glass substrate (560 μm). The thickness of the adhesive PDMS and glass substrate was measured using a digimatic micrometer (227 Series Adjustable Measurement Force Digimatic Micrometers, MISUMI Group Inc., Tokyo, Japan). The thickness of the adhesive layer was calculated by subtracting the sum of the thicknesses of the PDMS and the glass substrate (600 μm) from the value measured using the digimatic micrometer.
2.9. Ultraviolet-visible (UV-Vis) analysis
The UV-Vis spectra of the films of pristine PDMS (40 μm) and adhesive PDMS (PDMS: 40 μm) were obtained using a GENESYS 50 Spectrophotometer (Thermo Fisher Scientific K. K., Tokyo, Japan).
2.10. Tensile tests
Tensile tests were carried out on the films of pristine PDMS (40 μm) and adhesive PDMS (PDMS: 40 μm) using a tensile tester (EZ Test EZ-SX, SHIMADZU Co., Ltd) equipped with a 100 N load cell. Each film was stretched at a strain rate of 10 mm min−1. The elastic modulus was estimated from the slope of the stress–strain curve from 2% to 5% of the strain.
2.11. Tack separation tests with a custom jig
The adhesion performances of the films of the pristine PDMS (40 μm) and adhesive PDMS (PDMS: 40 μm, adhesive layer: 40 μm) were determined by tack separation tests using a tensile tester (EZ Test EZ-SX, SHIMADZU Co., Ltd) equipped with a 100 N load cell. Our method is based on a tack separation test11,25–27 and we modified this method to measure the adhesiveness of the adhesive-modified composite film rather than just the adhesive. Each film supported by a square frame (internal area: 20 × 20 mm2) comprising a polystyrene plate was attached to the surface of the skin model (10 × 10 × 5 mm3) wiped with 70% ethanol in advance. After applying a downward force of 0.1 N to the film against the skin model for 5 s, the force was decreased to 0 N and the film was kept to be attached to the skin model for 10 min at room temperature. After 10 min, the frame was lifted (5 mm min−1) until the film was detached from the skin model. From the obtained profile of the separation displacement versus the separation force, the adhesion energy of the film was calculated using the following equation:
where W, x, f, and s represent the adhesion energy, the displacement when the film was detached, the force required to lift the film against the skin model, and the displacement, respectively. The same tests were conducted by using pristine PDMS (9.4 or 133 μm) and adhesive PDMS (PDMS: 9.4 or 133 μm, adhesive layer: 40 μm) and adhesive PDMS (PDMS: 40 μm, adhesive layer: 4.5 or 10 μm).
2.12. Rheology measurements
The viscosity, storage modulus, and loss modulus were measured at 25 °C using a rheometer (Modular Compact Rheometer MCR102e, Anton Paar, Graz, Austria) with a laminator with a diameter of 20 mm and a cone angle of 2°. The flow sweep measurements (viscosity vs. shear rate) of the adhesive layer, which was synthesized from GA and EDC in ethanol, pure EDU, and a mixture of pure EGA and EDU (EGA + EDU), were conducted at shear rates of 10−2 to 102 s−1 at 25 °C. The temperature sweep measurements (viscosity vs. temperature) for each sample were conducted in the temperature range 15 to 50 °C at a shear rate of 10 s−1. The frequency sweep measurement (frequency vs. storage and loss moduli) of the adhesive layer was conducted in the frequency range from 10−1 to 102 rad s−1 at a shear strain of 1%.
3. Results and discussion
3.1. Structure analysis of modified PDMS films
We investigated the chemical structure of the film surface using FT-IR spectroscopy. We obtained FT-IR spectra of pristine PDMS, PDMS-NH2 (after reaction with APTMS), and PDMS-GA (after reaction with GA and EDC, and washing the adhesive layer with ethanol) to determine the chemical structure of the film surface. The FT-IR spectra confirmed the introduction of amino groups by the silane coupling reaction, and subsequently the introduction of gallol groups by reaction with GA and EDC on the PDMS film surface were achieved (Fig. S2, ESI†). We attributed the bands at 3200–3500 and 1570 cm−1 to N–H stretching vibrations and N–H bending vibrations,28 respectively, and confirmed the successful introduction of amino groups onto the PDMS films. The bands at 3100–3300 and 1610 cm−1 were attributed to the O–H stretching vibrations of gallol groups and the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibrations of amide bonds,14 respectively, and confirmed the successful introduction of GA on the surfaces of the PDMS films.
O stretching vibrations of amide bonds,14 respectively, and confirmed the successful introduction of GA on the surfaces of the PDMS films.
3.2. Structure analysis of the adhesive layer
We investigated the chemical structures of the compounds in the adhesive layer using ESI-MS and 1H NMR spectroscopy. The thickness and transparency of the adhesive layer were, respectively, evaluated using a digimatic micrometer and UV-Vis analysis. The adhesive layers on the films were analyzed by ESI-MS and 1H NMR spectroscopy to identify the specific synthesized compounds. The ESI-MS spectra of pure GA, pure EDC, and the compounds in the adhesive layer dissolved in methanol were measured (Fig. S3, ESI†). The peak (m/z = 193, 363) in GA solution is, respectively, attributed to GA monomer and GA dimer, which gained a sodium ion (Fig. S3a, ESI†), and the peak (m/z = 156) in an EDC solution is attributed to protonated EDC (Fig. S3b, ESI†). The peak derived from the cation (m/z = 174) which was observed in positive mode ESI-MS measurement of the adhesive layer was attributed to the protonated EDU (Fig. S3c, ESI†), and the peak derived from the anion (m/z = 197) which was observed in negative mode ESI-MS measurement of the adhesive layer was attributed to EGA, which lost a proton (Fig. S3d, ESI†). Considering the peaks of GA and EDC disappeared in the adhesive layer, we demonstrated GA and EDC are mostly reacted and converted to EDU and EGA. Next, the compounds in the adhesive layer, i.e. the products from GA and EDC, were separated into aqueous and organic layers by extraction, and the 1H NMR spectrum of each compound was obtained. The obtained spectra shown in Fig. 2 reveal that EGA and EDU were present in the organic and aqueous layers, respectively. Therefore, the adhesive layer comprised EGA and EDU, which were synthesized from GA and EDC in ethanol. In summary, the adhesive PDMS consisted of a PDMS layer, the surface of which had been modified with gallol, and an adhesive layer containing EGA and EDU. It was also shown that the adhesive layer had 40 ± 4.3 μm thickness and the same level of transparency as the pristine PDMS measured using a digimatic micrometer and UV-Vis measurement (Fig. S4, ESI†). Therefore, the modification method would be applicable to biomedical devices with light-emitting systems.4,11
 |
| | Fig. 2 Reaction scheme in the adhesive layer and the results of 1H NMR spectroscopy of the compounds in the adhesive layer after separation by extraction (1H NMR = proton nuclear magnetic resonance; EGA = ethyl gallate; EDU = 1-ethyl-3-(3-dimethylaminopropyl)urea). | |
3.3. Mechanical properties of adhesive PDMS
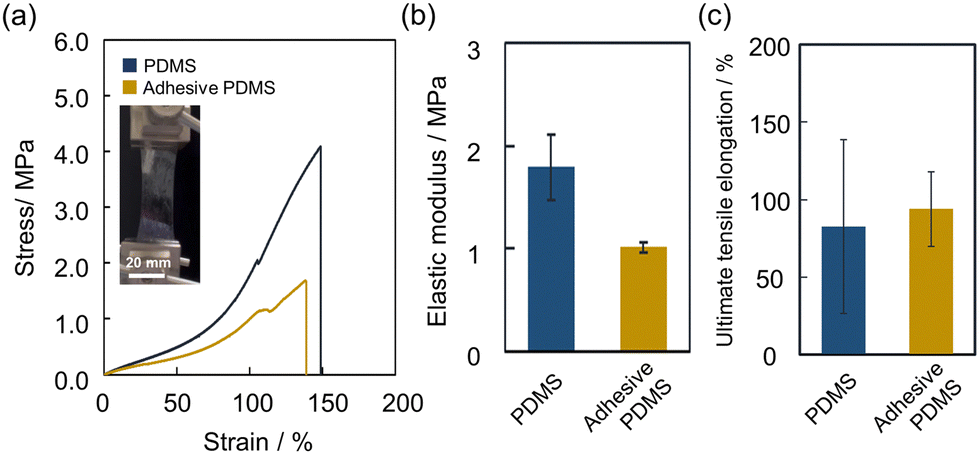
To confirm the stretchability of the adhesive based on abundant non-covalent bonds, the mechanical properties of the adhesive PDMS were evaluated by a tensile test. We used a pristine PDMS film (40 μm) and an adhesive PDMS film (80 μm (PDMS: 40 μm, adhesive layer: 40 μm)) for the measurement on a tensile tester. The films were stretched at a strain rate of 10 mm min−1, and their elastic moduli were estimated from their stress–strain curves. The results are shown in Fig. 3a and the calculated elastic moduli are shown in Fig. 3b. The elastic modulus of the adhesive PDMS film (1.01 MPa) was lower than that of the pristine PDMS film (1.79 MPa). The observed reduction in stress for the adhesive PDMS compared to the pristine PDMS can be attributed to accounting for the adhesive layer's thickness in the stress calculation, despite both films being subjected to the same tensile force (Fig. S5, ESI†). This result means the elastic modulus of the PDMS substrate is dominant over the adhesive layer. The ultimate tensile elongation of the adhesive PDMS film (94.0%) was as large as that of the pristine film (82.6%) (Fig. 3c). Therefore, we conclude that the adhesive modification does not impair the original mechanical properties of the PDMS film.
 |
| | Fig. 3 (a) Results of tensile tests on a PDMS film and an adhesive PDMS film, and the image of stretching adhesive PDMS. (b) Calculated elastic moduli and (c) ultimate tensile elongation values of each PDMS film (PDMS = polydimethylsiloxane). | |
3.4. Tack separation test of adhesive PDMS with a custom jig
Next, we conducted a tack separation test with a custom jig on a skin model to evaluate the adhesiveness of the adhesive-modified PDMS. First, the skin model (surface area: 10 × 10 mm2) was prepared on a plastic plate and pristine PDMS film (40 μm) or an adhesive PDMS film (PDMS: 40 μm, adhesive layer: 40 μm) was prepared and attached to the skin model for 10 min (Fig. 4a). Next, the PDMS film supported by a square frame was lifted until it detached from the skin model, and a profile of the separation displacement versus the separation force was obtained (Fig. 4b). The adhesion energy of each sample was obtained by calculating the integral value of the profile (Fig. 4c). The average adhesion energy value of the adhesive PDMS film was 27.4 ± 3.40 μJ cm−2, which was 130 times larger than that of pristine PDMS films ((2.11 ± 1.30) × 10−1 μJ cm−2). The order of the adhesion energy value was comparable to that of the value obtained using a previous method using polydopamine to enhance the adhesiveness of PDMS to chicken muscle11 This phenomenon can be attributed to the energy dissipation of the compounds in the adhesive layer, which facilitated an increase in the film's absolute adhesion area by effectively infiltrating the surface of the adherents. We prepared adhesive PDMS with different thicknesses of the adhesive layer (4.5 ± 3.0 and 10 ± 3.1 μm) by changing the concentration of the precursor solution of GA and EDC and a tack separation test of them was conducted (Fig. S6, ESI†). We found that the adhesion energy of adhesive PDMS increased as the thickness of the adhesive layer became thicker, and it hit the limit at the point around 10 μm thickness. This indicates that a thicker adhesive layer is required for efficient energy dissipation for strong adhesiveness but a too thick adhesive layer cannot contribute to strong adhesiveness because of cohesive failure.29 We also prepared adhesive PDMS with different thicknesses of the PDMS layer (9.4 ± 5.0, 40 ± 3.8, and 133 ± 5.9 μm) and confirmed that our adhesive modification can improve the adhesiveness of the PDMS films regardless of the thickness and mechanical properties (Fig. S7, ESI†). The adhesion energy as well as the data variation of adhesive PDMS (133 μm) was larger because the peeling behavior was considered inconsistent throughout the experiment. When using a thicker film, a larger force is required to pull up and deform the margin of the PDMS film in the phase before the peak of maximum force30 and the adhesion energy was calculated as a larger value. The increased data variation observed in the thicker film is attributed to the slight tilt of the adhesive PDMS film when attached to the skin model. This tilt causes a heterogeneous distribution of vertical pulling force. Although the thinner film is more stretchable and can remain attached on the skin model even if the pulling force is different on both sides of the tilt, the thicker film cannot remain attached and easily peels off, resulting in larger data variations due to the tilt of the film.
 |
| | Fig. 4 (a) The schematic image of the tack separation test of adhesive PDMS with a custom jig. (b) Results of the tack separation test and (c) calculated adhesion energy values of pristine PDMS and adhesive PDMS (PDMS = polydimethylsiloxane). | |
3.5. Viscoelasticity of the adhesive layer and the mechanism of adhesiveness
Next, we measured the viscoelasticity of the adhesive layer (Fig. 5a) using a rheometer to elucidate the mechanism underlying the adhesiveness of the film. For example, we sought to identify the chemical bonds formed by the functional groups that were required for proper cohesion performance and the energy dissipation required for a large adhesion area between the PDMS and the adherents. Adhesive layers comprising EGA and EDU, pure EDU, and a mixture of pure EGA and EDU (EGA + EDU) were prepared on a glass plate by each and investigated using a rheometer. The flow sweep measurements (viscosity versus shear rate) and temperature sweep measurements (viscosity versus temperature) are shown in Fig. 5b and Fig. S8 (ESI†), respectively. It is supposed that the larger decrease in the viscosity of EDU under an increasing shear rate and temperature was attributable to the dissociation of hydrogen bonds.18,31 However, it is suggested that the smaller decrease in the viscosity of the adhesive layer and the EGA + EDU layer compared with the EDU layer was due to stronger non-covalent bonds than hydrogen bonds. These bonds include electrostatic interactions between the cationic amino groups in the EDU and the anionic phenol groups in EGA or hydrophobic interactions, which contributed to the adhesiveness. However, further research on the relationships between functional groups and adhesiveness is required. Next, frequency sweep measurements (frequency vs. storage and loss moduli) revealed that G′ was larger than G′′ at low frequencies, but a gel–sol transition occurred at 7.09 rad s−1, after which G′′ exceeded G′ (tanδ > 1) at higher frequencies (Fig. 5c). Together with the adhesion mechanism, these phenomena suggest that the adhesive layer on the PDMS had high cohesive strength at smaller frequencies and high peel strength at higher frequencies.32 It is possible that a loss tangent close to 1 facilitated the proper diffusion of the compounds that contributed to a larger adhesion area between the skin model and the PDMS, as well as strong adhesiveness.33,34 This phenomenon could be attributed to energy dissipation by weak non-covalent bonds in the adhesive layer.35 It has been reported that a larger cohesion and a larger G′ are more important factors for stronger adhesiveness than a large G′′.36 Therefore, we conclude that the adhesive PDMS had stronger adhesiveness at lower frequencies and under slow retraction. Furthermore, the larger G′′ at higher frequencies meant that the adhesive layer could be easily detached by fast retraction. Therefore, it could be described as a pressure-sensitive adhesive. In short, we found that our adhesive layer applies to elastomer films in terms of balancing cohesion and molecular diffusion due to energy dissipation for effective adhesiveness and detachment.
 |
| | Fig. 5 (a) The compounds in the adhesive layer. (b) Flow sweep measurements (viscosity vs. shear rate) of the adhesive layer, EDU, and EGA + EDU and (c) frequency sweep measurements (frequency vs. storage (G′) and loss (G′′) moduli and loss tangent) of the adhesive layer (EDU = 1-ethyl-3-(3-dimethylaminopropyl)urea; EGA = ethyl gallate). | |
4. Conclusions
In conclusion, we demonstrated a facile method for fabricating an adhesive layer comprising the low-molecular-weight compounds; EGA and EDU for maintaining the original mechanical properties of PDMS. Our results suggested that abundant hydrogen bonds as well as many other kinds of non-covalent bonds among the gallol, urea, and tertiary amino groups contributed to both energy dissipation and strong cohesion for effective adhesiveness. To further strengthen the adhesive performance, the conjugation of low-molecular-weight compounds to the polymer backbone to improve cohesion for water resistance is also required. This versatile method could be applied not only to amine-bearing substrates but to a variety of elastomers to which amines can be introduced.37 Our method for modifying adhesives onto elastomer films does not sacrifice stretchability and could be applied to thicker films, despite thinner films being preferred to obtain larger adhesiveness.11,38 This method expands the possibility of the application of many kinds of thin film devices for electroencephalographic analysis,2 electromyographic analysis,3 measuring electrical signals in plants,6 and so on. The present research provides guidelines for the molecular design and modification of adhesives for biomedical devices and will contribute to their long-term use.
Data availability
The data that support the findings of this study are available from the corresponding author, Toshinori Fujie, upon reasonable request.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the Japan Agency for Medical Research and Development (AMED) (grant numbers JP22he2202018 and JP20he0322003), the Fusion Oriented Research for disruptive Science and Technology (FOREST) from the Japan Science and Technology Agency (JST) (grant number JPMJFR203Q), the Japan Society for the Promotion of Science (JSPS) (grant number 21H03815), and the Murata Science and Education Foundation. D. S. and T. F. acknowledge the support from JST ASPIRE for Rising Scientists (grant number JPMJAP2336). H. F. is supported by Funai Overseas Scholarship, Stanford Graduate Fellowship, JSPS Doctoral Fellowship (grant number 22J23773), and the Tokyo Tech Academy for Super Smart Society administered by MEXT, Japan. We thank Edanz (https://jp.edanz.com/ac) for editing a draft of this manuscript.
References
- A. Imai, S. Takahashi, S. Furubayashi, Y. Mizuno, M. Sonoda, T. Miyazaki, E. Miyashita and T. Fujie, Adv. Mater. Technol., 2023, 8(21), 2300300 CrossRef CAS
 .
.
- L. M. Ferrari, U. Ismailov, J.-M. Badier, F. Greco and E. Ismailova, Npj Flex. Electron., 2020, 4, 1–9 CrossRef .
- K. Yamagishi, T. Nakanishi, S. Mihara, M. Azuma, S. Takeoka, K. Kanosue, T. Nagami and T. Fujie, NPG Asia Mater., 2019, 11, 1–13 CrossRef .
- Z. Chen, R. T. Yin, S. N. Obaid, J. Tian, S. W. Chen, A. N. Miniovich, N. Boyajian, I. R. Efimov and L. Lu, Adv. Mater. Technol., 2020, 5(8), 2000322 CrossRef CAS PubMed .
- L. Lan, X. Le, H. Dong, J. Xie, Y. Ying and J. Ping, Biosens. Bioelectron., 2020, 165, 112360 CrossRef CAS PubMed .
- F. Meder, S. Saar, S. Taccola, C. Filippeschi, V. Mattoli and B. Mazzolai, Adv. Mater. Technol., 2021, 6, 2001182 CrossRef CAS .
- J. Deng, H. Yuk, J. Wu, C. E. Varela, X. Chen, E. T. Roche, C. F. Guo and X. Zhao, Nat. Mater., 2021, 20, 229–236 CrossRef CAS PubMed .
- Q. Yang, T. Wei, R. T. Yin, M. Wu, Y. Xu, J. Koo, Y. S. Choi, Z. Xie, S. W. Chen, I. Kandela, S. Yao, Y. Deng, R. Avila, T.-L. Liu, W. Bai, Y. Yang, M. Han, Q. Zhang, C. R. Haney, K. Benjamin Lee, K. Aras, T. Wang, M.-H. Seo, H. Luan, S. M. Lee, A. Brikha, N. Ghoreishi-Haack, L. Tran, I. Stepien, F. Aird, E. A. Waters, X. Yu, A. Banks, G. D. Trachiotis, J. M. Torkelson, Y. Huang, Y. Kozorovitskiy, I. R. Efimov and J. A. Rogers, Nat. Mater., 2021, 20, 1559–1570 CrossRef CAS PubMed .
- M. Takuma, H. Fujita, N. Zushi, H. Nagano, R. Azuma, T. Kiyosawa and T. Fujie, Biomater. Sci., 2024, 12, 3401–3410 RSC .
- M. Saito, E. Kanai, H. Fujita, T. Aso, N. Matsutani and T. Fujie, Adv. Funct. Mater., 2021, 31, 2102444 CrossRef CAS .
- K. Yamagishi, I. Kirino, I. Takahashi, H. Amano, S. Takeoka, Y. Morimoto and T. Fujie, Nat. Biomed. Eng., 2019, 3, 27–36 CrossRef CAS PubMed .
- H. Lee, S. M. Dellatore, W. M. Miller and P. B. Messersmith, Science, 2007, 318, 426–430 CrossRef CAS PubMed .
- M. A. Gwak, S. J. Lee, D. Lee, S. A. Park and W. H. Park, Int. J. Biol. Macromol., 2023, 227, 493–504 CrossRef CAS PubMed .
- S. N. Ramirez-Barron, S. Sanchez-Valdes, R. Betancourt, C. A. Gallardo, B. Puente-Urbina, O. S. Rodriguez-Fernández, M. G. Carneiro-da Cunha, M. T. dos Santos-Correia and Z. V. Sanchez-Martinez, Int. J. Adhes. Adhes., 2021, 104, 102749 CrossRef CAS .
- Q. Yang, L. Tang, C. Guo, F. Deng, H. Wu, L. Chen, L. Huang, P. Lu, C. Ding, Y. Ni and M. Zhang, Chem. Eng. J., 2021, 417, 127962 CrossRef CAS .
- J. Zhang, W. Wang, Y. Zhang, Q. Wei, F. Han, S. Dong, D. Liu and S. Zhang, Nat. Commun., 2022, 13, 5214 CrossRef CAS PubMed .
- X. Li, Y. Deng, J. Lai, G. Zhao and S. Dong, J. Am. Chem. Soc., 2020, 142, 5371–5379 CrossRef CAS PubMed .
- Q. Zhang, C.-Y. Shi, D.-H. Qu, Y.-T. Long, B. L. Feringa and H. Tian, Sci. Adv., 2018, 4, eaat8192 CrossRef CAS PubMed .
- C.-Y. Shi, Q. Zhang, H. Tian and D.-H. Qu, SmartMat, 2020, 1(1), e1012 CrossRef .
- Y. Liang, K. Wang, J. Li, Y. Zhang, J. Liu, K. Zhang, Y. Cui, M. Wang and C.-S. Liu, Mater. Horiz., 2022, 9, 1700–1707 RSC .
- K. Zhan, C. Kim, K. Sung, H. Ejima and N. Yoshie, Biomacromolecules, 2017, 18, 2959–2966 CrossRef CAS PubMed .
- G. P. Maier, M. V. Rapp, J. H. Waite, J. N. Israelachvili and A. Butler, Science, 2015, 349, 628–632 CrossRef CAS PubMed .
- D. Bodas and C. Khan-Malek, Sens. Actuators, B, 2007, 123, 368–373 CrossRef CAS .
- C.-C. Wu, C.-Y. Yuan and S.-J. Ding, Surf. Coat. Technol., 2011, 205, 3182–3189 CrossRef CAS .
-
D. Astm, ASTM D2979.
- B. S. Terry, A. C. Passernig, M. L. Hill, J. A. Schoen and M. E. Rentschler, J. Mech. Behav. Biomed. Mater., 2012, 15, 24–32 CrossRef PubMed .
- T. Horii, K. Yamashita, M. Ito, K. Okada and T. Fujie, NPG Asia Mater., 2024, 16, 33 CrossRef .
- H. Kim, H.-G. Kim, S. Kim and S. S. Kim, J. Memb. Sci., 2009, 344, 211–218 CrossRef CAS .
- L. F. M. da Silva, T. N. S. S. Rodrigues, M. A. V. Figueiredo, M. F. S. F. de Moura and J. A. G. Chousal, J. Adhesion, 2006, 82, 1091–1115 CrossRef CAS .
- J. P. Phillips, X. Deng, R. R. Stephen, E. L. Fortenberry, M. L. Todd, D. M. McClusky, S. Stevenson, R. Misra, S. Morgan and T. E. Long, Polymer, 2007, 48, 6773–6781 CrossRef CAS .
- J. Kajtna, B. Alič, M. Krajnc and U. Šebenik, Int. J. Adhes. Adhes., 2014, 49, 103–108 CrossRef CAS .
-
F. Mazzeo, TA Instruments report RH082.
- E. P. Chang, J. Adhes., 1991, 34, 189–200 CrossRef CAS .
- Y.-J. Wang, Y. He, S. Y. Zheng, Z. Xu, J. Li, Y. Zhao, L. Chen and W. Liu, Adv. Funct. Mater., 2021, 31, 2104296 CrossRef CAS .
- M. G. Mazzotta, A. A. Putnam, M. A. North and J. J. Wilker, J. Am. Chem. Soc., 2020, 142, 4762–4768 CrossRef CAS PubMed .
- M. Seong, S. Kondaveeti, G. Choi, S. Kim, J. Kim, M. Kang and H. E. Jeong, ACS Appl. Mater. Interfaces, 2023, 15, 11042–11052 CrossRef CAS PubMed .
- J. Song, B. Winkeljann and O. Lieleg, Adv. Mater. Interfaces, 2020, 7, 2000850 CrossRef CAS .
- T. Fujie, N. Matsutani, M. Kinoshita, Y. Okamura, A. Saito and S. Takeoka, Adv. Funct. Mater., 2009, 19, 2560–2568 CrossRef CAS .
Footnotes |
| † Electronic supplementary information (ESI) available: Detailed experimental and additional properties of LMWAs. See DOI: https://doi.org/10.1039/d4ma01102c |
| ‡ Present address: Department of Bioengineering, Stanford University, Stanford, CA 94305, USA. |
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Tatsuhiro
Horii
a,
Takeshi
Hata
a,
Tatsuhiro
Horii
a,
Takeshi
Hata