
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licenceperi-Diselenolo-substituted 1,8-naphthalimide derivatives as bipolar matrices for redox reactions in a non-aqueous electrolyte†
Delyana
Marinova
 *a,
Lyuben
Borislavov
a,
Silva
Stanchovska
a,
Rositsa
Kukeva
a,
Monika
Mutovska
b,
Natali
Simeonova
b,
Stanimir
Stoyanov
b,
Yulian
Zagranyarski
*b,
Mihail
Mondeshki
c,
Yanislav
Danchovski
ab,
Hristo
Rasheev
ab,
Alia
Tadjer
ab and
Radostina
Stoyanova
a
*a,
Lyuben
Borislavov
a,
Silva
Stanchovska
a,
Rositsa
Kukeva
a,
Monika
Mutovska
b,
Natali
Simeonova
b,
Stanimir
Stoyanov
b,
Yulian
Zagranyarski
*b,
Mihail
Mondeshki
c,
Yanislav
Danchovski
ab,
Hristo
Rasheev
ab,
Alia
Tadjer
ab and
Radostina
Stoyanova
a
aInstitute of General and Inorganic Chemistry, Bulgarian Academy of Sciences, 1113 Sofia, Bulgaria. E-mail: manasieva@svr.igic.bas.bg
bFaculty of Chemistry and Pharmacy, Sofia University “St. Kliment Ohridski”, 1164 Sofia, Bulgaria. E-mail: ohjz@chem.uni-sofia.bg
cDepartment of Chemistry, Johannes Gutenberg University, 55128 Mainz, Germany
First published on 17th December 2024
Abstract
In the search for bipolar organic materials as electrodes in rechargeable batteries, we report peri-diselenolo-substituted 1,8-naphthalimides (NIs). The molecular architecture consists of structural motifs comprising a naphthalimide core, the peri-diselenide bridge, hydrogen or halogens at positions 3 and 6, and an alkyl chain with a fixed length of 4 and 8. The resulting architecture is unprecedented in the naphthalimide chemistry and quantum chemical modelling was employed to rationalize the new design better. The NI-derivatives are prepared starting from tetra-halogenated naphthalic anhydride via nucleophilic substitution at both peri-positions in the respective imide. Non-covalent interactions facilitate the NIs self-organization into ordered nanostructures studied by SEM, PXRD, solid-state NMR spectroscopy and molecular modelling. The electrochemical properties of NI-derivatives are analysed in half lithium-ion cells with ionic liquid electrolytes. By comparing the experimentally determined potentials with the theoretically calculated ones, the mechanism of electrochemical oxidation and reduction is deduced. It is shown that below 2.0 V, NIs interact with a maximum of 6Li+ due to the sequential reduction of the diselenide bridge and the carbonyl groups, whereas above 4.0 V, the oxidation of NIs takes place with the participation of the electrolyte counterion TFSI− as a result of the involvement of Se atoms and carbons in the naphthalene unit. The hydrogen and halogen substituents affect both the self-organization, reduction and oxidation of NIs. The structural, morphological and compositional changes of the NIs after prolonged cycling are discussed based on ex situ XRD, SEM/EDS and EPR analyses. The data demonstrate that a molecular architecture based on peri-diselenolo-1,8-naphthalimide derivatives could be used to design new classes of organic bipolar electrodes.
1. Introduction
Organic electrode materials (OEMs) caught the research interest recently as they combine high-energy density with cost-effectiveness and environmental compatibility.1,2 The performance of OEMs relies on their specific redox reactions, which can be divided into three categories: n-, p- and bipolar types, based on their proclivity to be reduced or oxidized, along with the charge-compensating counterions from the electrolyte.3–5 The n-type OEMs interact reversibly with mono- or polyvalent ions (such as alkali, alkaline earth ions, etc.) because of their ability initially to be reduced, forming stable negatively charged species. The n-type reactions enable to reach high capacities, but at the expense of slow kinetics.6 In contrast, the p-type OEMs undergo oxidation, forming stable positively charged species that attract counter anions from the electrolyte (such as TFSI−, FSI−, PF6−). As a result, the p-type reactions take place at higher redox potentials and are characterized with faster kinetics than that of the n-type reactions.7 The bipolar OEMs combine the n- and p-type moieties into one molecule that undergoes reversible reduction and oxidation with the participation of both cation and anion counterparts from the electrolyte. Thus, bipolar OEMs demonstrate simultaneously high capacity, broad working potential range, and fast kinetics. Given the mechanism of the redox reactions, the n-type OEMs are applied in rechargeable metal-ion batteries (such as lithium, sodium, etc.),8 while p-type OEMs are mainly designed to be used in dual-ion batteries.9,10 Combining the characteristics of both n- and p-type materials, the bipolar OEMs are directed towards utilization in the newly emerged symmetric rechargeable batteries, where both the cathode and anode are made of the same organic substance.11 The advantage of symmetric batteries over metal-ion and dual-ion batteries is their lower manufacturing cost,12 but unlike n- and p-type materials, the lack of reliable bipolar materials is hampering the development of symmetric batteries.In the quest for bipolar organic materials, recent research has focused on the design of organic molecules with fusion of n- and p-type redox units.13–16 In this case, both the n- and p-type redox units will influence each other, allowing precise tuning of the bipolar redox activity.17 Thus, the binding of a n-type naphthalene diimide unit to two p-type viologen units leads to the formation of an organic compound that displays a six-electron redox reaction, making it possible to achieve a high specific capacity close to the theoretical one.18 This type of molecular architecture prevents unwanted dimerization decomposition of the molecules. In other recent examples, the multi-electron reactions are facilitated on the porphyrin macrocycle after the introduction of a ferrocene redox-based centre.19,20 Through a synergistic coupling of two redox centres of p-type dihydrophenazines and n-type anthraquinone in a single molecule, a bipolar-type indanthrene is proposed for the construction of a symmetric battery.21 In addition, the combination of n-type C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and p-type N–H redox centres in a molecule, such as indanthrone, results in a multi-electron electrode material with a significantly enhanced capacity.22 Among the different classes of bipolar OEMs, the 1,8-naphthalimide (NI) derivatives occupy a special place due to their intriguing redox-switchable properties.23,24 Recently, we have designed bipolar peri-dithiolo-1,8-naphthalimides and have demonstrated that they participate in n- and p-type redox reactions at around 2.0 V and above 4.0 V vs. Li/Li+.25 The organic architecture relies on several structural elements: (i) the aromatic character of the naphthalene core provides the reversibility of the process; (ii) the fixed dichalcogenide bridge offers a high degree of recombination; (iii) the peri-position of an imide acceptor motif to the disulfide bridge contributes to the inter/intra molecular electron transfer; (iv) by varying the type and length of the alkyl chain the physical and mechanical properties (solubility, crystallization ability, thermo- and chemo-stability, etc.) can be modified;26 (v) the inclusion of halogen atoms in positions 3 and 6 allows the electron-accepting properties of the naphthalimide core to be fine-tuned. The promising results for the bipolar peri-dithiolo-NIs encouraged us to further explore the synthesis-structure-redox properties relationships.
O and p-type N–H redox centres in a molecule, such as indanthrone, results in a multi-electron electrode material with a significantly enhanced capacity.22 Among the different classes of bipolar OEMs, the 1,8-naphthalimide (NI) derivatives occupy a special place due to their intriguing redox-switchable properties.23,24 Recently, we have designed bipolar peri-dithiolo-1,8-naphthalimides and have demonstrated that they participate in n- and p-type redox reactions at around 2.0 V and above 4.0 V vs. Li/Li+.25 The organic architecture relies on several structural elements: (i) the aromatic character of the naphthalene core provides the reversibility of the process; (ii) the fixed dichalcogenide bridge offers a high degree of recombination; (iii) the peri-position of an imide acceptor motif to the disulfide bridge contributes to the inter/intra molecular electron transfer; (iv) by varying the type and length of the alkyl chain the physical and mechanical properties (solubility, crystallization ability, thermo- and chemo-stability, etc.) can be modified;26 (v) the inclusion of halogen atoms in positions 3 and 6 allows the electron-accepting properties of the naphthalimide core to be fine-tuned. The promising results for the bipolar peri-dithiolo-NIs encouraged us to further explore the synthesis-structure-redox properties relationships.
Another feature of bipolar OEMs is their dependence on the correct choice of electrolyte,27,28 which not only ensures the transport of positively or negatively charged ions, but also must suppress the dissolution and/or diffusion of the organic molecules. For a non-aqueous electrolyte, ionic liquids (named Pyr1,3TFSI or Pyr1,3FSI) are the solvents-of-choice.29 The naphthalene diimide displays a better cycling stability in ether-based electrolytes (such as 1 M LiTFSI or 1 M LiClO4 in dioxolane/dimethoxyethane, vol 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) than in the conventional carbonate-based electrolytes (such as 1 M LiTFSI or 1 M LiPF6 in EC/DEC, vol 1:1),30 as a consequence of its interaction with EC from the electrolyte, leading to the formation of C–OH bonds. Solid-state electrolytes are also compatible with many OEMs.31
:1) than in the conventional carbonate-based electrolytes (such as 1 M LiTFSI or 1 M LiPF6 in EC/DEC, vol 1:1),30 as a consequence of its interaction with EC from the electrolyte, leading to the formation of C–OH bonds. Solid-state electrolytes are also compatible with many OEMs.31
Herein, we examine the reversible redox properties of peri-diselenolo-substituted 1,8-naphthalimide derivatives in a lithium-ion cell with non-aqueous electrolytes. In these molecules, sulphur is replaced by selenium driven by the fact that the latter is a better nucleophile and readily participates in one- and two-electron redox reactions due to its inability to form π bonds of any kind.32 Furthermore, the atoms at positions 3 and 6 vary from hydrogen to halogens (Cl and Br). The alkyl chain length is fixed at 4 and 8, respectively (Fig. 1A). The resulting architecture is unprecedented in the naphthalimide chemistry and quantum chemical modelling was employed to better rationalize the new design. The synthesis comprises an originally developed procedure starting from tetra-halogenated naphthalic anhydride via nucleophilic substitution at both peri-positions in the respective imide. The self-organization of the resulting architectures is investigated by SEM, PXRD and solid-state NMR spectroscopy. The NI-derivatives are used as electrodes in half lithium-ion cells with ionic liquid electrolytes. The mechanism of the redox reaction is analysed by comparing the experimentally determined potentials and the theoretically calculated ones. The structural, morphological and compositional changes of the naphthalimide derivatives after the electrochemical reaction are monitored by ex situ XRD, SEM/EDS and EPR analyses.
 | ||
| Fig. 1 (A) Molecular architecture of the naphthalimide derivatives (C – grey, O – red, N – blue). The models with R = CH3 are denoted as SeH, SeCl, and SeBr; (B) energy (in eV) of the highest occupied (HOMO) and the lowest unoccupied (LUMO) molecular orbitals of the modelled peri-diselenolo-naphthalimide derivatives. | ||
2. Experimental
2.1. Materials and methods
The model cells used for the testing were Swagelok-type two- and three-electrode cells. The electrolyte was a 1 M solution of (LiTFSI-Pyr1,3FSI (lithium bis(trifluoromethanesulfonyl)imide) in N-methyl, propyl(pyrrolidinium bis(fluorosulfonyl)imide), 1:9 by ratio). The assembly of the cells was accomplished in an MB-Unilab glovebox model Pro SP (1500/780). The electrochemical experiments were carried out in potentiostatic and galvanostatic mode on a multi-channel potentiostat/galvanostat Biologic VMP-3e, including an impedance meter and eight-channel system Arbin BT2000.
The scanning electron microscopy and EDS analysis were carried out on JEOL JSM 6390 (JEOL Ltd, Tokyo, Japan), equipped with an AZtec Oxford Instruments 6.0 energy dispersive X-ray spectrometer to observe the morphology and determine the chemical composition of the peri-substituted diselenides.
The EPR analyses was carried out in X band on a spectrometer Bruker EMX premium X working at frequency 9.4 GHz. For the measurements at temperature 100 K was used variable temperature unit VT ER 4141 VTM. Modulation amplitude (MA) 0.5 G and power attenuation (Att) 18 dB were applied when measuring charged and discharged electrodes, while for recording the EPR spectra of the electrolyte and soaked separator, the measurement parameters were MA = 0.05 G and Att = 50 dB.
The X-ray powder diffraction patterns within the range from 5 to 50 degree 2θ with a step of 0.02 degree 2θ and a counting time of 0.4 s per strip (total 70 s per step) were measured on a Bruker D8 Advance (Bruker Corporation, Karlsruhe, Germany) diffractometer with Cu Kα radiation and LynxEye detector.
The quantum chemical calculations are done with the hybrid DFT functional B3LYP33,34 utilizing the 6-31+G* basis set.35,36 The implicit solvent was simulated with the PCM scheme37–39 with ε = 15 for the ionic liquid. Dispersion is accounted for implementing the D3-BJ empirical correction.40,41 The population analysis used the NBO atomic charges.42,43 All calculations were carried out with the Gaussian 16 software package.44
All starting materials and solvents were commercially available and used without additional purification: Fluorochem (Glossop, UK), Across (Antwerpen, Belgium) and Fisher Scientific (Hampton, NH, USA).
Solution NMR spectra were recorded on a Bruker Avance 500 MHz instrument (Bruker, Karlsruhe, Germany) operating at 500 and 126 MHz for 1H and 13C, respectively. 1,1,2,2-Tetrachloroethane-d2 was used as solvent. Chemical shifts are reported in δ units (ppm) and referenced to the residual solvent signals (1H at 6.00 ppm and 13C at 73.78 ppm). Elemental analyses were carried out on a Leco CHNS-932 (Leco Europe, Geleen, The Netherlands). Thin layer chromatographic (TLC) analysis was performed on silica gel plates (Macherey-Nagel F60 254 40 × 80; 0.2 mm, Macherey-Nagel, Duren, Germany).
All solid-state NMR (ssNMR) spectra were recorded using a Bruker Avance DSX 400 MHz NMR spectrometer equipped with a three-channel 4 mm probe head at magic angle spinning (MAS) of 10 kHz. All 1H spectra were recorded with background suppression averaging 16 scans with 8 s recycle delay. The 13C cross-polarization (CP) NMR spectra were recorded with 20 ms and 2 ms contact times averaging 20k scans with 3.5 s recycle delay and TPPM proton decoupling scheme. The 77Se single pulse excitation spectra were recorded averaging between 20k and 40k scans with 20 s repetition time. Additional experiments at 8 kHz MAS were conducted to reveal the isotropic 77Se NMR shifts. The 1H and 13C NMR spectra were referenced to external adamantane as a secondary reference at 1.63 ppm and 38.5 ppm respectively. The 77Se NMR spectra were referenced to external aqueous H2SeO3 at 1288.1 ppm as a secondary reference.
2.2. Synthesis of compounds SeH8, SeCl4 and SeBr4
Purified by column chromatography. Yield 3.68 g (90%).
1H-NMR (δ (ppm), 1,1,2,2-tetrachloroethane-d2): 0.88 (t, 3H, 3JHH = 6.9 Hz); 1.27–1.42 (m, 10H); 1.65–1.71 (m, 2H); 4.10 (t, 3H, 3JHH = 7.7 Hz); 7.64 (d, 2H, 3JHH = 8.0 Hz); 8.29 (d, 2H, 3JHH = 8.0 Hz).
13C{1H}-NMR (δ (ppm), 1,1,2,2-tetrachloroethane-d2): 14.11; 22.57; 27.11; 27.97; 29.16; 29.27; 31.70; 40.42; 118.48; 121.57; 131.80; 132.05; 136.52; 152.31; 163.04.
77Se{1H}-NMR (δ (ppm), 1,1,2,2-tetrachloroethane-d2): 512.55.
Elemental analysis: calcd for C20H21NO2Se2: %C 51.63, %H 4.55, %N 3.01; found: %C 51.91, %H 4.45, %N 3.04.
DEI-MS: m/z = 465 [M]+.
Purified by recrystallization from DMF. Yield 4.45 g (93%).

1H-NMR (δ (ppm), 1,1,2,2-tetrachloroethane-d2): 0.98 (t, 3H, 3JHH = 7.3 Hz); 1.42 (sex, 2H, 3JHH = 7.5 Hz); 1.68 (quin, 2H, 3JHH = 7.6 Hz); 4.13 (t, 3H, 3JHH = 7.5 Hz); 8.29 (s, 2H).
13C{1H}-NMR (δ (ppm), 1,1,2,2-tetrachloroethane-d2): 14.84; 21.27; 30.89; 41.46; 121.77; 128.63; 128.90; 131.86; 137.86; 151.46; 163.11.
77Se{1H}-NMR (δ (ppm), 1,1,2,2-tetrachloroethane-d2): 544.47.
Elemental analysis: calcd for C16H11Cl2NO2Se2: %C 40.20, %H 2.32, %N 2.93; found: %C 40.45, %H 2.17, %N 3.17.
DEI-MS: m/z = 480.3 [MH]+.
Purified by recrystallization from DMF. Yield 4.99 g (88%).

1H-NMR (δ (ppm), 1,1,2,2-tetrachloroethane-d2): 1.01 (t, 3H, 3JHH = 7.3 Hz); 1.45 (sex, 2H, 3JHH = 7.4 Hz); 1.72 (quin, 2H, 3JHH = 8.0 Hz); 4.17 (t, 3H, 3JHH = 7.5 Hz); 8.48 (s, 2H).
13C{1H}-NMR (δ (ppm), 1,1,2,2-tetrachloroethane-d2): 13.67; 20.18; 29.93; 40.40; 116.21; 118.53; 120.78; 128.87; 133.55; 153.80; 161.96.
77Se{1H}-NMR (δ (ppm), 1,1,2,2-tetrachloroethane-d2): 548.62.
Elemental analysis: calcd for C16H11Br2NO2Se2: %C 33.89, %H 1.96, %N 2.47; found: %C 33.68, %H 2.11, %N 2.70.
3. Results and discussion
3.1. Design of bipolar derivatives
The ability of naphthalimide derivatives to participate in bipolar reactions is analysed using DFT calculations. The molecular architecture consists of structural motifs comprising the peri-diselenide bridge, halogens, and hydrogen at positions 3 and 6 (Fig. 1A). Since the hydrocarbon chain (C4 or C8) does not contribute to the redox properties,25 it was abridged to a CH3-group. The molecular structures were subject to geometry optimization, and their frontier molecular orbitals (MOs) were compared. The optimized structures retain the skeleton planarity and belong to the C2v symmetry group. In all cases HOMO and LUMO are asymmetric with respect to the mirror plane, lacking contributions from the atoms lying on the symmetry axis. Particularly, the nitrogen and the alkyl group bound to it are not involved in none of the frontier orbitals (HOMO−1, HOMO, LUMO, LUMO+1), which justifies the simplification of the side chain to a CH3-group in the models. The energies of the frontier orbitals are listed in Table 1. HOMO−1 is comparatively far from HOMO (1.4–1.5 eV), indicating that upon oxidation an electron from HOMO should be removed first. HOMO has the largest contribution from the Se–Se bridge, followed by the periphery of the naphthalene fragment, the imide moiety being almost excluded, so one can make a forecast of which part of the molecule will be the most affected one by an electron abstraction. Unlike the HOMOs, LUMO and LUMO+1 are fairly close (Table 1), particularly in the unsubstituted compound (SeH) where they are practically degenerate and inverted – LUMO is the one localized on the diselenide bridge (Fig. 1), while in the halogenated derivatives (SeCl and SeBr) this is LUMO+1. The degeneracy of LUMO and LUMO+1 in SeH infers that upon reduction, this compound may interact with 4Li atoms concurrently as it can accommodate 4 electrons simultaneously. The (quasi)degeneracy of LUMO and LUMO+1, one of them fully localized on the Se–Se fragment, is an indication that upon reduction, again, this moiety will be the most active one and the minor share of the halogens prompts that contrary to the expectations they will not be instrumental upon reduction. Irrespective of the variation in the energies of the HOMOs and LUMOs, the energy band gap is about 3.04 eV and is insensitive to atomic substitution at positions 3 and 6. These data indicate that the peri-diselenolo-NIs can participate in both oxidation and reduction reactions and that the voltage difference between them will be quite significant.| Compound | HOMO | HOMO−1 | ΔE(HOMO − HOMO−1) | LUMO | LUMO+1 | ΔE(LUMO − LUMO+1) | ΔE(HOMO − LUMO) |
|---|---|---|---|---|---|---|---|
| SeH | −5.87 | −7.40 | 1.53 | −2.83 | −2.79 | 0.04 | 3.04 |
| SeCl | −6.12 | −7.62 | 1.50 | −3.08 | −2.83 | 0.28 | 3.04 |
| SeBr | −6.09 | −7.51 | 1.41 | −3.06 | −2.79 | 0.27 | 3.03 |
3.2. Synthesis of peri-diselenolo-1,8-naphthalimides
The synthesis of the target diselenolo derivatives SeH8, SeCl4, and SeBr4, whose structures were based on the DFT calculations, is shown in Scheme 1. The imides 4–6 were synthesized from the corresponding anhydrides by a procedure recently reported by us.25 The imidization of tetrahaloanhydrides 2 and 3 with n-butylamine was carried out in a mixture of N-methylpyrrolidone (NMP) and acetic acid at 100 °C for two hours. After workup and purification by column chromatography, we isolated imides 4–5 with a yield of 93% and 90%, respectively. The imidization of dibromoanhydride 1 was also performed in a mixture of NMP and acetic acid, but at a significantly higher temperature (150 °C) for 1 hour. For the synthesis of the diselenolo-derivatives, we applied the already optimized conditions that we used for the preparation of dithiolo-Nis.25 The substitution reaction with elemental selenium proceeded smoothly in NMP at 175 °C for several hours, and the reaction was monitored by TLC. The yields were high (88–93%), and the products were isolated on a gram scale. | ||
| Scheme 1 Synthesis of the target diselenolo-NIs SeH8, SeCl4 and SeBr4. | ||
Only imide SeH8 had sufficient solubility to allow purification by column chromatography. The other target product SeCl4 and SeBr4 were purified by recrystallization from DMF. The final compounds have been characterized by means of 1H, 13C{1H}-NMR spectroscopic techniques as well as by elemental analysis and mass spectrometry (see the Experimental section).
3.3. Self-organization of SeCl4, SeBr4 and SeH8
 | ||
| Fig. 2 SEM images of pristine SeCl4, SeBr4 and SeH8 (A)–(C) and corresponding bare electrodes (D)–(F). The ex situ images of electrodes cycled for 50 times (G)–(I) in LiTFSI:Pyr1,3FSI electrolyte. The fibres correspond to the GF/A separator. | ||
 | ||
| Fig. 3 (A) XRD patterns of SeH8, SeCl4 and SeBr4; (B) ex situ XRD patterns of electrodes SeH8, SeCl4 and SeBr4 cycled in a lithium-ion cell for 50 cycles (*peaks of Al foil). | ||
The observation of multiple order reflections could be assigned to (00l) peaks (i.e. l = 1, 2 and 4), thus implying that SeBr4 adopts a layered structure. In contrast, SeH8 displays a splitting of the low-angle reflection into two components at 8.25° (10.70 Å) and 8.37° (10.55 Å), thus indicating the formation of at least two structural modifications. The most complex structure is observed for SeCl4 – there is a series of low-angle peaks between 5 and 10°, the most intensive being at 6.8° (13.0 Å) and 9.6° (9.2 Å).
Another structural feature of the new NIs is the molecular packing inside the unit cell lattice. This is a consequence of the π–π stacking interaction between naphthalimide rings, which is manifested by the reflection at around 25° in the XRD patterns (Fig. 3A).49,50 For SeBr4, the intense diffraction peak at 25.39° reveals a π–π stacking of naphthalimide rings at a distance of 3.51 Å. On the contrary, two different distances for π–π stacking interaction are observed for SeH8 – 3.53 Å and 3.51 Å (i.e., reflections at 25.22° and 25.30°, respectively). This agrees with the observation of two structural modifications having two interlayer spacings. SeCl4 displays extended π–π stacking distances: two low-intensity peaks at 24.99° and 25.16° can be distinguished, which correspond to distances of 3.56 Å and 3.54 Å. To rationalize the observed π–π stacking distances, previously reported data on compounds containing 1,8-naphthalimide groups joined by different alkyl linkers were taken into account.47 Detailed structural analysis shows that depending on the linker kind, the π–π stacking of adjacent naphthalimide rings varies between 3.40 and 3.58 Å. Our data also demonstrate that the π–π stacking covers distances in the range of 3.51–3.56 Å.
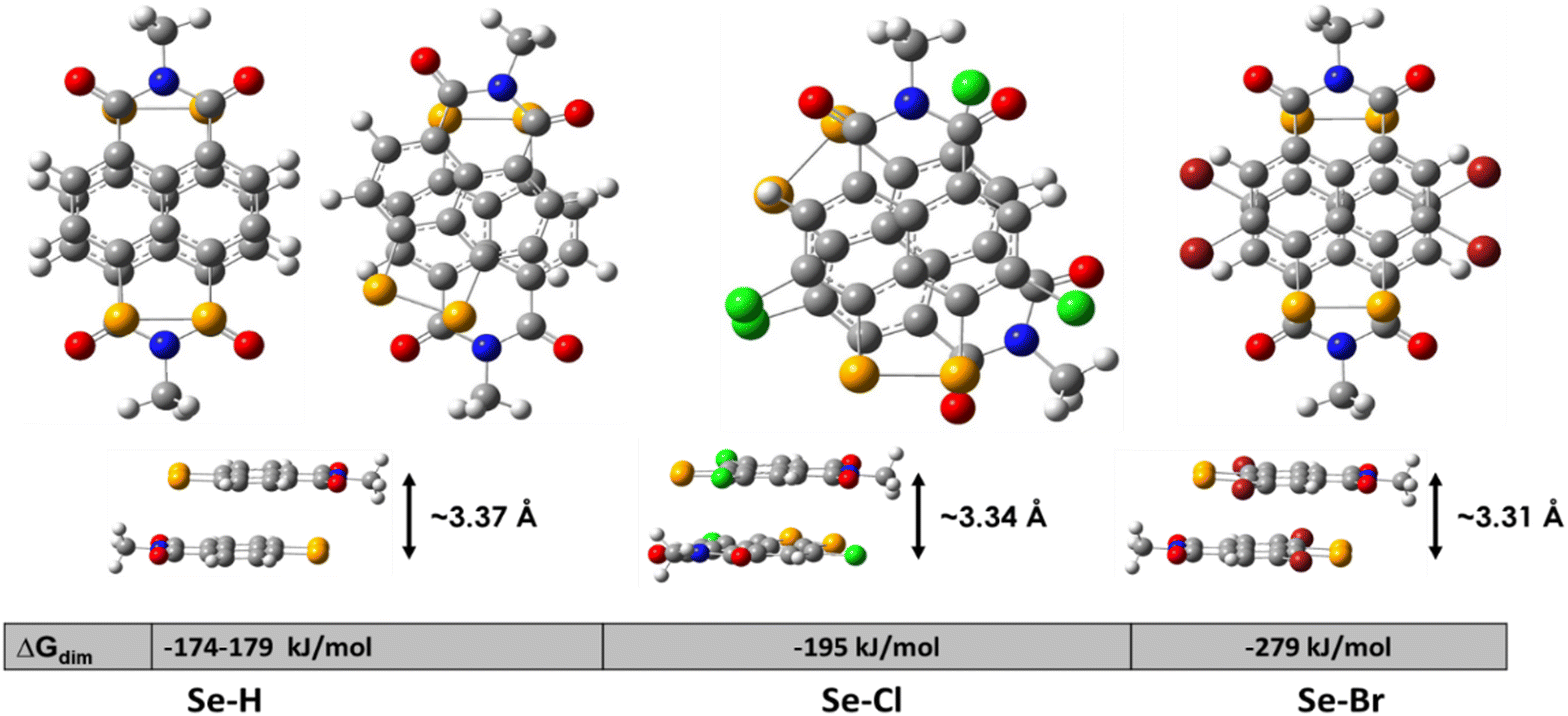 | ||
| Fig. 4 Optimized geometry of the dimers of the modelled molecules, with the calculated free energy of dimerization and intermolecular distances. | ||
In addition to the dipole–dipole coupling, the dispersion interactions contribute, being proportional to the molecular volume, and in this case, SeBr4 should feature the strongest attraction. As shown in Fig. 4, the free energy of molecular coupling grows in line with the molecular/substituent size and correspondingly decreases the intermolecular distance. The latter is the shortest possible in the absence of an environment (in the gas phase); in a multilayer structure, these distances will be larger.51 Significantly, the lowest π–π stacking distance is also experimentally observed for SeBr4 using XRD (Fig. 3).
 | ||
| Fig. 5 (A) 13C cross-polarization (CP) NMR spectra of SeBr4, SeCl4 and SeH8 samples recorded with 20 μs contact time and 10 kHz MAS; (B) 77Se ssNMR spectra recorded using single pulse excitation and 10 kHz MAS. The asterisks denote spinning sidebands. | ||
Further insight into the supramolecular organization provide the 77Se ssNMR spectra of the samples (Fig. 5B). In these spectra small differences in the selenium electronic environment lead to a more pronounced influence in the chemical shift due to the significantly larger selenium shift range (ca. 3000 ppm) compared to the 13C spectra. The arylselenides resonate in the 77Se shift range of 400–500 ppm. In the 77Se solution NMR spectra, a single 77Se resonance (due to symmetry) is detected for all samples – 548.6 ppm for SeBr4, 544.5 ppm for SeCl4, and 512.6 ppm for SeH8. In the solid state, however, due to π–π stacking, the samples self-organize, which results in different degrees to which the selenium nuclei experience the ring current of the neighbouring aromatic systems. Thus, two different 77Se shifts are observed for each sample. The 77Se NMR spectrum of SeBr4 reveals resonances at 582 and 560 ppm, the one of SeCl4 shows selenium shifts at 552 and 535 ppm, while the resonances of the SeH8 sample are observed at 476 and 435 ppm. In the sequence SeBr4:SeCl4:SeH8, the resonances are shifted to a higher field and the shift difference between both signals increases from 22 ppm to 27 ppm and finally to 41 ppm for the non-halogenated derivative. The isotropic shifts are detected by comparing 77Se ssNMR spectra recorded at two different (8 and 10 kHz) magic angle spinning (MAS) frequencies as the bands which do not change their position in the spectrum. The spinning sidebands are denoted with asterisks.
Summarising, for all samples the self-organization in the solid state due to π–π stacking leads to different electronic environments for both aromatic CH 13C and 77Se nuclei which results in splitting the respective solution signals (or resonance broadening for the 13C CH resonances of SeBr4 and SeH4).
3.4. Mechanism of the electrochemical reaction
 | ||
| Fig. 6 Cycling voltammetry curves of SeH8, SeCl4 and SeBr4 started with a catholic scan, theoretically calculated potentials and optimized geometries of the peri-diselenolo derivatives with different number of Li ions. | ||
To rationalize the observed reduction behaviours, the standard potentials of the interaction of each studied compound with Li+ are calculated and given on Fig. 6. In general, all naphthalimide-derivatives are prone to interact with 6Li+, in three consecutive steps. As expected from the charge distribution of the neutral molecules (Fig. S7, ESI†), the first step reflects the interaction of the peri-diselenide bridge with two Li+. For SeCl4 and SeBr4, the lithium is coordinated between Se and a halogen element, thus leading to an increase of the Se–Se distance: from 2.39 Å to 3.14 Å for SeCl4 and from 2.39 Å to 3.13 Å for SeBr4 (Table S2a, ESI†). Both Se and Cl/Br atoms remain attached in the molecular structure regardless of the increased Se–Se distance (Fig. 6). The calculated standard potential of the first reduction step reaches a magnitude of 1.71 V, which is identical for both halogenated representatives. The insensitivity of the calculated reduction potentials to the kind of halogen is in perfect agreement with the experimentally observed CV-curves (Fig. 6). It is worth mentioning that in the CV curves it is possible to differentiate two overlapping peaks corresponding to interaction of the organic molecule with each Li+ ion separately, while the molecular calculations give the average picture of the interaction of the organic molecule with a total of two Li+. The first reduction reaction proceeds at potentials of 1.86 V and 1.76 V for SeCl4, and 1.87 V and 1.72 V for SeBr4. The average potentials match the calculated ones: 1.81 V versus 1.71 V for SeCl4, and 1.80 V versus 1.71 V for SeBr4. The good agreement between experimental and calculated potentials corroborates that the peri-diselenide bridge is the first reduced during the interaction of the organic molecule with Li+.
For the H form, the degeneracy of the LUMO and LUMO+1 makes the reduction with Li+ different from that of the halogen-substituted forms. In this case, the first reduction step involves interactions with 4Li atoms, with a potential of 1.45 V. The calculated value is quite close to the experimentally determined one (i.e. 1.45 V). The accommodation of 4Li+ ions is accomplished by the peri-diselenide bridge and the two carbonyl groups (Fig. 6). Specifically, in this manner of lithium coordination in the H-form, the lithium serves to keep the Se atoms close together by bridging them into a single entity, but with a longer Se–Se bond length (Table S2a, ESI†). This is in contrast to the halogen-substituted form, where the Se–Se bond is also increased due to the involvement of halogen atoms in the lithium coordination (Table S2a, ESI†).
After the first reduction involving the peri-diselenide fragment, the next two Li+ ions attack the two carbonyl groups for the halogen-substituted NIs (Fig. 6). This leads to a decrease in the standard potentials: from 1.71 to 1.56 V for SeCl4 and from 1.71 to 1.63 V for SeBr4. Contrary to the reduction of the diselenide bridge, the reduction potential of carbonyl groups depends on whether H, Cl or Br appears in positions 3 and 6, following the order SeBr4 > SeCl4 ≫ SeH8. The same order in decreasing of the reduction potential is experimentally observed for the average potential of the second groups of peaks: 1.61 V for SeBr4; 1.54 V for SeCl4; and 1.45 V for SeH8. For the halogen-substituted representatives, the binding of Li+ ions around two carbonyl groups is accompanied by a rearrangement of Li coordination in the diselenide bridge: while one of the Li+ ions remains coordinated around Se and halogen atoms, the other is relocated, so that to bridge the 2Se atoms. This provokes a slight increase in the Se–Se bond length (Table S2a, ESI†). In contrast, in the H-form Li+ ions are coordinated around the carbonyl groups without affecting the Li+ in the diselenide fragment, which is manifested by a limited variation in the Se–Se bond length (Table S2a, ESI†). This is further computational evidence that the structure of the NI molecule is flexible enough to accommodate the changes induced by Li reduction.
The interaction of the next two lithium ions with the studied compounds is expressed in preferential coordination of Li+ around carbonyl groups again. This leads to a drastic decrease in the standard potential below 1 V following the order SeBr4 ∼ SeCl4 > SeH8. In the CV-curves, despite the difficulties in distinguishing the third groups of peaks, the average reduction potential also follows the order of the calculated standard potential: SeBr4 (1.03 V versus 0.86 V); SeCl4 (1.00 V versus 0.85 V) and SeH8 (0.73 V versus 0.36 V). Approaching full coordination of the carbonyl groups by Li+ ions induces a re-bridging of the Se atoms in the peri-diselenide fragment, which is realized via Li+ ions.
Based on the comparison between experimental and calculated results, it appears that the studied NI-derivatives interact with a maximum of 6Li+ through consecutive reduction of the diselenide bridge and both carbonyl groups. According to organic redox reactions nomenclature, this can be classified as an n-type interaction. Charge distribution proves that 6Li is inefficient, 8Li results in molecular destruction (Fig. S7, ESI†).
The lithiation has a fairly small effect over the electron distribution in the imide fragment: the first stage of reduction (2Li) does not change the charge distribution, the second step (4Li) accumulates about −0.7 charge on the fragment, and the next one (6Li) adds about −0.3 (see Table S2b, ESI†). The charge is localised predominately on the O-atoms of the imide fragment.
 | ||
| Fig. 7 (left) Cycling voltammetry curves of SeH8, SeCl4 and SeBr4 started with an anodic scan. (right) Theoretically calculated potentials for single and double oxidation of molecules without and in the presence of counterion TFSI− (full and dotted lines, respectively). | ||
To get insight into the electronic structure of the oxidized molecules, the oxidation reaction is modelled, and the calculated standard oxidation potentials are given on Fig. 7.
As one can see, the first oxidation potential falls between 4.2 V and 4.5 V, while the second oxidation potential is above 4.9 V. The comparison of the calculated and experimental data marks a relatively acceptable agreement of the first calculated oxidation potential with the experimentally observed oxidation peak: 4.28 V for SeH8, 4.49 V for SeCl4 and 4.48 V for SeBr4. It is worth mentioning that the CV experiments are carried out up to 4.7 V to avoid side reactions, including electrolyte degradation. To improve the agreement between experimental and calculated values, we also optimized neutral models, including the respective number of counterions (TFSI−) in the implicit solvent. In this case, the calculated oxidation potential decreases significantly and comes closer to the experimental data. Interestingly, the second oxidation potential is also diminished and becomes comparable to the additional oxidation peaks observed for SeH8 and SeBr4. This suggests that the oxidation of SeCl4 is predominantly a one-electron process, whereas two-electron oxidation reaction is most likely to occur for the other two compounds.
The variation of the charge distribution in the course of electron abstraction can serve as a marker to monitor the attacked part of the oxidized molecules (Table 2 and Fig. S7, ESI†). The data show that the charge distribution is insensitive to the type of the substituents at positions 3 and 6: in all cases, the positive charge is predominantly localized on the Se atoms (50% of the positive charge), on the same carbons from the naphthalene moiety where the spin density is high and negligibly on the oxygens in the imide fragment. Surprisingly, the remaining carbons in the naphthalene unit gain electron density, thus enhancing the charge polarization within the molecule. The substituents at positions 3 and 6 also acquire positive charges upon oxidation growing in the order H ≪ Br < Cl as seen in Table 2 and Fig. S7 (ESI†). Charge distribution analysis shows that Se atoms together with carbons in the naphthalene unit are involved in the oxidation of NI derivatives. It is worth mentioning that the specific linkage of Se atoms and the naphthalene unit in one molecule allows to achieve an oxidation potential (above 4.2 V), which is one of the highest reported in the literature for well-known p-type thioethers.54 To compare, the thianthrene, in which the two sulphur atoms are fixed in the core six-membered ring, displays an oxidation potential of 4.1 V.55 This is another indication of the suitability of the molecular architecture of the NI derivatives to participate in p-type redox reactions.
| SeH | SeCl | SeBr | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Neutral+TFSI | +1+TFSI | +2+2TFSI | Neutral+TFSI | +1+TFSI | +2+2TFSI | Neutral+TFSI | +1+TFSI | +2+2TFSI | |
| Sum of charges C (aromatic) | −1.579 | −1.311 | −1.093 | −1.226 | −1.025 | −0.839 | −1.582 | −1.158 | −0.994 |
| Se | 0.658 | 1.158 | 1.643 | 0.722 | 1.209 | 1.657 | 0.708 | 1.172 | 1.621 |
| TFSI | −0.996 | −0.985 | −1.845 | −0.987 | −0.986 | −1.883 | −0.830 | −0.992 | −1.895 |
| Molecule | −0.004 | 0.985 | 1.846 | 0.004 | 0.989 | 1.884 | 0.076 | 0.994 | 1.896 |
| H/Cl/Br | 0.528 | 0.566 | 0.601 | 0.051 | 0.183 | 0.330 | 0.105 | 0.180 | 0.268 |
Further information on the oxidation of SeCl4 at high voltages is provided by ex situ EPR spectroscopy (Fig. 8). After the first oxidation of SeCl4 at 4.15 V (Fig. 8E), the EPR spectrum displays an asymmetric narrow signal with a g-tensor of g⊥ = 2.0021 and g‖ = 2.0018. For the sake of comparison, the EPR spectra of partially reduced SeCl4 at 1.67 V, 1.5 V and 1.0 V are also shown on Fig. 8B–D. The reduction potentials reflect the stepwise interaction of the peri-diselenide bridge and carbonyl groups with lithium ions. Regardless of the fact that the EPR spectra of reduced SeCl4 are dominated by a signal due to the carbon black additives, the low-intensity narrow signal is enhanced with lowering the cathodic potential, culminating at 1.0 V in a nearly symmetrical signal with a g-value of around 2.002 and a line width of 0.2 mT. Going from the first cathodic scan to the next anodic scan and switching off the symmetric cell at a potential of 4.5 V, the EPR signal increases more than 30 times (Fig. 8F). In addition, the signal profile assumes an asymmetric shape with a g-tensor identical to the signal registered after the beginning of the oxidation processes (i.e., at 4.15 V). It is important that the signal detected at 4.5 V is about 15 times stronger than the signal detected at 4.15 V. The comparison of the EPR signals for the reduced and oxidized SeCl4 shows that the oxidation up to 4.5 V yields a strong asymmetric signal, while a low-intensity nearly symmetrical signal becomes visible after the reduction to 1.0 V. The radicals have a relatively long life – after 72 hours, the radicals disappear nearly completely, without changing the signal profile (Fig. S8, ESI†), meaning that they degrade by forming diamagnetic products.
 | ||
| Fig. 8 CV curves (A) and the respective ex situ EPR analysis of SeCl4 electrodes: after the first cathodic scan up to 1.67 V (B; black), then to 1.5 V (C; red) and to 1.0 V (D; magenta); after the first anodic scan to 4.15 V (E; blue), and after the cycle starting with cathodic scan to 1.0 V, followed with an anodic scan to 4.5 V (F; green). All spectra are recorded at T = 100 K. | ||
Contrary to the reduction with Li+ ions, the oxidation of SeCl4 requires the participation of the counter anion from the electrolyte salt. That is why, we further examined the oxidation and reduction stability of the lithium electrolyte, which contains LiTFSI dissolved in the ionic liquid Pyr1,3FSI (IL). The electrolyte stability is monitored by using a symmetric cell with Li working, reference and counter electrodes (details are given in Fig. S9, ESI†) the linear sweep voltammetry curves of the symmetric cell demonstrate that the IL-based electrolyte is stable in a broad potential range (i.e., from +6.5 to −6.5 V vs. Li/Li+, Fig. S9B, ESI†). Despite the electrochemical stability of the electrolyte, side reactions take place at high potentials, leading to the formation of radicals. To detect them, we analysed the soaked in electrolyte separator (without any washing) before the electrochemical experiment and after the symmetric cells switch-off at +6.5 and −6.5 V respectively. In addition, the soaked separator is measured at 100 K to improve the EPR sensitivity. As a reference, the EPR spectra of the unsoaked separator and the blank electrolyte are also given (Fig. S9D and E, ESI†). As one can see, both the blank electrolyte and the unsoaked separator show no signals. However, the EPR spectra of the reduced and oxidized electrolyte contain one symmetrical narrow signal with a g-value of 2.0021 and a line width of about 0.03 mT (Fig. S9A and C, ESI†). Moreover, the density of radicals for the reduced and oxidized electrolytes are comparable. This implies that the radicals are probably formed as secondary degradation products after the anodic and cathodic scans. This is not an unexpected result since the ILs are prone to easily form numerous types of radicals.56
The observation of radicals during the electrolyte oxidation and reduction allows to compare them with those detected on working SeCl4. The comparison of the EPR spectra reveals that the same radicals occur for oxidized SeCl4 and IL–electrolyte, but the different spectra profiles suggest that radicals reside on sites with different symmetry. This can be explained by the attachment of radicals on the surface of the oxidized SeCl4, thus losing some degree of mobility, resulting in an asymmetric signal profile. It is worth mentioning that the density of attached radicals is about two orders lower than that detected in the electrolyte. On the other hand, the attachment of the electrolyte-generated radicals to the electrode surface gives evidence for the participation of the electrolyte in the oxidation of SeCl4. During the reduction of SeCl4, the EPR signal preserves the symmetrical line shape, thus indicating the lack of interaction between reduced SeCl4 and the electrolyte.
After the first anodic and cathodic scans, the next cycles cause a change in the CV curve profile, as a result of which several peaks can be distinguished: in the low-voltage range between 1.0 and 2.5 V and in the high-voltage range between 3.5 and 4.5 V, respectively (Fig. 9). These two voltage ranges correspond to the regions, where the interaction of naphthalimide-derivatives with Li+, as well as their oxidation, take place. After the fifth scan, the CV curve profiles become identical, no matter whether the reaction begins with an anodic or a cathodic scan.
 | ||
| Fig. 9 CV curves after the fifth scan for SeH8 (green lines), SeCl4 (red lines), and SeBr4 (blue lines) used as electrodes in three-electrode cells started with a cathodic and anodic scan, respectively. | ||
3.5. Electrochemical characterization of SeH8, SeCl4 and SeBr4
Fig. 10 summarizes the cycling stability of SeH8, SeCl4 and SeBr4 electrodes. The charge–discharge curves adopt almost the same profiles after 5 cycles, regardless of how the test was started (Fig. S10, ESI†). This is consistent with the CV curves of the samples (Fig. 9). Among them, SeCl4 delivers the highest capacity, especially when the cell starts with a charge mode (about 250 mA h g−1). However, the capacity quickly decreases after 10 cycles, irrespective of whether the cell test starts with a charge or discharge mode: the capacity varies between 45 and 55 mA h g−1 after 25 cycles. The most stable performance is observed for SeBr4: the specific capacity reaches of about 150 mA h g−1 and remains nearly constant after 25 cycles for the cell starting with a charge mode, while the capacity is slightly lower (about 125 mA h g−1) for the counterpart cell. The H-form demonstrates the worst performance: for the cell starting with a discharge mode, the specific capacity decreases from 131 to 69 mA h g−1 after 25 cycles, while for the cell starting with a charge mode, there is a drastic decline in the capacity from 236 to 23 mA h g−1. During prolonged cycling, it seems that the capacities of the electrodes tend towards each other, irrespective of whether the test starts with charge or discharge. This is consistent with the CV curves after the 5th cycle (Fig. 9). On the other hand, the unsatisfactory cycling stability can be related with low electrical conductivity of naphthalimide derivatives.57 One approach to overcome this drawback is to fabricate the composite between naphthalimide derivatives and highly conductive additives.2,57 | ||
| Fig. 10 Cycling stability of SeH8, SeCl4, and SeBr4 used as electrodes in lithium half-cells starting with a charge (—○—) or discharge (—●—) mode. The scan rate is C/10. | ||
3.6. Ex situ analysis
To rationalize the cycling stability of the new compounds, ex situ XRD, SEM, and EDS analyses were performed. The subjects of study are SeH8, SeCl4 and SeBr4 electrodes after 25 cycles between 1.0 and 4.4 V and switched off at 1.0 V. The XRD patterns demonstrate that all compositions undergo amorphization during the cycling (Fig. 3B). Alongside, the SEM images display a greater or lesser increase in the electrode roughness in comparison with the pristine electrode (Fig. 2G–I). However, all electrodes remain stable after cycling. This is visible under high magnifications: in this case, the electrodes particles become blurry since they are covered by the electrolyte (Fig. S11, ESI†).EDS analyses were performed to compare the halogen/chalcogen content of the electrode materials before and after use. The abundance of C-atoms was altered during the experiments due to the use of a carbon containing underlay (SEM Conductive Double Sided Carbon Tape), but the ratios of Se-to-Cl/Br and Se-to-N were not affected and can provide valuable insight. The measured abundancies for the pristine compounds (Table S3, ESI†) comply with the nominal compositions and show a 1.00 ± 0.05 Se-to-Cl ratio for SeCl4 and 0.95 ± 0.05 Se-to-Br ratio for SeBr4. The EDS analysis of the used electrodes shows an increase to around 2.4 ± 0.4 for the former, while the latter remains approximately the same (0.7 ± 0.2) (Table S4, ESI†). This means that SeCl4 loses some of the Cl-substituents during cycling, which correlates with its worse cycling stability (Fig. 10). In comparison with SeCl4, the SeBr4 preserves, to a great extent, its composition during cycling and displays a better cycling stability. Contrary to the Se-to-Cl(Br) ratio, the Se-to-N ratio cannot be used to determine the composition of cycled electrodes, because of the deposition of electrolyte, which contains a significant amount of F, N and S elements. However, the F-to-S ratio can be used to unveil whether the electrolyte penetrates the electrodes or undergoes a side reaction of degradation. The calculated F-to-S ratio is as follows: 2.5 ± 0.3, 1.8 ± 0.2 and 1.7 ± 0.1 for SeCl4, SeBr4 and SeH8, respectively. These values are comparable to the nominal F-to-S ratio for the lithium salt LiTFSI (2-to-1) and the lithium solvent Pyr13FSI (1-to-1). This suggests that the electrolyte penetrates mainly into the electrodes, especially for SeBr4 and SeH8, whereas the different morphology of SeCl4 obstructs a sizable electrolyte penetration. In addition, partial electrolyte degradation cannot be excluded, especially for SeCl4. This is another sign for the observed different cycling performance for SeH8, SeCl4 and SeBr4.
4. Conclusions
This study reports on a novel naphthalimide-based bipolar OEMs consisting of three structural motifs: (i) peri-diselenide bridge; (ii) halogens or hydrogens at positions 3 and 6; and (iii) alkyl chain on imide nitrogen. The OEMs design was based on molecular modelling with DFT, and the simulated characteristics gave promising results encouraging the synthesis and further testing. The compounds tend to assemble into ordered nanostructures depending on the kind of the halogen substituents – the morphology is changed from nanowires for SeBr4, to belt-like aggregates for SeH8, via nanorods for SeCl4. This is a consequence of the π–π stacking of naphthalimide rings, with distance between them varying from 3.51 to 3.56 Å. The supramolecular architectures were characterized by SEM, PXRD and solid-state NMR spectroscopy. Despite the different morphology, electrodes with relatively smooth surfaces were fabricated by mixing the active materials with carbon black and CMC in a ratio of 70-20-10 wt%.Below 3.0 V vs. Li+/Li, the new NI derivatives interact with a maximum of 6Li+ through consecutive reduction of the peri-diselenide bridge and the carbonyl groups (n-type interaction). Above 4.0 V vs. Li+/Li, NI derivatives are oxidized due to the participation of the selenium atoms and the naphthalene unit carbons (p-type interaction). Although the oxidation of SeCl4 is predominantly a one-electron process, two-electron oxidation reaction occurs for SeBr4 and SeH8. The results are in good agreement with those predicted theoretically.
Among the new NI derivatives, the best performance is observed for the Br-substituted representative – it delivers a specific capacity of about 150 mA h g−1 and remains nearly constant during cycling. This is a consequence of the composition stability of SeBr4 in the electrolyte solution. Unlike it, SeCl4 has a higher capacity (around 250 mA h g−1), but poor cycling stability, together with partial extraction of Cl during cycling. The electrochemical testing mode determines the performance of SeH8 – the better performance is achieved when the cell begins with a discharge. These results indicate that further optimisation of the electrolyte composition is required to improve the electrochemical performance of the studied compounds.
In general, the reported peri-diselenolo-1,8-naphthalimide derivatives offer a bipolar molecular architecture that could initiate further design of organic bipolar electrode materials.
Author contributions
Conceptualization: DM, YZ, RS; methodology: DM, YZ, RS; software: YD, HR, LB; validation: DM, YZ, S. Stoyanov, AT; formal analysis: LB, S. Stanchovska, RK, M. Mutovska, NS, M. Mondeshki, YD; investigation: M. Mutovska, S. Stoyanov, DM, S. Stanchovska, LB, RK, YZ; resources: DM, S. Stoyanov, YZ, RS; data curation: DM, YZ, S. Stoyanov, YD, RS; writing – original draft: DM, RK, M. Mutovska, NS, S. Stoyanov, YZ, M. Mondeshki, AT; writing – review & editing: DM, S. Stoyanov, YZ, M. Mondeshki, AT, RS; visualization: DM, S. Stoyanov, YZ, M. Mondeshki, YD, RS; supervision: DM, YZ; project administration: DM, YZ; funding acquisition: DM, YZ.Data availability
All data generated or analysed during this study are included in this article and its ESI.† Software used and code: (a) OriginPro 8.6 (64 bit) Sr3, b99 (Academic). (b) EC-Lab Software, Biologic. (c) ChemDraw 8.0.3. (d) GaussView 6.0. (e) Gaussian 16, Gaussian Inc.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The Bulgarian National Science Fund funded this analytical and electrochemical characterization by grant number KΠ-06-H69/1 (8.12.2022). The DFT calculations were financed under project KΠ-06-ДB-6 (16.12.2019). S. Stoyanov and YZ would like to acknowledge the European Union-NextGenerationEU through the National Recovery and Resilience Plan of the Republic of Bulgaria, project no. BG-RRP-2.004-0008.Notes and references
- S. Lee, G. Kwon, K. Ku, K. Yoon, S. Jung, H. Lim and K. Kang, Adv. Mater., 2018, 30, 1704682, DOI:10.1002/adma.201704682.
- P. Poizot, F. Dolhem and J. Gaubicher, Curr. Opin. Electrochem., 2018, 9, 70–80, DOI:10.1016/j.coelec.2018.04.003.
- K. Deuchert and S. Hünig, Angew. Chem., Int. Ed. Engl., 1978, 17, 875–886, DOI:10.1002/anie.197808753.
- P. Poizot, J. Gaubicher, S. Renault, L. Dubois, Y. Liang and Y. Yao, Chem. Rev., 2020, 120, 6490–6557, DOI:10.1021/acs.chemrev.9b00482.
- Z. Yu, Y. Wang and Z. Luo, Ind. Eng. Chem. Res., 2024, 63, 9619–9630, DOI:10.1021/acs.iecr.4c00394.
- Z. Song and H. Zhou, Energy Environ. Sci., 2013, 6, 2280, 10.1039/c3ee40709h.
- J. Heiska, M. Nisula and M. Karppinen, J. Mater. Chem. A, 2019, 7, 18735–18758, 10.1039/C9TA04328D.
- J. Kim, Y. Kim, J. Yoo, G. Kwon, Y. Ko and K. Kang, Nat. Rev. Mater., 2022, 8, 54–70, DOI:10.1038/s41578-022-00478-1.
- H. Kye, Y. Kang, D. Jang, J. E. Kwon and B.-G. Kim, Adv. Energy Sustainability Res., 2022, 3, 2200030, DOI:10.1002/aesr.202200030.
- Y. Tong, Y. Wei, A. Song, Y. Ma and J. Yang, ChemSusChem, 2024, 17, e202301468, DOI:10.1002/cssc.202301468.
- C. Tang, B. Wei, W. Tang, Y. Hong, M. Guo, X. He, J. Hu, S. Jia and C. Fan, Chem. Eng. J., 2023, 474, 145114, DOI:10.1016/j.cej.2023.145114.
- Z. Tie, S. Deng, H. Cao, M. Yao, Z. Niu and J. Chen, Angew. Chem., Int. Ed., 2022, 61, e202115180, DOI:10.1002/anie.202115180.
- S. Y. An, T. B. Schon, B. T. McAllister and D. S. Seferos, EcoMat, 2020, 2, e12055, DOI:10.1002/eom2.12055.
- D. Chen, A. Avestro, Z. Chen, J. Sun, S. Wang, M. Xiao, Z. Erno, M. M. Algaradah, M. S. Nassar, K. Amine, Y. Meng and J. F. Stoddart, Adv. Mater., 2015, 27, 2907–2912, DOI:10.1002/adma.201405416.
- M. Veerababu and R. Kothandaraman, Electrochim. Acta, 2017, 232, 244–253, DOI:10.1016/j.electacta.2017.02.152.
- C. Chen, X. Zhao, H.-B. Li, F. Gan, J. Zhang, J. Dong and Q. Zhang, Electrochim. Acta, 2017, 229, 387–395, DOI:10.1016/j.electacta.2017.01.172.
- J. Kim, H. Kim, S. Lee, G. Kwon, T. Kang, H. Park, O. Tamwattana, Y. Ko, D. Lee and K. Kang, J. Mater. Chem. A, 2021, 9, 14485–14494, 10.1039/D1TA02059E.
- Y. Cho, D. Jang, J.-J. Park, H. Kye, J. E. Kwon and B.-G. Kim, ACS Appl. Energy Mater., 2023, 7(18), 7615–7623, DOI:10.1021/acsaem.3c02025.
- Y. Zhou, X. Huang, X. Chen, F. He, D. Chen, X. Sun, S. Tan and P. Gao, ACS Appl. Mater. Interfaces, 2022, 14, 40862–40870, DOI:10.1021/acsami.2c09649.
- V. Koleva, M. Kalapsazova, D. Marinova, S. Harizanova and R. Stoyanova, ChemSusChem, 2023, 16, e202201442, DOI:10.1002/cssc.202201442.
- C. Peng, F. Wang, Q. Chen, X. Yan, C. Wu, J. Zhang, W. Tang, L. Chen, Y. Wang, J. Mao, S. Dou and Z. Guo, Adv. Funct. Mater., 2024, 34, 2401001, DOI:10.1002/adfm.202401001.
- C. Hu, X. Yang, P. Liu, Z. Song, Y. Lv, L. Miao, M. Liu and L. Gan, J. Mater. Chem. A, 2024, 12, 11867–11874, 10.1039/D4TA00476K.
- A. Bhardwaj, Ch Mudasar Hussain, P. Dewangan and P. Mukhopadhyay, Chem. – Eur. J., 2024, 30, e202400208, DOI:10.1002/chem.202400208.
- M. Al Kobaisi, S. V. Bhosale, K. Latham, A. M. Raynor and S. V. Bhosale, Chem. Rev., 2016, 116, 11685–11796, DOI:10.1021/acs.chemrev.6b00160.
- M. Mutovska, N. Simeonova, S. Stoyanov, Y. Zagranyarski, S. Stanchovska and D. Marinova, Materials, 2023, 16(23), 7471–7484, DOI:10.3390/ma16237471.
- H. Abul-Futouh, Y. Zagranyarski, C. Müller, M. Schulz, S. Kupfer, H. Görls, M. El-Khateeb, S. Gräfe, B. Dietzek, K. Peneva and W. Weigand, Dalton Trans., 2017, 46, 11180–11191, 10.1039/c7dt02079a.
- Y. Liang, H. Dong, D. Aurbach and Y. Yao, Nat. Energy, 2020, 5, 646–656, DOI:10.1038/s41560-020-0655-0.
- F. Wang, E. Hu, W. Sun, T. Gao, X. Ji, X. Fan, F. Han, X.-Q. Yang, K. Xu and C. Wang, Energy Environ. Sci., 2018, 11, 3168–3175, 10.1039/C8EE01883A.
- X. Wang, Z. Shang, A. Yang, Q. Zhang, F. Cheng, D. Jia and J. Chen, Chem, 2019, 5, 364–375, DOI:10.1016/j.chempr.2018.10.018.
- Y. Shi, H. Tang, S. Jiang, L. V. Kayser, M. Li, F. Liu, F. Ji, D. J. Lipomi, S. P. Ong and Z. Chen, Chem. Mater., 2018, 30, 3508–3517, DOI:10.1021/acs.chemmater.8b01304.
- R. Shi, S. Jiao, Q. Yue, G. Gu, K. Zhang and Y. Zhao, Exploration, 2022, 2, 20220066, DOI:10.1002/EXP.20220066.
- H. J. Reich and R. J. Hondal, ACS Chem. Biol., 2016, 11, 821–841, DOI:10.1021/acschembio.6b00031.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652, DOI:10.1063/1.464913.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789, DOI:10.1103/PhysRevB.37.785.
- M. M. Francl, W. J. Pietro, W. J. Hehre, J. S. Binkley, M. S. Gordon, D. J. DeFrees and J. A. Pople, J. Chem. Phys., 1982, 77, 3654–3665, DOI:10.1063/1.444267.
- V. A. Rassolov, M. A. Ratner, J. A. Pople, P. C. Redfern and L. A. Curtiss, J. Comput. Chem., 2001, 22, 976–984, DOI:10.1002/jcc.1058.
- B. Mennucci and J. Tomasi, J. Chem. Phys., 1997, 106, 5151–5158, DOI:10.1063/1.473558.
- M. Cossi, G. Scalmani, N. Rega and V. Barone, J. Chem. Phys., 2002, 117, 43–54, DOI:10.1063/1.1480445.
- F. Lipparini, G. Scalmani, B. Mennucci, E. Cancès, M. Caricato and M. J. Frisch, J. Chem. Phys., 2010, 133, 014106, DOI:10.1063/1.3454683.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104, DOI:10.1063/1.3382344.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465, DOI:10.1002/jcc.21759.
- J. P. Foster and F. Weinhold, J. Am. Chem. Soc., 1980, 102, 7211–7218, DOI:10.1021/ja00544a007.
- E. D. Glendening, A. E. Reed, J. E. Carpenter and F. Weinhold, NBO Version 3.1 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision A.03, Gaussian Inc., Wallingford CT, 2016 Search PubMed.
- J. Lee, R. Singh, D. H. Sin, H. G. Kim, K. C. Song and K. Cho, Adv. Mater., 2016, 28, 69–76, DOI:10.1002/adma.201504010.
- J. Zhou, L. Xue, Y. Shi, X. Li, Q. Xue and S. Wang, Langmuir, 2012, 28, 14386–14394, DOI:10.1021/la303061r.
- D. L. Reger, A. Debreczeni, J. J. Horger and M. D. Smith, Cryst. Growth Des., 2011, 11, 4068–4079, DOI:10.1021/cg200636k.
- H. Li, Y. Lu, Y. Liu, X. Zhu, H. Liua and W. Zhu, Phys. Chem. Chem. Phys., 2012, 14, 9948–9955, 10.1039/C2CP41149K.
- S. Laschat, A. Baro, N. Steinke, F. Giesselmann, C. Hägele, G. Scalia, R. Judele, E. Kapatsina, S. Sauer, A. Schreivogel and M. Tosoni, Angew. Chem., Int. Ed., 2007, 46, 4832–4887, DOI:10.1002/anie.200604203.
- F. Zhang, Y. Ma, Y. Chi, H. Yu, Y. Li, T. Jiang, X. Wei and J. Shi, Sci. Rep., 2018, 8, 8208, DOI:10.1038/s41598-018-26502-5.
- D. Werner, D. H. Apaydin, D. Wielend, K. Geistlinger, W. D. Saputri, U. J. Griesser, E. Dražević, T. S. Hofer and E. Portenkirchner, J. Phys. Chem. C, 2021, 125, 3745–3757, DOI:10.1021/acs.jpcc.0c10778.
- R. Gonçalves, S. Lanceros-Méndez and C. M. Costa, Electrochem. Commun., 2022, 135, 107210, DOI:10.1016/j.elecom.2022.107210.
- N. Lingappan, L. Kong and M. Pecht, Renewable Sustainable Energy Rev., 2021, 147, 111227, DOI:10.1016/j.rser.2021.111227.
- Q. Zhang, Q. Ma, R. Wang, Z. Liu, Y. Zhai, Y. Pang, Y. Tang, Q. Wang, K. Wu, H. Wu, Y. Zhang, L. Zhang, C. Zhang, L. Fu, S. Eliseeva, V. Kondratiev and Y. Wu, Mater. Today, 2023, 65, 100–121, DOI:10.1016/j.mattod.2023.02.027.
- M. E. Speer, M. Kolek, J. J. Jassoy, J. Heine, M. Winter, P. M. Bieker and B. Esser, Chem. Commun., 2015, 51, 15261–15264, 10.1039/C5CC04932F.
- B. J. Mincher and J. F. Wishart, Solvent Extr. Ion Exch., 2014, 32, 563–583, DOI:10.1080/07366299.2014.925687.
- S. Kagatikar and D. Sunil, J. Mater. Sci., 2022, 57, 105–139, DOI:10.1007/s10853-021-06602-w.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1–S5 NMR spectra; Fig. S6 cycling stability; Fig. S7 NBO charges; Fig. S8 EPR spectra; Fig. S9 linear sweep voltammetry curves; Fig. S10 charge/discharge curves; Fig. S11 SEM images; Table S1 dipole moments of the neutral models; Table S2a Se–Se distance in Å as a function of the number of inserted Li+; Table S2b The electron distribution of the imide chain; Table S3 EDS data for the pristine compounds; Table S4 EDS data for the cycled electrodes. See DOI: https://doi.org/10.1039/d4ma01042f |
| This journal is © The Royal Society of Chemistry 2025 |