
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIn situ monitoring of barrier function on-chip via automated, non-invasive luminescence sensing†
Bryan G.
Schellberg
 a,
Abigail N.
Koppes
*abc and
Ryan A.
Koppes
*ab
a,
Abigail N.
Koppes
*abc and
Ryan A.
Koppes
*ab
aDepartment of Chemical Engineering, Northeastern University, Boston, Massachusetts, USA. E-mail: a.koppes@northeastern.edu; r.koppes@northeastern.edu
bDepartment of Bioengineering, Northeastern University, Boston, Massachusetts, USA
cDepartment of Biology, Northeastern University, Boston, Massachusetts, USA
First published on 4th April 2025
Abstract
Over the past 30 years, organs-on-a-chip (OOCs) have emerged as a robust alternative to address the technological challenges associated with current in vitro and in vivo options. Although OOCs offer improved bio-relevance and controlled complexity, broad adoption has remained limited. Most approaches to characterize on-chip structure and function require human intervention, limiting device translation and feasibility. Here, we introduce a new fiber optic-based sensing platform that enables automated, temporal luminescence sensing on-chip, validated for real-time readout of epithelial and endothelial barrier function under cytokine-induced inflammation. Our platform, capable of at least 1 μM resolution, tracked paracellular transport in situ for 9 days of culture under perfusion on-chip. These results offer an alternative sensing approach for continuous, non-invasive luminescence monitoring in OOCs.
Introduction
Organs-on-a-chip (OOCs) have significantly advanced human disease modelling and drug development, offering improved bio-relevance and controlled complexity by integrating physical and chemical stimuli matched to physiologically relevant conditions.1–4 Although significant innovations have been made toward recreating organ-specific physiology on chip, the methods available to study the structure and function of the cell microenvironment are still limited.5–7 Established analysis approaches, such as fluorescence microscopy, often rely on laborious offline workflows that yield acute or terminal time-point data.8,9 These complex techniques require substantial manual perturbations, frequently adding environmental and user-based variability to inherently inconsistent biological samples. As the OOC field continues to evolve, there is a unique opportunity to engineer improved in situ characterization methods into organ-chip devices. It is essential to develop novel approaches that address current limitations and enable real-time readout of relevant physiological parameters in OOCs to improve the reliability and robustness of these systems.Electrochemical and optical methods are the most common approaches for integrated sensing on-chip. Numerous demonstrations of electrochemical sensors have been integrated into OOC, showing high sensitivity and specificity for a range of analytes, including pH, oxygen concentration, glucose, and lactate.10–14 Electrochemical methods have also been adapted for structural characterization of the microenvironment, tracking transient changes in barrier permeability, cell health, and differentiation state throughout culture on-chip.15–17 Van der Helm et al. integrated plated electrodes on-chip for impedance spectroscopy measurements of Caco-2 monolayers differentiated on-chip.17 Although electrochemical methods offer continuous, real-time data from the on-chip microenvironment, these approaches suffer from high costs resulting from complicated fabrication workflows for sensor integration and relatively rapid sensor fouling or site saturation.5,18 Broad adoption of OOCs will require inexpensive consumable culture ware paired with reusable sensor technologies.
Optical interrogation methods have been the standard for characterizing the on-chip microenvironment, enabling direct visualization of cell structure and quantitative outputs, including viability and protein expression.7,19,20 Optical and fluorescence microscopy techniques offer non-invasive and non-destructive characterization of cellular dynamics on-chip, including oxygen consumption, calcium signalling, and barrier permeability.21–24 In a multi-organ chip system, Novak et al. tracked the distribution of inulin-FITC tracer dye with an automated fluorescence microscopy platform, demonstrating repeatable concentration profiles for each of eight organ models over 3 weeks.21 However, most experimental procedures are limited to single OOC setups and require human intervention for timepoint data collection. These requirements limit utility in drug development where high throughput is needed or requires expensive, high-content imaging systems.5,9
Recent work has attempted to address this issue by automating organ-chip handling or miniaturizing optical components for direct integration.18,21 Integrated sensing approaches are an attractive option for non-invasive characterization, combining the benefit of direct optical sensing with continuous, automated outputs.18,25,26 Despite the advantages of fully integrated optical sensing, most approaches are not amenable to on-chip integration due to the bulky equipment required for directing beam paths or the complexity of the organ-chip system used.
We sought to develop a fiber optic-based sensing system to enable fully integrated on-chip sensing, isolating optical components from the sample of interest by transmitting signals across fiber optic cables. Optical cables allowing for bidirectional light transmission are coupled to high-powered LEDs, simultaneously delivering excitation and recording emitted wavelengths to a CMOS camera or photomultiplier tube. The system collects signal as an aggregate sum of luminescence from the excited area, significantly increasing the signal-to-noise ratio but sacrificing single-cell resolution. Our technology presents the unique opportunity to integrate optical fibers on-chip for in situ characterization of the cellular microenvironment using luminescent probes. We are the first to translate this technology to OOCs for in situ optical sensing. While other attempts have been made to enable real-time, fiber-optic-based fluorescence sensing in an organ chip, these approaches are limited to readout of oxygen concentration and pH only, using commercial systems with limited spectral range.25,26 To our knowledge, our fiber-based system is the first to enable real-time, spatiotemporal sensing across the visible luminescence spectrum in OOCs.
Physiological barriers, for example, the intestinal epithelium and vascular endothelium, selectively regulate the flux of water, ions, and nutrients while preventing the passage of bacteria and pathogens.15,27 Disruption of barrier function in vivo leads to uninhibited, non-selective transport across the barrier and is associated with the pathophysiology of human diseases, such as inflammatory bowel disease.28 In OOC systems, barrier function is commonly assessed using trans-epithelial/endothelial electrical resistance (TEER) values.29–31 TEER is traditionally measured using an applied alternating current across a monolayer to determine the total ohmic resistance. Although TEER is a convenient method for characterizing barrier function on-chip, the current density applied across the barrier is dependent on electrode spatial configuration. Unequal distribution of current density across the barrier can lead to an over or underestimation of the actual TEER value. Further, with OOC platforms varying in size, shape, and orientation, it is difficult to translate measured values across systems without significant computational effort.17
Here, we report a novel optical sensing platform for automated, non-invasive sensing of barrier function in OOCs under baseline and perturbed conditions. Specifically, we validated our photometry-based platform by monitoring epithelial and endothelial cell responses to induced inflammation on-chip. This is the first report on applying photometry-based technology to OOC, offering a novel approach to study the on-chip environment in real-time, expanding on the current capabilities of optical microscopy and end-point analyses.
Experimental
Chip fabrication
Organ chips were assembled layer-by-layer using our established laser-cut-and-assembly method, adding fiber optic insertion channels for non-invasive fluorescence sensing.32,33 Each layer's design was created using computer-aided design software (SolidWorks, Adobe Illustrator) and converted to vector format. Layers were laser cut (Epilog Zing) from transparent, cast polymethyl methacrylate (PMMA, McMaster-Carr, 8560K211, and 8560K171) sheets. Polyethylene terephthalate (PET) semi-permeable membranes (It4Ip, ipCellCulture 2000M12/620M103) with 1 μm pore size were used to support the cell population and separate the apical and basal compartments. As previously described, layers were bonded with pressure-sensitive, double-sided adhesive tape (3M, 966). The chips were built on top of a no. 1 glass coverslip base layer (Fisher, 12-541-020), which sealed the bottom of the chip. Organ chips were assembled by hand, heat pressed at 55 °C for 90 seconds under moderate pressure and then placed in a vacuum oven for at least 5 days at 50 °C to allow the adhesive tape to off-gas and fully cure. Binder clips were used to apply additional pressure to the assembled chips while baking.Fiber photometry optical setup
Our fiber photometry platform consists of two CMOS cameras capable of imaging the proximal end of fiber optic patch cables, with filters and dichroic mirrors to isolate excitation and emission wavelengths. The optical setup was engineered for simultaneous dual-channel recording, enabling continuous monitoring of apical and basal organ-chip compartments by independent, duplicate optical components to isolate channel recordings.The platform was assembled as follows, according to the direction of the beam path. A high-powered 430 nm LED (M430F1, Thorlabs), controlled by a 4-channel LED driver (DC4104, Thorlabs), was coupled to a collimator (F671SMA-405, Thorlabs) by a fiber optic patch cable (M59L01, Thorlabs). The collimator directed excitation light through an excitation filter (430/10 nm, 65-682, Edmund) held in a quick-release filter holder (SM1-QP, Thorlabs). A dichroic mirror (510 nm, 86-332, Edmund) directed excitation light through a 20x magnification objective (1-U2B225, Olympus) and onto the bundled optical fiber (BF74LS01, Thorlabs) held in a 5-axis translator (KC1T, Thorlabs). Excitation light is propagated through the bundled fiber to individual low-autofluorescence patch cables (MAF3L1, Thorlabs) connected by SMA to an FC/PC adapter (ADAFCSMA1, Thorlabs). The distal ends of the fiber patch cables were connected to a quick-disconnect ferrule (ADAF2, Thorlabs) and a fiber stub (CFMC54L10, Thorlabs), which was inserted into the chip for optical recording. Emitted fluorescence was recovered to the dichroic mirror, passing through an emission filter (520/10 nm, FB520-10, Thorlabs) held in a quick-release filter holder (SM1-QP, Thorlabs). Filtered emission fluorescence was focused onto a CMOS camera (BFS-PGE-31S4M-C, FLIR) by an achromatic doublet (AC254-035-A-ML, Thorlabs) and recorded for analysis. Additional instructions and considerations for platform assembly and use are described in ESI† Methods.
Automated image capture and recording
A 4-channel LED driver and CMOS cameras were linked to an Arduino microcontroller (Mega 2560 Rev3, Arduino) to automate image capture and recording. Square-wave TTL triggers were sent at 10 minute intervals to excite lucifer yellow on-chip and subsequently record emitted fluorescence. Each LED pulse supplied 0.2–0.3 mW cm−2 with a 200 ms pulse width. A 1 second pause between apical and basal recordings was implemented to prevent spectral overlap across channels. Although this pause time may be reduced to 100 ms for improved temporal alignment across channels (system limitations), we found that a 1 second pause sufficiently captures the dynamics of barrier transport. Captured images were automatically saved to pre-named folders on an external laptop via camera software (SpinView, FLIR).Optical readout analysis
Fiber photometry recordings were analyzed, in real-time, using custom Python code to determine relative fluorescence intensity values, following each acquired image. The Python package scikit-image (https://scikit-image.org; version 0.21.0 and 0.22.0) was used to automatically detect and draw circular regions of interest (ROI) around each illuminated fiber end and measure mean pixel intensity within each ROI. Mean intensities were used to calculate dF/F for each fiber, with timestamped values exported to a CSV file for additional analysis. Additional analysis methods are described in detail in ESI† Methods.Caco-2 culture
Caco-2 epithelial cells were obtained from the American Type Culture Collection (ATCC) and cultured in phenol-free DMEM (Corning) supplemented with 10% fetal bovine serum (Gibco), 1% GlutaMAX™ Supplement (Gibco), 1% penicillin–streptomycin (Gibco), and 10 mM HEPES (Gibco). Cells were used between passages 40–57 for all experiments.Caco-2 chip seeding
In preparation for cell seeding, chips were UV sterilized (300 mJ cm−2) for 10 minutes on top and bottom sides (Spectrolinker XL-1000, Spectronics Corporation, Westbury NY), followed by treatment with oxygen plasma for 90 seconds (Harrick Plasma PDC-001). UV-sterilized fluidic connections and tubing were connected to the chips. The apical channel was treated with 200 μL of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 v/v solution of Matrigel™ (Corning) in ice-cold DPBS (Gibco) and incubated at 37 °C for 1 hour. Caco-2 cells were harvested from flasks via treatment with 0.25% trypsin–EDTA (Gibco) for 15 minutes at 37 °C. After cell detachment, the cell solution was diluted with an equal volume of cell culture media and centrifuged at 300g for 5 minutes at room temperature. The supernatant was removed, and the pelleted cells were resuspended at 5 × 106 cells per mL in fresh cell culture media.
:10 v/v solution of Matrigel™ (Corning) in ice-cold DPBS (Gibco) and incubated at 37 °C for 1 hour. Caco-2 cells were harvested from flasks via treatment with 0.25% trypsin–EDTA (Gibco) for 15 minutes at 37 °C. After cell detachment, the cell solution was diluted with an equal volume of cell culture media and centrifuged at 300g for 5 minutes at room temperature. The supernatant was removed, and the pelleted cells were resuspended at 5 × 106 cells per mL in fresh cell culture media.
The apical and basal fluidic inlets were clamped, and 200 μL of the cell suspension was infused into the apical channel using a sterile 1 mL pipette tip. The apical fluidic outlet was clamped, and the chips were incubated at 37 °C and 5% CO2 for 2 hours to allow cell attachment. After attachment, all clamps were removed, and pre-heated cell culture medium was perfused through apical and basal channels at 3 μL min−1via a syringe pump (NE-1600, New Era Pump Systems, Inc).
Dosing the pro-inflammatory cytokine cocktail on-chip
Caco-2 were cultured on organ-chips for 7 days until a confluent, differentiated monolayer formed. Perfusion was briefly paused to exchange growth media for media containing the pro-inflammatory cytokine cocktail. Lucifer yellow (Invitrogen, L453) at a concentration of 100 μM in appropriate cell culture media was perfused at 1 μL min−1 through the apical channel only. Proinflammatory cytokines TNF-α (PeproTech), IFN-γ (PeproTech), and IL-1β (PeproTech) were added to the basal channel culture media and perfused at 1 μL min−1. Cytokine concentrations for TNF-α and IFN-γ were 200 ng mL−1 and 100 ng mL−1 for IL-1β. The cytokine cocktail was perfused for 4 days to disrupt barrier function without causing irreversible damage. After 4 days, for barrier function recovery, the apical media contained 100 μM of lucifer yellow, and basal media was growth media only.Culture of Caco-2 monolayers on semi-permeable filter inserts
PET filter inserts in a 24-well plate were coated with 200 μL of a 10% Matrigel™ solution in DPBS and incubated at 37 °C for at least 1 hour. Caco-2 cells were harvested from a sub-confluent T-75 flask and processed as described for organ-chip seeding. The resulting cell suspension was diluted to a final concentration of 4 × 105 cells per mL. The Matrigel™ solution was removed, and 200 μL of cell suspension (80000 cells) was seeded on each filter insert. Culture media was exchanged after 6 hours to remove non-adhered cells and prevent cell stacking. Media was exchanged every 2–3 days until day 21. Inflammation was induced on day 21 by dosing with our pro-inflammatory cytokine cocktail. Briefly, media was removed, and monolayers were washed with DPBS for 5 minutes at room temperature. After washing, monolayers were dosed basally with media containing 200 ng mL−1 TNF-α, 200 ng mL−1 IFN-γ, and 100 ng mL−1 IL-1β.
HUVEC culture
Human umbilical vein endothelial cells (HUVECs) were obtained from the American Type Culture Collection (ATCC) and cultured in Vascular Cell Basal Medium (ATCC) supplemented with Endothelial Cell Growth Kit-BBE (ATCC) and 1% penicillin–streptomycin (Gibco). Cells were used between passages 4–7 for chip experiments and passage 4 for filter insert experiments.HUVEC chip seeding
Chips were prepared for seeding as previously described. HUVECs were harvested from flasks via treatment with 0.25% trypsin–EDTA (Gibco) for 10 minutes at 37 °C. After cell detachment, the cell solution was diluted with an equal volume of cell culture media and centrifuged at room temperature at 200g for 5 minutes. The supernatant was removed, and the pelleted cells were resuspended at 7.5 × 106 cells per mL in fresh cell culture media. Cells were seeded on-chip as previously described and perfused through apical and basal channels at 3 μL min−1.Dosing the TNF-α on-chip
HUVECs were cultured on organ-chips under 3 μL min−1 perfusion for 3 days until a confluent monolayer formed. Briefly, apical media was exchanged for 100 μM lucifer yellow media, and basal media was dosed with 100 ng mL−1 of TNF-α. Chips were perfused at 1 μL min−1 for 2 days to disrupt barrier function.Culture of HUVEC monolayers on semi-permeable filter inserts
PET filter inserts in a 24-well plate were coated with 200 μL of a 10% Matrigel™ solution in DPBS and incubated at 37 °C for at least 1 hour. HUVECs were harvested from a sub-confluent T-75 flask and processed as described for organ-chip seeding. The resulting cell suspension was diluted to a final concentration of 4 × 105 cells per mL. The Matrigel™ solution was removed, and 200 μL of cell suspension (80000 cells) was seeded on each filter insert. Media was exchanged every 2 days until a confluent monolayer was observed. Inflammation was induced by dosing with 100 ng mL−1 TNF-α.
Real-time barrier function monitoring
Fluorescence intensity in apical and basal channels was recorded via fiber photometry to track barrier function. Fluorescence intensity measurements were collected automatically from inserted optical fibers every 10 minutes during induced inflammation and recovery on-chip.Lucifer yellow permeability assays
For on-chip experiments, apical and basal effluent were collected every 24 hours and stored at 4 °C until analysis. All effluent samples were analyzed together to avoid temperature-dependent effects on measured fluorescence. Samples were analyzed via fluorescence plate reader (Synergy HT, Bio-Tek) with 485/20 excitation 528/20 emission filter set and gain set to 50.Monolayers on permeable filter supports were washed with DPBS for 5 minutes at room temperature. After washing, pre-heated phenol-free media was added to the basal compartment, and a solution of 100 μM lucifer yellow in media was added to the apical compartment (pH = 7.2). Monolayers were incubated at 37 °C for 3 hours. As previously described, samples from apical and basal compartments were collected and analyzed by a plate reader.
Immunocytochemistry
Samples were fixed with 4% paraformaldehyde in DPBS for 10 minutes at room temperature. After fixing, the solution was removed, and cells were washed three times with DPBS. Cells were permeabilized with 0.1% Triton X-100 in DPBS for 15 minutes, followed by another three DPBS washes. Samples were blocked with filtered 4% goat serum solution in DPBS overnight at 4 °C. Zonula occludens-1 (1:100, ZO-1, Invitrogen, 40-2200) primary antibodies in 4% goat serum were added to the samples, incubated overnight at 4 °C, and carefully rinsed with DPBS three times. Phalloidin Alexa Fluor 647 (1:1000, Invitrogen A22287) and DAPI (1:1000, Invitrogen, D1306) were diluted in 4% goat serum, incubated at room temperature for 2 hours, and carefully rinsed three times with DPBS. Samples were imaged immediately or stored in parafilm and aluminum foil at 4 °C.
For chips, monolayers were stained and imaged for tight junction protein expression to assess barrier function qualitatively. All steps were conducted via a syringe pump at a flow rate of 5 μL min−1 to minimize damage to the monolayer. Samples were fixed by perfusing 4% paraformaldehyde in DPBS for 30 minutes and permeabilized with 0.1% Triton X-100 in DPBS for 20 minutes at room temperature. Monolayers were blocked under perfusion with 4% goat serum in DPBS for 2 hours at room temperature. Chip tubing ends were clipped, all chips were covered with foil, and incubated in the fridge overnight at 4 °C. Following incubation, an antibody cocktail containing 1:200 ZO-1 conjugated antibody (Invitrogen, MA3-39100-A647), 1:1000 phalloidin (Invitrogen, A12379), and 1:1000 DAPI (Invitrogen, D1306) in 4% goat serum was perfused at 5 μL min−1 for 2 hours at room temperature, followed by static incubation overnight at 4 °C. Monolayers were washed with DPBS for at least 6 hours at 5 μL min−1 and imaged immediately.
Dual-color diffusion assay
The fiber photometry platform was engineered to enable sensing of fluorescein (494 ex./521 em.) and either Texas Red (595 ex./615 em.) or Alexa Fluor 647 (650 ex./668 em.) labelled dextrans. Optical components were exchanged for appropriate wavelength range delivery and recovery to enable dual-color sampling. A 470 nm LED (M470F4, Thorlabs) and 483/31 nm excitation filter (67-028, Edmund) delivered excitation light for fluorescein dextran. Either a 595 nm LED (M595F2, Thorlabs) and 590/10 nm excitation filter (FBH590-10, Thorlabs) or 625 nm LED (M625F2, Thorlabs) and 630/10 nm excitation filter (FBH630-10, Thorlabs) was used for Texas Red or Alexa Fluor 647 labelled dextrans, respectively. Dichroic mirrors (510 nm (86-332, Edmund) and either 552 nm (86-334, Edmund) or 660 nm (67-084, Edmund)) directed excitation light on-chip and emitted light to CMOS cameras.Dextrans were dosed apically onto untreated chips at a concentration of 25 μM using two combinations, varying size or charge. For varied charges, Texas Red (40 kDa, neutral, Invitrogen D1829) and fluorescein (40 kDa, anionic, Invitrogen D1844) were dosed. For varied sizes, Alexa Fluor 647 (10 kDa, anionic, Invitrogen D22914) and fluorescein (70 kDa, anionic, Invitrogen D1823) were dosed. The fluorescence intensity of each dextran was tracked by fiber photometry on-chip under static conditions for 48 hours.
The apparent permeability (Papp) of each fluorescent dextran was calculated using the following equation

Statistical analysis
Data are presented as mean ± standard deviation unless otherwise specified. All statistical analyses were conducted using GraphPad Prism 9 (GraphPad Software). One-way ANOVA with Tukey's multiple comparisons was applied, and p-values of <0.05 were considered significant. All chip experiments were reproduced at least three times across independent groups to confirm reliability.Results and discussion
Fiber photometry platform and optical fiber integration
Our previously established layer-by-layer gut-on-a-chip32,33 was adapted to add fiber optic insertion channels to the upper and lower fluidic layers (Fig. 1A and C). The 500 μm wide fiber channels were positioned at the outlets to detect the exit concentration of fluorophore from each fluidic layer. A 500 μm thick PMMA wall separates the fluidic channel from the fiber insertion channel for complete separation of the optical components from the culture volume. With the terminal end of the fiber optic cable in proximity (∼500 μm or less) to the culture volume of interest, excitation and emission luminescence may be sensed with a high signal-to-noise ratio, offering non-invasive delivery and collection of light with 92% visible light transmission.35 10 mm long fiber optic stubs were inserted into the channels for non-invasive sensing, delivering excitation light to the culture volume and receiving emitted light (Fig. 1B). Preliminary design testing found that orienting the fiber stubs perpendicular to the side of the culture flow channels was most effective when sensing both apical and basal fluorescence at the outlet of the chip without channel crosstalk. Other design configurations, such as positioning the excitation from the top-down, offered reasonable sensitivity despite increased measurement variability by attenuation through the cell monolayer and membrane. This optical sensing approach may be easily adapted to additional organ-chip designs and orientations, enabling optical sensing across numerous OOC models. Most OOC are fabricated from optically transparent materials for microscopy, commonly poly-dimethyl siloxane (PDMS) or thermoplastics, allowing direct adaptation to a photometry-based sensing approach. This approach enables the sensing components (ferrule coupled fiber stubs) to be reused over long lifespans and rapidly swapped between OOCs compared to embedded electrodes. | ||
| Fig. 1 Optical sensing organ-on-a-chip system. A) A fabricated chip with apical and basal fiber optical ports indicated. B) An assembled chip with optical fibers inserted for noninvasive real-time sensing. Both apical and basal fibers are illuminated with 470 nm light to exhibit the excitation volume. C) An exploded view of each layer used to fabricate platform. Optical fibers may be inserted in the apical and/or basal flow layer without disrupting cell culture or flow, as shown. | ||
Sensing setup and automation
The fiber photometry platform utilized for barrier sensing was assembled as a mirror image, enabling independent sensing of upper and lower fluidic compartments of each OOC (Fig. 2A and S1†)—bundled fiber optic cables allowed for sensing from up to 7 OOC simultaneously. Three OOCs were cultured concurrently (1 control, 2 experimental). For multiplexed sensing, excitation light for both λs was directed through the same fiber with a 3 second delay between channels to avoid spectral overlap. Collected emission was directed to independent cameras for λ-specific signal analysis (Fig. S2†). For both barrier sensing and multiplexed sensing, fiber optic cables were routed into an incubator at 37 °C and 5% CO2 for non-invasive sensing from up to three OOCs simultaneously (Fig. 2B).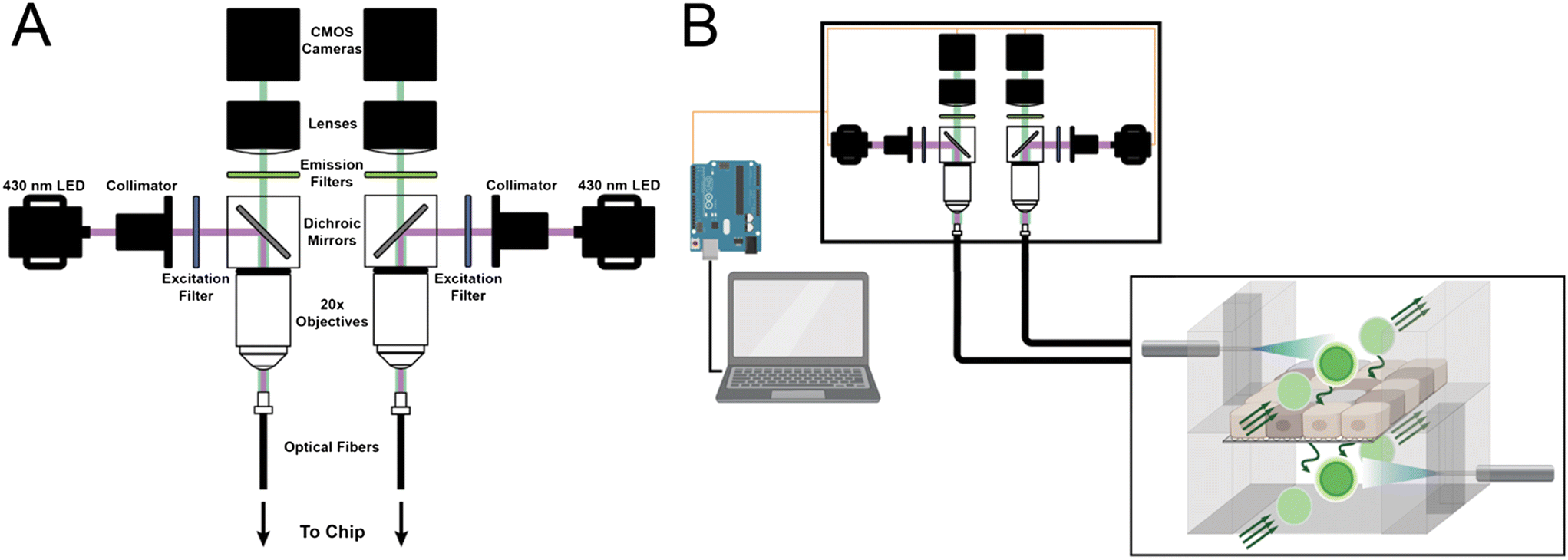 | ||
| Fig. 2 Fiber photometry system for optical sensing on-chip. A) Components consist of high-powered LEDs, optical filters, fiber optic cables, and CMOS cameras that simultaneously deliver excitation light and record emission from fluorophores on-chip. B) A laptop and Arduino microcontroller are used to automate LED power and image capturing. Fiber optic cables transmit excitation and emission wavelengths with minimal loss from within the incubator to the external hardware. | ||
To ensure cross-chip consistency, the intensity of light emitted from each fiber end was measured at 400 mA LED power, which was used for all sensing experiments. The measured intensity was 0.25 ± 0.04 mW cm−2, with a maximum difference of 20.3% from the mean intensity (Fig. S3†). Limiting intensity variation across individual fibers improves sensitivity and consistency when measuring across multiple OOCs since the excitation power is directly proportional to the collected emission signal. Maintaining output intensities within a reasonable range narrows the emission signal, allowing for optimization of the signal-to-noise ratio when first implementing the platform. In our experience, large variations in excitation power across fibers (>50%) may saturate some channels before acceptable signal-to-noise is achieved for other channels.
The platform was connected to an Arduino microcontroller to automate and synchronize image capture with fluorophore excitation. Our custom Arduino program sent 200 ms LED excitation pulses on-chip every 10 minutes to capture time-dependent diffusion data with minimal photobleaching. Timestamped images were automatically captured by a TTL trigger sent to the CMOS camera during excitation. Microcontroller-aligned LED and camera triggers enabled completely hands-free data collection of on-chip data throughout complete experimental timelines, requiring only initial setup and baseline image collection by hand. Our compact setup, consisting of the photometry optical equipment, an Arduino microcontroller, and a laptop, enables sensing on-chip in a small footprint essential for research labs with limited free space.
A preliminary calibration curve was constructed by dosing known concentrations of lucifer yellow in DPBS on blank chips to determine the relative sensing range of our optical sensing platform. Emitted fluorescence was recorded by our optical sensing platform and normalized to blanks chips containing only DPBS. The resulting calibration curve shows a robust linear relationship between fluorophore concentration and measured fluorescence, capable of detection to as low as 1 μM on-chip (R2 = 0.998, Fig. S4†).
Inflammation-induced disruption and recovery of Caco-2 cells on-chip
To establish the resolution of our system to measure small changes in barrier integrity, we aim to mimic an inflamed gut. A pro-inflammatory cytokine cocktail was added to Caco-2 monolayers, first on semi-permeable inserts, to increase permeability without fully disrupting the cell population. Caco-2 monolayers grown for at least 21 days on filter inserts were subjected to TNF-α (200 ng mL−1), IFN-γ (200 ng mL−1), and IL-1β (100 ng mL−1) for 4 days. Cytokines were removed, and monolayers were allowed to recover for up to 5 days. Compared to control monolayers on the same day of culture, monolayers exposed to the cytokine cocktail showed a significant (p = 0.0003) increase in permeability on day 4 of inflammation and significant (p = 0.0231) recovery of barrier function 2 days after cytokine removal (Fig. 4B).The described cytokine concentrations were used for inflammation of Caco-2 on-chip. Caco-2 cells were cultured on-chip for 7 days under continuous perfusion at 3 μL min−1 (0.076 dyne per cm2), mimicking physiological shear stress in the intestine.3,36 Our previous work has demonstrated epithelial maturity on-chip at this timepoint. We then dosed a pro-inflammatory cytokine cocktail composed of TNF-α (200 ng mL−1), IFN-γ (200 ng mL−1), and IL-1β (100 ng mL−1) to induce inflammation on-chip (Fig. 3A and B).
 | ||
| Fig. 3 Experimental overview for real-time barrier function sensing on-chip. A) Fiber optic cables detect fluorescence from apical and basolateral chambers in our gut-on-a-chip. Inflammation was induced in mature epithelial monolayers (left), resulting in barrier disruption and increased fluorophore transport (right). B) Timeline for cytokine-induced inflammation in our gut-on-a-chip. Cells formed a mature monolayer after 7 days of culture under continuous flow. On day 0, the paracellular permeability marker lucifer yellow was added to apical media and proinflammatory cytokines (TNF-a, IFN-g, IL-1b) were dosed via basal media. Barrier function was tracked by our sensing platform. | ||
Barrier function was continuously assessed by our novel sensing platform and by daily collection of chip effluent (Fig. 4A). Comparison of sensed and sampled fluorescence shows good agreement, with similar response time to the addition and removal of the pro-inflammatory cocktail (Fig. 4C). The mean increase in barrier permeability across experimental replicates (n = 5) is relatively consistent despite some distinct peaks caused by effluent sampling and syringe exchange on day 4 (Fig. 4D). Our sensing platform is robust and capable of automated data collection over at least 9 days with consistent outputs from independent experimental replicates.
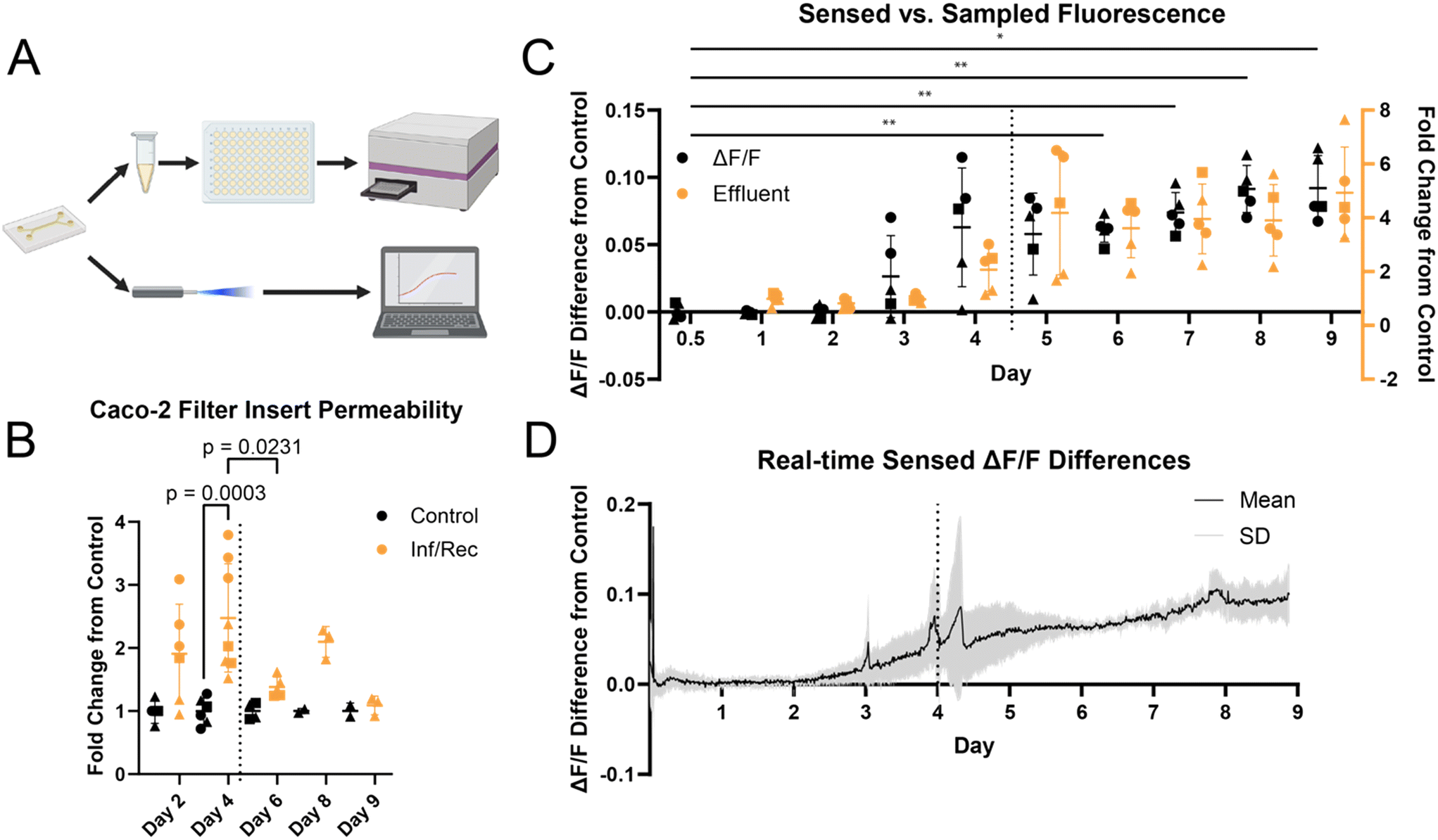 | ||
| Fig. 4 A) Graphical description of effluent- and photometry-based barrier function assays. Effluent was collected from chips daily and fluorescence was measured via plate reader. Fiber photometry readings were collected automatically by our novel platform. B) Relative permeability of Caco-2 monolayers exposed to the cytokine cocktail (TNF-a, IFN-g, IL-1b) on day 0. Dotted vertical line represents the removal of cytokine cocktail. One-way ANOVA with Tukey's multiple comparisons. Circles, squares, and triangles represent biological replicates 1, 2, and 3, respectively. C) Comparison of barrier function characterization methods. dF/F (black) sensed by photometry shows a similar time course increase in barrier permeability compared to effluent (orange). One-way ANOVA with Tukey's multiple comparisons (*p < 0.05, **p < 0.01). n = 3, m = 3 control, 5 experimental replicates. Circles, squares, and triangles represent biological replicates 1, 2, and 3, respectively. Dotted vertical line represents the removal of the cytokine cocktail. D) Basal sensed fluorescence difference from control sensed by fiber photometry during Caco-2 monolayer inflammation and recovery. Sensed fluorescence intensity trends increasing compared to controls as exposure time to the cytokine cocktail increases. Data is shown as mean (black) ± standard deviation (grey). n = 3, m = 3 control, 5 experimental replicates. Dotted vertical line represents the removal of the cytokine cocktail. | ||
Further, data collected using our platform enabled real-time readout of barrier function over the 9 day experimental time course. Real-time, optical readout of barrier function in OOC has not previously been reported. Compared to daily effluent sampling, we significantly increased the temporal resolution of cellular responses to induced inflammation on-chip, observing a continuous, gradual increase in barrier permeability earlier than daily effluent sampling (Fig. 4C). Further, a change in permeability is observed as early as shortly after the day 2 sampling timepoint (Fig. 4D), while no discernible change in permeability presents till day 4 for effluent sampling. Application of this approach to time-dependent assays on shorter experimental timescales may significantly improve temporal resolution, increase sensitivity, and data quality.
Effluent and sensed barrier function corroborate that the Caco-2 monolayer underwent inflammation in response to the dosed cytokine cocktail. Still, the level and kinetics of recovery did not match that observed in filter inserts; however, prolonged barrier integrity disruption measured on-chip, effluent, and optical measures matched. Qualitatively, immunofluorescence imaging suggests that the barrier had recovered partially, indicated by qualitative reorganization of ZO-1 expression to cell–cell junctions (Fig. 5).
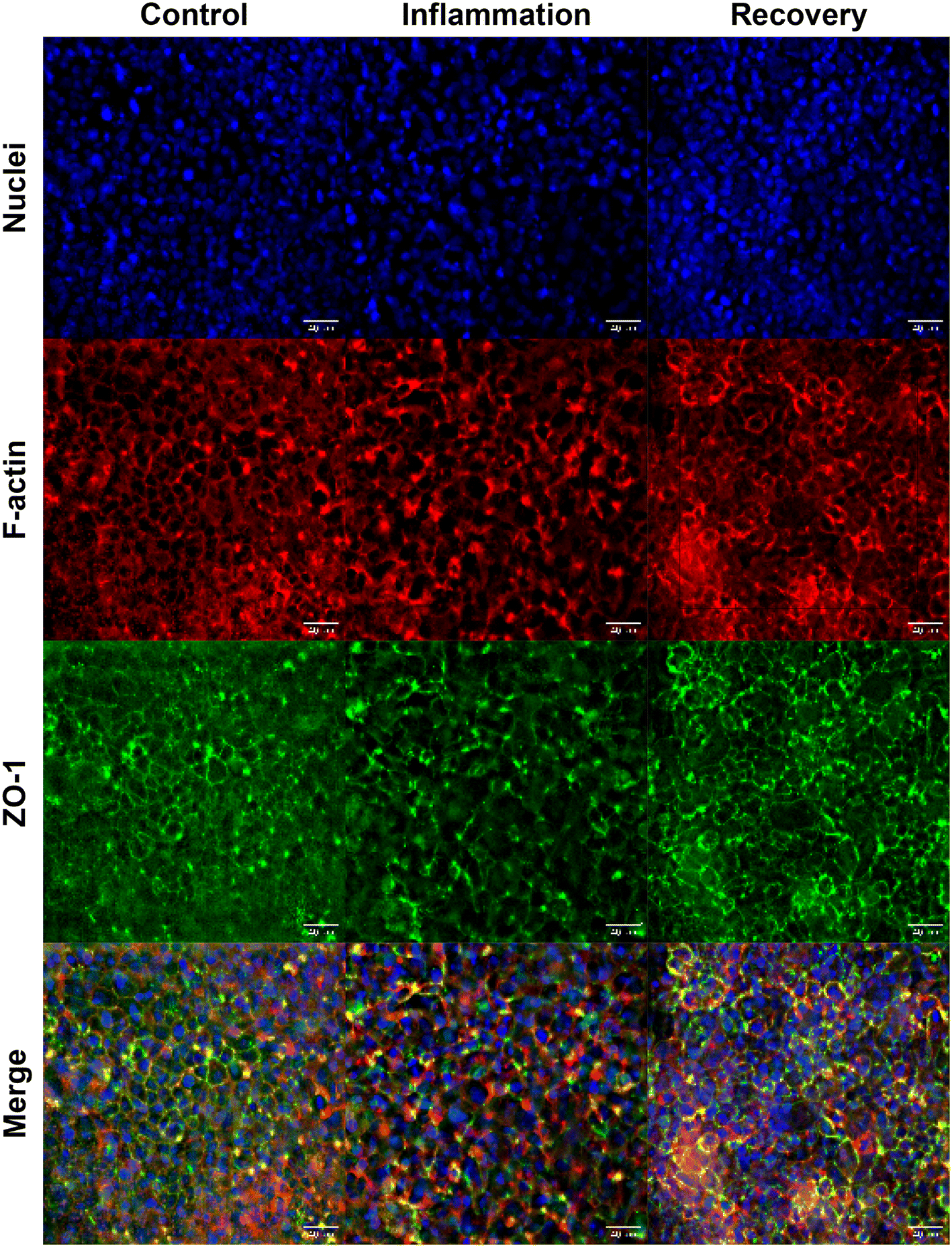 | ||
| Fig. 5 Representative immunocytochemistry results on-chip from control (left column), 96 hour cytokine exposure (center column), and 120 hour recovery (right column) groups. Comparison of tight-junction expression (ZO-1) shows more diffuse expression in inflamed monolayers compared to control and recovery. Images taken at 40× magnification, maximum intensity projection of 10-slice z-stack. | ||
Quantitative analysis of collected effluent and sensed data show that barrier permeability was significantly increased compared to controls from day 6 until the experiment ended. The absence of native stem cells and immune cells in this gut-on-a-chip model may prevent complete recovery from induced inflammation.37–39 Our lab has previously demonstrated that Caco-2 cells cultured on our gut-on-a-chip differentiate in 5 days, exhibiting polarization and brush borders comparable to mature epithelium.30,32 Continuous perfusion provides physiological shear stress, continuous nutrient supply, and waste removal, whereas static cultures receive fresh media intermittently. Mechanical shear stress may influence proliferation and the restructuring of tight-junctions, resulting in the differences in recovery observed across filter insert and on-chip systems (Fig. 4B and C).40–42 After cytokine-induced inflammation of differentiated Caco-2 monolayers, tight junctions must undergo replication and reorganization to return to a healthy, functional morphology.40,41 Caco-2 cultured on filter inserts may high a higher propensity to proliferate and restructure more rapidly compared to on-chip due to the lack of fluidic shear stress, as reflected in our experiments. Although previous work indicates that tight-junction reorganization and recovery on-chip is possible in less than 24 hours, these studies induced acute barrier dysfunction by the chelating agent EGTA.43,44 To our knowledge, we are the first to report on the recovery response of Caco-2 on-chip following chronically induced barrier inflammation.
Additionally, excess cytokines may have remained present during the recovery phase due to the diffusion-dependent clearing of cytokines from the on-chip microenvironment. Unlike typical culture approaches that allow washing the monolayer, the cytokines could not be thoroughly flushed from the system due to the risk of destroying the fragile, inflamed monolayer under higher shear stress. The differences between filter inserts and on-chip experiments further support this hypothesis: monolayers cultured on filter inserts showed significant recovery only 48 hours after cytokine removal. In comparison, the OOC monolayers showed no significant decrease in permeability after 96 hours (Fig. 4B and C).
A shift from increasing barrier permeability to constant, elevated permeability was observed following the removal of the cytokine cocktail, demonstrating the capability of this platform to observe the transient shift in cell response with high resolution. Of particular importance is that the fiber photometry platform aligns well with offline effluent analysis, the current standard approach. The platform may more accurately align temporally observed outputs with accurate on-chip cellular responses. Collection of effluent from OOC typically requires hours due to the low volumetric flow rates used during culture. Standard analysis equipment, such as fluorescence plate readers, usually needs at least 50 μL for reliable reading from a standard 96-well plate.45,46
Inflammation of HUVECs on filter inserts
To assess the inflammation response of HUVECs, we dosed TNF-α at 100 ng mL−1 on HUVEC monolayers grown on semi-permeable filter inserts. Significant barrier inflammation was observed by lucifer yellow permeability assay at 1 day (p = 0.0002) and 2 days (p < 0.0001) post-dose (Fig. S5†). Morphological changes in monolayer structure were observed by immunocytochemistry, showing elongation of actin filaments in response to inflammation. Additionally, the expression of tight-junction protein ZO-1 is more diffuse at cell borders compared to controls (Fig. S6†).Inflammation of HUVECs on-chip
To demonstrate the broad applicability of our novel sensing platform to cell agnostic barrier function assays, we extended our investigation to include sensed barrier formation and inflammation of HUVECs cultured on our OOC. Our established gut-on-a-chip was adapted to culture HUVECs under continuous perfusion, highlighting the versatility of our layer-by-layer design. HUVECs have been used to mimic the endothelial barrier of human vasculature, and chronic vascular inflammation is a risk factor for hypertension, kidney disease, and atherosclerosis.47–50 Continuous monitoring of endothelial barrier function may help elucidate the transient effects of pro-inflammatory cytokines with higher granularity compared to standard trans-endothelial electrical resistance or permeability assays.Our platform tracked HUVEC monolayer formation and the time-dependent increase in barrier permeability in response to dosed TNF-α (Fig. 6A and B). Significant increases in sensed fluorescence compared to control samples were observed at 24 hours (p = 0.0357) and 48 hours (p = 0.0072) after dosing TNF-α on-chip. Similar to results obtained for the inflammation and recovery of Caco-2 on a chip, a comparison of sensed and sampled fluorescence shows identical response time to the addition of TNF-α (Fig. 6A). Immunofluorescence shows more diffuse expression of ZO-1 in HUVEC monolayers exposed to TNF-α for 48 hours compared to controls cultured for the same period, qualitatively supporting the disruption of barrier function due to induced inflammation (Fig. S7†).
 | ||
| Fig. 6 A) Comparison of barrier function characterization methods for HUVECs on-chip. dF/F (black) sensed by photometry shows a similar time course increase in barrier permeability compared to effluent (orange). One-way ANOVA with Tukey's multiple comparisons (*p < 0.05, **p < 0.01). n = 3, m = 3 control, 6 experimental replicates. Circles, squares, and triangles represent biological replicates 1, 2, and 3, respectively. Dotted vertical line represents the addition of TNF-a. B) Basal sensed fluorescence difference from control sensed by fiber photometry during HUVEC monolayer formation and inflammation. Sensed fluorescence intensity increases compared to controls as exposure time to the cytokine cocktail increases. Data is shown as mean (black) ± standard deviation (grey). n = 3, m = 3 control, 6 experimental replicates. Dotted vertical line represents the addition of TNF-a. | ||
The photometry platform is adaptable for barrier function from multiple barrier-forming cell types. Importantly, our sensing platform required no change or optimization when transitioning from Caco-2 cells to HUVECs. This technology is easily integrated into our laser cut and assemble method for fabricating OOCs. This method keeps the manufacturing costs of culture platforms down and allows fiber stubs to be swapped from one experimental OOC to another without signal degradation. Sensing is based on detected luminescence and is not dependent on the particular cell type used; however, some troubleshooting may be required for OOC systems with particularly high flow rates or fluidic channels susceptible to occlusion with cell debris or other obstructions.
Multiplexed sensing
Barrier function is commonly characterized with TEER. Investigating passive vs. active transport or permeability-specific analytes in vitro cannot be accomplished with TEER. To demonstrate the versatility of our novel on-chip sensing platform in combating these challenges, we investigated the diffusion rate of fluorescently labelled dextrans across our OOC's semi-permeable membrane. The platform was adapted to enable dual-sensing, simultaneously collecting fluorescence from two distinct fluorophores present on-chip. Specifically, the chosen dextrans differed either in size (10 kDa vs. 70 kDa) or in net charge (neutral vs. anionic) to detect the dependency of tracer structure on observed transport.Our sensing platform enabled direct estimation of dextran apparent permeability in our OOC system. When tracked simultaneously, the 10 kDa (2.45 × 10−4 cm s−1) dextran had a higher apparent permeability compared to the 70 kDa dextran (1.62 × 10−4 cm s−1). As expected, the larger 70 kDa dextran diffused slower through the on-chip membrane, as shown by a gradual increase in sensed fluorescence (Fig. 7A). The net charge had no discernible effect on apparent permeability across neutral (2.08 × 10−4 cm s−1) and anionic (2.09 × 10−4 cm s−1) dextrans of equivalent size (Fig. 7B).
 | ||
| Fig. 7 Optical sensing allows real-time multiplexing of fluorescently tagged analyte. A) Size-dependent diffusion rate of fluorescently tagged dextran sensed simultaneously. A 10 kDa dextran (tagged with Alexa Fluor 647) diffused more rapidly through the semi-permeable membrane compared to the 70 kDa dextran (tagged with fluorescein). B) Charge-dependent diffusion rate of dextran of similar size (neutral: Texas Red; anionic: fluorescein). | ||
Although no differences were observed in our empty OOC system, the presence of binding sites with high ionic affinity in other biological systems may influence diffusion kinetics, revealing system-specific transport differences.51 By utilizing our platform for multiplexed sensing, observed differences in diffusive rate kinetics may help elucidate process-specific transport in biological systems, especially where both modes of transport are of interest. For example, transport across the blood–brain barrier (BBB) has been extensively studied on-chip, where both passive and active transport influence observed barrier transport.52–56 Genetic and chemical tools may be further leveraged to monitor the transport of specific analytes, data required to yield insight into pharmacokinetics in vitro.
Additionally, the hardware and utility applied to enable simultaneous, dual-channel sensing may be readily adapted to achieve multi-channel luminescence sensing, expanding on the technology demonstrated in this work. Our dual-color sensing platform recorded apical and basal channel fluorescence from a single camera, offering dual-channel detection of two distinct fluorophores. While this approach utilized only two of the seven available fibers, the described platform may be expanded to collect independent spatiotemporal luminescence from up to seven total channels in OOCs, offering multi-channel capabilities beyond the demonstrated dual-channel recordings.
Our fiber optic-based platform has the potential to sense and record any luminescence, provided the appropriate excitation and emission waveguides are implemented.57 We have demonstrated our platform's versatility by sensing from four different fluorophores (lucifer yellow, fluorescein, Texas Red, Alexa Fluor 647) in this manuscript. By simple exchange of LEDs, filters, and optical waveguides, we may adapt the platform to sense across the visible range, allowing for direct use with commercial probes. As a result, our sensing capabilities may be broadened to applications beyond barrier function, such as fluorescent protein expression or calcium sensing.
Further, our system has the potential for high-throughput, real-time readouts of luminescence intensity for temporal tracking of cellular responses from multiple OOCs simultaneously. We have demonstrated our platform's capability for multi-OOC sensing by monitoring barrier functions in three OOCs simultaneously. Although our platform may be capable of sensing from up to seven OOCs simultaneously using commercially available fiber bundles, our throughput is inherently limited to three OOCs by syringe-driven physiological flow in our gut-on-a-chip. Bulky syringe pumps and the routing of fluidic tubing introduce significant barriers to OOC throughput, limiting available incubator space while introducing multiple rate limiting steps in culture setup. With ongoing advancement in fluidic handling for OOCs, we hope to significantly expand our sensing throughput to approach levels comparable to effluent sampling. A recent demonstration of similar technology in freely moving animals has shown that high-density recording of transient calcium flux in up to 48 individual brain areas is possible.58 Applying custom high-density fiber arrays to organ chips may allow for whole-chip spatial sensing and high throughput data collection across numerous OOCs simultaneously, offering high-content, spatiotemporal optical sensing to address current throughput limitations.
Conclusions
In summary, we have developed a novel optical sensing platform for automated, non-invasive barrier function measurements in OOCs. The platform has been validated for sensing small changes in barrier integrity in real-time, improving on the current standard of effluent sampling and analysis or TEER measurements. Our platform does not interact directly with the on-chip sample, offering advantages over integrated electrochemical sensors susceptible to electrode fouling or site saturation. We have demonstrated that, by exchanging optical filters and dichroic mirrors, luminescence across the visible wavelength range may be measured. Further, the system may be utilized for multiplexed sensing of multiple fluorophores simultaneously, broadening applications to size- and process-specific transport across barriers.Data availability
The data supporting this article have been included as part of the ESI.† The code for data analysis and platform automation can be found at https://github.com with DOI: https://doi.org/10.5281/zenodo.14200150. The version of the code employed for this study is version 1.0.0.Author contributions
B. G. S.: conceptualization, formal analysis, investigation, methodology, writing – original draft, writing – review & editing A. N. K.: conceptualization, funding acquisition, investigation, methodology, resources, supervision, writing – review & editing R. A. K.: conceptualization, funding acquisition, investigation, methodology, resources, supervision, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Christina Aniolek for help with HUVEC culture and filter insert protocol optimization in the Department of Chemical Engineering. This work was supported by NASA (80ARC023CA005) and the National Institute of General Medical Sciences (R35GM142741).References
- C. A. Paggi, J. Hendriks, M. Karperien and S. Le Gac, Emulating the chondrocyte microenvironment using multi-directional mechanical stimulation in a cartilage-on-chip, Lab Chip, 2022, 22(9), 1815–1828 RSC.
- D. Huh, B. D. Matthews, A. Mammoto, M. Montoya-Zavala, H. Y. Hsin and D. E. Ingber, Reconstituting organ-level lung functions on a chip, Science, 2010, 328(5986), 1662–1668 CrossRef CAS.
- H. J. Kim, D. Huh, G. Hamilton and D. E. Ingber, Human gut-on-a-chip inhabited by microbial flora that experiences intestinal peristalsis-like motions and flow, Lab Chip, 2012, 12(12), 2165–2174 RSC.
- A. Marsano, C. Conficconi, M. Lemme, P. Occhetta, E. Gaudiello and E. Votta, et al., Beating heart on a chip: a novel microfluidic platform to generate functional 3D cardiac microtissues, Lab Chip, 2016, 16(3), 599–610 Search PubMed.
- J. Kieninger, A. Weltin, H. Flamm and G. A. Urban, Microsensor systems for cell metabolism–from 2D culture to organ-on-chip, Lab Chip, 2018, 18(9), 1274–1291 RSC.
- V. Allwardt, A. J. Ainscough, P. Viswanathan, S. D. Sherrod, J. A. McLean and M. Haddrick, et al., Translational roadmap for the organs-on-a-chip industry toward broad adoption, Bioengineering, 2020, 7(3), 112 Search PubMed.
- C. M. Leung, P. de Haan, K. Ronaldson-Bouchard, G.-A. Kim, J. Ko and H. S. Rho, et al., A guide to the organ-on-a-chip, Nat. Rev. Methods Primers, 2022, 2(1), 33 CrossRef CAS.
- D. Marrero, F. Pujol-Vila, D. Vera, G. Gabriel, X. Illa and A. Elizalde-Torrent, et al., Gut-on-a-chip: Mimicking and monitoring the human intestine, Biosens. Bioelectron., 2021, 181, 113156 CrossRef CAS.
- D. Caballero, R. L. Reis and S. C. Kundu, Boosting the clinical translation of organ-on-a-Chip technology, Bioengineering, 2022, 9(10), 549 CrossRef PubMed.
- A. Weltin, K. Slotwinski, J. Kieninger, I. Moser, G. Jobst and M. Wego, et al., Cell culture monitoring for drug screening and cancer research: a transparent, microfluidic, multi-sensor microsystem, Lab Chip, 2014, 14(1), 138–146 RSC.
- J. Dornhof, J. Kieninger, H. Muralidharan, J. Maurer, G. A. Urban and A. Weltin, Microfluidic organ-on-chip system for multi-analyte monitoring of metabolites in 3D cell cultures, Lab Chip, 2022, 22(2), 225–239 RSC.
- M. Oliveira, P. Conceição, K. Kant, A. Ainla and L. Diéguez, Electrochemical Sensing in 3D Cell Culture Models: New Tools for Developing Better Cancer Diagnostics and Treatments, Cancers, 2021, 13(6), 1381 Search PubMed.
- K. Nemčeková and J. Labuda, Advanced materials-integrated electrochemical sensors as promising medical diagnostics tools: A review, Mater. Sci. Eng., C, 2021, 120, 111751 CrossRef PubMed.
- J. R. McKenzie, A. M. Palubinsky, J. E. Brown, B. McLaughlin and D. E. Cliffel, Metabolic Multianalyte Microphysiometry Reveals Extracellular Acidosis is an Essential Mediator of Neuronal Preconditioning, ACS Chem. Neurosci., 2012, 3(7), 510–518 CrossRef CAS PubMed.
- J. F. Wong, M. D. Mohan, E. W. Young and C. A. Simmons, Integrated electrochemical measurement of endothelial permeability in a 3D hydrogel-based microfluidic vascular model, Biosens. Bioelectron., 2020, 147, 111757 CrossRef CAS PubMed.
- J. R. McKenzie, A. C. Cognata, A. N. Davis, J. P. Wikswo and D. E. Cliffel, Real-Time Monitoring of Cellular Bioenergetics with a Multianalyte Screen-Printed Electrode, Anal. Chem., 2015, 87(15), 7857–7864 CrossRef CAS PubMed.
- M. W. van der Helm, O. Y. Henry, A. Bein, T. Hamkins-Indik, M. J. Cronce and W. D. Leineweber, et al., Non-invasive sensing of transepithelial barrier function and tissue differentiation in organs-on-chips using impedance spectroscopy, Lab Chip, 2019, 19(3), 452–463 RSC.
- Y. S. Zhang, J. Aleman, S. R. Shin, T. Kilic, D. Kim and S. A. Mousavi Shaegh, et al., Multisensor-integrated organs-on-chips platform for automated and continual in situ monitoring of organoid behaviors, Proc. Natl. Acad. Sci. U. S. A., 2017, 114(12), E2293–E2302 CAS.
- M. Gori, M. C. Simonelli, S. M. Giannitelli, L. Businaro, M. Trombetta and A. Rainer, Investigating nonalcoholic fatty liver disease in a liver-on-a-chip microfluidic device, PLoS One, 2016, 11(7), e0159729 CrossRef PubMed.
- N. Senutovitch, L. Vernetti, R. Boltz, R. DeBiasio, A. Gough and D. L. Taylor, Fluorescent protein biosensors applied to microphysiological systems, Exp. Biol. Med., 2015, 240(6), 795–808 CrossRef CAS PubMed.
- R. Novak, M. Ingram, S. Marquez, D. Das, A. Delahanty and A. Herland, et al., Robotic fluidic coupling and interrogation of multiple vascularized organ chips, Nat. Biomed. Eng., 2020, 4(4), 407–420 CrossRef PubMed.
- T. Schulze, K. Mattern, E. Früh, L. Hecht, I. Rustenbeck and A. Dietzel, A 3D microfluidic perfusion system made from glass for multiparametric analysis of stimulus-secretioncoupling in pancreatic islets, Biomed. Microdevices, 2017, 19, 1–11 CrossRef PubMed.
- F. Mazzarda, A. D'Elia, R. Massari, A. De Ninno, F. R. Bertani and L. Businaro, et al., Organ-on-chip model shows that ATP release through connexin hemichannels drives spontaneous Ca2+ signaling in non-sensory cells of the greater epithelial ridge in the developing cochlea, Lab Chip, 2020, 20(16), 3011–3023 RSC.
- Z. Jian, H. Han, T. Zhang, J. Puglisi, L. T. Izu and J. A. Shaw, et al., Mechanochemotransduction During Cardiomyocyte Contraction Is Mediated by Localized Nitric Oxide Signaling, Sci. Signaling, 2014, 7(317), ra27 CrossRef PubMed.
- J. Grant, E. Lee, M. Almeida, S. Kim, N. LoGrande and G. Goyal, et al., Establishment of physiologically relevant oxygen gradients in microfluidic organ chips, Lab Chip, 2022, 22(8), 1584–1593 RSC.
- H. Zirath, S. Spitz, D. Roth, T. Schellhorn, M. Rothbauer and B. Müller, et al., Bridging the academic–industrial gap: application of an oxygen and pH sensor-integrated lab-on-a-chip in nanotoxicology, Lab Chip, 2021, 21(21), 4237–4248 RSC.
- M. M. France and J. R. Turner, The mucosal barrier at a glance, J. Cell Sci., 2017, 130(2), 307–314 CrossRef CAS.
- Y. B. Arık, M. W. Van Der Helm, M. Odijk, L. I. Segerink, R. Passier and A. Van Den Berg, et al., Barriers-on-chips: Measurement of barrier function of tissues in organs-on-chips, Biomicrofluidics, 2018, 12(4), 042218 CrossRef PubMed.
- M. Bartosova, D. Ridinger, I. Marinovic, J. Heigwer, C. Zhang and E. Levai, et al., An experimental workflow for studying barrier integrity, permeability, and tight junction composition and localization in a single endothelial cell monolayer: proof of concept, Int. J. Mol. Sci., 2021, 22(15), 8178 CrossRef CAS PubMed.
- I. Hubatsch, E. G. Ragnarsson and P. Artursson, Determination of drug permeability and prediction of drug absorption in Caco-2 monolayers, Nat. Protoc., 2007, 2(9), 2111–2119 CrossRef CAS PubMed.
- A. M. Ross, D. R. Walsh, R. M. Cahalane, L. Marcar and J. J. Mulvihill, The effect of serum starvation on tight junctional proteins and barrier formation in Caco-2 cells, Biochem. Biophys. Rep., 2021, 27, 101096 CAS.
- S. Hosic, A. J. Bindas, M. L. Puzan, W. Lake, J. R. Soucy and F. Zhou, et al., Rapid Prototyping of Multilayer Microphysiological Systems, ACS Biomater. Sci. Eng., 2021, 7(7), 2949–2963 CrossRef CAS PubMed.
- J. A. Hayes, A. W. Lunger, A. S. Sharma, M. T. Fernez, R. L. Carrier and A. N. Koppes, et al., Engineered bacteria titrate hydrogen sulfide and induce concentration-dependent effects on the host in a gut microphysiological system, Cell Rep., 2023, 42(12), 113481 CrossRef CAS PubMed.
- H.-H. Hsu, J.-K. Kracht, L. E. Harder, K. Rudnik, G. Lindner and K. Schimek, et al., A method for determination and simulation of permeability and diffusion in a 3D tissue model in a membrane insert system for multi-well plates, J. Visualized Exp., 2018, 132, e56412 Search PubMed.
- W. Wu, Q. Ouyang, L. He and Q. Huang, Optical and thermal properties of polymethyl methacrylate (PMMA) bearing phenyl and adamantyl substituents, Colloids Surf., A, 2022, 653, 130018 CrossRef CAS.
- J. R. Soucy, A. J. Bindas, R. Brady, T. Torregrosa, C. M. Denoncourt and S. Hosic, et al., Reconfigurable Microphysiological Systems for Modeling Innervation and Multitissue Interactions, Adv. Biosyst., 2020, 4(9), 2000133 CrossRef.
- M. Belaid, J. Javorovic, G. Pastorin and D. Vllasaliu, Development of an in vitro co-culture model using Caco-2 and J774A.1 cells to mimic intestinal inflammation, Eur. J. Pharm. Biopharm., 2024, 197, 114243 CrossRef CAS.
- R. C. van der Wath, B. S. Gardiner, A. W. Burgess and D. W. Smith, Cell Organisation in the Colonic Crypt: A Theoretical Comparison of the Pedigree and Niche Concepts, PLoS One, 2013, 8(9), e73204 CrossRef CAS.
- S. Kozar, E. Morrissey, M. Nicholson Anna, M. van der Heijden, I. Zecchini Heather and R. Kemp, et al., Continuous Clonal Labeling Reveals Small Numbers of Functional Stem Cells in Intestinal Crypts and Adenomas, Cell Stem Cell, 2013, 13(5), 626–633 CrossRef CAS PubMed.
- J. A. Espina, M. H. Cordeiro, M. Milivojevic, I. Pajić-Lijaković and E. H. Barriga, Response of cells and tissues to shear stress, J. Cell Sci., 2023, 136(18), jcs260985 CrossRef CAS.
- C.-Y. Huang, J.-K. Chen and W.-T. Kuo, Glutamine induces remodeling of tight junctions in Caco-2 colorectal cancer cell, Med. Oncol., 2022, 40(1), 32 CrossRef.
- P. Arumugam, K. Saha and P. Nighot, Intestinal Epithelial Tight Junction Barrier Regulation by Novel Pathways, Inflammatory Bowel Dis., 2025, 31(1), 259–271 CrossRef.
- D. Marrero, A. Guimera, L. Maes, R. Villa, M. Alvarez and X. Illa, Organ-on-a-chip with integrated semitransparent organic electrodes for barrier function monitoring, Lab Chip, 2023, 23(7), 1825–1834 RSC.
- E. G. B. M. Bossink, M. Zakharova, D. S. De Bruijn, M. Odijk and L. I. Segerink, Measuring barrier function in organ-on-chips with cleanroom-free integration of multiplexable electrodes, Lab Chip, 2021, 21(10), 2040–2049 RSC.
- E. Csibra and G.-B. Stan, Absolute protein quantification using fluorescence measurements with FPCountR, Nat. Commun., 2022, 13(1), 6600 CrossRef CAS PubMed.
- J. Petersen and J. Nguyen, Comparison of Absorbance and Fluorescence Methods for Determining Liquid Dispensing Precision, SLAS Technol., 2005, 10(2), 82–87 CAS.
- H. Ehlers, A. Nicolas, F. Schavemaker, J. P. M. Heijmans, M. Bulst and S. J. Trietsch, et al., Vascular inflammation on a chip: A scalable platform for trans-endothelial electrical resistance and immune cell migration, Front. Immunol., 2023, 14, 1118624 CrossRef CAS PubMed.
- A. Junaid, J. Schoeman, W. Yang, W. Stam, A. Mashaghi and A. J. van Zonneveld, et al., Metabolic response of blood vessels to TNFα, eLife, 2020, 9, e54754 CrossRef CAS PubMed.
- K. Miyazaki, K. Hashimoto, M. Sato, M. Watanabe, N. Tomikawa and S. Kanno, et al., Establishment of a method for evaluating endothelial cell injury by TNF-α in vitro for clarifying the pathophysiology of virus-associated acute encephalopathy, Pediatr. Res., 2017, 81(6), 942–947 CrossRef CAS PubMed.
- S. Huveneers, M. J. A. P. Daemen and P. L. Hordijk, Between Rho(k) and a Hard Place, Circ. Res., 2015, 116(5), 895–908 CrossRef CAS PubMed.
- S. B. Hladky and M. A. Barrand, Fluid and ion transfer across the blood–brain and blood–cerebrospinal fluid barriers; a comparative account of mechanisms and roles, Fluids Barriers CNS, 2016, 13(1), 19 CrossRef PubMed.
- J. A. Brown, S. G. Codreanu, M. Shi, S. D. Sherrod, D. A. Markov and M. D. Neely, et al., Metabolic consequences of inflammatory disruption of the blood-brain barrier in an organ-on-chip model of the human neurovascular unit, J. Neuroinflammation, 2016, 13(1), 1–17 CrossRef PubMed.
- J. A. Brown, V. Pensabene, D. A. Markov, V. Allwardt, M. D. Neely and M. Shi, et al., Recreating blood-brain barrier physiology and structure on chip: A novel neurovascular microfluidic bioreactor, Biomicrofluidics, 2015, 9(5), 054124 CrossRef PubMed.
- S. I. Ahn, Y. J. Sei, H.-J. Park, J. Kim, Y. Ryu and J. J. Choi, et al., Microengineered human blood–brain barrier platform for understanding nanoparticle transport mechanisms, Nat. Commun., 2020, 11(1), 175 CAS.
- S. Jeong, S. Kim, J. Buonocore, J. Park, C. J. Welsh and J. Li, et al., A three-dimensional arrayed microfluidic blood–brain barrier model with integrated electrical sensor array, IEEE Trans. Biomed. Eng., 2017, 65(2), 431–439 Search PubMed.
- C. E. Staicu, F. Jipa, E. Axente, M. Radu, B. M. Radu and F. Sima, Lab-on-a-chip platforms as tools for drug screening in neuropathologies associated with blood–brain barrier alterations, Biomolecules, 2021, 11(6), 916 CrossRef CAS.
- A. Formozov, A. Dieter and J. S. Wiegert, A flexible and versatile system for multi-color fiber photometry and optogenetic manipulation, Cells Rep. Methods, 2023, 3(3), 100418 CrossRef PubMed.
- Y. Sych, M. Chernysheva, L. T. Sumanovski and F. Helmchen, High-density multi-fiber photometry for studying large-scale brain circuit dynamics, Nat. Methods, 2019, 16(6), 553–560 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4lc01090f |
| This journal is © The Royal Society of Chemistry 2025 |