
LEADS – a comprehensive human liver-on-a-chip for non-alcoholic steatohepatitis (NASH) drug testing†
Gowri Manohari
Balachander
 ab,
Inn Chuan
Ng
b,
Roopesh R.
Pai‡
bc,
Kartik
Mitra‡
b,
Farah
Tasnim§
d,
Yee Siang
Lim§
e,
Royston
Kwok
e,
Yoohyun
Song
b,
Lai Ping
Yaw
e,
Clarissa Bernice
Quah
b,
Junzhe
Zhao
f,
Wahyunia L.
Septiana
g,
Vishnu Goutham
Kota
b,
Yao
Teng
b,
Kexiao
Zheng
h,
Yan
Xu
b,
Sei Hien
Lim
i,
Huck Hui
Ng
ejkl and
Hanry
Yu
*bmn
ab,
Inn Chuan
Ng
b,
Roopesh R.
Pai‡
bc,
Kartik
Mitra‡
b,
Farah
Tasnim§
d,
Yee Siang
Lim§
e,
Royston
Kwok
e,
Yoohyun
Song
b,
Lai Ping
Yaw
e,
Clarissa Bernice
Quah
b,
Junzhe
Zhao
f,
Wahyunia L.
Septiana
g,
Vishnu Goutham
Kota
b,
Yao
Teng
b,
Kexiao
Zheng
h,
Yan
Xu
b,
Sei Hien
Lim
i,
Huck Hui
Ng
ejkl and
Hanry
Yu
*bmn
aSchool of Biomedical Engineering, Indian Institute of Technology (BHU) Varanasi, Varanasi-221005, India
bDepartment of Physiology, The Institute for Digital Medicine (WisDM), Yong Loo Lin School of Medicine, MD9-04-11, 2 Medical Drive, Singapore 117593, Singapore
cBioprinting Lab, Department of Dermatology, Dr. D.Y. Patil Medical College, Hospital & Research Centre, Sant Tukaram Nagar, Pimpri, Pune, Maharashtra, India
dBiomedical Sciences Industry Partnership Office (BMSIPO), A*STAR, 31 Biopolis Way, 138669, Singapore
eGenome Institute of Singapore, Agency for Science, Technology and Research (A*STAR), Singapore
fDuke-NUS Medical School, 8 College Road, Singapore 169857, Singapore
gDepartment of Histology, Faculty of Medicine, Gunadarma University, Depok, Indonesia
hNano-Bio-Chem Centre and Organoid Innovation Center, Suzhou Institute of Nano-tech and Nano-bionics, Chinese Academy of Sciences, 398 Ruoshui Road, Suzhou 215123, China
iAIM Biotech Pte. Ltd., 21 Biopolis Road, #01-24 Nucleos, 138567, Singapore
jDepartment of Biological Sciences, National University of Singapore, Singapore
kDepartment of Biochemistry, Yong Loo Lin School of Medicine, National University of Singapore, Singapore
lSchool of Biological Sciences, Nanyang Technological University, Singapore
mMechanobiology Institute, National University of Singapore, T-Lab, #05-01, 5A Engineering Drive 1, Singapore 117411, Singapore. E-mail: phsyuh@nus.edu.sg
nCAMP, Singapore-MIT Alliance for Research and Technology, 1 CREATE Way, Level 4 Enterprise Wing, Singapore 138602, Singapore
First published on 14th May 2025
Abstract
Metabolic dysfunction associated steatohepatitis (MASH), also known as non-alcoholic steatohepatitis (NASH), is a progressive form of steatotic liver disease (SLD). It is an emerging healthcare threat due its high prevalence, accelerated and non-linear progression, and final culmination as decompensated liver failure and/or hepatocellular carcinoma (HCC). The pathogenesis of NASH is complex with strong ethnic influences and genetic predispositions, underscoring the need for preclinical models that utilize patient-derived cells to enhance our understanding of the disease. Current models face three major limitations: (i) reliance on primary cells with limited reproducibility, high cost, short culture duration and ethical considerations, (ii) failure to recapitulate all key features of NASH, and (iii) inadequate drug testing data and/or data did not correlate with clinical responses. Therefore, there is a pressing need for robust and relevant preclinical models that faithfully recapitulate human NASH, allow generation of patient-specific models and provide quantitative responses for mechanistic studies and drug testing. We have developed a functional liver tissue-on-a-chip by co-culturing human adult liver stem cell (haLSC)-derived hepatobiliary organoids, induced pluripotent stem cell (iPSC)-derived Kupffer cells (iKCs) and iPSC-derived hepatic stellate cells (iHSCs). We simulated the metabolic microenvironment of hyper nutrition and leaky gut by treating the cells with a concoction of free fatty acids (FFAs), fructose, gut-derived lipopolysaccharides (LPS) and a gut-derived metabolite, phenyl acetic acid (PAA). Through optimization of co-culture media and induction regimens, we were able to stably induce steatosis, hepatocellular ballooning, inflammation, and activation of iHSC and fibrosis—all key hallmarks of NASH. Our LEADS (liver-on-a-chip for NASH drug testing) model also recapitulated the pathological types of steatosis and allowed for quantification of the key features via microscopic evaluation and secretome profiling to score for disease severity. Notably, treatment with saroglitazar, pioglitazone, cenicriviroc (CVC), obeticholic acid (OCA) and resmetirom produced responses similar to those observed in clinical trials. Taken together, our LEADS model is the first model developed using patient-derived hepatic stem cells which recapitulated all key features used for comprehensive drug testing, with results matching to clinical responses.
 Gowri M. Balachander | Gowri Balachander is an Assistant Professor in the School of Biomedical Engineering at the Indian Institute of Technology (BHU) Varanasi. She pursued her MSc and PhD from the Department of Bioengineering at the Indian Institute of Science (IISc) (2018) where she developed 3D organotypic models for studying breast cancer metastasis. She then joined the National University of Singapore (NUS) and developed a morphogenetic model for the development of intrahepatic bile ducts and the LEADS liver-on-a-chip model. She is passionate about the unsolved mysteries of liver regeneration in NASH and is setting up her lab at IIT to pursue her dream. |
 Ng Inn Chuan | Ng Inn Chuan was a Research Fellow at the National University of Singapore (Singapore), specializing in using quantitative imaging methods to understand cellular/tissue behaviour and mechanisms. He completed his Bachelor of Science with Honours in Microbiology from Universiti Kebangsaan Malaysia (Malaysia) in 2007. He was then awarded an NGS Scholarship and pursued his PhD degree in Integrative Sciences and Engineering from the National University of Singapore (Singapore), which he completed in 2015. His main research interests are mechanobiology and liver diseases. |
 Roopesh Ramesh Pai | Roopesh R. Pai is a Principal Scientist in the 3D Bioprinting lab at Dr D. Y. Patil Medical College, Hospital & Research Centre, Pune, India. He obtained his PhD in Biological Sciences from Sree Chitra Tirunal Institute for Medical Sciences & Technology, Trivandrum in 2023 and pursued his post-doctoral studies at the National University of Singapore. He has worked extensively on liver tissue engineering with an emphasis on devising novel methods for liver organoid generation and formulating tissue-specific bioinks for liver organoid bioprinting. At present his research focuses on developing mechanobiology-based strategies for tissue bioprinting and organ regeneration. |
 Kartik Mitra | Kartik Mitra is a post-doctoral research fellow at the Translational Mechanobiology Laboratory, National University of Singapore. He completed his Ph.D. jointly at IIT-M, India, and NUS, Singapore. His research interests lie at the interface of integrative and translational biology, with a focus on bridging basic science and clinical pathophysiology to uncover disease mechanisms. He is currently investigating early mechano-chemical events driving metabolic dysregulation in steatotic fatty liver disease (MASLD). |
 Hanry Yu | Hanry Yu is a Professor in the Department of Physiology, Yong Loo Lin School of Medicine, and NUS College, National University of Singapore (NUS). He is also a Principal Investigator in the Mechanobiology Institute in NUS. He was trained as a Cell Biologist in the US (Duke) and Europe (EMBL). He ventured into biomaterials, tissue engineering, biomedical optics, lab-on-chips, computation and systems biology, mechanobiology and recently into food science, focusing on the translation of various expertise into applications such as drug or toxin testing, AI-enhanced telepathology, cell therapy and cell-based meat manufacturing. |
1. Introduction
Metabolic dysfunction-associated steatotic liver disease (MASLD), previously known as non-alcoholic fatty liver disease (NAFLD), is the hepatic manifestation of the metabolic syndrome which presents as more than 5% lipid accumulation in the liver, a condition known as steatosis.1 NAFLD is currently the most prevalent chronic liver disease (CLD) affecting about 25–30% of the adult population worldwide. It was considered a chronic condition without severe consequences. However, in about 12–15% of patients, NAFLD driven by lipotoxicity and inflammation may progress to a more severe form of disease called metabolic dysfunction-associated steatohepatitis (MASH), also known as non-alcoholic steatohepatitis (NASH).2 While early stages of NASH, up to stage 2 fibrosis, are reversible with weight loss, the progression beyond that is irreversible, culminating in stage 4 fibrosis or cirrhosis, decompensated liver failure, and or hepatocellular carcinoma (HCC).3 The progression was once calculated to be one stage per decade; however, recent data indicate faster progression of one stage per 7 years.4 NASH is now the leading cause of liver transplantation and the fastest-growing cause of HCC in the United States.5 The burden of end-stage liver disease caused by NAFLD is expected to increase by two- to threefold by 2030.6The pathogenesis of NASH progression is multifactorial, but while not fully understood, is widely accepted to involve insulin resistance (IR) in the adipose tissue. IR diminishes adipose tissue expandability and lipogenic capacity, leading to accumulation of lipids such as triglycerides (TGs) and non-esterified free fatty acids (FFAs) in the bloodstream. These lipids are then taken up by hepatocytes and stored as lipid droplets. Excess hepatic lipid accumulation disrupts normal triglyceride balance and increases mitochondrial fatty acid oxidation (mtFAO). An increase in mtFAO without a concomitant increase in the mitochondrial respiratory chain activity (MRC) results in the production of reactive oxygen species (ROS) which damages the hepatocytes via inducing endoplasmic reticulum (ER) stress.7 The damaged and dying hepatocytes release inflammatory and pro-fibrogenic signals such as IL-6, IL-8, CXCL10 and sonic hedgehog (SHH) ligands, which activate the Kupffer cells (KCs) and hepatic stellate cells (HSCs). Bacterial metabolites and lipopolysaccharides (LPS) from the gut can exacerbate this damage by activating the liver sinusoidal vascular endothelial cells (LSECs) and the KCs.8 The persistent inflammation further promotes HSC activation, leading to deposition and remodeling of the extracellular matrix and the progression of fibrosis to cirrhosis and eventually decompensated liver failure.9 Additionally, factors like the lipid types, fructose intake, gut microbiota, and genetic polymorphism in the genes such as PNPLA3, TM6SF2 and MBOT are also implicated in the NASH pathogenesis.10 The definitive diagnosis and staging of NASH are done via histopathology of a liver biopsy specimen using the NAFLD activity score (NAS). NAS is evaluated based on the presence and the extent of steatosis, hepatocellular ballooning, lobular and portal inflammation and fibrosis. NAS ≥5 is widely accepted to distinguish steatohepatitis from simple steatosis. Apart from NAS, the staging of fibrosis is done as follows: stage 1 – zone 3 pericellular or “chicken-wire” fibrosis, stage 2 – periportal fibrosis, stage 3 – bridging fibrosis and stage 4 – cirrhosis.11
Over the past decade, several drug candidates have been evaluated, that target a specific mechanism within the spectrum of the pathogenesis of NASH, from energy intake and disposal to liver lipotoxicity to anti-inflammatory.12–14 The primary endpoints in clinical trials are mostly an improvement in the liver histology as denoted by a decrease in NAS by 2 points without worsening of fibrosis15 or improvement in ≥1 stage of liver fibrosis and no worsening of steatohepatitis, meaning no increase in NAS scores.16 Several drugs have struggled to clear phase II trials, and some have failed in large-scale phase III trials even after good performance in the phase IIb trials. Recently resmetirom (Rezdiffra™), an oral thyroid hormone receptor-β (THR-β) agonist, was given accelerated approval in the United States for treatment of adults with noncirrhotic NASH with moderate to advanced liver fibrosis, but only in combination with diet and exercise.17 As for the other drug candidates, quite a few have struggled to clear phase II trials, while several have failed in large-scale phase III trials even after promising phase IIb results.
One of the major hurdles in screening and testing for drugs for NASH is that different etiologies such as diet, genetic polymorphisms, and gut microbiome could exert their effects differently in different individuals based on the presence and the extent of contribution from other factors. This fact is completely overlooked in animal models wherein NASH is induced via manipulation of one of these causes such as diet or genetics. While this is good for reproducibility, this is one of the major reasons why drugs fail in large-scale clinical trials even after spectacular results from animal models. The essence of recapitulating human NASH not only lies in reproducing the features of human NASH in animal models like hepatocellular ballooning but also in being able to individually manipulate the disease-causing factors and testing the candidate drug against all these different types of NASH.
Organoids and organ-on-a-chip recapitulate human biology and organ level functions in vitro and can be valuable for mechanistic studies and drug testing.18–20 Organ-on-a-chip models allow for the compartmentalization of different cell and tissue types and integration of biophysical signals and fluid flow. We aimed to develop a liver-on-a-chip for human NASH to address the limitations discussed earlier. We have developed a system wherein we can co-culture patient-derived hepatobiliary organoids with non-parenchymal cells to recapitulate human NASH in a format relevant for preclinical drug testing. Considering the pathogenesis of NASH and the clinical assessment of the disease severity, we designed a liver-on-a-chip model – LEADS – that incorporated the hepatocytes, KCs and HSCs in separate compartments. The compartments allow for paracrine interactions between the cells while facilitating independent and detailed microscopy-based evaluation for mechanistic studies and scoring of disease severity. By modulating the metabolic microenvironment and the induction factors, we simulated various histopathological features of NASH performed drug testing and matched our results to data from clinical trials.
2. Materials and methods
2.1. Differentiation of iPSC into Kupffer cells and hepatic stellate cells
Human induced pluripotent stem cells (iPSCs) IMR90-P1 were obtained from WiCell® (Lot# WB66756). The IMR90-P1 iPSCs were cultured in a feeder-independent method as prescribed by WiCell®, in Matrigel-coated plates with mTeSR1 medium (StemCell™ Technologies, Cat# 85850). The pluripotency of the iPSCs were confirmed by Nanog, Oct4 and SOX expression. The human IMR90 iPSCs were grown to 70% confluency and the colonies were digested with Accutase™ (StemCell™, Cat# 07920) to obtain single cells. Then the single cells were seeded into growth factor reduced-Matrigel (Corning® Matrigel® growth factor reduced (GFR) basement membrane matrix, Cat# 354230) coated plates at a density of 1.2 × 105 cells per cm2 and cultured under hypoxic conditions (5% CO2, 90% N2, 5% O2) for 24 h in the presence of Y-27632 (10 μM).The single cells are further differentiated to Kupffer cells and hepatic stellate cells following previously established protocols described elsewhere. The iPSC-derived Kupffer cells (iKCs) were developed following our previously established protocol described elsewhere.21 The embryoid bodies were formed by simultaneous differentiation of IMR90 single cells to hemogenic and meso-endothelial fate by supplementing with BMP4 (50 ng mL−1), SCF (50 ng mL−1), VEGF (20 ng mL−1) and Y-27632 (10 μM) in mTeSR1 medium from days 1 to 3. Then, erythro-myeloid fate induction was triggered using M-CSF (100 ng mL−1) and IL3 (25 ng mL−1) from days 3 to 25 in X-Vivo™ medium (Lonza, Cat# 02-060Q). Primitive macrophages (pre-macrophages) were produced and seen as floating cells from day 25, which were then harvested and further conditioned with KC maturation medium (KCM) supplemented with M-CSF (50 ng mL−1) for 7 days from day 25 to 32 to promote maturation of liver-resident macrophage. The KC phenotype of the differentiated cells was ascertained via immunostaining for KC markers CD68 and CD163 (Fig. S1A†). The hepatic stellate cell differentiation comprises four stages directed by addition of stage-specific growth factors to HSC basal medium:22 days 1–4: BMP4 (20 ng mL−1); day 5–6: BMP4 (20 ng mL−1), FGF1 (20 ng mL−1) and FGF3 (20 ng mL−1); days 7–8: FGF1 (20 ng mL−1), FGF3 (20 ng mL−1), retinol (5 μM) and palmitic acid (100 μM); days 9–12: retinol (5 μM) and palmitic acid (100 μM). The media was changed daily. The mature HSC-like cells were harvested at day 13 and used for further experiments. The HSC phenotype of the differentiated cells was ascertained via immunostaining for HSC markers alpha smooth muscle actin (αSMA) and glial fibrillary acidic protein (GFAP) (Fig. S1A†). All the growth factors were purchased from R&D Systems, and the reconstituted growth factor stock solutions were stored at −20 °C and used within 6 months.
2.2. Differentiation of liver stem cells into hepatobiliary organoids
Liver stem cells were isolated from human liver biopsy tissues and differentiation into hepatobiliary organoids using our patented technology23 (https://patents.google.com/patent/US20200199538A1/en). Briefly, liver biopsies were collected from healthy donors with no previous history of diabetes, hypertension or dyslipidemia after due review and approval from the Institutional Review Boards (IRBs) and Human Research Ethics Committee. Adult liver stem cells were isolated and propagated and were differentiated into hepatobiliary organoids following a patented protocol which yielded compact organoids. More than 80% of cells in these organoids were mature hepatocytes which was ascertained via immunostaining for albumin and CYP3A4. About 10–15% of cells displayed cholangiocyte features with positive staining for cytokeratin 7 (CK7) (Fig. S1A†).2.3. Seeding organoids and non-parenchymal cells onto the chip
Hepatobiliary organoids and NPCs were co-cultured in a microfluidic chip, idenTx 3 from AIM Biotech Pte. Ltd. The microfluidic chip consists of a central channel for the 3D culture of cells embedded in the hydrogel, flanked by two side channels for introducing medium and/or secondary cell types. Both sides of the central channel are bordered by vertical pillars with a triangular base. The pillars are positioned at regular distances from each other, ensuring the interstitial flow of medium from the side channels towards the central channel. In this study, hepatobiliary organoids were cultured in the central channel and the side channels were used for NPC culture. To initiate the coculture in chips, three idenTx 3 chips were assembled on the chip holder plate provided by the manufacturer. The organoids were collected in a centrifuge tube, washed twice with fresh H3 medium and resuspended in an appropriate volume of cold H3 medium. Following this, the organoid suspension was mixed with an equal volume of growth factor-reduced Matrigel solution (Corning, Cat #356230) and was placed on ice until loading into chips. About 10 μL of the organoid suspension containing 6 to 8 organoids was loaded into the central channel of the chip by carefully pipetting in through either one of the inlet ports without generating any air bubbles. The chips were observed under a microscope to ensure uniform distribution of organoids prior to incubating at 37 °C for 15 min to allow for complete gelation of Matrigel. After crosslinking, the troughs surrounding the medium inlet ports on the left and right side of the side channels were filled with 70 μL and 50 μL co-culture medium, respectively. Thereafter, monolayer cultures of iHSC and iKC were trypsinized and cell suspensions containing 1 × 106 cells per mL and 4 × 106 cells per mL, respectively, were prepared. About 10 μL of the iKC suspension was loaded into the top side channel and 10 μL of the iHSC suspension was loaded into the bottom side channel. Sterile deionized water was filled in the reservoir on the chip holder and the plate was covered with a lid to prevent medium evaporation. The microfluidic chips were carefully incubated inside a 5% CO2 incubator set at 37 °C for the entire culture duration. Co-culture medium was replaced at regular intervals by carefully aspirating the medium out from the troughs without reaching into the medium inlet. Three different co-culture media, C1, C2 and C3, were tested. The C1, C2 and C3 media are volumetric mixtures of hepatocyte culture medium (HCM), Kupffer cell medium (KCM) and hepatic stellate cell medium (SCM) in the ratios 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1, 10:4:1 and 2:2:1. HCM is Lonza HCM™ Hepatocyte Culture Medium Bullet Kit® without hydrocortisone. KCM is Advanced DMEM with Cocktail B from GIBCO™ containing penicillin–streptomycin, ITS+ (insulin, transferrin, selenium complex, BSA, and linoleic acid), GlutaMAX™, and HEPES. SCM is DMEM low glucose and MCDB-201 with supplements (2.5 μM dexamethasone, 0.1 μM β-mercaptoethanol, retinol and 100 μM palmitic acid).
:1:1, 10:4:1 and 2:2:1. HCM is Lonza HCM™ Hepatocyte Culture Medium Bullet Kit® without hydrocortisone. KCM is Advanced DMEM with Cocktail B from GIBCO™ containing penicillin–streptomycin, ITS+ (insulin, transferrin, selenium complex, BSA, and linoleic acid), GlutaMAX™, and HEPES. SCM is DMEM low glucose and MCDB-201 with supplements (2.5 μM dexamethasone, 0.1 μM β-mercaptoethanol, retinol and 100 μM palmitic acid).
2.4. Functional assays
2.5. Preparation of NASH induction reagents
Induction of NASH was achieved using oleic acid (OA), palmitic acid (PA), fructose, lipopolysaccharides (LPS) and phenylacetic acid (PAA). A 2.5 M stock of fructose was prepared in water and filter sterilized and stored at −20 °C. A 5 mM stock of PA was prepared as follows: a 150 mM solution of sodium palmitate (in ethanol) (Sigma, Cat# P9767) was heated to 80 °C and was mixed with 0.1 N sodium hydroxide (NaOH) also at 80 °C to obtain a 50 mM PA. This was then added to prewarmed advanced DMEM F12 containing 12% fatty acid-free bovine serum albumin (FAF BSA) and 1% penicillin and streptomycin at 42 °C to a final concentration of 5 mM. The mixture was incubated at RT for 30 min for conjugation with BSA, after which it was filter sterilized and stored at −20 °C. A 150 mM stock of oleic acid (sodium oleate; Sigma, Cat# O7501) was prepared in 100% ethanol and stored at −20 °C. A 0.5 M stock solution of PAA was prepared in 100% ethanol and stored at −20 °C. 1 mg mL−1 LPS was prepared in phosphate buffered saline (PBS) and stored at 4 °C. The stock solutions of each of the four reagents were diluted to their final working concentration in the co-culture medium just before use. Medium containing an equivalent amount of the vehicle was used as control.2.6. ELISA
ELISA for albumin was performed using goat anti-human albumin antibody (Thermo Fisher Scientific, Cat# A80-129A) using a sandwich ELISA protocol from Bethyl Laboratories. The conditioned medium was collected by gently aspirating the medium from the troughs and immediately stored at −20 °C. Conditioned media from two chips from the same experiment were pooled to minimize intra-experiment errors. Conditioned media from a minimum of 4 experiments were used for an assay. On the day of the ELISA, the frozen conditioned medium was thawed on ice and centrifuged at max RPM for 10 min to remove any cellular debris. A total of 100 μL of conditioned medium was used as prescribed by the manufacturer. A 4-parameter logistic (4PL) was used to fit the standard curve for the albumin ELISA.2.7. Staining on the chip
The medium in the chips was removed and the side channels were gently flushed with 1× PBS. The PBS was removed and replaced with 4% formaldehyde. About 250 μL of formaldehyde was added to all the ports to make sure that the channels and the ports were filled. As the middle channel with the organoids is not directly accessible and the media from the side channels slowly diffuse into the middle channel, the chips were placed on a gentle rocker for 2 h for complete fixation. After fixation, the formaldehyde was removed and replaced with 1× PBS. At this stage, the chips can be stored for up to a week at 4 °C. For staining, the chips are washed with 1× PBS twice for 15 min each. The incubation for the washing and staining steps was done at room temperature under mild rocking conditions. After washes, the chips were permeabilized with 1% Triton X-100 in PBS for 2 h. The permeabilization solution was then replaced with a blocking buffer containing 5% BSA, 0.02% sodium azide and 0.5% Triton X-100 in PBS for 2 h. Primary antibodies were diluted in blocking buffer and 60 μL of primary antibody was added to each port and incubated overnight. Every chip requires 240 μL of primary antibody. The primary antibodies were removed, and the chips were washed with 0.2% Tween in PBS (PBST) for 15 min, thrice. After three washes, the corresponding secondary antibodies were added and incubated overnight. After incubation, the chips were washed with PBST twice for 15 min and with PBS once for 10 min. A 300 nM DAPI solution in 1× PBS was used to stain the nucleus and incubated for 15 min. This was followed by a PBS wash for 10 min with gentle rocking. Lipid and actin staining was performed using 1 μg mL−1 BODIPY™ (Thermo Fisher Scientific, Cat# D3922) and 1 μM SiR-actin, respectively, which was added along with the secondary antibodies. The list of antibodies is given in the ESI.†2.8. Confocal microscopy and image analyses
Fluorescence images of the stained organoids and NPCs on the chip were captured with an Olympus FluoView FV3000 (Olympus, Japan) laser scanning confocal microscope. The Z-stack images (step size = 0.9 μm) of individual organoids was captured using a 10× or 20× objective. The iKCs and iHSCs were imaged using the multi-area time-lapse (MATL) module. A 10× objective was used to capture the Z-stack images (step size = 3.9 μm) of the cells. Image analysis was performed using IMARIS 9.5 software (Oxford Instruments, UK). To determine the total nuclear count, nuclear segmentation was done using Spots creation wizard by setting 5 μm as the XY diameter. Surface creation wizard (surface grain size = 0.2 μm and manual threshold value = 800) was used to segment and analyse the lipid droplets within organoids. The features of the lipid droplets such as size, number and total volume, obtained from the analysis were normalized to the total nuclear count (Fig. S2B†). For the side channel cells, to quantify the expression of cytoskeletal protein α-SMA and extracellular matrix proteins such as type 1 collagen and fibronectin, the total intensity of the respective laser channel was recorded and normalized to the total nuclear count.2.9. Viability assays
The viability of the hepatobiliary organoids, iKC and iHSCs were assessed on days 2, 5, 8 and 10 using the Invitrogen™ LIVE/DEAD™ Viability/Cytotoxicity Kit (Thermo Fisher Scientific, Cat# L3224) as per manufacturer's instructions. As the signal intensity in the organoids was very low, we first treated the cells with verapamil, which is an effective inhibitor of ABC transporters to prevent the efflux of the dyes. Firstly, the conditioned medium was collected from the chips and replaced with fresh medium containing 1 μM verapamil and incubated for 45 min. After incubation, the medium was replaced with fresh medium containing two drops of NucBlue™ Live ReadyProbes™ Reagent (Hoechst 33342), 2 μM calcein AM and 4 μM ethidium homodimer-1 (EthD-1). After 15 min of incubation with the dyes, the chips were taken for imaging. The chips with the holder were mounted onto the live cell incubation chamber of the Olympus FV3000 microscope. The images were taken using a 10× objective. The iKCs and iHSCs in the side channels were imaged using multi-area time-lapse (MATL) module to cover the entire area of the side channels. The organoids were imaged individually, and a stack of the z sections was made to visualize them. For iKCs and iHSCs, the cells with high calcein signal and low EthD-1 signal were considered as live cells and the reverse was used to count dead cells. For organoids, due to less cell spreading and significant overlap in 3D and dead cells appearing as fragmented nuclei, the nuclei with high intensity of EthD-1 was considered dead and the nuclei with low intensity of EthD-1 were considered alive (Fig. S8†). The live cells were compared against the total cell numbers to get a ratio of live cells for every time point (days 2, 5, 8 and 10). A total of 2 chips were used per condition per time point. The number of live cells in the control were taken as 100% and the relative ratio of live cells in the NASH-induced chips were calculated for every time point. This was repeated for 3 biological experiments. The average was calculated and the ratio of live cells in induced versus control was used as the normalising ratio for the cytokine data from multiplex assay or ELISA (Fig. S9†).2.10. Multiplex cytokine assessment
The conditioned medium was collected at the following time points as follows: 48 h (2 days) post seeding which serves as the baseline, 5 days post seeding, 8 days post seeding and 10 days post seeding. Four sets of experiments performed on different days served as four biological replicates. Within an experiment, medium was collected from 2 chips and pooled to account for variability in organoid number. The medium was frozen immediately upon collection. On the day of the experiment, the conditioned medium was thawed on ice and centrifuged at high speed for 10 min to remove any debris. The multiplex assay was performed using a customised ProcartaPlex 14 plex assay from Life Technologies (Thermo Fisher Scientific) and run on the Luminex system as per manufacturer's instructions. The panel had the capture antibodies and standards for the following molecules: IL-10, IL-6, IL-8, TNF-α, MIP-1β, MIP-3α, MCP-1, MMP-1, MMP-2, osteopontin, VEGFA, TIMP-1, tPA and PAI. The net mean fluorescence intensity (MFI) and the test result data were analysed. Amongst the analytes, TIMP-1 and MMP-2 values were above the saturation limits. The values were normalized to the organoid count, percentage of viable iKCs, percentage of viable iHSCs or a combination thereof depending on whether the analyte is secreted by the hepatobiliary organoids, iKCs or iHSCs. The normalising ratio was obtained via viability assay as mentioned in section 2.9. The normalising ratios are given in Fig. S9G.†2.11. Drug testing
Stock solutions of 10 mM saroglitazar (MedChem Express, Cat# HY-19937) and pioglitazone (Sigma, Cat# E6910) were prepared in DMSO. Stock solutions of 10 mM cenicriviroc (Selleck Chemicals, Cat# TAK-652) and 100 mM obeticholic acid (Adipogen, Cat# 6-ECDCA) were prepared in 100% ethanol. The drugs were supplemented at a dilution of 1:1000 along with the co-culture medium with the NASH induction factors and added to the chips 48 h post seeding to allow for cell attachment and stabilization. The drug treatment followed the induction protocol and was performed at days 2, 5 and 8 during every medium change for a total duration of 8 days. The control for drug treatment was medium with the induction factors and the vehicle control specific to the drug. At the end of the treatment the chips were fixed with formaldehyde for immunostaining. For testing the effects of resmetirom, the drug treatment was done after the induction for NASH. Stock solutions of the drug resmetirom (MGL3196; Selleck Chemicals, Cat# S6663) were prepared in DMSO. The stock solutions were then diluted either in NASH induction medium or coculture medium to prepare the working solutions having a final concentration of 0.2 μM, 1 μM, 5 μM, 20 μM, and 80 μM. The final concentration of DMSO in the working solution was 0.2%. To initiate the drug testing experiment, the chips were first categorized into four groups: (1) healthy, (2) NASH, (3) vehicle (DMSO) and (4) drug. The healthy group was treated with coculture medium for 12 days with intermittent medium change every 2 days. For the NASH group, the chips were treated with induction medium until day 10 and was then changed to coculture medium for another 2 days. The vehicle and drug groups were treated with NASH induction medium on days 2 and 4 post-cell seeding. Thereafter, on days 6 and 8, induction medium containing either DMSO (0.2%) or drug was added. Finally, on day 10, coculture medium containing DMSO (0.2%) or drug was added and treated for another 2 days.
2.12. Statistical analysis
Statistical analysis was performed using GraphPad Prism 5 (GraphPad Software Inc., California, USA) and all data were expressed as mean ± standard error of the mean or mean ± standard deviation as mentioned. Unpaired two-tailed Student's t-test was used to compare the data sets from different groups and a p value < 0.05 was considered as significant.3. Results
3.1. Setting up the LEADS model
The iKCs and iHSCs were differentiated from iPSCs, while hepatobiliary organoids were differentiated from adult liver stem cells, as described in Materials and Methods. Successful differentiation was ascertained by immunostaining for the appropriate cell type markers (Fig. S1A†). We adopted the idenTx 9 chip for the co-culture of hepatobiliary organoids, iKCs and iHSCs, as explained in section 2.3. A total of nine chips can be assembled on a chip holder plate with dimensions (X–Y) matching those of a standard multi-well plate. A single plate can screen nine samples in a single experimental run. The layout of the chips is depicted in Fig. 1. The design and dimensions of the chip are shown in Fig. S1B.† Hepatobiliary organoids were seeded in the central gel channel, embedded in 50% growth factor reduced (GFR) Matrigel, while iKCs and iHSCs were seeded in separate side channels (Fig. S1C†). Preliminary data showed that Matrigel was more conducive to maintaining the integrity of hepatobiliary organoids and their hepatic functions, such as albumin and urea secretion, when compared to type I collagen (Fig. S1D and E†). Type I collagen gels induced tubular outgrowths of biliary cells (data not shown). The spacing between the triangular barriers and the porous 50% Matrigel allowed diffusion of paracrine factors between the different cell types, while the compartmentalization of these cell types allowed for independent microscopic evaluation and prevented the overgrowth and dominance of a single cell type, which is often observed in direct contact co-cultures. We estimated that each organoid contained approximately 4 × 103 cells through image-based assessment. Initially, the number of organoids in every chip was calculated, and a proportional number of iKCs and iHSCs that mimics the 10:4:1 ratio of the cells in the liver24–29 were seeded on the side channels. Although the attachment of iHSCs to the side channels was poor (Fig. S1C†), they proliferate upon co-culture with the hepatobiliary organoids. Regretfully, this method yielded a high variability in the final numbers of iHSCs in each chip. A high number of iHSCs will lead to degradation of the Matrigel, migration of iHSCs toward the organoids, and subsequent deterioration of organoid integrity and outgrowths of ductular structures. Therefore, the numbers of iKCs and iHSCs were fixed at 4 × 104 cells and 1 × 104 cells per channel, respectively. This seeding density gave consistent responses for 2–8 organoids per chip.
 | ||
| Fig. 1 Illustration of the liver-on-a-chip for the NASH drug testing (LEADS) platform. (A) Layout of microfluidic chips assembled on the chip holder plate. (B) Zoomed-in view of a microfluidic chip showing hepatobiliary organoids and non-parenchymal cells in different channels before and after NASH induction. FFAs, free fatty acids; LPS, lipopolysaccharides. | ||
3.2. Optimization of growth conditions for induction of NASH on the LEADS chip – steatosis
The next critical consideration is the selection of medium for the co-culture of the different cell types. The iKCs and iHSCs are derived from iPSC, while the hepatobiliary organoids are derived from the patient-derived liver stem cells using a specific concoction of growth factors and signaling molecules. The co-culture medium must provide sufficient signaling molecules for preserving the functions of these cells in the chip, which in turn influences the intercellular communication between the cell types when treated with lipogenic factors and the consequent induction of NASH. On a preliminary assessment, the hepatocyte maturation medium (H3) was found to be incompatible with the iKCs as it contains dexamethasone, a corticosteroid used to induce maturation in hepatocytes, due to its potent anti-inflammatory activity. In the co-culture, dexamethasone can suppress the activation of iKCs in response to stimulation with LPS, which is required for the inflammatory conditions in NASH. Hence, it is critical to select the co-culture medium that maintains a functional liver microenvironment on the chip and allow manifestation of NASH features upon induction. The commonly adopted strategies for the co-culture medium for liver-on-a-chip models involve using either a special co-culture medium, distinct from the medium for each of the cell types, or a mixture of the medium used for each of the cell types, blended in ratios reflecting the ratio of these cells in the native liver tissue or on the chip30–32 (comparison with state-of-the-art models shown in ESI† Table S2). We selected three different combinations of the hepatocyte culture medium (HCM), KC medium (KCM) and the HSC medium (SCM) at different ratios. The C1, C2, and C3 media had a 1:1:1, 10:4:1, and 2:2:1 ratio of the HCM, KCM and SCM medium, respectively. The C2 medium reflected the ratio of hepatocytes:KCs:HSCs found in the native tissue. No significant differences were observed in albumin expression in the hepatobiliary organoids on the chip in the three types of media (Fig. S2A†).
We aimed to induce NASH using free fatty acids, namely oleic acid (OA) and palmitic acid (PA), along with fructose, bacterial lipopolysaccharides (LPS) and phenylacetic acid (PAA). Dietary fatty acids directly contribute to hepatic steatosis.33 PA, with 16 carbon atoms, is the most common saturated fatty acid present in the diet and serum. OA is a monounsaturated 19-carbon fatty acid. Fructose is a highly lipogenic nutrient; chronic consumption of high-fructose corn syrup has been associated with the development of dyslipidemia, insulin resistance and metabolic syndrome; it reduces leptin expression, inhibits fatty acid oxidation, and causes oxidative stress and inflammation.34,35 The gut microbiome of NASH patients shows significant alterations with low microbial gene richness compared to healthy controls; gene signatures from the gut indicated increased biosynthesis of LPS and increased biosynthesis and metabolism of branched-chain amino acids (BCAAs).36 LPS from the gut activates iKCs via toll-like receptor (TLR) signaling, which leads to the secretion of an array of chemokines and cytokines such as TNF-α and IL-6.8 PAA is a metabolic product of phenylalanine and is the strongest microbial metabolite associated with steatosis in metabolome-wide association studies and has been experimentally shown to induce steatosis in animal models.36 We performed a small-scale experiment to first understand the effects of OA, PA, LPS, fructose and PAA on the cells on the chip and assessed their ability to induce steatosis. Treatment with 50 and 100 ng mL−1 LPS led to a significant decline in iKC viability; therefore we used 10 ng mL−1 for further experiments (Fig. S2C†). We treated the co-culture of hepatobiliary organoids with the iHSCs on the chip with varied concentrations of OA (0, 150, 300 and 600 μM) and PA (0, 300, 450 and 600 μM) and constant concentrations of PAA (10 mM), LPS (10 ng mL−1) and fructose (100 mM) (Fig. 2A). Quantification of the lipid droplets showed that lipid accumulation per cell increased with increasing levels of OA (Fig. 2B). This was mainly due to the increase in the size of the lipid droplets (Fig. 2C) and increasing the concentration of OA beyond 300 μM had no further increase in the number of lipid droplets (Fig. 2D). Interestingly, treatment with 450 μM PA showed distended hepatocytes with foamy-appearing cytoplasm with a large number of uniform small-sized lipid droplets, characteristic of microvesicular steatosis (MiS)37 (Fig. 2E). This contrasts with organoids treated with equal and higher doses of OA, where non-uniform sized small and large droplets were observed (Fig. 2F), as reported for small-droplet macrovesicular steatosis (Sd-MaS).38
 | ||
| Fig. 2 Optimization of induction and co-culture conditions on the LEADS chip for steatosis. (A) Table showing the concentration of PA and OA used in combination with 100 mM fructose and 10 mM PAA. (B)–(D) Graphs showing the average lipid droplet volume per cell, average size of lipid droplets and the average number of lipid droplets per cell, respectively. Error bars indicate standard deviation, N = 9–11 organoids (pooled from 3 experiments, with 3–4 organoids per experiment). Representative images of organoid displaying (E) true microvesicular steatosis (Mi-S) when treated with 450 μM PA and 150 μM OA and (F) small-droplet macrovesicular steatosis (Sd-MaS) when treated with 0 μM PA and 600 μM OA. Organoids were stained with BODIPY (green) and DAPI (blue). Imaging was performed using a confocal microscope and maximum intensity projections are shown. Scale bar: 50 μm in (E); 50 μm and 30 μm in the left and right images in (F), respectively. (G)–(I) Graphs showing the average lipid droplet volume per cell, average size of lipid droplets and the average number of lipid droplets per cell, respectively, under control and induced samples in different media and co-culture conditions; error bars represent standard error of the mean. N = 4 experiments. Paired two-tailed t-tests were performed. For each combination of medium and culture condition, induced (blue) was compared against control (orange). *p < 0.05; **p < 0.01; ***p < 0.001. | ||
We selected equimolar concentrations of 300 μM OA and PA, 100 mM fructose, 10 ng mL−1 LPS and 10 mM PAA and used this to assess the different media for NASH induction for steatosis, hepatocellular ballooning (HCB) and fibrosis. Quantification of lipid accumulation showed that organoids exhibited increased levels of steatosis upon tri-culture with iHSC and iKC compared to monoculture, and the maximum steatosis was observed in the C1 medium (Fig. 2G and S3†). This was due to a significant increase in the number of lipid droplets and the increase in the size of the lipid droplets (Fig. 2H and I).
3.3. Assessment of hepatocellular ballooning (HCB) upon induction on the LEADS chip
Hepatocellular ballooning and lobular inflammation are two key defining features of NASH that drive fibrotic progression. While simple steatosis is rarely associated with fibrosis progression, histological confirmation of NASH which includes the presence of steatosis, HCB and lobular inflammation, is associated with a higher prevalence of cirrhosis, overall liver-related mortality and death.39 The ballooned hepatocytes are identified with larger size with cytoplasmic inclusion of ubiquitinated cytokeratins, named Mallory–Denk bodies (MDBs), formed due to unfolded protein response (UPR) dysregulation and autophagy suppression as a result of excessive ER stress.39 HCB indicates liver cell injury or damage and these injured ballooned hepatocytes secrete paracrine pro-fibrogenic signals such as Sonic Hedgehog (SHH), which in turn activates the hepatic stellate cells.40 HCB is scored by assessing the histological sections by assessing for large hepatocytes with pale staining and reticulated cytoplasm.39 The organoids treated with NASH induction factors showed large ballooned hepatocytes characteristic of HCB (Fig. 3A). However, the evaluation of HCB based on histology alone has shown large discrepancies even amongst expert pathologists.41 We tested M30, an antibody that recognises the neoepitope on the caspase-cleaved cytokeratin 18, to quantify HCB in the LEADS chip. First, we validated the suitability of M30. We observed that co-immunostaining of M30 with CK8/18 and ubiquitin showed M30 localising with the CK8/18 and ubiquitin aggregates (Fig. 3B). We treated the organoids with increasing doses (0.0625, 0.125, 0.25, 0.5, 1 and 2 μg mL−1) of tunicamycin, a natural antibiotic that interrupts protein maturation and induces ER stress,40 and observed a proportional increase in M30 levels (Fig. S4†). We quantified M30 immunostaining as a readout for hepatocellular ballooning and observed that the organoids in C1 medium had the highest levels of M30 (Fig. 3C and S5†). The LEADS chip showed the highest levels of steatosis and ballooning injury upon induction in the C1 medium. The hepatobiliary organoids, iKCs and iHSCs maintained their differentiation status (Fig. 4A) and a functional liver microenvironment on the chip in the C1 medium (Fig. 4B–D). These data suggested that C1 medium fostered a conducive environment for optimal liver functions, induction of steatosis and hepatocellular ballooning. We performed all further experiments with the C1 medium. | ||
| Fig. 3 Assessment of hepatocellular ballooning injury on the LEADS chip. (A) Representative images of organoids showing ballooned cells. Imaging was performed using a confocal microscope and maximum intensity projections are shown. Scale bar: 50 μm. (B) Representative images of organoids immunostained for CK8/18, ubiquitin and M30 and counterstained with DAPI. Imaging was performed using a confocal microscope and maximum intensity projections are shown. Scale bars: 50 μm (top), 20 μm (bottom). (C) Graph depicting the quantification of M30 per cell under control and induced conditions for C1, C2 and C3 media. Error bars indicate standard error of the mean, N = 4 experiments. Paired two-tailed t-tests were performed. For each combination of medium and culture condition, induced (blue) was compared against control (orange). However, none reached statistical significance. | ||
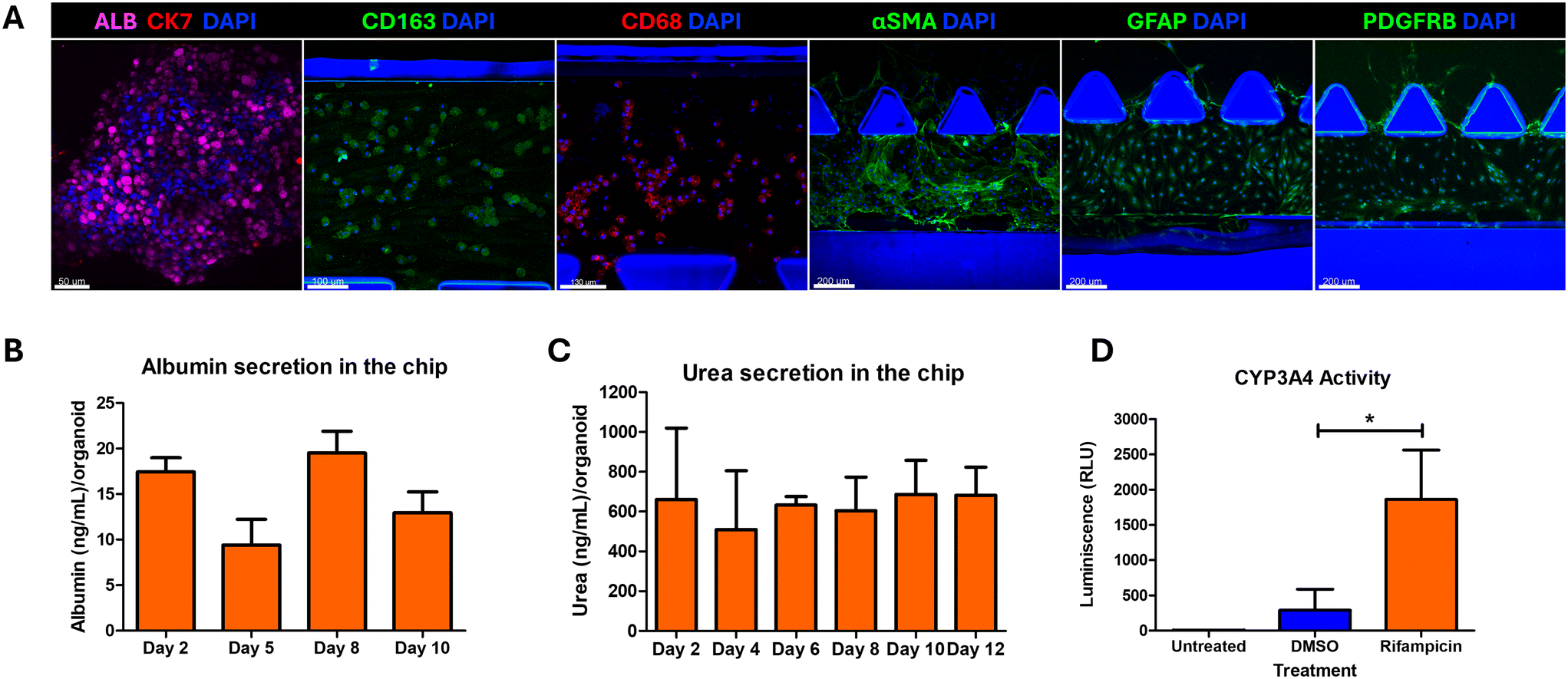 | ||
| Fig. 4 Optimized growth conditions for co-culture of hepatobiliary organoids, iKCs and iHSCs on the LEADS chip. (A) Immunostaining of hepatobiliary organoids, iKCs and iHSCs on the LEADS chip in C1 medium after 10 days of co-culture on the chip. Markers indicating the differentiation and functional status of the different cell types were chosen for immunostaining. Albumin for hepatocytes; CK7 for biliary cells; CD163 and CD68 for iKCs; αSMA, GFAP and PDGFRB for iHSCs. (B) Graph showing the amount of albumin secreted from the LEADS chip at the indicated days of co-culture quantified by ELISA. Error bars represent standard error of the mean. N = 3 experiments. (C) Graph showing the quantification of urea secretion from the LEADS chip at the indicated days of co-culture. Error bars represent standard deviation. N = 3 experiments. (D) Graph indicating the assessment of CYP3A4 enzyme activity. Error bars represent standard deviation. Paired t-test was performed. Rifampicin (induced) was compared against DMSO treatment (vehicle control). *p < 0.05. | ||
3.4. Refining the metabolic microenvironment on the LEADS chip
The most accepted pathogenesis of NASH development in humans is the primary development of steatosis due to hypernutrition, which could either be benign or progress to steatohepatitis based on the presence of secondary insults such as leaky gut and altered gut-microbiome and their metabolites that can cause inflammation and ER stress and activate the non-parenchymal cells.42–44 To mimic this natural progression and to decipher the involvement of the non-parenchymal cells in NASH progression, we performed the following experiment. We treated the LEADS chip with OA, PA and fructose for the first 3 days to mimic conditions of hypernutrition, followed by addition of LPS and PAA to mimic leaky and altered gut microbiome. This was performed under both co-culture and monoculture conditions (without the iKCs and the iHSCs). As OA is critical for lipid accumulation (Fig. 2B) and excess PA could cause hepatocellular injury,45–47 we used 450 μM OA and 150 μM PA. We assessed the development of steatosis, HCB and fibrosis at the start of the induction (day 0) and at days 3, 6 and 8. We named this model of induction as progression model and compared this to the endpoint model, wherein all the induction factors OA, PA, fructose, LPS and PAA were introduced together and assessment for steatosis, HCB and fibrosis were performed at the end of 10 days. The experimental scheme is depicted in Fig. S6.† Interestingly, very little lipid accumulation was observed when the organoids were treated with only OA, PA and fructose in the monoculture conditions and even lower when co-cultured with iKCs and iHSCs (Fig. 5A). Appreciable steatosis was observed only after the introduction of LPS and PAA as shown by the drastic increase in the size and the number of lipid droplets (Fig. 5B–D). The levels of steatosis were higher in the endpoint model than in the progression model, although both these systems were treated with the same levels of FFA and fructose but only varying in the levels of PAA and LPS. Taken together, these data suggest that hypernutrition alone might not cause steatosis; ER stress and inflammation are required and they cooperate with hypernutrition to increase the levels of steatosis. In the endpoint model, we observed a clear distinction in the pattern of steatosis in the organoids under mono-cultured and co-cultured conditions. Organoids induced for NASH without the iKCs and iHSCs had large lipid droplets which displaced the nucleus to the periphery characteristic of large droplet macrovesicular steatosis (Ld-MaS)38 (Fig. 6). This is in contrast with the co-cultured conditions which exhibited Sd-MaS (Fig. 6). This data indicated that the iHSCs and the iKCs influenced the pattern of lipid droplet accumulation in the hepatocytes. Further experimentation is required to understand the mechanisms and the consequence of different patterns of lipid accumulation in hepatocytes and the involvement of non-parenchymal cells in this phenomenon. Interestingly, the levels of M30, a surrogate marker for hepatocellular ballooning, were higher in the co-cultured conditions (Fig. 5D). In order to assess fibrosis, we immunostained for type I collagen and imaged the entire side channel and normalized the intensity of collagen signal to the number of nuclei in the channel. The data showed a gradual increase in the levels of type I collagen with induction and the highest levels of collagen were observed in the endpoint model (Fig. 5E and F). Under the same conditions of the endpoint model, we found a substantial increase in the expression of iHSC activation markers, GFAP and αSMA, and other fibrous matrix such as fibronectin upon induction (Fig. 5G). Taken together, the progression model of induction did not follow the two/multiple hit hypothesis of the NASH model; steatosis was observed only after induction with LPS and PAA. The endpoint model of induction showed higher levels of steatosis, HCB and fibrosis compared to the progression model. | ||
| Fig. 5 Progression versus endpoint models of NASH induction and assessment of steatosis, hepatocellular ballooning injury and fibrosis. Graphs showing the average lipid droplet volume per cell (A), average size of lipid droplets (B), the average number of lipid droplets per cell (C) and the quantification of M30 per cell (D) in organoids under monoculture and co-culture conditions in the progression and endpoint models. Error bars represent standard error of the mean. N = 3 experiments. Paired two-tailed t-tests were performed. Days 3, 6 and 8 were separately compared against day 0 of their respective culture condition. *p < 0.05; **p < 0.01. (E) Multi-area time lapse images of the side channel harbouring the iHSCs, immunostained for type I collagen. Stitched image of the multi-area 10× images with the maximum intensity projection is shown. (F) Graph showing the levels of collagen per iHSC in the side channels under co-culture conditions in the progression and endpoint models. Error bars represent standard error of the mean. N = 3 experiments. Paired two-tailed t-tests were performed. Days 3, 6 and 8 were separately compared against day 0. **p < 0.01. (G) Graph showing the levels of fibronectin, GFAP and αSMA per iHSC in the side channels under control and induced conditions following the endpoint model of induction. Error bars represent standard error of the mean. N = 3 experiments. Paired two-tailed t-tests were performed. For each marker, induced (blue) was compared against control (orange). *p < 0.05. | ||
 | ||
| Fig. 6 Recapitulation of pathological types of steatosis on the LEADS chip. Representative images of the organoids displaying (A) large-droplet macrovesicular (Ld-MaS), (B) small-droplet macrovesicular (Sd-MaS) and (C) true microvesicular steatosis (Mi-S). (A′), (B′) and (C′) are the digitally magnified images of the regions shown in (A), (B) and (C) respectively. Scale bars: 50 μm (A–C), 20 μm (A′–C′). (D) Immunostaining of iHSC on the chip for the indicated markers for HSC activation and ECM deposition. Scale bar: 100 μm. Imaging was performed using a confocal microscope and maximum intensity projections are shown. | ||
3.5. Secretome profiling – deciphering the paracrine interactions on the LEADS chip
Induction of NASH using the endpoint model led to a significant increase in steatosis, HCB and fibrosis; more strikingly the pattern of steatosis was altered under co-cultured conditions compared to monoculture conditions. To understand the paracrine interactions amongst the different cell types, we profiled the conditioned medium on the LEADS chip and assessed for an array of chemokines, cytokines, signaling molecules, matrix modulators, profibrogenic and angiogenic factors using a multiplex assay. These molecules are secreted by either the hepatocytes, iKCs or iHSCs or a combination thereof and are known to play crucial roles in paracrine activation in NASH progression as shown in Table 1. The levels of these markers were normalized to the number of viable cells, which is explained in sections 2.9 and 2.10 and Fig. S9.†| S. no. | Name | Hepatocytes | Hepatic stellate cells | Kupffer cells | Role in NASH | References |
|---|---|---|---|---|---|---|
| 1 | IL-6 | + | — | ++ | Promotes inflammation and fibrosis by inducing acute phase response and hepatic stellate cell activation | 48–50 |
| 2 | IL-8 | + | — | + | Mediates LPS induced hepatic inflammation and injury by acting as a chemokine attracting neutrophils to sites of inflammation | 51, 52 |
| 3 | CXCL-10 | + | — | + | Involved in T-cell recruitment and activation, associated with increased inflammatory responses in liver tissues during NASH. Promotes fibrosis by preventing NK cell mediated hepatic stellate cell inactivation | 53, 54 |
| 4 | MCP-1α | — | + | + | Mediates immune cell recruitment, promotes inflammation by inducing the expression of pro-inflammatory cytokines such as TNF-α, IL-1β, and IL-6 and contributes to fibrosis development by activation of hepatic stellate cells | 55, 56 |
| 5 | MIP-1β | — | — | ++ | Contribute to chronic inflammation and insulin resistance in liver by recruiting monocytes and M1-polarized macrophages. Involved in hepatic stellate cell activation further promoting fibrosis | 57–59 |
| 6 | MIP-3α | — | ++ | + | Exerts proinflammatory and profibrogenic effects in liver by recruiting immature dendritic cells and lymphocytes, and modulation of ECM secretion in stellate cells, respectively | 60–62 |
| 7 | MMP-1 | + | — | — | Involved in extracellular matrix remodelling; elevated levels can indicate fibrosis progression in NASH | 63, 64 |
| 8 | OPN | + | + | + | Acts as a chemoattractant promoting migration of Kupffer cells and macrophages into hepatic necrotic areas. Promotes fibrosis through activation of hepatic stellate cells | 65–67 |
| 9 | PAI-1 | ++ | — | + | Inhibits fibrinolysis and promotes fibrosis; high levels are associated with increased fibrosis risk in NASH patients | 68, 69 |
| 10 | TNF-α | — | — | + | Contributes to hepatic steatosis in NAFLD. Promotes hepatic inflammation and insulin resistance by stimulating the production of pro-inflammatory cytokines like IL-6 and IL-1β and suppressing anti-inflammatory cytokine adiponectin | 70–72 |
| 11 | t-PA | + | — | — | Involved in fibrinolysis; impaired in NASH causing hypo fibrinolysis and increased risk of thrombotic diseases | 73, 74 |
| 12 | VEGF-A | + | — | — | Contributes to fibrosis and HCC development by activating hepatic stellate cells and hepatic progenitor cells (ductular reaction) via VEGF–VEGFR signalling | 75, 76 |
We observed a significant increase in the levels of pro-inflammatory cytokines IL-6, IL-8 and TNF-α upon NASH induction (Fig. 7). IL-8 levels peaked on day 5, which is 72 h after induction. The levels of MCP-1α, MIP-1β, MIP-3α and CXCL10, although they had shown a slight increase at certain time points, did not change significantly upon induction. VEGF-A, which has been shown to increase progressively with NASH progression, showed a significant increase upon induction at all time points. Osteopontin (OPN) was significantly increased after 72 h after induction and then faded at later time points. A significant increase in MMP-1 was also observed after 72 h after induction. The levels of plasminogen activator inhibitor-1 (PAI-1) steadily increased and a significant increase was observed on day 10, while the levels of tissue plasminogen activator (tPA) steadily and significantly decreased upon induction. PAI-1, a major regulator of the plasminogen pathway, inhibits the activity of tPA and formation of plasmin and the downstream fibrin degradation. Injured cells activate TGF-β, which promotes the expression of PAI-1, and sustained PAI-1 activity leads to inhibition of plasmin-dependent MMPs, excess ECM deposition, fibrosis and epithelial-to-mesenchymal transition (EMT). The sustained increase in PAI-1 levels indicate a possible activation of the TGF-β pathway on the LEADS chip. The sharp increase in MMP-1 and OPN upon induction, the sustained increase in OPN, VEGF-A, PAI-1 and the strong decline in tPA levels indicate a strong pro-fibrogenic microenvironment on the LEADS chip. Taken together, the increased levels of IL-6, IL-8 and TNF-α and the increase in pro-fibrogenic markers indicate a multitude of paracrine interactions that lead to a sustained inflammatory and fibrogenic microenvironment on the LEADS chip. Interrogation into the specific signaling pathways that cause the activation of these markers could be done if a particular pathway needs to be investigated during NASH development and progression.
 | ||
| Fig. 7 LEADS chip harbours a sustained pro-inflammatory and pro-fibrogenic microenvironment. Graphs showing the levels of the mentioned cytokine secreted in the LEADS chip at 0, 3, 6 and 8 days of induction. Paired t-test was performed, values of induced were compared against control at the particular time point. Error bars represent standard error of the mean. N = 4 experiments. Paired t-tests were performed. For each time point, induced (blue) was compared against control (orange). *p < 0.05; **p < 0.01; ***p < 0.001. | ||
3.6. Drug testing
The endpoint model had shown significant induction of steatosis, HCB, a sustained inflammatory and pro-fibrogenic microenvironment, activation of iHSC and a significant increase in the deposition of type I collagen and fibronectin. This indicated that the LEADS chip was a good model for NASH. We tested four different types of NASH drugs on the LEADS chip to evaluate its potential as a preclinical model (Fig. 8). These drugs target different signaling pathways in NASH and are at different stages of clinical trials, as shown in Table 2. | ||
| Fig. 8 Drug testing on the LEADS chip. Graphs showing the average lipid droplet volume per cell (A), the average size of lipid droplets (B), the average number of lipid droplets per cell (C) and the quantification of M30 per cell (D) in organoids under the effects of the drugs and the vehicle controls. Error bars represent standard error of the mean. N = 3 experiments. Paired two-tailed t-tests were performed. Drug treatments were compared against their vehicle control, with saroglitazar (Sar) and pioglitazone (Pio) against DMSO (orange), and obeticholic acid (OCA) and Cenicriviroc (CVC) against ethanol (blue). ns, not significant; *p < 0.05; **p < 0.01; ***p < 0.001. (E) Graph indicates the levels of type I collagen per iHSC under the effects of the drugs and the vehicle controls. Error bars represent standard error of the mean. N = 4 experiments. Paired two-tailed t-tests were performed. Drug treatments were compared against their vehicle control, with saroglitazar (Sar) and pioglitazone (Pio) against DMSO (orange), and obeticholic acid (OCA) and Cenicriviroc (CVC) against ethanol (blue). ns, not significant; **p < 0.01. (F) Table compiles the effects of the drugs on the NASH features assessed on the LEADS chip. | ||
| Drug | Mechanism of action | Status | Effects observed in clinical trials |
|---|---|---|---|
| Saroglitazar | Dual peroxisome proliferator-activated receptor (PPAR) α/γ agonist | Met endpoints on phase II trials (NCT03061721); currently in phase 2b (EVIDENCES X NCT05011305); currently in phase IV clinical EVIDENCES XI (NCT05872269) trial in India; approved for use in India | Compared to placebo, it significantly reduced ALT levels, improved liver fat content, reduced triglycerides, reduced ELF scores77 |
| Obeticholic acid (OCA) | Farnesoid X receptor (FXR) agonist | Met endpoints in phase II (FLINT NCT01265498) and III (REGENERATE NCT02548351) trials;78 approval for clinical use declined due to safety concerns | Compared to the placebo, the drug-treated group showed marked improvement in lobular inflammation and significant improvement in ≥1 stage in fibrosis79 |
| Pioglitazone | PPAR γ agonist | PIVENS study (NCT00063622) showed significant effects on steatosis, inflammation and ballooning, but not on NAS scores; subjects gained weight. No large phase III trials have been conducted | The primary outcome of reduction in NAS score was achieved in a double-blind placebo-controlled study (NCT00994682); reduction in fibrosis and increase in adipose insulin sensitivity observed in patients with diabetes80,81 |
| Cenicriviroc (CVC) | C–C chemokine receptor types 2 (CCR2) and 5 (CCR5) antagonist | Phase III AURORA study failed to show improvement in fibrosis;82 discontinued due to lack of histological efficacy | Showed potent antifibrotic effects in phase IIb CENTAUR study83 |
| Resmetirom | Thyroid hormone receptor β (THR-β) agonist | US FDA has approved resmetirom (Rezdiffra) for the treatment of noncirrhotic metabolic dysfunction-associated steatohepatitis (MASH) in adults | Exhibited NASH resolution (including a reduction in NAFLD activity score by ≥2 points) with no worsening of fibrosis, and reduction in fibrosis by at least one stage with no worsening of the NAFLD activity score84 |
In a phase II double-blind study, patients treated with saroglitazar exhibited a marked improvement in steatosis and hepatocellular ballooning, but an underwhelming effect on fibrosis was observed in the liver biopsies. On the LEADS chip, treatment with 10 μM saroglitazar showed a significant reduction in steatosis and hepatocellular ballooning, but a moderate reduction in the levels of fibrosis. The effects on steatosis were due to a significant reduction in the number of lipid droplets; however, the size of the droplets was unaffected. Obeticholic acid (OCA) has shown improvements in hepatocellular ballooning, lobular inflammation and a significant improvement in ≥1 stage in fibrosis at high doses. On the LEADS chip, we observed a significant reduction in steatosis, contributed by a decrease in the size and number of lipid droplets; however, modest reduction in hepatocellular ballooning and fibrosis were observed. Pioglitazone has been studied extensively in small-scale phase II trials with promising results; however, the drug is not recommended due to its side effects on weight gain. PXL065, a deuterium-stabilized R-enantiomer of pioglitazone, showed in a phase II randomized placebo-controlled trial (DESTINY-1) a significant reduction in liver fat content, improvement in fibrosis and a reduction in NAS score, without the side effects of weight gain and oedema.85 On the LEADS chip, we observed a significant reduction in hepatocellular ballooning and fibrosis upon treatment with 10 μM pioglitazone; however, there was no change in the levels of steatosis. Cenicriviroc (CVC) is an antagonist of C–C chemokine receptor types 2 (CCR2) and 5 (CCR5) and exerts anti-NASH effects by targeting liver inflammation.83 Although it showed promising anti-fibrotic effects in phase II trials, it was discontinued due to a lack of efficacy in phase III trials. Treatment with 10 μM on the LEADS chip showed a marked reduction of steatosis and hepatocellular ballooning but did not reduce fibrosis. Amongst the four drugs tested, we observed the largest reduction in steatosis and hepatocellular ballooning upon treatment with saroglitazar, which is currently being tested in phase IV clinical trials.
Recently, treatment with resmetirom or MGL-3196, a highly selective small-molecule agonist of THR-β (thyroid hormone receptor β) that plays a key role in regulating the lipid metabolism of hepatocytes, had shown a significant reduction in haptic fat content in patients with NASH in phase II trials84 and had shown safety and tolerance in phase III trials.86 Following the excellent response in the clinical trials, the Food and Drug Administration (FDA) has provided accelerated approval for use in non-cirrhotic NASH patients along with diet and exercise. We observed the expression of THR-β in the hepatobiliary organoids (Fig. 9B). We tested the effects of resmetirom on the LEADS chip following a modified protocol as depicted in Fig. 9A. Treatment with resmetirom at doses of 5 μM and above exhibited a significant reduction in the number of lipid droplets and total lipid droplet volume compared to the vehicle control (Fig. 9D and E). However, the levels of collagen remain unchanged (Fig. 9F). Taken together, these data show the applications of the LEADS chip for NASH drug testing.
 | ||
| Fig. 9 Testing of resmetirom on the LEADS chip. (A) Scheme for NASH induction and drug treatment for testing resmetirom on the LEADS chip. (B) Representative images of organoid immunostained with THR-β. Scale bar: 100 μm. (C) Representative images of the organoids stained with BODIPY to visualize the lipid droplets. Scale bar: 100 μm. Imaging was performed using a confocal microscope and maximum intensity projections are shown. Graphs showing the average number of lipid droplets per cell (D), the average lipid droplet volume per cell (E) and the expression of type I collagen per HSC (F). Error bars represent standard deviation. N = 3 experiments. Paired t-tests were performed. Drug treatments at were compared against vehicle control. ns, not significant; *p < 0.05; **p < 0.01; ***p < 0.001. (G) The table compiles the effects of the drug resmetirom on the NASH features assessed on the LEADS chip. | ||
4. Discussion
Organ-on-a-chip technology offers two critical advantages that are valuable for preclinical drug testing.87,88 Firstly, it provides compartmentalization for the culture of different cell/tissue types connected via fluidics for communication via paracrine interactions. Based on the chip design, each compartment can be incorporated with cell/tissue-specific biochemical signals and subjected to independent microscopic assessment of cell behaviour in the different compartments. This is advantageous over direct coculture techniques wherein the cell types might eventually segregate into different regions within the co-culture based on cell–cell adhesion partners, or lead to gross differences in cellular behaviour due to uncontrolled cell–cell interactions. These advantages are reflected in the ability of organ-on-a-chip models to faithfully recapitulate events during organ development, normal physiology and diseased conditions. This directly translates into a very important aspect of the ability to test a wide variety of factors that can cause a particular disease, particularly for a multifactorial disease such as NASH.NASH is caused by a multitude of factors such as a hypercaloric diet, insulin resistance, gut microbiome dysbiosis and genetics.89 More often, a combination of these factors could be present in an individual with each of these factors contributing to a varying extent. Typically, animal models are developed by inducing NASH using a specific set of factors like diet – methionine and choline-deficient (MCD) diet or high-fat diet (HFD), genetic changes – leptin deficiency (ob/ob) mice and toxins – carbon tetrachloride (CCl4).90 Even with the STAM model which is a combination of a toxin (streptozotocin) and HFD that boasts of a better recapitulation of human NASH, these animal models are still incompetent for preclinical drug testing for NASH as drugs that have performed well in animal models have failed in clinical trials.90 This is the second largest advantage of the organ-on-a-chip model. We could induce different types of NASH via different combinations of disease-causing factors and test the efficacy of drugs against the different types of NASH as what would be observed in a clinical setting. Given these advantages, most of the liver-on-a-chip models developed for NASH struggle with limitations such as the use of primary cells which lead to high costs, loss of functions within a short duration, severe batch-to-batch variations and issues with reproducibility, and complex chip design that make it difficult without microfluidics expertise and have poorly demonstrated drug testing capabilities. Liver-on-chip systems also offer significant advantages in drug development and toxicity screening by providing improved sensitivity and predictive accuracy.91 High-throughput liver-on-chip platforms can accommodate multiple devices (e.g., 96 devices per plate), facilitating the simultaneous testing of numerous compounds.92 In addition, integrated sensors allow for real-time assessment of liver function and drug metabolism, enhancing data collection and analysis during drug screening.92 These systems enable the early identification of lead compounds and toxic compounds, allowing for more informed decision-making in the drug development pipeline.93 Conversely, while high-throughput liver-on-chip systems present numerous advantages, challenges remain in standardizing these technologies for widespread adoption in pharmaceutical research.
Our LEADS chip is a liver-on-chip model that demonstrates the critical advantages of organ-on-chip models and overcomes the abovementioned limitations with the usage of primary cells and serves as an excellent preclinical model for NASH drug testing. We have built the liver-on-a-chip using cells differentiated from hepatic stem cells and iPSCs via standard protocols. These renewable cell sources allow extensive experimentation and provide robust and reproducible phenotypes. We adopted a commercially available and a simple microfluidic chip for our model so that this could be easily tested in labs without the need for expensive microfluidics set-up and expertise. We carefully optimized the co-culture conditions and the induction factors to select conditions that support the hepatic functions to establish a functional liver microenvironment and allow for NASH induction. We observed that the levels of induction factors – OA, PA, fructose, PAA and LPS – and the coculture medium affected the levels of steatosis by affecting the size and number of lipid droplets (Fig. 2 and S3†). Interestingly we observed different pathological states of steatosis, Sd-MaS and true MiS, under different induction conditions (Fig. 2). These two states of steatosis have been clinically correlated to the severity of the disease, with macrovesicular and microvesicular steatosis representing the early and the advanced stages of NAFLD, respectively.30,31 Several liver-on-a-chip models that have been developed for NAFLD/NASH have not shown clear imaging data, from where the size and the distribution of the lipid droplets can be appreciated and the status of microvesicular or macrovesicular steatosis could be inferred.30 The microphysiological system (MPS) developed using primary cells report Oil Red O staining for neutral lipids and have provided low-magnification images of the entire organoids; the size and the numbers of lipid droplets in the organoids are indistinguishable.94 Similarly, in the liver lobule chip with dual blood supply, high-magnification images of a small area of the chip are shown.95 From the liver-on-chip or other organoid-based in vitro models that have reported clear staining images for lipid droplets, more often a mixed distribution of lipid droplets is observed32,96 and fall under the category of small-droplet macrovesicular steatosis (Sd-MaS). In a NASH model developed using spheroids from primary cells clear images of lipid droplets in the microtissues were shown. Although the authors claim microvesicular and macrovesicular steatosis, this is based only on the size of the lipid droplets.97 The definition of large-droplet macrovesicular (Ld-MaS), small-droplet macrovesicular (Sd-MaS) and true microvesicular steatosis (MiS) depends on the size and distribution of the lipid droplets, the position of the nucleus and the size of the hepatocyte. The histopathological definition of microvesicular steatosis encompasses distended hepatocytes with numerous small and uniformly sized lipid droplets which give the cytoplasm a foamy appearance and a centrally located nucleus. This is in contrast with the Ld-MaS, where there is a large fat vacuole displacing the nucleus to the periphery. Sd-MaS is considered when there is a mixture of different sizes of lipid droplets in a normal-sized hepatocyte without the discernible features of microvesicular steatosis.37 Our LEADS chip is able to recapitulate these pathological states of steatosis with clear distinguishable features under the conditions specified in Table 3. This demonstrated the capability of our LEADS chip in modelling different stages of NAFLD.
| Type of steatosis | Non-parenchymal cells | Type of NASH induction | Levels of OA and PA at constant levels of fructose at 100 mM, LPS at 10 ng mL−1 and PAA at 10 mM |
|---|---|---|---|
| Macrovesicular steatosis (Ld-MaS) | Absent | Endpoint model | OA – 450 μM |
| PA – 150 μM | |||
| Small droplet macrovesicular steatosis (Sd-MaS) | Present | Endpoint/progression model | OA – 300 μM and higher |
| PA – 300 μM and lower | |||
| Microvesicular steatosis (MiS) | Present | Endpoint model | OA – 150 μM and lower |
| PA – 450 μM and higher |
HCB is a definitive feature of NASH; however, observation and enumeration of HCB in vitro remains a challenge. Previously, Freag et al. (2021) showed the development of HCB in their NASH-on-chip model based on the accumulation of ubiquitinated proteins and displaced nuclei within ballooned hepatocytes.30 However, this study did not evaluate the presence of cytokeratin in ballooned hepatocytes, a major constituent of MDB. To address this limitation, we used an M30 antibody to probe the caspase-cleaved cytokeratin 18 which was found to be colocalized with CK8/18 deposits and ubiquitin. Further based on this observation we validated an imaging-based method for HCB quantification (Fig. 3, S4 and S5†). These experiments established a suitable co-culture medium for optimal hepatic functions and NASH induction (Fig. 4). While refining the induction protocol, we observed that activation of iKCs using LPS and PAA, that are known to contribute to inflammation and ER stress, significantly increased the levels of steatosis (Fig. 5). We also observed that the absence of iKCs and iHSCs led to a different pattern of steatosis, which is Ld-MaS, showing for the first time the involvement of non-parenchymal cells in modulating the pattern of steatosis (Fig. 6). Assessment of the secretome via cytokine profiling showed the various paracrine signaling molecules that are secreted in response to NASH induction (Fig. 7). The patterns of cytokine release during NASH induction in the LEADS chip were consistent with previous clinical investigations and research on in vitro NASH models.30,98–100 Moreover, we could provide a comprehensive overview of cytokine dynamics associated with NASH.
Upon successful establishment of NASH in the chip, we proceeded towards the testing of anti-NASH drugs. The drug testing experiments revealed that the LEADS chip was able to recapitulate some of the observations for these drugs in clinical trials particularly reduction in steatosis and HCB (Fig. 8) despite some indifferences in the resolution of fibrosis, which could be attributed to a strong activation of iHSC upon induction (Fig. 6D). Treatment with pioglitazone on the LEADS chip showed a significant reduction on HCB and fibrosis but had no appreciable effects on steatosis, indicating that these phenomena are not always coupled as conceived by the conventional narrative of NASH pathogenesis in the literature. Concurrently, the LEADS chip recapitulated the clinical response of resmetirom, the first drug approved by the U.S. FDA for treating patients with NASH. Treatment with resmetirom on the chip showed a dose-dependent reduction in steatosis (Fig. 9), which was one of the primary endpoints in the MAESTRO-NASH clinical trial.101,102 These experiments and the data observed on the LEADS chip corroborate with several independent mechanistic studies on NASH as well as offer several new insights into NASH pathogenesis and drug testing.
The LEADS chip is a first-of-a-kind model that is most suited for preclinical drug testing for NASH. The adoption of a simple microfluidics chip, renewable stem cell sources, independent modulation of a variety of NASH induction factors, clear microscopy-based assessment of NASH features and quantification and recapitulation of drug responses observed in clinical trials demonstrate the suitability of the chip for NASH modelling and drug testing. Another advantage of the model is that the adult liver stem cell lines used for producing the hepatobiliary organoids can be generated from donors of different ethnic groups and from donors with specific SNPs implicated in NASH. This would allow the development of personalized treatment strategies across the globe. While we have demonstrated drug testing with a specific set of induction factors that show a good NASH phenotype, the model could be tested at varying levels of NASH induction by different combinations of induction factors to emulate the disease at a clinical setting. Our rationale for optimizing the model parameters offers clear insights into developing such organ-on-chip models for preclinical drug testing.
5. Conclusion
We developed a human liver-on-a-chip – the LEADS chip – via co-culture of adult liver stem cell-derived hepatobiliary organoids, iPSC-derived Kupffer cells and iPSC-derived hepatic stellate cells. Via optimization of co-culture conditions, we obtained a stable and functional liver microenvironment. We induced NASH on the LEADS chip by mimicking conditions of hypernutrition and leaky gut using free fatty acids, fructose, LPS and PAA. Upon induction, we observed steatosis, hepatocellular ballooning injury, inflammation and fibrosis, and quantified using image-based assays and multiplex cytokine assays. We were able to recapitulate pathological states of steatosis and observed that the presence of iKCs and iHSCs had a pronounced effect on the type of steatosis. We tested anti-NASH drugs with different targets and with varied outcomes in clinical trials and observed that the model could recapitulate the responses observed in clinical trials. Taken together, the LEADS chip is an excellent preclinical model for studying NASH and NASH drug testing.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Gowri Manohari Balachander: conceptualization, formal analysis, investigation, methodology, project administration, writing – original draft. Inn Chuan Ng: conceptualization, formal analysis, investigation, methodology, writing – original draft. Roopesh R. Pai: investigation, methodology, writing – original draft. Kartik Mitra: investigation, writing – original draft. Farah Tasnim: investigation, methodology. Yee Siang Lim: investigation, methodology. Royston Kwok: investigation. Yoohyun Song: investigation. Lai Ping Yaw: investigation. Clarissa Bernice Quah: investigation. Junzhe Zhao: investigation. Wahyunia L. Septiana: investigation. Vishnu Goutham Kota: investigation. Yao Teng: investigation. Kexiao Zheng: investigation. Yan Xu: investigation. Sei Hien Lim: resources. Huck Hui Ng: resources. Hanry Yu: conceptualization, funding acquisition, resources, supervision.Conflicts of interest
All authors declare no conflict of interest.Acknowledgements
This work is supported in part by the Institute of Bioengineering & Bioimaging (IBB), Biomedical Research Council, Agency for Science, Technology and Research (A*STAR), A*STAR; IAF (H18/01/a0/017); NMRC (CIRG21nov-0032, CIRG22jul-0018)); SMART CAMP; The Institute for Digital Medicine (WisDM); and Mechanobiology Institute of Singapore (NRF-MSG-2023-0001) funding to HYU. Support from the Science and Engineering Research Board (SERB), Government of India (SRG/2023/001183) to GMB is gratefully acknowledged. ZKX wishes to acknowledge support from the China Scholarship Council (CSC) for his visiting scholarship No. 202004910165. All imaging was performed at the Confocal Microscopy Unit, Yong Loo Lin School of Medicine (YLLSOM), National University of Singapore (NUS). We thank Lee Shu Ying from the Confocal Microscopy Unit, YLLSOM, NUS, for assistance with microscopy. The multiplexing measurements were carried out at the Stem Cell Core Facility which is supported by the Healthy Longevity Translational Research Programme at the YLLSOM, NUS. We thank Dr. Marek Kukumberg and Sabrina Adam from the Stem Cell Core Facility, Healthy Longevity Translational Research Programme, YLLSOM, NUS, for assistance with the multiplex assay.References
- M. Eslam, P. N. Newsome and S. K. Sarin, et al., A New Definition for Metabolic Dysfunction-Associated Fatty Liver Disease: An International Expert Consensus Statement, J. Hepatol., 2020, 73(1), 202–209, DOI:10.1016/j.jhep.2020.03.039.
- Z. Younossi, Q. M. Anstee and M. Marietti, et al., Global burden of NAFLD and NASH: trends, predictions, risk factors and prevention, Nat. Rev. Gastroenterol. Hepatol., 2018, 15(1), 11–20, DOI:10.1038/nrgastro.2017.109.
- Q. M. Anstee, H. L. Reeves and E. Kotsiliti, et al., From NASH to HCC: Current Concepts and Future Challenges, Nat. Rev. Gastroenterol. Hepatol., 2019, 16(7), 411–428, DOI:10.1038/s41575-019-0145-7.
- S. Singh, A. M. Allen and Z. Wang, et al., Fibrosis Progression in Nonalcoholic Fatty Liver vs Nonalcoholic Steatohepatitis: A Systematic Review and Meta-analysis of Paired-Biopsy Studies, Clin. Gastroenterol. Hepatol., 2015, 13(4), 643–654.e9, DOI:10.1016/J.CGH.2014.04.014.
- Z. Younossi, M. Stepanova and J. P. Ong, et al., Nonalcoholic Steatohepatitis Is the Fastest Growing Cause of Hepatocellular Carcinoma in Liver Transplant Candidates, Clin. Gastroenterol. Hepatol., 2019, 17(4), 748–755, DOI:10.1016/j.cgh.2018.05.057 , e3.
- S. K. Asrani, H. Devarbhavi and J. Eaton, et al., Burden of liver diseases in the world, J. Hepatol., 2019, 70(1), 151–171, DOI:10.1016/j.jhep.2018.09.014.
- K. Begriche, J. Massart and M. A. Robin, et al., Mitochondrial Adaptations and Dysfunctions in Nonalcoholic Fatty Liver Disease, Hepatology, 2013, 58(4), 1497–1507, DOI:10.1002/hep.26226.
- Z. Mokhtari, D. L. Gibson and A. Hekmatdoost, Nonalcoholic Fatty Liver Disease, the Gut Microbiome, and Diet, Adv. Nutr., 2017, 8(2), 240–252, DOI:10.3945/an.116.013151.
- R. F. Schwabe, I. Tabas and U. B. Pajvani, Mechanisms of Fibrosis Development in Nonalcoholic Steatohepatitis, Gastroenterology, 2020, 158(7), 1913–1928, DOI:10.1053/j.gastro.2019.11.311.
- G. Parthasarathy, X. Revelo and H. Malhi, Pathogenesis of Nonalcoholic Steatohepatitis: An Overview, Hepatol. Commun., 2020, 4(4), 478–492, DOI:10.1002/hep4.1479.
- D. G. Tiniakos, M. B. Vos and E. M. Brunt, Nonalcoholic Fatty Liver Disease: Pathology and Pathogenesis, Annu. Rev. Pathol.: Mech. Dis., 2010, 5, 145–171, DOI:10.1146/annurev-pathol-121808-102132.
- J. M. Fraile, S. Palliyil and C. Barelle, et al., Non-Alcoholic Steatohepatitis (NASH) - A Review of a Crowded Clinical Landscape, Driven by a Complex Disease, Drug Des., Dev. Ther., 2021, 15, 3997–4009, DOI:10.2147/DDDT.S315724.
- S. A. M. Albhaisi and A. J. Sanyal, New drugs for NASH, Liver Int., 2021, 41(S1), 112–118, DOI:10.1111/liv.14844.
- S. A. Harrison, R. Loomba and J. Dubourg, et al., Clinical Trial Landscape in NASH, Clin. Gastroenterol. Hepatol., 2023, 21(8), 2001–2014, DOI:10.1016/j.cgh.2023.03.041.
- B. A. Neuschwander-Tetri, R. Loomba and A. J. Sanyal, et al., Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): A multicentre, randomised, placebo-controlled trial, Lancet, 2015, 385(9972), 956–965, DOI:10.1016/S0140-6736(14)61933-4.
- Q. M. Anstee, B. A. Neuschwander-Tetri and V. W. S. Wong, et al., Cenicriviroc for the treatment of liver fibrosis in adults with nonalcoholic steatohepatitis: AURORA Phase 3 study design, Contemp. Clin. Trials, 2020, 89, DOI:10.1016/j.cct.2019.105922.
- S. J. Keam, Resmetirom: First Approval, Drugs, 2024, 84(6), 729–735, DOI:10.1007/s40265-024-02045-0.
- S. E. Park, A. Georgescu and D. Huh, Organoids-on-a-chip, Science, 2019, 364(6444), 960–965 CrossRef CAS PubMed.
- F. Kaluthantrige Don and M. Huch, Organoids, Where We Stand and Where We Go, Trends Mol. Med., 2021, 27(5), 416–418, DOI:10.1016/j.molmed.2021.03.001.
- K. Karnawat, R. Parthasarathy and M. Sakhrie, et al., Building in Vitro Models for Mechanistic Understanding of Liver Regeneration in Chronic Liver Diseases, J. Mater. Chem. B, 2024, 12(32), 7669–7691, 10.1039/d4tb00738g.
- F. Tasnim, J. Xing, X. Huang, S. Mo, X. Wei, M.-H. Tan and H. Yu, Generation of mature kupffer cells from human induced pluripotent stem cells, Biomaterials, 2019, 192, 377–391, DOI:10.1016/j.biomaterials.2018.11.016.
- M. Coll, L. Perea, R. Boon, S. B. Leite, J. Vallverdú, I. Mannaerts, A. Smout, A. El Taghdouini, D. Blaya, D. Rodrigo-Torres, I. Graupera, B. Aguilar-Bravo, C. Chesne, M. Najimi, E. Sokal, J. J. Lozano, L. A. van Grunsven, C. M. Verfaillie and P. Sancho-Bru, Generation of Hepatic Stellate Cells from Human Pluripotent Stem Cells Enables In Vitro Modeling of Liver Fibrosis., Cell Stem Cell, 2018, 23(1), 101–113.e7, DOI:10.1016/j.stem.2018.05.027.
- H. H. Ng, Y. S. Chan and W. J. TNG, Derivation of Liver Organoids from Human Pluripotent Stem Cells, 2020 Search PubMed.
- K. A. Rose, N. S. Holman and A. M. Green, et al., Co-culture of Hepatocytes and Kupffer Cells as an in Vitro Model of Inflammation and Drug-Induced Hepatotoxicity, J. Pharm. Sci., 2016, 105(2), 950–964, DOI:10.1016/S0022-3549(15)00192-6.
- J. W. Park, G. Jeong and S. J. Kim, et al., Predictors reflecting the pathological severity of non-alcoholic fatty liver disease: Comprehensive study of clinical and immunohistochemical findings in younger Asian patients, J. Gastroenterol. Hepatol., 2007, 22(4), 491–497, DOI:10.1111/j.1440-1746.2006.04758.x.
- G. Baffy, Kupffer Cells in Non-Alcoholic Fatty Liver Disease: The Emerging View, J. Hepatol., 2009, 51(1), 212–223, DOI:10.1016/j.jhep.2009.03.008.
- J. H. Lefkowitch, J. H. Haythe and N. Regent, Kupffer cell aggregation and perivenular distribution in steatohepatitis, Mod. Pathol., 2002, 15(7), 699–704, DOI:10.1097/01.MP.0000019579.30842.96.
- M. R. Ebrahimkhani, J. A. S. Neiman and M. S. B. Raredon, et al., Bioreactor Technologies to Support Liver Function in Vitro, Adv. Drug Delivery Rev., 2014, 69–70, 132–157, DOI:10.1016/j.addr.2014.02.011.
- D. E. Malarkey, K. Johnson and L. Ryan, et al., New Insights into Functional Aspects of Liver Morphology, Toxicol. Pathol., 2005, 33(1), 27–34, DOI:10.1080/01926230590881826.
- M. S. Freag, B. Namgung and M. E. Reyna Fernandez, et al., Human Nonalcoholic Steatohepatitis on a Chip, Hepatol. Commun., 2021, 5(2), 217–233, DOI:10.1002/hep4.1647.
- R. E. Feaver, B. K. Cole and M. J. Lawson, et al., Development of an in vitro human liver system for interrogating nonalcoholic steatohepatitis, JCI Insight, 2016, 1(20), 18, DOI:10.1172/jci.insight.90954.
- M. Duriez, A. Jacquet and L. Hoet, et al., A 3D human liver model of nonalcoholic steatohepatitis, J. Clin. Transl. Hepatol., 2020, 8(4), 359–370, DOI:10.14218/JCTH.2020.00015.
- M. J. Gómez-Lechón, M. T. Donato and A. Martínez-Romero, et al., A human hepatocellular in vitro model to investigate steatosis, Chem.-Biol. Interact., 2007, 165(2), 106–116, DOI:10.1016/j.cbi.2006.11.004.
- M. J. Dekker, Q. Su and C. Baker, et al., Fructose: a highly lipogenic nutrient implicated in insulin resistance, hepatic steatosis, and the metabolic syndrome, Am. J. Physiol., 2010, 299, 685–694, DOI:10.1152/ajpendo.00283.2010.-As.
- L. Zhao, X. Guo and O. Wang, et al., Fructose and glucose combined with free fatty acids induce metabolic disorders in HepG2 cell: A new model to study the impacts of high-fructose/sucrose and high-fat diets in vitro, Mol. Nutr. Food Res., 2016, 60(4), 909–921, DOI:10.1002/mnfr.201500635.
- L. Hoyles, J. M. Fernández-Real and M. Federici, et al., Molecular phenomics and metagenomics of hepatic steatosis in non-diabetic obese women, Nat. Med., 2018, 24(7), 1070–1080, DOI:10.1038/s41591-018-0061-3.
- S. Tandra, M. M. Yeh and E. M. Brunt, et al., Presence and significance of microvesicular steatosis in nonalcoholic fatty liver disease, J. Hepatol., 2011, 55(3), 654–659, DOI:10.1016/j.jhep.2010.11.021.
- E. M. Brunt, Pathology of fatty liver disease, Mod. Pathol., 2007, 20(1), 40–48, DOI:10.1038/modpathol.3800680.
- C. Lackner, Hepatocellular Ballooning in Nonalcoholic Steatohepatitis: The Pathologist's Perspective, Expert Rev. Gastroenterol. Hepatol., 2011, 5(2), 223–231, DOI:10.1586/egh.11.8.
- F. Rangwala, C. D. Guy and J. Lu, et al., Increased production of sonic hedgehog by ballooned hepatocytes, J. Pathol., 2011, 224(3), 401–410, DOI:10.1002/path.2888.
- E. M. Brunt, A. D. Clouston and Z. Goodman, et al., Complexity of ballooned hepatocyte feature recognition: Defining a training atlas for artificial intelligence-based imaging in NAFLD, J. Hepatol., 2022, 76(5), 1030–1041, DOI:10.1016/j.jhep.2022.01.011.
- E. Buzzetti, M. Pinzani and E. A. Tsochatzis, The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD), Metabolism, 2016, 65(8), 1038–1048, DOI:10.1016/j.metabol.2015.12.012.
- J. K. Dowman, J. W. Tomlinson and P. N. Newsome, Pathogenesis of Non-Alcoholic Fatty Liver Disease, QJM, 2009, 103(2), 71–83, DOI:10.1093/qjmed/hcp158.
- Y.-L. Fang, H. Chen and C.-L. Wang, et al., Pathogenesis of non-alcoholic fatty liver disease in children and adolescence: From “two hit theory” to “multiple hit model”, World J. Gastroenterol., 2018, 24(27), 2974–2983, DOI:10.3748/wjg.v24.i27.2974.
- M. Ricchi, M. R. Odoardi and L. Carulli, et al., Differential effect of oleic and palmitic acid on lipid accumulation and apoptosis in cultured hepatocytes, J. Gastroenterol. Hepatol., 2009, 24(5), 830–840, DOI:10.1111/j.1440-1746.2008.05733.x.
- X. Zeng, M. Zhu and X. Liu, et al., Oleic acid ameliorates palmitic acid induced hepatocellular lipotoxicity by inhibition of ER stress and pyroptosis, Nutr. Metab., 2020, 17, 11, DOI:10.1186/s12986-020-0434-8.
- X. Chen, L. Li and X. Liu, et al., Oleic acid protects saturated fatty acid mediated lipotoxicity in hepatocytes and rat of non-alcoholic steatohepatitis, Life Sci., 2018, 203, 291–304, DOI:10.1016/j.lfs.2018.04.022.
- D. Schmidt-Arras and S. Rose-John, IL-6 pathway in the liver: From physiopathology to therapy, J. Hepatol., 2016, 64(6), 1403–1415, DOI:10.1016/j.jhep.2016.02.004.
- D. M. Xiang, W. Sun and B. F. Ning, et al., The HLF/IL-6/STAT3 feedforward circuit drives hepatic stellate cell activation to promote liver fibrosis, Gut, 2018, 67(9), 1704–1715, DOI:10.1136/gutjnl-2016-313392.
- P. Kagan, M. Sultan and I. Tachlytski, et al., Both MAPK and STAT3 signal transduction pathways are necessary for IL-6-dependent hepatic stellate cells activation, PLoS One, 2017, 12(5), e0176173, DOI:10.1371/journal.pone.0176173.
- M. Mühlbauer, T. S. Weiss and W. E. Thasler, et al., LPS-mediated NFκB activation varies between activated human hepatic stellate cells from different donors, Biochem. Biophys. Res. Commun., 2004, 325(1), 191–197, DOI:10.1016/j.bbrc.2004.10.020.
- A. S. Khazali, A. M. Clark and A. Wells, Inflammatory cytokine IL-8/CXCL8 promotes tumour escape from hepatocyte-induced dormancy, Br. J. Cancer, 2018, 118(4), 566–576, DOI:10.1038/bjc.2017.414.
- Y. Saiman and S. L. Friedman, The Role of Chemokines in Acute Liver Injury, Front. Physiol., 2012, 3, 213, DOI:10.3389/fphys.2012.00213.
- E. Hintermann, M. Bayer and J. M. Pfeilschifter, et al., CXCL10 promotes liver fibrosis by prevention of NK cell mediated hepatic stellate cell inactivation, J. Autoimmun., 2010, 35(4), 424–435, DOI:10.1016/j.jaut.2010.09.003.
- J. Xie, L. Yang and L. Tian, et al., Macrophage Migration Inhibitor Factor Upregulates MCP-1 Expression in an Autocrine Manner in Hepatocytes during Acute Mouse Liver Injury, Sci. Rep., 2016, 6, 27665, DOI:10.1038/srep27665.
- L. G. Alexopoulos, J. Saez-Rodriguez and B. D. Cosgrove, et al., Networks inferred from biochemical data reveal profound differences in toll-like receptor and inflammatory signaling between normal and transformed hepatocytes, Mol. Cell. Proteomics, 2010, 9(9), 1849–1865, DOI:10.1074/mcp.M110.000406.
- T. Zhang, C. J. Guo and Y. Li, et al., Interleukin-1β induces macrophage inflammatory protein-1β expression in human hepatocytes, Cell. Immunol., 2003, 226(1), 45–53, DOI:10.1016/j.cellimm.2003.10.005.
- Y. Koyama and D. A. Brenner, Liver inflammation and fibrosis, J. Clin. Invest., 2017, 127(1), 55–64, DOI:10.1172/JCI88881.
- E. Seki, S. De Minicis and C. H. Österreicher, et al., TLR4 enhances TGF-β signaling and hepatic fibrosis, Nat. Med., 2007, 13(11), 1324–1332, DOI:10.1038/nm1663.
- H. Miao, Y. Zhang and Z. Lu, et al., FOXO1 increases CCL20 to promote NF-κB-dependent lymphocyte chemotaxis, Mol. Endocrinol., 2012, 26(3), 423–437, DOI:10.1210/me.2011-1233.
- X. Chu, Q. Jin and H. Chen, et al., CCL20 is up-regulated in non-alcoholic fatty liver disease fibrosis and is produced by hepatic stellate cells in response to fatty acid loading, J. Transl. Med., 2018, 16(1), 108, DOI:10.1186/s12967-018-1490-y.
- A. Hanson, I. S. Piras and D. Wilhelmsen, et al., Chemokine ligand 20 (CCL20) expression increases with NAFLD stage and hepatic stellate cell activation and is regulated by miR-590-5p, Cytokine, 2019, 123, 154789, DOI:10.1016/j.cyto.2019.154789.
- I. Okazaki, S. Shibata and W. Ando, et al., Sequential Matrix Metalloproteinase-1 Expression Triggered by Infiltrating Monocytic Lineage Cells Modulates Pathophysiological Aspects of Human Nonalcoholic Steatohepatitis</p>, Metalloproteinases Med., 2020, 7, 1–13, DOI:10.2147/MNM.S252991.
- O. Zbodakova, K. Chalupsky and J. Tureckova, et al., Metalloproteinases in liver fibrosis: current insights, Metalloproteinases Med., 2017, 4, 25–35, DOI:10.2147/mnm.s124363.
- W.-K. Syn, K. M. Agboola and M. Swiderska, et al., NKT-associated hedgehog and osteopontin drive fibrogenesis in non-alcoholic fatty liver disease, Gut, 2012, 61(9), 1323–1329, DOI:10.1136/gutjnl-2011-301857.
- Y. Wen, S. Jeong and Q. Xia, et al., Role of Osteopontin in Liver Diseases, Int. J. Biol. Sci., 2016, 12(9), 1121–1128, DOI:10.7150/ijbs.16445.
- W.-K. Syn, S. S. Choi and E. Liaskou, et al., Osteopontin is induced by hedgehog pathway activation and promotes fibrosis progression in nonalcoholic steatohepatitis, Hepatology, 2011, 53(1), 106–115, DOI:10.1002/hep.23998.
- S. Olivares and A. S. Henkel, Endoplasmic reticulum stress induces hepatic plasminogen activator inhibitor 1 in murine nonalcoholic steatohepatitis, FASEB BioAdv., 2020, 2(12), 695–704, DOI:10.1096/fba.2020-00056.
- G. Rivas, B. Hummer-Bair and D. Bezinover, et al., Plasminogen activator inhibitor is significantly elevated in liver transplant recipients with decompensated NASH cirrhosis, BMJ Open Gastroenterol., 2021, 8(1), e000683, DOI:10.1136/bmjgast-2021-000683.
- F. Wandrer, S. Liebig and S. Marhenke, et al., TNF-Receptor-1 inhibition reduces liver steatosis, hepatocellular injury and fibrosis in NAFLD mice, Cell Death Discovery, 2020, 11(3), 212, DOI:10.1038/s41419-020-2411-6.
- S. Lu, Y. Wang and J. Liu, Tumor necrosis factor-α signaling in nonalcoholic steatohepatitis and targeted therapies, J. Genet. Genomics, 2022, 49(4), 269–278, DOI:10.1016/j.jgg.2021.09.009.
- J. M. Hui, A. Hodge and G. C. Farrell, et al., Beyond insulin resistance in NASH: TNF-α or adiponectin?, Hepatology, 2004, 40(1), 46–54, DOI:10.1002/hep.20280.
- Z. Zheng, K. Nakamura and S. Gershbaum, et al., Interacting hepatic PAI-1/tPA gene regulatory pathways influence impaired fibrinolysis severity in obesity, J. Clin. Invest., 2020, 130, 4348–4359, DOI:10.1172/JCI135919.
- P. L. Eriksen, K. L. Thomsen and M. Sørensen, et al., Impaired fibrinolysis without hypercoagulability characterises patients with non-alcoholic fatty liver disease, Thromb. Res., 2022, 213, 9–15, DOI:10.1016/j.thromres.2022.02.023.
- H. Shen, H. Yu and L. Q. Yu, et al., Hepatocyte-derived VEGFA accelerates the progression of non-alcoholic fatty liver disease to hepatocellular carcinoma via activating hepatic stellate cells, Acta Pharmacol. Sin., 2022, 43(11), 2917–2928, DOI:10.1038/s41401-022-00907-5.
- H. Siddiqui, P. Rawal and C. Bihari, et al., Vascular Endothelial Growth Factor Promotes Proliferation of Epithelial Cell Adhesion Molecule–Positive Cells in Nonalcoholic Steatohepatitis, J. Clin. Exp. Hepatol., 2020, 10(4), 275–283, DOI:10.1016/j.jceh.2019.11.011.
- S. Gawrieh, M. Noureddin and N. Loo, et al., Saroglitazar, a PPAR-α/γ Agonist, for Treatment of NAFLD: A Randomized Controlled Double-Blind Phase 2, Trial, 2021, 74(4), 2021, DOI:10.1002/hep.31843/suppinfo.
- Z. M. Younossi, V. Ratziu and R. Loomba, et al., Obeticholic acid for the treatment of non-alcoholic steatohepatitis: interim analysis from a multicentre, randomised, placebo-controlled phase 3 trial, Lancet, 2019, 394(10215), 2184–2196, DOI:10.1016/S0140-6736(19)33041-7.
- B. A. Neuschwander-Tetri, R. Loomba and A. J. Sanyal, et al., Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): A multicentre, randomised, placebo-controlled trial, Lancet, 2015, 385(9972), 956–965, DOI:10.1016/S0140-6736(14)61933-4.
- F. Bril, S. Kalavalapalli and V. C. Clark, et al., Response to Pioglitazone in Patients With Nonalcoholic Steatohepatitis With vs Without Type 2 Diabetes, Clin. Gastroenterol. Hepatol., 2018, 16(4), 558–566.e2, DOI:10.1016/j.cgh.2017.12.001.
- A. J. Sanyal, N. Chalasani and K. V. Kowdley, et al., Pioglitazone, Vitamin E, or Placebo for Nonalcoholic Steatohepatitis, N. Engl. J. Med., 2010, 362(18), 1675–1685, DOI:10.1056/nejmoa0907929.
- Q. M. Anstee, B. A. Neuschwander-Tetri and V. W. S. Wong, et al., Cenicriviroc for the treatment of liver fibrosis in adults with nonalcoholic steatohepatitis: AURORA Phase 3 study design, Contemp. Clin. Trials, 2020, 89, 105922, DOI:10.1016/j.cct.2019.105922.
- V. Ratziu, A. Sanyal and S. A. Harrison, et al., Cenicriviroc Treatment for Adults With Nonalcoholic Steatohepatitis and Fibrosis: Final Analysis of the Phase 2b CENTAUR Study, Hepatology, 2020, 72(3), 892–905, DOI:10.1002/hep.31108/suppinfo.
- S. A. Harrison, M. R. Bashir and C. D. Guy, et al., Resmetirom (MGL-3196) for the treatment of non-alcoholic steatohepatitis: a multicentre, randomised, double-blind, placebo-controlled, phase 2 trial, Lancet, 2019, 394(10213), 2012–2024, DOI:10.1016/S0140-6736(19)32517-6.
- V. Jacques, S. Bolze and S. Hallakou-Bozec, et al., Deuterium-Stabilized (R)-Pioglitazone (PXL065) Is Responsible for Pioglitazone Efficacy in NASH yet Exhibits Little to No PPARγ Activity, Hepatol. Commun., 2021, 5(8), 1412–1425, DOI:10.1002/hep4.1723.
- S. A. Harrison, R. Taub and G. W. Neff, et al., Resmetirom for nonalcoholic fatty liver disease: a randomized, double-blind, placebo-controlled phase 3 trial, Nat. Med., 2023, 29(11), 2919–2928, DOI:10.1038/s41591-023-02603-1.
- C. Ma, Y. Peng and H. Li, Organ-on-a-Chip: A New Paradigm for Drug Development, Trends Pharmacol. Sci., 2021, 42(2), 119–133, DOI:10.1016/j.tips.2020.11.009.
- B. Zhang, A. Korolj and B. F. L. Lai, et al., Advances in Organ-on-a-Chip Engineering, Nat. Rev. Mater., 2018, 3(8), 257–278, DOI:10.1038/s41578-018-0034-7.
- A. M. Diehl and C. Day, Cause, Pathogenesis, and Treatment of Nonalcoholic Steatohepatitis, N. Engl. J. Med., 2017, 377(21), 2063–2072, DOI:10.1056/NEJMra1503519.
- A. M. Diehl, Lessons from animal models of NASH, Hepatol. Res., 2005, 33(2), 138–144, DOI:10.1016/j.hepres.2005.09.022.
- S. R. Meyer, C. J. Zhang, M. A. Garcia, M. C. Procario, S. Yoo, A. L. Jolly, S. Kim, J. Kim, K. Baek, R. D. Kersten, R. J. Fontana and J. Z. Sexton, A High-Throughput Microphysiological Liver Chip System to Model Drug-Induced Liver Injury Using Human Liver Organoids, Gastro Hep Advances, 2024, 3(8), 1045–1053, DOI:10.1016/j.gastha.2024.08.004.
- H. Azizgolshani, J. R. Coppeta and E. M. Vedula, et al., High-throughput organ-on-chip platform with integrated programmable fluid flow and real-time sensing for complex tissue models in drug development workflows, Lab Chip, 2021, 21(8), 1454–1474, 10.1039/d1lc00067e.
- J. Fu, H. Qiu and C. S. Tan, Microfluidic Liver-on-a-Chip for Preclinical Drug Discovery, Pharmaceutics, 2023, 15(4), 1300, DOI:10.3390/pharmaceutics15041300.
- T. Kostrzewski, P. Maraver and L. Ouro-Gnao, et al., A Microphysiological System for Studying Nonalcoholic Steatohepatitis, Hepatol. Commun., 2019, 4(1), 2020, DOI:10.1002/hep4.1450/suppinfo.
- K. Du, S. Li and C. Li, et al., Modeling nonalcoholic fatty liver disease on a liver lobule chip with dual blood supply, Acta Biomater., 2021, 134, 228–239, DOI:10.1016/j.actbio.2021.07.013.
- R. Ouchi, S. Togo and M. Kimura, et al., Modeling Steatohepatitis in Humans with Pluripotent Stem Cell-Derived Organoids, Cell Metab., 2019, 30(2), 374–384.e6, DOI:10.1016/j.cmet.2019.05.007.
- S. Ströbel, R. Kostadinova and K. Fiaschetti-Egli, et al., A 3D primary human cell-based in vitro model of non-alcoholic steatohepatitis for efficacy testing of clinical drug candidates, Sci. Rep., 2021, 11(1), 22765, DOI:10.1038/s41598-021-01951-7.
- T. C. M. Fontes-Cal, R. T. Mattos and N. I. Medeiros, et al., Crosstalk Between Plasma Cytokines, Inflammation, and Liver Damage as a New Strategy to Monitoring NAFLD Progression, Front. Immunol., 2021, 12, 708959, DOI:10.3389/fimmu.2021.708959.
- I. C. Bocsan, M. V. Milaciu and R. M. Pop, et al., Cytokines Genotype-Phenotype Correlation in Nonalcoholic Steatohepatitis, Oxid. Med. Cell. Longevity, 2017, 2017(1), 4297206, DOI:10.1155/2017/4297206.
- T. Kostrzewski, S. Snow and A. L. Battle, et al., Modelling human liver fibrosis in the context of non-alcoholic steatohepatitis using a microphysiological system, Commun. Biol., 2021, 4(1), 1080, DOI:10.1038/s42003-021-02616-x.
- S. A. Harrison, P. Bedossa and C. D. Guy, et al., A Phase 3, Randomized, Controlled Trial of Resmetirom in NASH with Liver Fibrosis, N. Engl. J. Med., 2024, 390(6), 497–509, DOI:10.1056/NEJMoa2309000.
- M. Kokkorakis, C. Boutari and M. A. Hill, et al., Resmetirom, the First Approved Drug for the Management of Metabolic Dysfunction-Associated Steatohepatitis: Trials, Opportunities, and Challenges, Metabolism, 2024, 154, 155835, DOI:10.1016/j.metabol.2024.155835.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5lc00221d |
| ‡ Co-third authors. |
| § Co-fourth authors. |
| This journal is © The Royal Society of Chemistry 2025 |