
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Machine learning-driven antiviral libraries targeting respiratory viruses†
Gabriela
Valle-Núñez
 ,
Raziel
Cedillo-González
,
Juan F.
Avellaneda-Tamayo
,
Fernanda I.
Saldívar-González
,
Diana L.
Prado-Romero
and
José L.
Medina-Franco
*
,
Raziel
Cedillo-González
,
Juan F.
Avellaneda-Tamayo
,
Fernanda I.
Saldívar-González
,
Diana L.
Prado-Romero
and
José L.
Medina-Franco
*
DIFACQUIM Research Group, Department of Pharmacy, School of Chemistry, Universidad Nacional Autónoma de México, Avenida Universidad 3000, Mexico City 04510, Mexico. E-mail: medinajl@unam.mx; Tel: +52-55-5622-3899
First published on 4th April 2025
Abstract
Viral infections represent a significant global health concern. Viral diseases can range from mild symptoms to life-threatening conditions, and the impact of these infections has grown due to increased contagious rates driven by globalization. A prime example is the SARS-CoV-2 pandemic, which emphasized the urgent need to design and develop new antiviral drugs. This study aimed to generate a curated data set of compounds relevant to respiratory infections, focusing on predicting their antiviral activity. Specifically, the study leverages ML classification models to evaluate focused and on-demand compound libraries targeting pathways associated with viral respiratory infections. ML models were trained based on the antiviral biological activity related to respiratory diseases deposited on a major public compound database annotated with biological activity. The models were validated and retrained to classify and design antiviral-focused libraries on seven respiratory targets.
1 Introduction
Viral infections can range from mild, self-limited illnesses to severe, human life-threatening diseases.1 In an era of increased global interdependence, climate change, forced migration and intensified globalization, the rapid replication and high contagion rates of viruses have become a critical concern. The resurgence of infections once believed to be under control, driven by viral genetic mutations and anti-vaccine movements, has further heightened the threat of deadly pandemics.2 To combat this global health crisis, there has been a renewed focus on drug development strategies. The World Health Organization (WHO) has emphasized the challenge posed by limited resources for disease research and development, particularly given the vast array of potential pathogens. WHO has established a prioritized list of diseases with the greatest public health impact, based on their epidemic potential and the lack of effective countermeasures.3 This list is further detailed by the WHO's document “Pathogens Prioritization” which outlines the viral families and their respective members considering the risk of causing Public Health Emergencies of International Concern (PHEICs) or a pandemic (Table S1†).4–6Despite ongoing efforts, developing effective antivirals for most viruses remains a significant challenge due to several key obstacles in antiviral discovery. These include the identification of specific targets, narrow treatment windows, vector spread and control, and the emergence of mutations that contribute to antiviral resistance.7 In response, there has been a growing focus on developing structurally diverse antivirals with enhanced safety profiles, as well as those that retain efficacy against drug-resistant strains. This shift in focus has led to renewed interest in compounds with novel mechanisms of action.8
Acute respiratory disease (ARD) represents a significant portion of acute illnesses and fatalities worldwide. Acute viral respiratory tract infections alone are responsible for approximately 80% of ARD cases.9 Key viral pathogens in this category include influenza, respiratory syncytial virus (RSV), coronaviruses, adenovirus, and rhinovirus, all of which are related to some of the most highlighted diseases on the WHO's prioritized list (Table S1†). While viruses like adenovirus and rhinovirus typically result in lower mortality rates, they contribute substantially to morbidity and place a significant economic burden on healthcare systems.10
The emergence of highly pathogenic coronaviruses, such as the SARS-CoV-2 virus, responsible for the COVID-19 pandemic, has highlighted the severe threat posed by these pathogens. Other coronavirus strains, including those that caused the Severe Acute Respiratory Syndrome (SARS) and Middle East Respiratory Syndrome (MERS) outbreaks, persist as significant public health risks, and place substantial pressure on healthcare systems, especially in regions with high comorbidity rates and limited financial resources.11,12
The COVID-19 pandemic represented one of the most significant threats to global health and stability in recent history, triggering an unprecedented surge in antiviral drug and vaccine research, alongside broader innovations in healthcare and daily life. Antiviral development integrates a diverse array of strategies, spanning well-established therapeutic approaches and emerging targeted interventions.13 This field draws upon both synthetic and natural sources, yielding compounds that exhibit a wide range of chemical structures and mechanisms of action, including direct inhibition of viral replication, immune system modulation, and disruption of host–virus interactions.14–16
Guo et al. reviewed recent advances in natural products (NPs) for antiviral research, with a particular focus on addressing drug resistance.16 Various NPs target essential viral enzymes such as integrase, reverse transcriptase, and protease.17 Flavonoids and polyphenols constitute the largest group of antiviral NPs, followed by diterpenes and triterpenes, with fewer examples found among alkaloids.18 Examples of plant-derived compounds with demonstrated antiviral properties are quercetin, curcumin, and baicalein. Quercetin has shown effectiveness against RSV, MERS-CoV, influenza, and rhinoviruses through inhibition of viral entry and replication.19 Curcumin has been proven to inhibit the SARS-CoV-2 spike glycoprotein, ACE2 receptor, and proteases.20Scutellaria baicalensis root extract is traditionally used in Asia, as an antiviral, antioxidant, and anti-inflammatory. This extract contains baicalein, which has demonstrated inhibition of SARS-CoV-2 main protease (Mpro) activity and viral replication in vitro (Fig. 1).21,22
 | ||
| Fig. 1 Compounds with antiviral activity against targets associated with respiratory infections, identified or developed from different sources. (A) Inhibit viral RNA-dependent RNA polymerase (RdRp), preventing viral replication. (B) Block the viral neuraminidase enzyme, preventing viral release from infected cells. (C) Inhibit viral proteases required for processing viral polyproteins. (D) Plant-derived compounds with antiviral activity through various mechanisms. (E) Inhibits viral cap-dependent endonuclease, blocking viral mRNA synthesis. (F) Blocks the JAK-STAT signaling pathway, reducing excessive immune responses. | ||
Drug repositioning of approved drugs and advanced stages developing molecules has also played a key role in the development of novel antivirals with known molecules, as is the case of SARS-CoV-2.23,24
Computer-aided drug design (CADD) has significantly advanced antiviral discovery. Liao et al. identified five natural compounds – narcissoside, kaempferol-3-O-gentiobioside, rutin, vicin-2, and isoschaftoside – as potential SARS-CoV-2 Mpro inhibitors.25 Generative topographic mapping (GTM) has aided in identifying antiviral motifs and screening virtual chemical libraries, as demonstrated in the design of anti-herpes compounds (herpes simplex virus type 1).26,27 CADD methods have also identified several promising antiviral compounds. These include baricitinib, galidesivir, and molnupiravir. Baricitinib was predicted by artificial intelligence (AI)-driven analysis to inhibit viral entry and inflammation in SARS-CoV-2.28 Galidesivir, an antiviral for Ebola and Zika, was evaluated through structural modeling as a potential inhibitor of SARS-CoV-2 RdRp.29 Molnupiravir (EIDD-2801), a prodrug of β-D-N4-hydroxycytidine, was optimized through docking and molecular dynamics to interfere with SARS-CoV-2 replication (Fig. 1).30 Approved antivirals such as remdesivir, favipiravir, and ritonavir have been repurposed for respiratory viruses through virtual screening (VS), further confirming their potential to inhibit RNA polymerases (Fig. 1).31
Focused virtual libraries of compounds are valuable resources for bioactive compound discovery. These libraries compile data on molecules with potential biological activity, identified through ligand-based and structure-based drug discovery approaches. They play a crucial role in prioritizing candidates for synthesis, biological evaluation, and efficient allocation of resources. Notable recent examples of disease-specific focused virtual libraries include those targeting neglected infectious diseases,32 SARS-CoV-2,33,34 Sirtuin-1 dysregulation,35 and type 2 diabetes mellitus.36
Given the ongoing demand for respiratory-focused antivirals, extensive research has generated a wealth of structure–activity data available in public repositories such as ChEMBL.37,38 This data serves as a crucial input for machine learning (ML) models to design focused libraries for further experimental screening.
The main goal of this study was to design antiviral libraries focused on molecular targets related to respiratory diseases. To achieve this, we trained, retrained and validated ML classification models using bioactivity data from ChEMBL 33.37,38 The predictive models were used to filter compound libraries from diverse sources. As part of the data preparation to train the ML models, the chemical data sets were analyzed and characterized in terms of chemical diversity and coverage in chemical space using chemoinformatics methods. The resulting antiviral-focused chemical libraries, which are freely available in the public domain, offer valuable starting points for further computational and/or experimental screening, which is the next logical step of this study.
2 Methods
The methodology followed in this study is outlined schematically in Fig. 2 and is detailed in the subsequent sections. | ||
| Fig. 2 Workflow followed in this study to design antiviral focused libraries. | ||
2.1 Data acquisition and preparation
Using the ChEMBL Application Programming Interface (API),38 we retrieved all compounds from ChEMBL 33 (updated June 2023) associated with 13 viral targets linked to respiratory diseases. Molecular structures of these compounds were encoded in Simplified Molecular Input Line Entry System (SMILES) format.39 A specific acronym was assigned to each virus with a target of interest, based on its strain, ensuring consistent and accurate identification throughout the analysis. Table 1 lists the names and target IDs for each of the 13 targets.| Family | Virus | Acronym | ChEMBL target ID |
|---|---|---|---|
| Coronaviridae | Feline coronavirus | FCoV | CHEMBL612744, CHEMBL4295624 |
| Human coronavirus 229E | HCoV-229E | CHEMBL613837, CHEMBL4888440 | |
| Human coronavirus NL63 | HCoV-NL63 | CHEMBL3232683 | |
| Middle East respiratory syndrome-related coronavirus | MERS-CoV | CHEMBL4296578, CHEMBL4295557 | |
| Severe acute respiratory syndrome coronavirus | SARS-CoV | CHEMBL4802007 | |
| Severe acute respiratory syndrome coronavirus 2 | SARS-CoV-2 | CHEMBL4888460, CHEMBL5169223, CHEMBL4303835 | |
| Picornaviridae | Enterovirus A71 | HEV-71 | CHEMBL612436, CHEMBL4295606, CHEMBL4295525 |
| Human rhinovirus | HRV | CHEMBL613760, CHEMBL2857, CHEMBL612470 | |
| Paramyxoviridae | Human parainfluenza virus 1 | HPIV-1 | CHEMBL1764934 |
| Pneumoviridae | Human respiratory syncytial virus | HRSV | CHEMBL4635143, CHEMBL2364165, CHEMBL4630897 |
| Orthomyxoviridae | Influenza A virus | IAV | CHEMBL613740, CHEMBL612610, CHEMBL2367089 |
| Influenza B virus | IBV | CHEMBL613129, CHEMBL4295840, CHEMBL2028641 | |
| Paramyxoviridae | Henipavirus nipahense | NiV | CHEMBL6047, CHEMBL615055 |
For compounds with multiple recorded biological activity values (“standard value”) against a target, we ranked these values from smallest to largest to ensure consistency in the data. The pIC50 was calculated for each compound, and compounds were classified according to the following criteria:
(a) IC50 ≤ 10 μM: were labeled as “Inhibitor”.
(b) 10 μM < IC50 < 20 μM: were labeled as “Unknown”.
(c) IC50 ≥ 20 μM: were labeled as “No_Activity”.
If a single category represented at least 80% of the recorded data for a compound, that category was used; otherwise, the label “Mixed” was assigned. Compounds with fewer than five data points retained their original classification, with “Mixed” assigned if they were labeled across multiple categories.
Additionally, one supplementary compound database was assembled, containing approved antivirals from DrugBank 5.1.12.40 Compounds from ChEMBL associated with each viral target, along with those from DrugBank, were compiled into two collections.
A comprehensive data curation process was applied to data sets to ensure data integrity. Compounds with null values, empty entries, or duplicates were removed, resulting in a final count of 4521 compounds from ChEMBL 33, and 92 approved antivirals from DrugBank. Molecular structures were standardized using RDKit version 2024.03.5,41 and MolVS,42 following a well-established and used standardization protocol.43 Data sets and code notebooks are publicly accessible through DIFACQUIM's GitHub repository at https://github.com/DIFACQUIM/antiviral_ML.
2.2 Data modelability and target selection
To assess the feasibility of developing binary classification models, we calculated the modelability index (MODI), proposed by Golbraikh et al.44 MODI measures the proportion of compounds in a data set whose nearest neighbor belongs to the same class within a defined feature space. We calculated the MODI values for the ChEMBL data sets using Molecular ACCess System (MACCS) keys (166 bits)45 and Morgan Chiral of radius 2 (2048 bits) fingerprints,46 using the RDKit, NumPy, pandas, and SciPy libraries for Python 3. For each target in the ChEMBL data sets, MODI was calculated using two approaches: (1) including compounds classified as “Mixed” in the overall classification, and (2) excluding them (Table S2†).Target selection for predictive model development was guided by the criteria established by Sánchez-Cruz and Medina-Franco.47 According to these guidelines, a target was deemed suitable for predictive modeling if it included at least 30 active and 30 inactive compounds, and if it had a MODI score of 0.7 or higher for at least one molecular representation. Based on these criteria, we selected the seven targets listed in Table S2† for the construction of predictive models.
2.3 Chemoinformatic characterization of training data sets of selected targets
For each training data set of selected targets, active compounds were collected to perform a chemoinformatic characterization, detailed hereunder.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P), topological polar surface area (TPSA), molecular weight (MW), number of saturated rings, fraction of sp3 carbon atoms (CSP3), number of heavy atoms, number of rings systems, number of alicyclic rings formed by carbon atoms, number of alicyclic rings that include heteroatoms, number of heteroatoms, rotatable bond fraction, number of aromatic rings formed by carbon atoms, number of aromatic rings that include heteroatoms. Additionally, 11 descriptors were calculated using MOE: the number of acid atoms, aromatic atoms, basic atoms, nitrogen, oxygen, bromine, chlorine, fluorine, iodine, the fraction of rotatable bonds, and the number of chiral centers.
P), topological polar surface area (TPSA), molecular weight (MW), number of saturated rings, fraction of sp3 carbon atoms (CSP3), number of heavy atoms, number of rings systems, number of alicyclic rings formed by carbon atoms, number of alicyclic rings that include heteroatoms, number of heteroatoms, rotatable bond fraction, number of aromatic rings formed by carbon atoms, number of aromatic rings that include heteroatoms. Additionally, 11 descriptors were calculated using MOE: the number of acid atoms, aromatic atoms, basic atoms, nitrogen, oxygen, bromine, chlorine, fluorine, iodine, the fraction of rotatable bonds, and the number of chiral centers.
2.4 Machine learning models
To transform the activity data into a binary format, compounds labeled as “Mixed” and “Unknown” in the ChEMBL data set were discarded, yielding two classes: “Inhibitor” = 1 and “No_Activity” = 0. Then, we computed Morgan Chiral of radius 2 (2048 bits) fingerprint with RDKit,46 and 19 drug-likeness descriptors from the Datamol library.51 These descriptors included Lipinski-related parameters52 and other descriptors of pharmaceutical relevance: MW, CSP3, HBA, HBD, number of rings, number of heteroatoms, number of heavy atoms, number of rotatable bonds, TPSA, logP, number of aliphatic carbocycles, number of aliphatic heterocycles, number of aliphatic rings, number of aromatic carbocycles, number of aromatic heterocycles, number of aromatic rings, number of saturated carbocycles, number of saturated heterocycles, and number of saturated rings.
Only data corresponding to the seven selected targets (Table S2†) was filtered for model building. To evaluate and reduce multicollinearity, the Pearson correlation between descriptors was computed. The second descriptor was discarded if any pair showed a correlation above 0.8, prioritizing drug-likeness relevance.
For supervised binary classification modeling, we employed PyCaret version 3.3.2 for Python to develop models using 15 different ML algorithms (Table S3 in the ESI†).53 Each model was trained on ChEMBL data for the selected targets, associated with a binary activity label (active/inactive). Morgan Chiral of radius 2 (2048 bits) fingerprint and physicochemical descriptors were used as molecular representation, with PyCaret's default hyperparameter settings.
Normalization, fold generation, and imbalance correction were achieved using z-score normalization, stratified k-fold cross-validation, and the Adaptive Synthetic Sampling (ADASYN) algorithm, respectively.54 Additionally, we mitigated the risk of overfitting by enabling an early stopping mechanism to ensure that the models remain capable of generalizing well to new data points.
2.5 Training, test, and validation data sets
The predictive models generated for each target were evaluated by internal and external validations. Data sets were divided into training (80%) and test (20%) sets, using DeepChem library for Python,55,56 selecting Morgan Chiral of radius 2 (1024 bits) fingerprint. The internal validation of all the models was conducted by cross-validation, in which a new training/testing split multiple times from the available data is chosen. The external validation of all the models was conducted with the 20 percent of unseen testing properties. For both validations, the following metrics were calculated with PyCaret: accuracy, Area Under the Curve (AUC), recall, precision, F1 score, kappa, Matthew's Correlation Coefficient (MCC) and Balanced Accuracy (BA). All seven metrics are statistical indicators of quality, used for model evaluation, offering insight into prediction accuracy with a focus on the active class.57For both validations, we obtained the MCC and calculated its average for all models, so as to select the three best architectures for the data sets studied in this analysis (Table S4†). MCC is a robust metric for assessing the quality of binary classification models, ranging from −1 to 1. A value of 1 indicates perfect classification, 0 corresponds to random predictions, and −1 represents completely inverse predictions. The architectures selected for modeling were retrained on the complete data set corresponding to each target, aiming to significantly enhance the MCC and improve model generalization by optimizing performance. This retraining process utilized the same hyperparameters as those in the initial model construction and was applied to predict the final antiviral activity class of the data set assembled for VS, as described in Section 2.7. Cross validation was performed to assess the effectiveness of this retraining by obtaining the MCC retraining value.
2.6 Consensus ML models
For each target, the three top-performing ML models selected and retrained as outlined in Section 2.5, were combined to generate a consensus model. These consensus models underwent internal validation through cross-validation to calculate the MCC value using Scikit-Learn50 functions, enabling performance comparison with each individual model.2.7 Classification and design of antiviral focused libraries
In order to classify and design antiviral-focused libraries, first a total of 22 diverse focused and commercial chemical libraries from various online sources (Table 2) were compiled. The assembled database contained 339040 compounds after curation (Table 2), that were classified and filtered using the predictive models developed for each target to assign a final antiviral activity class (see Section 2.5).
| Database | Acronym | Description | Compounds after curation |
|---|---|---|---|
|
a Number of compounds before curation: 396595.
|
|||
| ChemDiv coronavirus library58 | ChD_covL | Collection of small molecules with potential antiviral activity against coronavirus | 20750 |
| ChemDiv antiviral library59 | ChD_AvL | Collection of small molecules with potential antiviral activity, targeting over 50 key proteins in viruses | 64958 |
| OTAVA drug-like green collection60 | OT_DLGC | Drug-like green collection compound library, curated based on screening compounds for prompt delivery and pre-formatted according to Lipinski's rule of five | 169356 |
| Enamine antiviral library61 | Ena_AvL | Collection of molecules designed for discovery of new nucleoside-like antivirals | 3200 |
| ChemSpace discovery diversity set62 | ChE_DDS | Collection of small molecules that are synthesized from in-house building blocks using carefully developed and optimized reactions | 10000 |
| LifeChemicals helicase targeted library63 | LC_HTL | Collection of structurally diverse molecules with potential activity against key helicase-related drug targets, selected by a chemoinformatics team through in silico molecular docking | 3291 |
| LifeChemicals helicase focused library63 | LC_HFL | A curated collection of compounds targeting helicases, including viral and genetic disorder-related enzymes like hepatitis C NS3 and Werner syndrome helicases. Compounds were selected based on structural similarity (84% Tanimoto threshold) | 3665 |
| LifeChemicals 2019-nCoV papain-like protease (PLP) targeted library64 | LC_plpL | A curated collection of drug-like compounds designed to target the PLP of SARS-CoV-2, using docking-based screening without constraints. Compounds were filtered for binding accuracy and removed if they were PAINs, toxic, or reactive | 1736 |
| LifeChemicals DNA polymerase targeted library65 | LC_dptL | Library of structurally diverse compounds targeting DNA polymerase-related drug targets, developed using pharmacophore-driven screening | 628 |
| LifeChemicals polymerase focused library 15 polymerase assays65 | LC_polL | A library of molecules identified for potential polymerase inhibition, created by screening 4567 active compounds from 15 polymerase assays targeting RNA and DNA polymerases. Compounds were selected using Tanimoto similarity from the life chemicals HTS compound collection and ranked by predicted activity | 15676 |
| LifeChemicals polymerase focused library similarity to ChEMBL database65 | LC_polsL | A library of drug-like screening compounds selected through a 2D fingerprint similarity search (Tanimoto index > 85%) against a reference set of 20000 compounds from the ChEMBL database, all with reported activity against DNA and RNA polymerase targets |
13608 |
| LifeChemicals SARS coronavirus focused library66 | LC_covL | A curated collection of small-molecule compounds selected through a 2D fingerprint similarity search, targeting key SARS-CoV proteins. The compounds were chosen based on activity criteria from a reference set of 300 known SARS inhibitors | 436 |
| LifeChemicals 2019-nCoV main protease targeted library67 | LC_mproL | A curated collection of drug-like compounds designed to target the main protease of SARS-CoV-2, using docking-based screening without constraints. Toxicophore filters were applied, while peptide-like structures were retained to enhance binding potential | 2338 |
| LifeChemicals antiviral targeted library68 | LC_AVL | A curated library of diverse compounds identified through structure-based screening, targeting antiviral proteins like hepatitis B core protein and influenza A PA endonuclease. Developed using phase modeling and life chemicals' HTS collection, with customization options available | 1350 |
| LifeChemicals merged antiviral screening superset69 | LC_MASS | Data set of small-molecule compounds consolidated into individual screening subsets for various viral diseases, providing a comprehensive resource in one collection | 45546 |
| LifeChemicals antiviral library combined ligand-based and structure-based approaches70 | LC_ALCLBSBA | A curated library of potential antiviral agents, designed using protein crystal structures of key viral targets. Selected through glide docking and UNITY pharmacophore searches, with PAINs and reactive compounds excluded | 3514 |
| LifeChemicals antiviral screening compound library 2D similarity70 | LC_ASCL2DS | The antiviral screening compound library was designed using a 2D fingerprint similarity search against a reference set of 46518 biologically active compounds from therapeutically relevant viral assays, covering various virus species and their target proteins |
15455 |
| LifeChemicals bioactive compound library71 | LC_BCL | A collection of structurally diverse screening compounds, each with confirmed biological activity against approximately 600 pharmaceutical targets | 9897 |
| LifeChemicals EF1A targeted library GDP site72 | LC_gdp | Library that includes compounds selected through docking-based VS of GDP site on the eEF1A protein. The compounds have high predicted affinity, are Ro5-compliant, and exclude PAINS, toxic, or reactive groups. Subsets for each binding site are provided with docking scores | 1267 |
| LifeChemicals EF1A targeted library EF1B site73 | LC_ef1b | Library that includes compounds selected through docking-based VS of EF1B site on the eEF1A protein. The compounds have high predicted affinity, are Ro5-compliant, and exclude PAINS, toxic, or reactive groups. Subsets for each binding site are provided with docking scores | 1544 |
| LifeChemicals pre-plated coronavirus COVID-19 screening set-384 well73 | LC_cov19 | Screening set consists of drug-like compounds from the 2019-nCoV Mpro targeted library, designed to support anti-coronavirus drug discovery efforts | 2300 |
| LifeChemicals preplated helicase screening set 6080 cmpds 384 well73 | LC_PHSS384 | Screening sets that include drug-like small-molecule compounds with potential helicase-related activity for drug discovery targeting infectious diseases and cancer. Alternatively, two smaller, non-overlapping subsets of 3520 and 2560 helicase-focused molecules are also available for separate purchase | 6080 |
| General screening antiviral data seta | VS data set | Data set containing only unique structures from all chemical libraries | 339040 |
2.8 Distance to model
To establish a quantitative measure that relates to the applicability domain of the classification models, the similarity or distance for each predicted molecule to the training set was computed. Since the models were constructed using Morgan fingerprints and physicochemical properties, two types of similarity metrics were calculated. Jaccard distance was calculated using Morgan Chiral of radius 2 (2048 bits) fingerprint. While physicochemical properties' distance to the model was calculated with Euclidean distance, based on the preserved drug-like descriptors for each model architecture during the construction process, as outlined in Section 2.4. All predictions were categorized into four quartiles based on their mean Jaccard or Euclidean distance, as appropriate, from the compounds in the retraining set.74 To assess this quartile, the mean distance between the predicted molecule and all compounds in the retraining set was compared with the resulting quartiles from the intraset distances of the corresponding retraining set. If the resulting distance fell into the distances from the training set, the same quartile was assigned; otherwise, the predicted compound was labeled as “Out.”2.9 ADMET properties calculation
Absorption, distribution, metabolism, excretion, and toxicity (ADMET) properties were calculated for the VS data set, generated by the assembly of focused and commercial libraries (see Section 2.7). This was accomplished using ADMET-AI,75,76 utilizing the Python library for local predictions. ADMET-AI computes eight physicochemical properties with RDKit and predicts forty-one ADMET properties through its Chemprop-RDKit graph neural networks.3 Results and discussion
3.1 Data acquisition and preparation
Fig. 3 presents an overview of the initial data set, which includes 4521 compounds retrieved from ChEMBL 33. These compounds are associated with 32 distinct viral targets across 13 viruses implicated in respiratory infections. Among these, IAV is the most represented, with 1968 compounds, followed by SARS-CoV-2, with 1139 compounds. This distribution underscores a predominant research focus on these viruses, likely driven by their significant global impact, recurrent outbreaks, and the prioritization of pandemic-related research efforts. Indeed, the data set highlights a strong emphasis on highly studied targets such as proteases and polymerases, which play critical roles in viral replication and are central to current antiviral strategies. Conversely, targets with minimal representation, such as helicases and spike glycoproteins, represent potential gaps in current research and may offer promising avenues for future drug discovery. Interestingly, the inclusion of compounds targeting less-studied (or reported in ChEMBL) viruses, such as HRV (487 compounds) and IBV (228 compounds), indicates a growing interest in broad-spectrum antiviral strategies. This trend suggests a shift towards addressing a wider range of respiratory viruses, which could enhance preparedness for emerging infections. | ||
| Fig. 3 Overview of the viral target multiverse as reported in ChEMBL 33 (updated June 2023). | ||
3.2 Data modelability and target selection
As summarized in Table 3, seven out of 32 targets related to respiratory diseases were selected according to the criteria detailed in Section 2.2. Although the IAV M2 proton channel target did not meet the criterion of having at least 30 inactive compounds, it was selected as a test case to explore the implications of this criterion. This decision was based on the observation that many targets in the data set faced similar limitations—failing to meet the required number of active or inactive compounds—yet still achieved a MODI score of 0.7 or higher. The premise was that models built for such targets might exhibit different performance characteristics. The details of this analysis and its implications are further discussed in Section 3.4.| Target | Organism | Count | pIC50 median | Active | Inactive | MODI MACCS keys (166 bits) | MODI Morgan Chiral of radius 2 (2048 bits) |
|---|---|---|---|---|---|---|---|
| M2 proton channel | IAV | 92 | 5.45 | 68 | 24 | 0.82 | 0.83 |
| Mpro | SARS-CoV | 197 | 4.52 | 77 | 120 | 0.66 | 0.77 |
| SARS-CoV-2 | 815 | 6.35 | 651 | 164 | 0.88 | 0.91 | |
| Neuraminidase | IAV | 1123 | 5.72 | 733 | 390 | 0.88 | 0.91 |
| IBV | 202 | 5.47 | 132 | 70 | 0.72 | 0.71 | |
| Polymerase (PA) | IAV | 256 | 5.40 | 151 | 105 | 0.84 | 0.88 |
| Protease | HRV | 389 | 5.96 | 298 | 91 | 0.83 | 0.85 |
Targets such as SARS-CoV-2 Mpro and IAV neuraminidase exhibited the highest MODI scores (0.88 for MACCS keys (166 bits) and 0.91 for Morgan Chiral of radius 2 (2048 bits)), indicating strong modelability. In contrast, targets like SARS-CoV Mpro, which was selected for its relatively higher MODI score with Morgan Chiral of radius 2 compared to MACCS keys, or IBV neuraminidase, which had lower MODI scores (0.72 for MACCS keys and 0.71 for Morgan Chiral of radius 2), suggest potentially more challenging modelability. Notably, when comparing the performance of the two fingerprints, most targets showed slightly better modelability with Morgan Chiral of radius 2, suggesting that these fingerprints capture relevant chemical features more effectively for these targets.
The ratio of active to inactive compounds also appears to influence the MODI score. For instance, IAV neuraminidase, with a high number of both active (733) and inactive (390) compounds, had a correspondingly high MODI score. Additionally, the relationship between the median pIC50 values and the MODI scores is worth highlighting. For example, SARS-CoV-2 Mpro, with the highest median pIC50 value (6.35), aligned with its strong modelability, whereas targets with lower pIC50 values may exhibit more variable performance. Similarly, the total number of compounds may serve as another important indicator of modelability. For example, SARS-CoV-2 Mpro, with the second largest data set, likely benefits from a richer data set for model training. Conversely, smaller data sets, such as IAV M2 proton channel, may result in reduced predictive performance due to limited data diversity, as discussed further in the next sections. It is also notable that targets, such as IBV neuraminidase, had lower MODI scores despite a reasonably balanced data set. This discrepancy could be attributed to factors such as structural complexity or compound heterogeneity, which may pose additional challenges for predictive modeling.
3.3 Chemoinformatic characterization of training data sets
For the seven selected data sets, we performed a characterization of the structural content, diversity and coverage in chemical space using different structural representations and descriptors (e.g., chemical multiverse).It is important to emphasize that the ML models were constructed using different sets of physicochemical properties tailored to each target. This variation could significantly influence the data modelability, as certain properties might be more relevant for specific viral targets. For future research, exploring the impact of these individual properties on the performance of the models could provide deeper insights and help refine predictive frameworks.
 | ||
| Fig. 4 Absolute and relative percentage frequencies of the most frequent scaffolds in compounds with known activity for seven selected antiviral targets. Prominent scaffolds are categorized by target, highlighting key structural features in the data set used for ML model development. | ||
More details about the most frequent scaffolds in the training data sets for each target are illustrated in Fig. S2† after following the curation and standardization processes.
Antiviral drugs commonly exhibit diverse structural scaffolds, including adenine derivatives and privileged frameworks that facilitate interactions with multiple viral mechanisms. These compounds frequently feature key atoms such as nitrogen, oxygen, and carbon, which play critical roles in their biological activity and contribute to their structural diversity.78
As presented in the ESI, Fig. S3† shows the most frequent ring systems in the training data sets, while Fig. S4 and S5† depict the predominant scaffolds and ring systems in the VS data set, respectively. Not surprisingly, the benzene ring was the most frequent scaffold and ring system, reflecting its ubiquitous presence across the chemical data set. Notably, the top ring systems in the VS data set has a high prevalence of nitrogen-containing rings, a feature that holds significant promise for enhancing antiviral activity due to their well-established role in molecular recognition and binding to viral targets.79,80
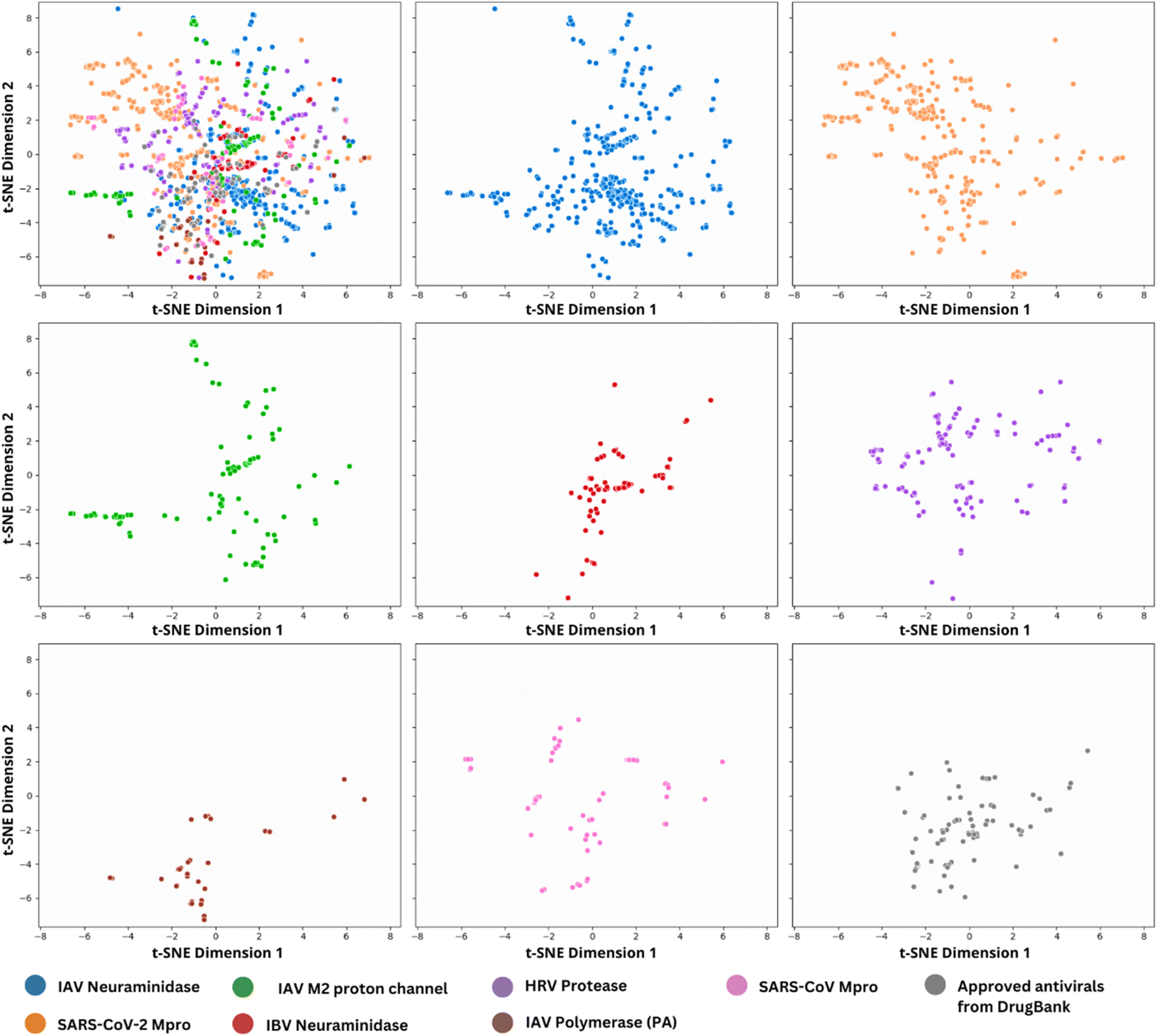 | ||
| Fig. 5 Chemical multiverse visualization of seven antiviral data sets focused on different targets as compared with approved antivirals from DrugBank. The visualization is done with t-SNE using Morgan Chiral of radius 2 (2048 bits) fingerprint. On the upper left are illustrated superimposed data sets, followed by individual data sets using the same coordinates for all of them. | ||
3.4 Machine learning models
Table S6 in the ESI† summarizes the MCC values from each validation phase for the top three best-performing architectures selected for each target. To determine the optimal model, we prioritized the MCC values obtained during the retraining phase (refer to Methods Section 2.4 for further details). As shown in Table S6,† MCC values improved in general after the retraining phase, with the AdaBoost model for IBV neuraminidase standing out as a particularly strong performer. These results showed the capability of the models to accurately identify promising compounds, underscoring their potential utility in antiviral drug discovery.According to the discussion of data modelability in Section 3.2, as expected, the IAV M2 proton channel target had the lowest MCC values for all three top models in each validation phase. This result emphasized the importance of the minimum amount of compounds and reinforced the relevance of implementing several criteria while selecting and analyzing molecular targets to develop reliable and useful ML models.
As illustrated in Fig. 6, some models were robust for a few targets at different validation phases but not in all of them. For instance, the AdaBoost classifier performed best for IBV neuraminidase during retraining while others like the Support Vector Machine (SVM – linear kernel) performed best for IAV polymerase (PA) during the internal validation of the training process. Moreover, this model showed consistent MCC values across training and retraining. This consistency suggests robust model performance and generalizability.
 | ||
| Fig. 6 Comparison of MCC per target, best-fit model (highlighted in yellow in Table S6†), and validation phase. | ||
3.5 Consensus machine learning models
The three best-performing models discussed in Section 3.4 were combined to create a consensus model for each target. Table S6 in the ESI† summarizes the MCC values calculated for the consensus model in comparison with those obtained from each of the top three models individually. In general, the consensus models improved the MCC value or maintained it at a comparable level. This result underscored the robustness of combining predictions from diverse algorithms, which likely reduces individual model biases. Indeed, combining outcomes from complementary methodologies has shown advantages in several areas of chemoinformatics.82 A notable improvement was obtained for SARS-CoV Mpro, where the consensus model surpassed the performance of any single model, further validating this approach as a reliable strategy for enhancing predictive accuracy. However, a few exceptions were noted. For example, the IAV M2 proton channel exhibited the lowest MCC value for the consensus model. This result aligns with previously discussed limitations for the IAV M2 proton channel, including the small and imbalanced data set (see Section 3.2 for further details). Such cases highlight that while consensus models generally provide stability and enhancement, their effectiveness depends on the quality and representativeness of the input data.3.6 Classification of antiviral-focused libraries
The prediction of antiviral activity for the compounds from the assembled general screening data set (VS data set) (Table 2) led to the design of seven individual antiviral-focused libraries for each target. The design consisted in the calculation of two outputs using PyCaret's prediction function: binary values (0/1) for labeling, indicating whether a compound is predicted (classified) to be active (1) or inactive (0); and a prediction score, which represents the probability of the compound belonging to the active class (for further details, see Section 2.5). Table 4 summarizes the results of these predictions from the VS data set for the seven antiviral targets. The table includes the number of compounds predicted to be active by one, two, and three models and for the top, second, and third best models, as well as its relative frequency.| Target | Number of predicted active compounds bya | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 3 models | 2 models | 1 model | Top best model | Active | Second best model | Active | Third best model | Active | |
| a Number in parenthesis is the relative percentage frequency. | |||||||||
| IAV_polymerase (PA) | 9234 (2.72) | 11272 (3.32) |
26409 (7.79) |
SVM – linear kernel | 22295 (6.58) |
Extra trees classifier | 26444 (7.80) |
Gradient boosting classifier | 27916 (8.23) |
| HRV_protease | 225751 (66.59) |
69364 (20.46) |
24034 (7.09) |
Gradient boosting classifier | 302149 (89.12) |
Extreme gradient boosting | 284152 (83.81) |
AdaBoost classifier | 253714 (74.83) |
| IAV_M2 proton channel | 13296 (3.92) |
174286 (51.41) |
146457 (43.20) |
Quadratic discriminant analysis | 326809 (96.39) |
K neighbors classifier | 26209 (7.73) |
Dummy classifier | 181899 (53.65) |
| SARS-CoV_Mpro | 16660 (4.91) |
46894 (13.83) |
103702 (30.59) |
Linear discriminant analysis | 49992 (14.75) |
K neighbors classifier | 112286 (33.12) |
SVM – linear kernel | 85192 (25.13) |
| SARS-CoV-2_Mpro | 169434 (49.97) |
79272 (23.38) |
52692 (15.54) |
Light gradient boosting machine | 245950 (72.54) |
Extreme gradient boosting | 244792 (72.20) |
Decision tree classifier | 228796 (67.48) |
| IAV_neuraminidase | 8101 (2.39) | 21190 (6.25) |
120020 (35.40) |
Logistic regression | 133100 (39.26) |
Random forest classifier | 33079 (9.76) |
Extra trees classifier | 20524 (6.05) |
| IBV_neuraminidase | 1054 (0.31) | 32091 (9.47) |
137600 (40.59) |
AdaBoost classifier | 162085 (47.81) |
K neighbors classifier | 3167 (0.93) | Extra trees classifier | 39692 (11.71) |
| Range | IAV_polymerase (PA) | SARS-CoV-2_Mpro | ||||
|---|---|---|---|---|---|---|
| Total | Best model (actives) | Consensus (actives) | Total | Best model (actives) | Consensus (actives) | |
| Out | 4855 | 310 | 125 | 1162 | 881 | 593 |
| Q1 | 11 | — | — | 3 | 2 | 2 |
| Q2 | 660 | 44 | 16 | 3 | 2 | 1 |
| Q3 | 14083 |
926 | 410 | 19210 |
13975 |
9622 |
| Q4 | 319431 |
21015 |
8683 | 318662 |
231090 |
159216 |
| Range | HRV_protease | IAV_neuraminidase | ||||
|---|---|---|---|---|---|---|
| Total | Best model (actives) | Consensus (actives) | Total | Best model (actives) | Consensus (actives) | |
| Out | 2883 | 2569 | 1936 | 1162 | 476 | 35 |
| Q1 | 404 | 371 | 268 | 3 | 1 | — |
| Q2 | 3552 | 3224 | 2469 | 20 | 8 | 1 |
| Q3 | 28978 |
25835 |
19322 |
28109 |
10786 |
611 |
| Q4 | 303223 |
270150 |
201756 |
309746 |
121829 |
7454 |
| Range | IAV_M2 proton channel | IBV_neuraminidase | ||||
|---|---|---|---|---|---|---|
| Total | Best model (actives) | Consensus (actives) | Total | Best model (actives) | Consensus (actives) | |
| Out | 5277 | 5086 | 3537 | 320156 |
152989 |
997 |
| Q1 | 2879 | 2776 | 1981 | — | — | — |
| Q2 | 119392 |
115160 |
79248 |
— | — | — |
| Q3 | 149663 |
144277 |
99610 |
2 | 1 | — |
| Q4 | 61829 |
59510 |
41375 |
18882 |
9095 | 57 |
| Range | SARS-CoV_Mpro | ||
|---|---|---|---|
| Total | Best model (actives) | Consensus (actives) | |
| Out | 1506 | 210 | 55 |
| Q1 | 83 | 12 | 2 |
| Q2 | 11376 |
1676 | 597 |
| Q3 | 195827 |
28807 |
9526 |
| Q4 | 130248 |
19287 |
6480 |
| Range | IAV_polymerase (PA) | SARS-CoV-2_Mpro | ||||
|---|---|---|---|---|---|---|
| Total | Best model (actives) | Consensus (actives) | Total | Best model (actives) | Consensus (actives) | |
| Out | 1360 | 95 | 34 | 85 | 56 | 44 |
| Q1 | 78691 |
5356 | 2298 | 102608 |
74364 |
51313 |
| Q2 | 83268 |
5609 | 2276 | 54992 |
39935 |
27668 |
| Q3 | 97040 |
6262 | 2614 | 85659 |
62136 |
42693 |
| Q4 | 78681 |
4973 | 2012 | 95696 |
69459 |
47716 |
| Range | HRV_rotease | IAV_neuraminidase | ||||
|---|---|---|---|---|---|---|
| Total | Best model (actives) | Consensus (actives) | Total | Best model (actives) | Consensus (actives) | |
| Out | 909 | 817 | 628 | 846 | 332 | 17 |
| Q1 | 74326 |
66105 |
49283 |
58267 |
22808 |
1386 |
| Q2 | 87034 |
77425 |
57813 |
86483 |
33667 |
2054 |
| Q3 | 90649 |
80835 |
60465 |
101979 |
40126 |
2428 |
| Q4 | 86122 |
76967 |
57562 |
91465 |
36167 |
2216 |
| Range | IAV_M2 proton channel | IBV_neuraminidase | ||||
|---|---|---|---|---|---|---|
| Total | Best model (actives) | Consensus (actives) | Total | Best model (actives) | Consensus (actives) | |
| Out | 7 | 7 | 4 | 13563 |
6500 | 37 |
| Q1 | 46163 |
44539 |
30710 |
84952 |
40397 |
262 |
| Q2 | 56607 |
54556 |
37522 |
55476 |
26488 |
176 |
| Q3 | 74969 |
72264 |
49778 |
87531 |
42181 |
257 |
| Q4 | 161294 |
155443 |
107737 |
97518 |
46519 |
322 |
| Range | SARS-CoV_Mpro | ||
|---|---|---|---|
| Total | Best model (actives) | Consensus (actives) | |
| Out | 1709 | 274 | 77 |
| Q1 | 66411 |
9898 | 3346 |
| Q2 | 85126 |
12495 |
4255 |
| Q3 | 90323 |
13285 |
4407 |
| Q4 | 95471 |
14040 |
4575 |
| Quartile | HRV_protease | IAV_M2 proton channel | IAV_neuraminidase | IAV_polymerase (PA) | IBV_neuraminidase | SARS-CoV_Mpro | SARS-CoV-2_Mpro |
|---|---|---|---|---|---|---|---|
| Q1 | 268 | 126 | — | — | — | 2 | 2 |
| Q2 | 2469 | 4484 | 1 | 16 | — | 597 | 1 |
| Q3 | 19317 |
5948 | 611 | 410 | — | 9526 | 9622 |
| Q4 | 201730 |
2518 | 7454 | 8683 | 57 | 6480 | 159213 |
| Out | 1934 | 220 | 35 | 125 | 997 | 55 | 592 |
In general, fewer compounds are considered “Out” when the distance is calculated with physicochemical properties. This could be due to the preservation of drug-like properties for the commercial and focused libraries, and the ChEMBL compounds, as observed in Section 3.3.1. The results for IBV neuraminidase had the greatest number of compounds labeled as “Out” for both distances. This is aligned with the MODI results since IBV neuraminidase was the target with the lowest value for fingerprints.
3.7 ADMET properties profiling of antiviral-focused libraries
Among the main reasons for antiviral drug failure are issues related to pharmacokinetics (PK) and pharmacodynamics (PD). To this end, ADMET profiling serves as a critical step in reducing attrition rates during preclinical and clinical stages.83 ADMET property profiling also provides critical insights into the PK and safety profiles of compounds identified as potentially active against the selected respiratory viral targets. ADMET profiling provides early indications of potential safety issues, including hepatotoxicity, cardiotoxicity, and drug–drug interactions. These factors are particularly relevant for antiviral therapies, which are often administered in combination with other treatments. Maintaining sufficient plasma concentrations within the therapeutic window is critical for inhibiting viral replication effectively while minimizing toxicity and preventing resistance. Key PK parameters, such as the volume of distribution (VD) and clearance (Cl) significantly influence a compound's antiviral effectiveness.84For all 339040 compounds in the VS data set (see Table 2) forty-one ADMET properties were calculated using ADMET-AI (see Methods Section 2.9 for further details). This detailed profiling is included in the structure file of the VS data set to facilitate a comprehensive evaluation of candidate molecules. The user of the newly assembled and designed libraries is free to use other tools to estimate the ADMET profile of the newly assembled and designed libraries (see Section 3.9).
3.8 Machine learning-driven antiviral libraries targeting respiratory viruses
The classification step enabled the identification of 398 promising compounds with a high likelihood of antiviral activity, which were subsequently organized into seven focused libraries for each target. These libraries aim to streamline the prioritization of candidates for further experimental validation or biological testing and are publicly accessible at https://github.com/DIFACQUIM/antiviral_ML.To provide a comprehensive overview of the compounds and take all analysis and predictions together, eight distinct libraries focused on respiratory viruses were designed: the VS data set in conjunction with its predictions and seven subsets for each target. Each library is annotated with: “Canonical SMILES, Murcko SMILES, identifier (ID), database (DB), number of repetitions, prediction label (for each model), prediction score (for each model), Quartile Pairsim (structural), Quartile (structural), Quartile Distance (physicochemical properties), Quartile (physicochemical properties)” plus the ADMET profile in the VS data set library.
Fig. 7 shows the chemical structures of representative compounds included in the newly generated antiviral-focused libraries. Specifically, the figure highlights predicted active compounds for four selected antiviral targets, with high predictive confidence (e.g., predictions within the first quartile (Q1) of the distance to model as described in Section 3.6). Notably, the presence of nitrogen atoms across all illustrated compounds aligns with the findings discussed in Section 3.3.2, emphasizing its relevance to antiviral activity.
 | ||
| Fig. 7 Chemical structures of Q1 compounds (top five for targets HRV_protease and IAV_M2 proton channel) attired by Morgan Chiral of radius 2 (2048 bits) fingerprint. The label below each structure represents the acronym of each library as stated in Table 2. | ||
Fig. S8 in the ESI† illustrates the chemical space distribution of top predicted active compounds, specifically those with the highest prediction confidence (Q1, as detailed in Table 7), across the newly designed target-focused libraries. These visualizations, generated using t-SNE based on the Morgan Chiral of radius 2 (2048 bits) fingerprint, highlight the structural diversity of Q1 compounds.
Notably, certain libraries lack Q1 compounds entirely, while libraries such as HRV protease and IAV M2 proton channel exhibit broader structural diversity, as reflected by the dispersed distribution of Q1 compounds, moreover, SARS-CoV-2 Mpro library, shows clustering, indicating structural similarity among predicted actives. This analysis underscores variability in structural diversity across libraries, suggesting that improvements in library design and model training could enhance the identification of high-confidence active compounds.
4 Conclusions
Herein we designed seven compound libraries with prospective antiviral activity, targeting respiratory viruses. The libraries were assembled and designed using available data in ChEMBL and predictive ML models. The designed libraries target specific antiviral targets: M2 proton channel of IAV, Mpro of SARS-CoV and SARS-CoV-2, neuraminidase of IAV and IBV, polymerase of IAV, and protease of HRV. Additionally, we report the results of a chemoinformatics analysis of the training compounds to assess their drug-like properties and scaffold diversity, comparing these features with those of approved antiviral drugs. The analysis revealed that training compounds exhibited favorable drug-like physicochemical properties. Scaffold analysis of the most frequent scaffolds from the construction of the ML models data set indicated that compounds targeting HRV protease and IAV M2 proton channel share at least one scaffold with the set of approved antivirals. Furthermore, compounds targeting neuraminidases of influenza A and B exhibited common scaffolds, predominantly single-ring structures. Analysis of the chemical space based on different fingerprint representations emphasized the large diversity of compounds with activity against SARS-CoV-2 Mpro and IAV neuraminidase.For the seven antiviral target data sets with high modelability, we developed ML predictive models, which showed improved MCC values after retraining. Among these, the AdaBoost model for IBV neuraminidase demonstrated the best performance. Overall, consensus ML models outperformed individual models, particularly for targets with larger and more balanced data sets. Compounds within the top confidence predictions from the seven newly designed antiviral-focused libraries represent strong candidates for further screening, including biological testing, which is the next step of this study from the wet lab experimental point of view. All seven antiviral-focused libraries developed in this study are freely available at https://github.com/DIFACQUIM/antiviral_ML for the scientific community to select, acquire, and biologically test the chemical libraries. To facilitate the use of these databases, each compound is annotated with confidence predictions and ADMET property profiles.
Abbreviations
| ADASYN | Adaptive synthetic sampling |
| ADMET | Absorption, distribution, metabolism, excretion, and toxicity |
| AI | Artificial intelligence |
| API | Application programming interface |
| ARD | Acute respiratory disease |
| AUC | Area under the curve |
| BA | Balanced accuracy |
| CADD | Computer-aided drug design |
| Cl | Clearance |
| CSP3 | Fraction of sp3 carbon atoms |
| ECFP | Extended connectivity fingerprint |
| FCoV | Feline coronavirus |
| GTM | Generative topographic mapping |
| HBA | H-bond acceptors |
| HBD | H-bond donors |
| HEV-71 | Enterovirus A71 |
| HCoV-229E | Human coronavirus 229E |
| HCoV-NL63 | Human coronavirus NL63 |
| HIV | Human immunodeficiency virus |
| HPIV-1 | Human parainfluenza virus 1 |
| HRV | Human rhinovirus |
| HRSV | Human respiratory syncytial virus |
| IAV | Influenza A virus |
| IBV | Influenza B virus |
| JAK | Janus kinase |
| logP | Partition coefficient octanol/water |
| MACCS | Molecular ACCess system |
| MCC | Matthew's correlation coefficient |
| MERS | Middle East respiratory syndrome |
| MERS-CoV | Middle East respiratory syndrome coronavirus |
| ML | Machine learning |
| MODI | Modelability index |
| MOE | Molecular operating environment |
| Mpro | Main protease |
| MW | Molecular weight |
| NiV | Henipavirus nipahense |
| NPs | Natural products |
| PD | Pharmacodynamics |
| PK | Pharmacokinetics |
| PLP | Papain like-protease |
| PHEIC | Public health emergencies of international concern |
| R&D | Research and development |
| RDKit | Rational discovery kit |
| RdRp | RNA dependent RNA polymerase |
| RSV | Respiratory syncytial virus |
| RVs | Rhinoviruses |
| SARS | Severe acute respiratory syndrome |
| SARS-CoV | Severe acute respiratory syndrome coronavirus |
| SARS-CoV-2 | Severe acute respiratory syndrome coronavirus 2 |
| SMILES | Simplified molecular input line entry system |
| TPSA | Topological polar surface area |
| t-SNE | t-Distributed stochastic neighbor embedding |
| VD | Volume of distribution |
| VS | Virtual screening |
| WHO | World Health Organization |
Data availability
The antiviral focused libraries generated in this study and the project code are available in ZENDO: https://doi.org/10.5281/zenodo.15131233. Supplementary figures and tables are included in a ESI file.†Author contributions
Gabriela Valle-Núñez: methodology, software, validation, formal analysis, investigation, visualization, writing – original draft, writing – review & editing. Raziel Cedillo-González: conceptualization, data acquisition and curation, methodology, visualization, software, formal analysis, writing – original draft, writing – review & editing. Juan F. Avellaneda-Tamayo: conceptualization, methodology, software, supervision, formal analysis, writing – original draft, writing – review & editing. Fernanda I. Saldívar-González: conceptualization, methodology, software, formal analysis, writing – original draft, writing – review & editing. Diana L. Prado-Romero: conceptualization, methodology, software, formal analysis, writing – original draft, writing – review & editing. José L. Medina-Franco: conceptualization, formal analysis, investigation, resources, writing – review & editing, supervision, project administration, funding acquisition.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
We are grateful for the support of DGAPA, UNAM, Programa de Apoyo a Proyectos de Investigación e Innovación Tecnológica (PAPIIT), grant no. IV200121. R. C.-G., J. F. A.-T., F. I. S.-G. and D. L. P.-R. thank Consejo Nacional de Humanidades, Ciencias y Tecnologías (CONAHCyT), Mexico, for the postgraduate scholarships 1099206, 1270553, 848061, and 888207, respectively. Helpful discussions with Felipe Victoria-Muñoz are greatly acknowledged. We also thank Pedro A. Laurel-García for sharing the code to use ADMET-AI.References
- S. Kausar, F. S. Khan, M. I. M. Ur Rehman, M. Akram, M. Riaz, G. Rasool, A. H. Khan, I. Saleem, S. Shamim and A. Malik, Int. J. Immunopathol. Pharmacol., 2021, 35, 20587384211002621, DOI:10.1177/20587384211002621.
- R. E. Baker, A. S. Mahmud, I. F. Miller, M. Rajeev, F. Rasambainarivo, B. L. Rice, S. Takahashi, A. J. Tatem, C. E. Wagner, L.-F. Wang, A. Wesolowski and C. J. E. Metcalf, Nat. Rev. Microbiol., 2022, 20, 193–205, DOI:10.1038/s41579-021-00639-z.
- Prioritizing Diseases for Research and Development in Emergency Contexts, https://www.who.int/activities/prioritizing-diseases-for-research-and-development-in-emergency-contexts, accessed 15 May 2024 Search PubMed.
- Pathogens Prioritization. A Scientific Framework for Epidemic and Pandemic Research Preparedness, https://cdn.who.int/media/docs/default-source/consultation-rdb/prioritization-pathogens-v6final.pdf?sfvrsn=c98effa7_7%26download=true, accessed 17 December 2024 Search PubMed.
- S. Katz, Overview of Viral Respiratory Infections, https://www.msdmanuals.com/professional/infectious-diseases/respiratory-viruses/overview-of-viral-respiratory-infections, accessed 21 January 2025 Search PubMed.
- About Respiratory Illnesses, https://www.cdc.gov/respiratory-viruses/about/index.html#:%7E:text=Everyyearrespiratoryvirusessuch,fallandwintervirusseason, accessed 21 January 2025 Search PubMed.
- G. Mathez and V. Cagno, Int. J. Mol. Sci., 2023, 24, 13500, DOI:10.3390/ijms241713500.
- T. Majerová and J. Konvalinka, Mol. Aspects Med., 2022, 88, 101159, DOI:10.1016/j.mam.2022.101159.
- J. B. Mahony, A. Petrich and M. Smieja, Crit. Rev. Clin. Lab Sci., 2011, 48, 217–249, DOI:10.3109/10408363.2011.640976.
- A. Fendrick, A. Monto, B. Nightengale and M. Sarnes, Arch. Intern. Med., 2003, 163, 487–494, DOI:10.1001/archinte.163.4.487.
- N. Zhang, L. Wang, X. Deng, R. Liang, M. Su, C. He, L. Hu, Y. Su, J. Ren, F. Yu, L. Du and S. Jiang, J. Med. Virol., 2020, 92, 408–417, DOI:10.1002/jmv.25674.
- A. Fitero, S. G. Bungau, D. M. Tit, L. Endres, S. A. Khan, A. F. Bungau, I. Romanul, C. M. Vesa, A.-F. Radu, A. G. Tarce, M. A. Bogdan, A. C. Nechifor and N. Negrut, Int. J. Clin. Pract., 2022, 2022, 1571826, DOI:10.1155/2022/1571826.
- L. A. De Jesús-González, M. León-Juárez, F. I. Lira-Hernández, B. Rivas-Santiago, M. A. Velázquez-Cervantes, I. M. Méndez-Delgado, D. I. Macías-Guerrero, J. Hernández-Castillo, X. Hernández-Rodríguez, D. N. Calderón-Sandate, W. S. Mata-Martínez, J. M. Reyes-Ruíz, J. F. Osuna-Ramos and A. C. García-Herrera, Pathogens, 2024, 14, 20, DOI:10.3390/pathogens14010020.
- D. Chattopadhyay and T. N. Naik, Mini Rev. Med. Chem., 2007, 7, 275–301, DOI:10.2174/138955707780059844.
- S. K. Kim, T. S. Vo and D. H. Ngo, in Advances in Food and Nutrition Research, ed. S.-K. Kim, Academic Press, 2011, vol. 64, pp. 245–254, DOI:10.1016/B978-0-12-387669-0.00019-3.
- Y. Guo, A. Ma, X. Wang, C. Yang, X. Chen, G. Li and F. Qiu, Front. Chem., 2022, 10, 1005360, DOI:10.3389/fchem.2022.1005360.
- K. A. El Sayed, in Studies in Natural Products Chemistry, ed. A. ur-Rahman, Elsevier, 2000, part E, vol. 24, pp. 473–572, DOI:10.1016/S1572-5995(00)80051-4.
- S. Peng, H. Wang, Z. Wang and Q. Wang, Molecules, 2022, 27, 7370, DOI:10.3390/molecules27217370.
- P. Mehrbod, D. Hudy, D. Shyntum, J. Markowski, M. J. Łos and S. Ghavami, Biomolecules, 2021, 11, 10, DOI:10.3390/biom11010010.
- K. Liu, Y. Zhu, X. Cao, Y. Liu, R. Ying, Q. Huang, P. Gao and C. Zhang, Heliyon, 2023, 9, e21648 CrossRef CAS PubMed.
- H. Liu, F. Ye, Q. Sun, H. Liang, C. Li, S. Li, R. Lu, B. Huang, W. Tan and L. Lai, J. Enzyme Inhib. Med. Chem., 2021, 36, 497–503, DOI:10.1080/14756366.2021.1873977.
- E. Gayozo and L. Rojas, Rev. Soc. Cient. del Parag., 2022, 27, 101–121, DOI:10.32480/rscp.2022.27.2.101.
- N. A. Ashour, A. A. Elmaaty, A. A. Sarhan, E. B. Elkaeed, A. M. Moussa, I. A. Erfan and A. A. Al-Karmalawy, Drug Des., Dev. Ther., 2022, 16, 685–715, DOI:10.2147/DDDT.S354841.
- D. A. Winkler, J. Mater. Chem., 2024, 62, 2844–2879, DOI:10.1007/s10910-023-01568-3.
- Q. Liao, Z. Chen, Y. Tao, B. Zhang, X. Wu, L. Yang, Q. Wang and Z. Wang, Sci. Rep., 2021, 11, 22796, DOI:10.1038/s41598-021-02266-3.
- K. Klimenko, G. Marcou, D. Horvath and A. Varnek, J. Chem. Inf. Model., 2016, 56, 1438–1454, DOI:10.1021/acs.jcim.6b00192.
- J. V. de Julián-Ortiz, J. Gálvez, C. Muñoz-Collado, R. García-Domenech and C. Gimeno-Cardona, J. Med. Chem., 1999, 42, 3308–3314, DOI:10.1021/jm981132u.
- P. Richardson, I. Griffin, C. Tucker, D. Smith, O. Oechsle, A. Phelan, M. Rawling, E. Savory and J. Stebbing, Lancet, 2020, 395, e30–e31, DOI:10.1016/S0140-6736(20)30304-4.
- P. Ranjan, J. Rani, U. Sahoo, A. Nayak and P. Kumar, Pharm. Innov., 2023, 12, 1070–1073 Search PubMed.
- E. S. Istifli, N. Okumus, C. Sarikurkcu, E. R. Kuhn, P. A. Netz and A. S. Tepe, J. Biomol. Struct. Dyn., 2023, 42, 8202–8214, DOI:10.1080/07391102.2023.2267696.
- K. Srivastava and M. K. Singh, Metab. Open, 2021, 12, 100121, DOI:10.1016/j.metop.2021.100121.
- C. Wilson, J. M. F. Gardner, D. W. Gray, B. Baragana, P. G. Wyatt, A. Cookson, S. Thompson, C. Mendoza-Martinez, M. J. Bodkin, I. H. Gilbert and G. J. Tarver, PLoS Neglected Trop. Dis., 2023, 17, e0011799, DOI:10.1371/journal.pntd.0011799.
- F. Potlitz, A. Link and L. Schulig, Expert Opin. Drug Discovery, 2023, 18, 303–313, DOI:10.1080/17460441.2023.2171984.
- J. Kuan, M. Radaeva, A. Avenido, A. Cherkasov and F. Gentile, Wiley Interdiscip. Rev.:Comput. Mol. Sci., 2023, 13, e1678, DOI:10.1002/wcms.1678.
- A. Gryniukova, F. Kaiser, I. Myziuk, D. Alieksieieva, C. Leberecht, P. P. Heym, O. O. Tarkhanova, Y. S. Moroz, P. Borysko and V. J. Haupt, J. Med. Chem., 2023, 66, 10241–10251, DOI:10.1021/acs.jmedchem.3c00128.
- F. I. Saldívar-González, G. Navarrete-Vázquez and J. L. Medina-Franco, Front. Pharmacol, 2023, 14, 1276444, DOI:10.3389/fphar.2023.1276444.
- B. Zdrazil, E. Felix, F. Hunter, E. J. Manners, J. Blackshaw, S. Corbett, M. de Veij, H. Ioannidis, D. M. Lopez, J. F. Mosquera, M. P. Magarinos, N. Bosc, R. Arcila, T. Kizilören, A. Gaulton, A. P. Bento, M. F. Adasme, P. Monecke, G. A. Landrum and A. R. Leach, Nucleic Acids Res., 2024, 52, D1180–D1192, DOI:10.1093/nar/gkad1004.
- M. Davies, M. Nowotka, G. Papadatos, N. Dedman, A. Gaulton, F. Atkinson, L. Bellis and J. P. Overington, Nucleic Acids Res., 2015, 43, W612–W620, DOI:10.1093/nar/gkv352.
- D. Weininger, J. Chem. Inf. Comput. Sci., 1988, 28, 31–36, DOI:10.1021/ci00057a005.
- D. S. Wishart, Y. D. Feunang, A. C. Guo, E. J. Lo, A. Marcu, J. R. Grant, T. Sajed, D. Johnson, C. Li, Z. Sayeeda, N. Assempour, I. Iynkkaran, Y. Liu, A. Maciejewski, N. Gale, A. Wilson, L. Chin, R. Cummings, D. Le, A. Pon, C. Knox and M. Wilson, Nucleic Acids Res., 2018, 46, D1074–D1082, DOI:10.1093/nar/gkx1037.
- G. Landrum, RDKit, https://www.rdkit.org/, accessed 7 November 2024 Search PubMed.
- MolVS: Molecule Validation and Standardization — MolVS 0.1.1 documentation, https://molvs.readthedocs.io/en/latest/, accessed 7 November 2024 Search PubMed.
- N. Sánchez-Cruz, B. A. Pilón-Jiménez and J. L. Medina-Franco, F1000Research, 2020, 8, 2071, DOI:10.12688/f1000research.21540.2.
- A. Golbraikh, E. Muratov, D. Fourches and A. Tropsha, J. Chem. Inf. Model., 2014, 54, 1–4, DOI:10.1021/ci400572x.
- J. L. Durant, B. A. Leland, D. R. Henry and J. G. Nourse, J. Chem. Inf. Comput. Sci., 2002, 42, 1273–1280, DOI:10.1021/ci010132r.
- D. Rogers and M. Hahn, J. Chem. Inf. Model., 2010, 50, 742–754, DOI:10.1021/ci100050t.
- N. Sánchez-Cruz and J. L. Medina-Franco, J. Med. Chem., 2021, 64, 8208–8220 CrossRef PubMed.
- Molecular Operating Environment (MOE), 2024.0601, Chemical Computing Group ULC, Sherbrooke St. W., Montreal, QC H3A 2R7, 2025, pp. 910–1010, https://www.chemcomp.com/en/Research-Citing_MOE.htm Search PubMed.
- G. W. Bemis and M. A. Murcko, J. Med. Chem., 1996, 39, 2887–2893, DOI:10.1021/jm9602928.
- F. Pedregosa, G. Varoquaux, A. Gramfort, V. Michel, B. Thirion, O. Grisel, M. Blondel, A. Müller, J. Nothman, G. Louppe, P. Prettenhofer, R. Weiss, V. Dubourg, J. Vanderplas, A. Passos, D. Cournapeau, M. Brucher, M. Perrot and É. Duchesnay, arXiv [cs.LG], 2012, preprint, 1201.0490, DOI:10.48550/arXiv.1201.0490.
- H. Mary, E. Noutahi, DomInvivo, L. Zhu, M. Moreau, S. Pak, D. Gilmour, S. Whitfield, t., Valence-JonnyHsu, H. Hounwanou, I. Kumar, S. Maheshkar, S. Nakata, K. M. Kovary, C. Wognum, M. Craig and D. Bot, datamol-io/datamol: 0.12.3, Zenodo, 2024, DOI:10.5281/zenodo.10535844.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Adv. Drug Delivery Rev., 1997, 23, 3–25, DOI:10.1016/S0169-409X(96)00423-1.
- M. Ali, PyCaret: an open source, low-code machine learning library in Python, https://pycaret.org/, accessed 7 November 2024 Search PubMed.
- H. He, Y. Bai, E. A. Garcia and S. Li, in 2008 IEEE International Joint Conference on Neural Networks (IEEE World Congress on Computational Intelligence), IEEE, 2008, pp. 1322–1328, DOI:10.1109/IJCNN.2008.4633969.
- B. Ramsundar, P. E. P. Walters, and V. Pande, Deep Learning for the Life Sciences, O'Reilly Media, Sebastopol, CA, 2019 Search PubMed.
- Splitters — Deepchem 2.8.1.dev Documentation, https://deepchem.readthedocs.io/en/latest/api_reference/splitters.html#fingerprintsplitter, accessed 14 November 2024 Search PubMed.
- C. Miller, T. Portlock, D. M. Nyaga and J. M. O'Sullivan, Front. Bioinform., 2024, 4, 1457619, DOI:10.3389/fbinf.2024.1457619.
- CORONAVIRUS Library, https://www.chemdiv.com/catalog/focused-and-targeted-libraries/coronavirus-library/, accessed 30 August 2024 Search PubMed.
- Antiviral Library, https://www.chemdiv.com/catalog/focused-and-targeted-libraries/antiviral-library/, accessed 30 August 2024 Search PubMed.
- Drug-Like Green Collection, https://www.otavachemicals.com/products/compound-libraries-for-hts/drug-like-green-collection, accessed 28 November 2024 Search PubMed.
- Antiviral Library, https://enamine.net/compound-libraries/targeted-libraries/antiviral-library?highlight=WyJhbnRpdmlyYWwiXQ==, accessed 28 November 2024 Search PubMed.
- Discovery Diversity Set, https://chem-space.com/compounds/discovery_diversity_set, accessed 31 August 2024 Search PubMed.
- Helicase Screening Libraries, https://lifechemicals.com/screening-libraries/targeted-and-focused-screening-libraries/helicase-focused-library, accessed 28 November 2024 Search PubMed.
- Antiviral Screening Compound Libraries, https://lifechemicals.com/screening-libraries/targeted-and-focused-screening-libraries/antiviral-libraries, accessed 28 November 2024 Search PubMed.
- DNA and RNA Polymerase Screening Libraries, https://lifechemicals.com/screening-libraries/targeted-and-focused-screening-libraries/polymerase-focused-libraries, accessed 28 November 2024 Search PubMed.
- Coronavirus Inhibitor Screening Libraries, https://lifechemicals.com/screening-libraries/targeted-and-focused-screening-libraries/coronavirus-screening-libraries, accessed 28 November 2024 Search PubMed.
- Coronavirus Inhibitor Screening Libraries, https://lifechemicals.com/screening-libraries/targeted-and-focused-screening-libraries/coronavirus-screening-libraries, accessed 28 November 2024 Search PubMed.
- Antiviral Screening Compound Libraries, https://lifechemicals.com/screening-libraries/targeted-and-focused-screening-libraries/antiviral-libraries, accessed 28 November 2024 Search PubMed.
- Antiviral Screening Compound Libraries, https://lifechemicals.com/screening-libraries/targeted-and-focused-screening-libraries/antiviral-libraries, accessed 28 November 2024 Search PubMed.
- Antiviral Screening Compound Libraries, https://lifechemicals.com/screening-libraries/targeted-and-focused-screening-libraries/antiviral-libraries, accessed 28 November 2024 Search PubMed.
- Pre-plated Focused Libraries, https://lifechemicals.com/screening-libraries/pre-plated-focused-libraries, accessed 28 November 2024 Search PubMed.
- Coronavirus Inhibitor Screening Libraries, https://lifechemicals.com/screening-libraries/targeted-and-focused-screening-libraries/coronavirus-screening-libraries, accessed 28 November 2024 Search PubMed.
- Coronavirus Inhibitor Screening Libraries, https://lifechemicals.com/screening-libraries/targeted-and-focused-screening-libraries/coronavirus-screening-libraries, accessed 28 November 2024 Search PubMed.
- I. Sushko, S. Novotarskyi, R. Körner, A. K. Pandey, A. Cherkasov, J. Li, P. Gramatica, K. Hansen, T. Schroeter, K.-R. Müller, L. Xi, H. Liu, X. Yao, T. Öberg, F. Hormozdiari, P. Dao, C. Sahinalp, R. Todeschini, P. Polishchuk, A. Artemenko, V. Kuz'min, T. M. Martin, D. M. Young, D. Fourches, E. Muratov, A. Tropsha, I. Baskin, D. Horvath, G. Marcou, C. Muller, A. Varnek, V. V. Prokopenko and I. V. Tetko, J. Chem. Inf. Model., 2010, 50, 2094–2111, DOI:10.1021/ci100253r.
- K. Swanson, P. Walther, J. Leitz, S. Mukherjee, J. C. Wu, R. V. Shivnaraine and J. Zou, Bioinformatics, 2024, 40, btae416, DOI:10.1093/bioinformatics/btae416.
- admet_ai: Training and Prediction Scripts for Chemprop Models Trained on ADMET Datasets, Github, https://github.com/swansonk14/admet_ai, accessed 28 November 2024 Search PubMed.
- Y. A. Shtyrya, L. V. Mochalova and N. V. Bovin, Acta Naturae, 2009, 1, 26–32, DOI:10.32607/20758251-2009-1-2-26-32.
- C. Wang, Z. Song, H. Yu, K. Liu and X. Ma, Acta Pharm. Sin. B, 2015, 5, 431–441, DOI:10.1016/j.apsb.2015.07.002.
- G. Ahmad, M. Sohail, M. Bilal, N. Rasool, M. U. Qamar, C. Ciurea, L. G. Marceanu and C. Misarca, Molecules, 2024, 29, 2232, DOI:10.3390/molecules29102232.
- A. Mermer, T. Keles and Y. Sirin, Bioorg. Chem., 2021, 114, 105076, DOI:10.1016/j.bioorg.2021.105076.
- J. J. Naveja and J. L. Medina-Franco, Front. Chem., 2019, 7, 510, DOI:10.3389/fchem.2019.00510.
- J. L. Medina-Franco, J. R. Rodríguez-Pérez, H. F. Cortés-Hernández and E. López-López, Artif. Intell. Life Sci., 2024, 6, 100117, DOI:10.1016/j.ailsci.2024.100117.
- L. Strasfeld and S. Chou, Infect. Dis. Clin. North Am., 2010, 24, 413–437, DOI:10.1016/j.idc.2010.01.001.
- T. Chaira, C. Subramani and T. K. Barman, Pharmaceutics, 2023, 15, 1212, DOI:10.3390/pharmaceutics15041212.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5dd00037h |
| This journal is © The Royal Society of Chemistry 2025 |