
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Improving reaction prediction through chemically aware transfer learning†
Angus
Keto
 a,
Taicheng
Guo
b,
Nils
Gönnheimer
c,
Xiangliang
Zhang
b,
Elizabeth H.
Krenske
a and
Olaf
Wiest
*c
a,
Taicheng
Guo
b,
Nils
Gönnheimer
c,
Xiangliang
Zhang
b,
Elizabeth H.
Krenske
a and
Olaf
Wiest
*c
aSchool of Chemistry and Molecular Biosciences, The University of Queensland, Brisbane, QLD 4072, Australia
bDepartment of Computer Science and Engineering, University of Notre Dame, USA
cDepartment of Chemistry and Biochemistry, University of Notre Dame, USA. E-mail: owiest@nd.edu
First published on 28th March 2025
Abstract
Practical applications of machine learning (ML) to new chemical domains are often hindered by data scarcity. Here we show how data gaps can be circumvented by means of transfer learning that leverages chemically relevant pre-training data. Case studies are presented in which the outcomes of two classes of pericyclic reactions are predicted: [3,3] rearrangements (Cope and Claisen rearrangements) and [4 + 2] cycloadditions (Diels–Alder reactions). Using the graph-based generative algorithm NERF, we evaluate the data efficiencies achieved with different starting models that we pre-trained on datasets of different sizes and chemical scope. We show that the greatest data efficiency is obtained when the pre-training is performed on smaller datasets of mechanistically related reactions (Diels–Alder, Cope and Claisen, Ene, and Nazarov) rather than >50× larger datasets of mechanistically unrelated reactions (USPTO-MIT). These small bespoke datasets were more efficient in both low re-training and low pre-training regimes, and are thus recommended alternatives to large diverse datasets for pre-training ML models.
Introduction
One of the most important bottlenecks for applications of machine learning (ML) in chemistry is the lack of access to reaction data. Even for widely used reactions such as amide, Suzuki, and SNAr reactions,1 reaction datasets can be considered small by machine learning (ML) standards2,3 or in comparison to datasets of molecules and their properties.4–7 Even with popular reaction datasets such as USPTO,8 Pistachio,9 and Reaxys,10 data filtering is required and for the latter two, commercial restrictions apply. The problem of data scarcity is especially acute for novel reactions, where only limited (small and/or homogeneous) reaction data are available for use in the training of predictive models. Experimentally generating datasets of significant size and diversity is a non-trivial task. We have recently shown that new generative ML algorithms featuring built-in “chemical-awareness” can efficiently predict chemical reactivity in low-data regimes.11 Alternative, computational, strategies include data augmentation12–14 and transfer learning.15–19Transfer learning20,21 involves retraining an existing ML model on a new domain of chemistry (Fig. 1A) and in many cases improves model accuracy while reducing training costs by not necessitating a brand new model. It requires two related datasets: one pre-training dataset that is used to train an initial model and another to re-train (fine-tune) the model on the target reactions/domain. In theory, the shared principles between these two domains can be learned during pre-training and leveraged during fine-tuning to produce a more effective model than training alone. However, the ideal relationship between the pre-training and re-training datasets in reaction prediction is not clear: Should the emphasis be on the size of the pre-training dataset, on molecular structure or on similarity of the reaction mechanisms? Chemical intuition would posit the mechanism, specifically the electron flow, contains the most applicable information but this requires a model that properly encodes this information. In contrast, the data hungry nature of neural networks would suggest that a significantly larger (one or more orders of magnitude) and more diverse dataset be more effective.
 | ||
| Fig. 1 (A) General transfer learning workflow. (B) Pericyclic reactions and their mechanistic similarities. | ||
We investigate the following question: in situations where data are scarce, do models pre-trained on mechanistically related reactions require less data than models trained on diverse reaction data? We address this question through studies of two target pericyclic reactions: [3,3] rearrangements (Cope22,23 and Claisen24,25 rearrangements) and [4 + 2] cycloadditions (Diels–Alder26,27 reactions) (Fig. 1B). These reactions were chosen not just for their synthetic utility of atom-economy efficient transformations,28–31 but crucially because they share a common mechanistic feature: the shuffling of electrons around a six-membered cyclic transition state. These reactions are compared and pre-trained with datasets from the Ene reaction, which shares the cyclic movement of six electrons, and the Nazarov cyclization, a 4-electron electrocyclic reaction. Our work examines whether ML models can recognize these shared mechanistic principles, in this case when predicting the major product of these reactions.
Methods
Dataset curation
Pericyclic reaction datasets were generated by Reaxys10 database searches and were curated using workflows that filtered based on atom-economy, bonding patterns, atom-mapping, and reaction templates (see ESI for more details†). The two target reaction datasets were: 3289 Cope and Claisen (CC) rearrangements and 9537 Diels–Alder (DA) reactions.11For transfer learning, we also generated pre-training datasets representing different sizes and chemistry: (1) 80%-of-∼480![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 diverse reactions from the USPTO-MIT database,8,32 (2) 80%-of-9537 Diels–Alder reactions (DA1), (3) 40%-of-9537 Diels–Alder reactions (DA2), (4) 80%-of-3289 Cope and Claisen rearrangements, (5) 80%-of-2322 Ene reactions, (6) 80%-of-1029 Nazarov cyclizations where the reactant and product were represented as their charge-neutral forms (Naz1), (7) 80%-of-1029 Nazarov cyclizations with the reactant and product represented as their protonated forms (Naz2). The Jupyter notebooks to regenerate these datasets using a Reaxys license are available as described in the ESI.†
000 diverse reactions from the USPTO-MIT database,8,32 (2) 80%-of-9537 Diels–Alder reactions (DA1), (3) 40%-of-9537 Diels–Alder reactions (DA2), (4) 80%-of-3289 Cope and Claisen rearrangements, (5) 80%-of-2322 Ene reactions, (6) 80%-of-1029 Nazarov cyclizations where the reactant and product were represented as their charge-neutral forms (Naz1), (7) 80%-of-1029 Nazarov cyclizations with the reactant and product represented as their protonated forms (Naz2). The Jupyter notebooks to regenerate these datasets using a Reaxys license are available as described in the ESI.†
Machine learning architectures
Machine learning models were trained with:(1) NERF (non-autoregressive electron redistribution framework) algorithm.33 NERF predicts the changes in edges of a molecular graph (corresponding to the changes in bond order that define a chemical reaction) using connectivity and nodes characterised by atom type, aromaticity, charge, and positional and segment embeddings. Its design principles33 and performance11 have been previously documented.
(2) Chemformer,17 a natural language processing (NLP) model built on the Bidirectional Auto-Regressive Transformers (BART)34 architecture.
Results and discussion
Comparing pericyclic models
Before conducting transfer learning, NERF models were pretrained on the pericyclic datasets. It was possible to exceed 90% Top-1 accuracy when using at least 80% of the Diels–Alder (DA1, 94.7% (ref. 11)) and Cope and Claisen (90.1%) datasets (see Fig S1†). Ene reactions were close behind at 89.2% while the neutral (Naz1) and protonated (Naz2) Nazarov cyclizations had respective accuracies of 84.7% and 85.1%. Despite the data efficiency of the NERF architecture, the size of the datasets affects the model performance and this can subsequently also effect transfer learning.Cope and Claisen reactions
We first developed baseline (non-pre-trained) models for Cope and Claisen (CC) reactions using the NERF architecture where the task was to generate the major product, including the site selectivity (location of reaction center) given only the structure of the reactant as the input. It should be noted however that a generative prediction, as opposed to a deterministic prediction (e.g. through a template model), is a more flexible but also more complex task. Multiple possible outcomes are most common when aromatic systems supply one of the double bonds (68.4% of dataset), but this can also occur in aliphatic systems (see ESI†).For each pre-training dataset, 10 separate pretrained NERF models were created, using 10 random splits. To reduce computational cost, only one USPTO model, trained on the split used in Jin et al.,32 was created. The model with the highest accuracy from each set of 10 was then fine-tuned on CC training data. This fine-tuning occurred on 10 random splits of five different ratios of CC training data (between 10% and 85%). With 5 fine-tuning splits investigated, there were 50 transfer learned models per pretraining dataset. Top-1 accuracy (i.e. accuracy according to the most confident prediction) was used.
The black line in Fig. 2A depicts the baseline situation where no pre-training was undertaken before training the CC model. Without any pre-training, the NERF model only achieves predictive accuracies of >90% when 80% of the CC dataset (2795 reactions) is used for training. This shows the CC dataset is sufficiently large to develop an effective NERF model (>90% accuracy) without pre-training but only if a large percentage of the dataset is used for training.
 | ||
| Fig. 2 Performance of NERF models generated by transfer learning for predicting the outcomes of (A) Cope and Claisen rearrangements and (B) Diels–Alder reactions, as a function of (i) the chemistry of the pretrained model and (ii) the amount of training data. The models were trained using the following training datasplits of the 3289-reaction Cope and Claisen dataset or the 9537-reaction Diels–Alder dataset (training:validation:testing): 10:45:45, 40:30:30, 60:20:20, 80:10:10, 85:5:10. The performance of models built without pre-training are also shown for comparison. Top-1 prediction accuracies are shown and are the average of ten runs. | ||
Next, we investigated the effect of pre-training using different reactions. Fig. 2A shows the effect of pre-training by Diels–Alder (orange), USPTO-MIT (red), Ene (purple), and Nazarov (green) reactions as the pre-training datasets. For the pretrained models, any result above the baseline indicates that the pre-training step has enhanced the model's predictive accuracy. The most relevant data split to be considered is the lowest training regime where only 10% (328 training reactions) of the CC dataset is used as this most closely mirrors the low-data scenarios common in developing areas of chemistry. All six pre-training datasets prove beneficial here but the greatest benefit came from pre-training with Diels–Alder data: DA1 and DA2 achieved accuracies of 76.0% and 73.1%, respectively, compared to the baseline of 62.7%. Pre-training on the USPTO-MIT dataset had a moderate benefit (68.9%), while pre-training on the Ene and Nazarov datasets were least beneficial (64.1–66.7%). These results demonstrate the balance between mechanistically similar versus using larger but more general pre-training data sets. Even though the Diels–Alder pre-training datasets were 48 times smaller than the USPTO-MIT dataset, the mechanistically related Diels–Alder reactions were more efficient pre-training sources. The difference of the effectiveness of the DA1 and DA2 datasets (which is half the size of DA1) for pre-training shows, in analogy to the results in Fig. S1† discussed above, that pretraining dataset size affects accuracy, in line with the observed the lower performance of the smaller Nazarov datasets. The standard deviations (Table S1†) range between 0.6–2.3% for all approaches, indicating that the model performances and pretraining benefits are robust.
The benefit of pre-training drops off as more training data is introduced. When 85% of the CC dataset is used as training, the highest performing pretrained model (DA1) has a Top-1 accuracy of 92.3% compared to the baseline of 90.7%. All other models are within 0.9% of the baseline. In high training regimes, there may be fewer areas of chemical space that these pre-training datasets can help elucidate.
Diels–Alder reactions
To investigate the effect of pre-training on a reaction with a larger available dataset, we built NERF models for Diels–Alder reactions, where the dataset was approximately three times larger than the one available for the Cope and Claisen reactions. Here, prediction of the major product is a more complicated challenge as it requires differentiation between site- and regio-selective outcomes (connectivity of the reaction centre). The baseline performance of NERF for Diels–Alder reactions was discussed previously.11 Here, we compared the performance of Cope and Claisen, USPTO-MIT, Ene, and Nazarov reactions as the pre-training datasets.Similar to the Cope and Claisen predictions, the lowest data regime for Diels–Alder reactions is the most relevant and displays the greatest performance improvement from pre-training. All pre-training approaches were beneficial. Pre-training on USPTO-MIT gave the highest accuracy (82.9%) when 10% of the Diels–Alder dataset was used, while pre-training on Cope and Claisen rearrangements was next, with an accuracy of 78.5%. This latter result is noteworthy given that the CC pre-training dataset is ∼145× smaller than the USPTO-MIT used for pre-training. As the amount of training data increases, the effect of pre-training drops off noticeably. When 40% or more of the Diels–Alder reactions are used for training, only USPTO-MIT pre-training delivers a noticeable increase in accuracy relative to the baseline. Model performance is again robust, with standard deviations of between 0.5–2.3% (Table S2†).
All pericyclic pre-training datasets are smaller (≤3289 reactions) than the dataset for the Diels–Alder reaction used in the baseline model, and consequently better accuracy is obtained from pre-training with the large USPTO-MIT dataset even though it largely contains unrelated reactions. While the pericyclic pre-training reactions impart specific reactivity information, the USPTO-MIT imparts a broad and general understanding of reactivity due to its large size and the diversity of reactions it contains.
To understand what knowledge gaps in the training data the pre-training was helping with, the performance increase across different Diels–Alder sub-categories were investigated relative to a baseline of no-pretraining (Fig. 3). For further comparison, the pre-training approaches were also compared with a non-pre-training approach that simply used selected additional training examples (reactions with 17 triazines and 32 oxazoles) reported previously.11 This would illustrate not only what area of chemical space benefits the most, but also whether pretraining or manual data mining is more effective. Analysis was conducted on the 80:10:10 split for comparability. The pre-training and additional training approaches all increased the Top-1 accuracies overall. Underrepresented sub-categories, including intramolecular, aromatic, and hetero-Diels–Alder reaction centres, showed the highest improvement. Pre-training with the USPTO-MIT dataset is the most beneficial approach but the focused datasets (here Cope and Claisen pretraining) can be a small fraction of the size of a generalized dataset and be effective. This assumes that the needed mechanistic information is contained in the small dataset and the model is capable of using this information. The alternative approach using key additional training examples shows that extracting from the literature, or even carrying out these experiments, can also be effective. However, this may not be feasible for some chemistries. Overall, the best approach for a new dataset will depend on factors including training and pre-training dataset sizes as well as whether there is possible untapped literature data.
 | ||
| Fig. 3 Increase in Top-1 accuracy when pre-training on USPTO-MIT or Cope and Claisen data is compared a no pre-training approach that includes an additional 49 select examples of inverse electron demand Diels–Alder reactions on triazines and oxazoles. | ||
Equal dataset size comparison
A potential caveat for the interpretation of the results is the significantly different size of the pre-training datasets used. We thus quantitatively examined the question of size vs. mechanistic similarity of the pre-training datasets. NERF models for CC reaction predictions were trained using models pretrained on identically-sized 1000-reaction subsets of the USPTO, Diels–Alder, Ene, and Nazarov datasets. To account for the random distribution of pre-training data for these examples, 10 pretrained NERF models were each tested on 10 different subsets of Cope and Claisen rearrangements.In Fig. 4, the performance of each NERF model is compared against the non-pretrained NERF baseline of 62.7% Top-1 accuracy. When pre-trained using equally sized pre-training datasets, the most accurate Cope and Claisen predictions were obtained from models pretrained on Diels–Alder reactions (+3.7%, Fig. 4A). Pre-training on Ene and Nazarov reactions also increased performance but to a lesser extent (+0.7 to 1.9%). USPTO-MIT pre-training had a negative impact on accuracy, suggesting that the benefits seen for pre-training by USPTO-MIT data in Fig. 2A were the result of the general chemical understanding provided by the entire dataset and could not be replicated by selecting only a small subset of its reactions. This confirms again the hypothesis that the more mechanistically similar the dataset, the more effective the pretraining.
 | ||
| Fig. 4 Effects of pre-training chemistry on prediction accuracies for NERF models where each pre-training dataset comprised 1000 reactions. The height of each bar indicates how the Top-1 accuracy compares to the baseline (non-pretrained model). (A) Models were made for the Cope and Claisen rearrangement using 328 reactions (10%) of the dataset as training. (B) Models were made for the Diels–Alder dataset also using 328 reactions (3%) of the dataset as training. | ||
This same equal size pretraining set approach was then applied to predicting Diels–Alder reactions and the same number of training reactions (328), which represents 3% of the Diels–Alder dataset, were used (Fig. 4B). This was for comparability and to further investigate the impact of pretraining when the baseline Top-1 accuracy (39.5%) is very low. Pretraining approaches had an outsized impact here because of this low baseline accuracy. In agreement with Fig. 4A, mechanistically related pretraining data proved more effective, as seen by the +13.1% increase in Top-1 accuracy when pretraining on Cope and Claisen reactions. USPTO-MIT pretraining however resulted in a decrease in accuracy of −10.5%, reinforcing that the entire USPTO-MIT dataset is needed for effective pretraining. Pretraining on Ene reactions had a beneficial effect of +5.4% while Nazarov reactions saw only extremely minor changes (+0.2%). In order to get the improvement in accuracy with USPTO-MIT data, it appears the entire dataset needs to be used as pretraining.
To further understand the effectiveness of pericyclic vs. non-pericyclic pre-training data, we visualized reaction fingerprints (rxnfp35) of these reactions in two dimensions using UMAP36 (Fig. 5). The clear separation between the pericyclic datasets and the USPTO-MIT dataset reinforces the distinctness of the reactions in these datasets. Meanwhile, the pericyclic reactions are positioned closer together and in some cases even overlap. Diels–Alder reactions occupy large areas of chemical space near and between Cope and Claisen rearrangements. This suggests why Diels–Alder reactions are the most effective form of pretraining for Cope and Claisen reaction: Diels–Alder reactions are diverse and mechanistically relevant.
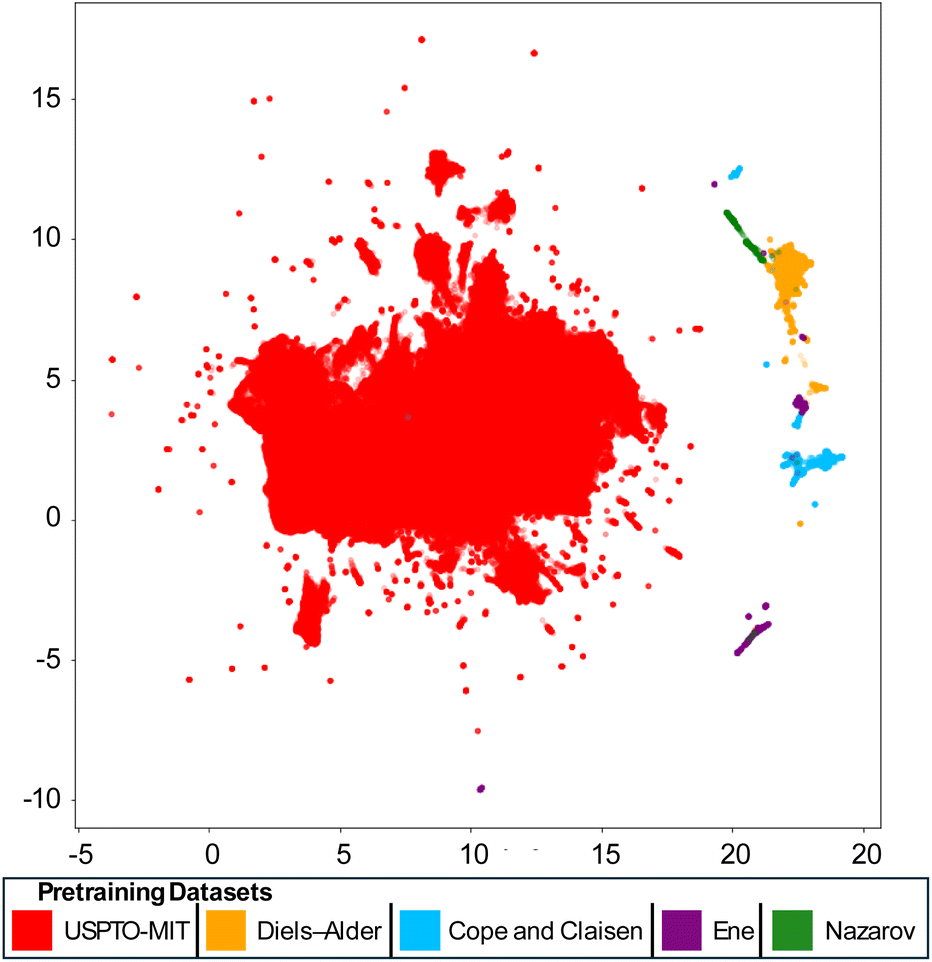 | ||
| Fig. 5 UMAP of rxnfp fingerprints of USPTO-MIT, Diels–Alder, Cope and Claisen, Ene, and Nazarov reaction datasets. | ||
Artificial data
We also explored an alternative approach to pre-training on experimentally reported data that is relevant to situations where the quantity of such data is insufficient for effective pre-training. An artificial pre-training dataset of 1000 randomly generated Cope and Claisen reactions was created using a workflow combining RDKit and random SMILES generation (see ESI† for details). No attempt was made to ensure the chemical reasonableness of the reactant structures in the artificial dataset; rather, the motive was to teach a model the fundamentals of the target reaction before fine-tuning it on actual experimental data for that same reaction type. The results in Fig. 4 (pink bar) show that the artificial dataset was similarly beneficial as the experimentally-derived Nazarov reaction datasets but not the Diels–Alder and Ene datasets, while outperforming USPTO-MIT pretraining. This suggests that in certain circumstances, artificial data could be a viable and easily executed alternative to the use of experimental data for pretraining. Identifying the best strategies for designing artificial or augmented data will require further study.Transfer learning with other models
To complement our studies with NERF, we also investigated a second type of ML algorithm, Chemformer.17 We developed Chemformer-based predictive models of Cope and Claisen rearrangements by pre-training with the DA1 and DA2 datasets. The larger DA1 dataset gave generally better prediction accuracies (Top-1 accuracies 24.1–71.7%) than the smaller DA2 dataset (5.1–41.8%). However, the accuracies never exceeded 80%, even when using as much as 85% of the Cope and Claisen dataset for training (Table ESI S5†). In comparison, pre-training with the USPTO-MIT dataset gave accuracies comfortably exceeding 90% even when trained on as little as 40% of the Cope and Claisen reactions. Transformer models can be data-intensive37 and we speculate that pre-training with USPTO-MIT outperformed DA1 or DA2 because it provided more opportunities for Chemformer to be trained on SMILES syntax in addition to general reactivity principles. Interestingly, when the USPTO-MIT pretrained Chemformer was subjected to a second pre-training step with the DA1 and DA2 datasets before training on Cope and Claisen reactions, the predictive accuracy increased by 5.0% and 4.8%, respectively. The closeness of these two values suggests that there may be a limit to what Chemformer can learn from the additional pericyclic pre-training data. It is clear that Chemformer does not learn the mechanistic similarities between the reactions the way the “chemistry aware”11 NERF does.Compared with Chemformer, NERF appears to be more efficient at utilizing pre-training data and derives greater improvements in accuracy from pre-training. The major cause of this difference is the learning target set for these two models, i.e., ‘what should be learned’. Although both Chemformer and NERF are implemented as neural networks based on Transformer architecture, Chemformer is trained to learn the sequence (input) to sequence (output) correlation, while NERF is trained to learn the difference between reactant (input) and product (output). As a result, NERF is more likely to capture the mechanistic similarities that can be transferred between different reactions.
Conclusions
The prediction of reaction outcomes using transfer learning can be accomplished in a more data-efficient way, and with better accuracy, by using mechanistically relevant, high-quality pre-training datasets that mirror the chemical principles of the target reaction type if the ML model chosen makes effective use of this information. This allows the development of NERF models that predict the outcome of two widely used pericyclic reactions with Top-1 accuracies of >90% and illustrates that pre-training on small mechanistically relevant datasets can lead to comparable or better performance than pre-training with a large and diverse dataset that is many times the size. The results also suggest that for reactions where the available training sets are too small to build reliable models, pre-training on a dataset of mechanistically similar reactions can be effective at instilling a “mechanistic understanding” of reactivity into the pre-trained model. While this is also possible using general datasets such as the USPTO, in such cases much larger datasets are needed to achieve similar improvements in performance. Transfer learning is effective by filling in knowledge gaps present in the training data, particularly for underrepresented categories. Artificially generated datasets represent a promising alternative source of pre-training data that merit further investigation. Overall, the results reported here show the importance of using chemically-relevant training data, designed to capture specific reactivity knowledge, as a complement to general reactivity knowledge in the applications of ML to low-data problems. Aside from mechanism, other factors such as catalysts and reaction conditions could be considered in future applications of insight-driven transfer learning.Data availability
Additional figures, explanations, link to Github with the NERF and Reaxys reaction IDs are contained in the ESI.† Due to copyright limitations from Reaxsys, the full dataset cannot be released completely. As is widely accepted in the community, we made instead the Reaxys reaction ID and the scripts that are needed to regenerate the dataset (assuming the availability of a Reaxys license) available at: Cope and Claisen rearrangements, Ene reactions, and Nazarov cycloadditions: https://github.com/angusketo/PericyclicTL/, Diels–Alder reactions (previous work2): https://github.com/angusketo/DA_DataExtraction. A sample dataset containing the Reaxys IDs and reaction SMILES of 100 randomly selected, representative reactions is included in the ESI.† These Jupyter notebooks, Conda environment information, and datasets (artificial data and Reaxys IDs), can also be found on Zenodo: https://doi.org/10.5281/zenodo.15056997.Author contributions
A. K., E. H. K., and O. W. provided the original concept. A. K. and T. C. conducted the machine learning experiments. A. K. and N. G. created datasets. All authors contributed to the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the United States National Science Foundation under the NSF Center for Computer Assisted Synthesis (C-CAS), grant number CHE-2202693, and by the Australian Research Council (DP180103047). AK acknowledges the support of an Australian Government Research Training Program Scholarship and an American Australian Association Graduate Education Fund Scholarship. NG was a participant in the ISAP program funded by the German Academic Exchange Service (DAAD). Computer resources were provided by the Notre Dame Center for Research Computing, Australian National Computational Infrastructure, and University of Queensland Research Computing Centre.References
- D. G. Brown and J. Bostrom, J. Med. Chem., 2016, 59, 4443–4458 CrossRef CAS PubMed.
- A. Bender and I. Cortes-Ciriano, Drug Discovery Today, 2021, 26, 1040–1052 Search PubMed.
- B. Dou, Z. Zhu, E. Merkurjev, L. Ke, L. Chen, J. Jiang, Y. Zhu, J. Liu, B. Zhang and G. W. Wei, Chem. Rev., 2023, 123, 8736–8780 Search PubMed.
- J. J. Irwin, K. G. Tang, J. Young, C. Dandarchuluun, B. R. Wong, M. Khurelbaatar, Y. S. Moroz, J. Mayfield and R. A. Sayle, J. Chem. Inf. Model., 2020, 60, 6065–6073 CrossRef CAS PubMed.
- T. Sterling and J. J. Irwin, J. Chem. Inf. Model., 2015, 55, 2324–2337 CrossRef CAS PubMed.
- REAL database, https://enamine.net/compound-collections/real-compounds/real-database, accessed 4/7/2024, 2024 Search PubMed.
- D. Mendez, A. Gaulton, A. P. Bento, J. Chambers, M. De Veij, E. Felix, M. P. Magarinos, J. F. Mosquera, P. Mutowo, M. Nowotka, M. Gordillo-Maranon, F. Hunter, L. Junco, G. Mugumbate, M. Rodriguez-Lopez, F. Atkinson, N. Bosc, C. J. Radoux, A. Segura-Cabrera, A. Hersey and A. R. Leach, Nucleic Acids Res., 2019, 47, D930–D940 CAS.
- D. Lowe, Chemical reactions from US patents (1976-Sep2016), figshare Dataset, 2017, DOI:10.6084/m9.figshare.5104873.v1.
- J. Mayfield, D. Lowe and R. Sayle, Pistachio. 2.0, 2022 Search PubMed.
- Reaxys Elsevier Information Systems GmbH, 2024 Search PubMed.
- A. Keto, T. Guo, M. Underdue, T. Stuyver, C. W. Coley, X. Zhang, E. H. Krenske and O. Wiest, J. Am. Chem. Soc., 2024, 146, 16052–16061 Search PubMed.
- Z. Wu, X. Cai, C. Zhang, H. Qiao, Y. Wu, Y. Zhang, X. Wang, H. Xie, F. Luo and H. Duan, J. Chem. Inf. Model., 2022, 62, 4579–4590 Search PubMed.
- Y. Zhang, L. Wang, X. Wang, C. Zhang, J. Ge, J. Tang, A. Su and H. Duan, Org. Chem. Front., 2021, 8, 1415–1423 CAS.
- M. Wen, S. M. Blau, X. Xie, S. Dwaraknath and K. A. Persson, Chem. Sci., 2022, 13, 1446–1458 CAS.
- P. Schwaller, T. Laino, T. Gaudin, P. Bolgar, C. A. Hunter, C. Bekas and A. A. Lee, ACS Cent. Sci., 2019, 5, 1572–1583 Search PubMed.
- J. Lu and Y. Zhang, J. Chem. Inf. Model., 2022, 62, 1376–1387 CrossRef CAS PubMed.
- R. Irwin, S. Dimitriadis, J. He and E. J. Bjerrum, Mach. Learn.: Sci. Technol., 2022, 3, 015022 Search PubMed.
- S. G. Espley, E. H. E. Farrar, D. Buttar, S. Tomasi and M. N. Grayson, Digital Discovery, 2023, 2, 941–951 RSC.
- C. Cai, S. Wang, Y. Xu, W. Zhang, K. Tang, Q. Ouyang, L. Lai and J. Pei, J. Med. Chem., 2020, 63, 8683–8694 CrossRef CAS PubMed.
- S. J. Pan and Q. Yang, IEEE Trans. Knowl. Data Eng., 2010, 22, 1345–1359 Search PubMed.
- W. Zhang, L. Deng, L. Zhang and D. Wu, IEEE/CAA J. Autom. Sin., 2023, 10, 305–329 Search PubMed.
- A. C. Cope and E. M. Hardy, J. Am. Chem. Soc., 1940, 62, 441–444 Search PubMed.
- N. Graulich, Wiley Interdiscip. Rev. Comput. Mol. Sci., 2011, 1, 172–190 Search PubMed.
- L. Claisen, Chem. Ber., 1912, 45, 3157–3166 Search PubMed.
- A. M. M. Castro, Chem. Rev., 2004, 104, 2939–3002 Search PubMed.
- O. Diels and K. Alder, Liebigs Ann. Chem., 1928, 460, 98–122 Search PubMed.
- K. N. Houk, F. Liu, Z. Yang and J. I. Seeman, Angew. Chem., Int. Ed., 2021, 60, 12660–12681 Search PubMed.
- J. Nowicki, Molecules, 2000, 5, 1033–1050 CrossRef CAS.
- J. A. Funel and S. Abele, Angew. Chem., Int. Ed., 2013, 52, 3822–3863 Search PubMed.
- M. Gregoritza and F. P. Brandl, Eur. J. Pharm. Biopharm., 2015, 97, 438–453 CrossRef CAS PubMed.
- B. Briou, B. Ameduri and B. Boutevin, Chem. Soc. Rev., 2021, 50, 11055–11097 CAS.
- W. Jin, C. W. Coley, R. Barzilay and T. Jaakkola, Adv. Neural Inf. Process. Syst., 2017, 30, 2607 Search PubMed.
- H. Bi, H. Wang, C. Shi, C. Coley, J. Tang and H. Guo, Presented in part at the Proc. 38th Int. Conf. Mach. Learn., 2021, vol. 139, p. 904 Search PubMed.
- M. Lewis, Y. Liu, N. Goyal, M. Ghazvininejad, A. Mohamed, O. Levy, V. Stoyanov and L. Zettlemoyer, arXiv, 2019, DOI:10.48550/arXiv.1910.13461.
- P. Schwaller, D. Probst, A. C. Vaucher, V. H. Nair, D. Kreutter, T. Laino and J.-L. Reymond, Nat. Mach. Intell., 2021, 3, 144–152 Search PubMed.
- L. McInnes, J. Healy, N. Saul and L. Großberger, J. Open Source Softw., 2018, 3, 861 Search PubMed.
- J. W. Rae, S. Borgeaud, T. Cai, K. Millican, J. Hoffmann, F. Song, J. Aslanides, S. Henderson, R. Ring, S. Young, E. Rutherford, et al., arXiv, 2021, 2112.11446v11442 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional figures, explanations, link to Github, Reaxys reaction, IDs and link to Jupyter notebook to regenerate dataset. See DOI: https://doi.org/10.1039/d4dd00412d |
| This journal is © The Royal Society of Chemistry 2025 |