
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
DOPtools: a Python platform for descriptor calculation and model optimization
Said
Byadi
 a,
Philippe
Gantzer
a,
Timur
Gimadiev
b and
Pavel
Sidorov
*a
a,
Philippe
Gantzer
a,
Timur
Gimadiev
b and
Pavel
Sidorov
*a
aInstitute for Chemical Reaction Design and Discovery (WPI-ICReDD), Hokkaido University, Kita 21 Nishi 10, Kita-ku, Sapporo, 001-0021, Japan. E-mail: pavel.sidorov@icredd.hokudai.ac.jp
bA.M. Butlerov Institute of Chemistry, Kazan Federal University, 18 Kremlyovskaya Str., Kazan, 420008, Russia
First published on 17th March 2025
Abstract
The DOPtools (Descriptors and Optimization tools) platform is a Python library for the calculation of chemical descriptors, hyperparameter optimization, and building and validation of QSPR models. In addition to the Python code that can be integrated in custom scripts, it provides a command line interface for the automatic calculation of various descriptors and for eventual hyperparameter optimization of statistical models, enabling its use in server applications for QSPR modeling. It is especially suited for modeling reaction properties via functions that calculate descriptors for all reaction components. While a variety of existing tools and libraries can calculate various molecular descriptors, their output format is often unique, which complicates their integration with standard machine learning libraries. DOPtools provides a unified API for the calculated descriptors as input for the scikit-learn library. The modular nature of the code allows easy addition of algorithms if required by the end user. The code for the platform is freely available at GitHub and can be installed through PyPI.
1 Introduction
Quantitative Structure–Property Relationship (QSPR) modeling is, to this day, one of the largest application fields of chemoinformatics.1 In a classical QSPR approach, to enable the prediction of properties, molecules must be encoded by numerical parameters – molecular descriptors, which are then used to train machine learning (ML) algorithms. The choice of the optimal molecular descriptors, as well as the ML algorithm, is often left to rigorous benchmarking prior to modeling itself. However, the lack of an open, unified pipeline to go from molecules to descriptors to model optimization necessitates piecing together disjointed steps produced by different software or scripts.Numerous software tools have been specifically tailored for the generation of molecular descriptors, providing user-friendly platforms via graphical user interfaces (GUIs) for inputting chemical structures and extracting a diverse array of molecular information. Commercial tools developed by OpenEye Scientific Software,2 Molecular Operating Environment (MOE),3 and ChemAxon4 provide a variety of functions, including the calculation of molecular properties and descriptors. On the other hand, programming libraries such as Chemistry Development KIT (CDK),5 RDKit,6 and OpenBabel7 give access to a wide range of chemoinformatics functions, with RDKit being a de facto standard for chemoinformatics applications in most areas of research. Other software kits such as PaDEL,8 ISIDA (In Silico Design and Data Analysis) platform,9 GUIDEMOL,10etc., implement GUI or command line interfaces (CLI) for the calculation of specific types of descriptors. Python libraries CGRtools11 and Chython12 provide an open-access code for managing chemical data, including the calculation of descriptors (e.g., Chython implements its own Morgan-like and linear fingerprints). Recently reported packages, MolPipeline13 and Scikit-Mol,14 provide an array of functions that facilitates integrating chemical information into ML modeling through automatic SMILES-to-descriptor calculations that include important curation steps.
Once the descriptors are calculated, a ML algorithm needs to be trained, and many solutions are available for this task. Statistical software such as WEKA,15 XLSTAT, Statistica,16etc., give access to a wide array of ML models that can be applied to precomputed descriptors. More specialized chemoinformatics software, like MOE, combines the calculation of descriptors and the building of models in one package. The commercial KNIME Analytics Platform17 is an open-source platform with workflow-driven cheminformatics capabilities, implementing the most commonly used descriptor types and ML algorithms, and providing bindings for external chemoinformatics and molecular modeling tools and libraries. QSAR-Co18 is an open source tool written in Java that is capable of robust data analysis and the development of classification models, including multi-task ones. However, models often require optimization of parameters, which can be complicated with the above tools. Often, researchers create in-house scripts tailored to the third-party tools they have access to.19 Some commercial solutions exist for this problem, too; for example, Schrödinger provides a platform with access to both calculation of descriptors and modeling (CANVAS), as well as tools for the automatization of model optimization and building (AutoQSAR,20 which has recently evolved into DeepAutoQSAR). Auto-Sklearn has been introduced recently21,22 as an ML automation (AutoML) platform, implementing a Bayesian optimization algorithm for data set preparation, feature calculation and preprocessing, and model hyperparameter optimization.
The alternative that gives the most freedom for customization of descriptors and models is writing custom scripts using the abundance of tools available to researchers these days. Specifically, the Python programming language has an extraordinary level of community support for ML-related tasks, with open-source libraries like scikit-learn23 for ML algorithms, pandas24 for data processing, as well as RDKit, OpenBabel, or Chython for chemoinformatics-related tasks. Scikit-learn has limited capabilities for model parameters optimization using grid search, and other libraries such as Optuna25 provide an expanded selection of optimization algorithms. However, there is still an issue of compatibility between application programming interfaces (API) of these libraries, especially chemical ones, as their outputs often cannot directly serve as inputs for ML algorithms. Still, some recently reported tools, such as ROBERT,26 QSPRPred,27 QSARTuna,28 and PREFER,29 allow building a complete workflow from descriptor generation to modeling of molecular properties.
The field of reaction modeling is rapidly gaining traction in chemoinformatics; however, a notable gap remains in the availability of comprehensive, ready-to-use programming libraries capable of seamlessly performing reaction modeling tasks. Since reactions involve multiple molecular entities (reactants, products, catalysts, and additives), the most common approach to their representation is concatenating the descriptors of different species into a single table. Addressing this challenge with existing solutions typically requires significant customization, integration of multiple tools, or the development of bespoke algorithms.
An alternative approach is the Condensed Graph of Reaction (CGR),30 which simplifies reaction representation by encoding it as a single graph with explicit annotations for dynamic bonds and atoms – those that change during the reaction. The CGR concept has been successfully applied in numerous studies to model various reaction properties.31–33 However, most Python-based chemical libraries lack support for CGR structures. To our knowledge, the only libraries capable of handling CGRs are CGRtools11 and Chython.12
In this work, we present a new Python library, DOPtools, with the capabilities to calculate an extensive array of molecular descriptors, encompassing physico–chemical, structural, and fragment-based descriptors, within an API tailored to most ML libraries. DOPtools are especially tailored for reaction modeling, providing functions for the calculation of descriptors both in a classical way (concatenation of species) and using Condensed Graphs of Reactions. Moreover, the library provides a CLI for automatic descriptor calculation and optimization of hyperparameters for QSPR models, suitable for server applications. While only three major statistical methods – Support Vector Machine (SVM),34 XGBoost,35 and Random Forest (RF)36 – are available out of the box, the modular structure of the library and the simplicity of Python itself allow for easy extension to other methods or descriptor types. Moreover, this versatile tool extends its utility by facilitating the visualization of atomic contributions within the developed models. We present several examples of functions to demonstrate the capabilities of the library, which are also available as tutorials in the GitHub repository.37
2 Implementation
DOPtools (current version 1.2) is a Python library that provides functions to calculate a variety of molecular descriptors in a unified manner, compatible with common ML libraries, as well as scripts to prepare and optimize regression and classification models. The library uses Python version 3.9 (or higher, as long as the compatibility of other packages is not compromised) and the widely used computation and machine learning packages such as Numpy (v.1.25), Pandas (v.2.1), Scikit-learn (v.1.5), and XGBoost (v.2.0). The handling of chemical structures is ensured by the packages Chython (v.1.7) and RDKit (v.2023.9.6). Molecular descriptors are calculated using RDKit, the Mordred39 library (v.1.2), or built-in functions within DOPtools. Model's hyperparameter optimization is implemented using the Optuna library (v.3.5).The library can be installed from the PyPI repository:  . It is recommended to install it in an Anaconda environment or similar, to have easy access to executable scripts provided in the library. Alternatively, the source code and setup files for the version in development are also available on GitHub.37 The library has been tested and validated on ×86 platforms, but its performance on arm64 is not guaranteed due to certain modules dependencies.
. It is recommended to install it in an Anaconda environment or similar, to have easy access to executable scripts provided in the library. Alternatively, the source code and setup files for the version in development are also available on GitHub.37 The library has been tested and validated on ×86 platforms, but its performance on arm64 is not guaranteed due to certain modules dependencies.
The main features provided by DOPtools are as follows:
• Reading of chemical structures (both molecules and reactions) in the SMILES format and standardization of structures are performed by the Chython library.
• Preparation of a wide array of descriptors from chemical structures – structural (fingerprints from RDKit and newly implemented molecular fragments) and physico–chemical (Mordred library). Reaction fragments can be calculated via the use of CGRs. Concatenation of different types of features or features for several structures is implemented out of the box.
• Physico–chemical descriptors for 152 solvents.
• Model hyperparameter optimization, including the selection of the best descriptor set.
• Interpretation of models built on molecular fragments using the ColorAtom methodology.40
• A command-line interface is provided for descriptor preparation, model optimization and plotting.
The following sections explain in detail the structure and the functionality of various modules within the library. A brief comparison of the features provided by DOPtools with other similar tools is given in Table 1.
| Feature | DOPtools | ROBERT | QSPRpred | QSARtuna | PREFER |
|---|---|---|---|---|---|
| a Abbreviations for ML algorithms: SVM – Support Vector Machines, RF – Random Forest, NN – Neural Networks, DNN – Deep Neural Networks, and GP – Gaussian Processes | |||||
| Reaction/mixture modeling | Yes | No | No | No | No |
| CLI for automation | Yes | No | Yes | Yes | No |
| GUI support | In development | Yes | No | No | No |
| Hyperparameter optimization | Optuna | hyperopt38 | Customizable | Optuna | Python AutoML libraries and Optuna |
| Uncertainty estimation | No | Yes | No | Yes | No |
| Explainability features | ColorAtom | Yes | Yes | Yes | Yes |
| Descriptor types | Fragments, fingerprints, physico–chemical (2D) | Topological, quantum, empirical | Physico–chemical, fingerprints, graph | Structural (RDKit), physico–chemical | 2D physico-chemical, RDKit descriptors |
| ML algorithms | SVM, XGBoost, RF | Auto-selected (RF, SVM, NN, GP) | SVM, RF, NN | SVM, RF, GP | AutoML DL (DNN, RF, XGBoost) |
2.1
 module
module

 submodule.
The DOPtools platform, in its current implementation, allows the calculation of 2D molecular descriptors of several types. The broadest category includes the molecular fingerprints provided by the RDKit library, which include Morgan,41 Avalon,42 atom pairs,43 topological torsion,44 and native RDKit fingerprints. In addition, 2D Mordred descriptors are available through the import of the Mordred library.39 Finally, two types of fragment descriptors are implemented natively via Chython functions – circular fragments CircuS (first introduced in ref. 31) and linear fragments ChyLine (a new implementation). The brief information on these descriptors and details of their implementation are discussed in this section. The Python objects that calculate each type of descriptors are coded as scikit-learn transformers and realize the standard
submodule.
The DOPtools platform, in its current implementation, allows the calculation of 2D molecular descriptors of several types. The broadest category includes the molecular fingerprints provided by the RDKit library, which include Morgan,41 Avalon,42 atom pairs,43 topological torsion,44 and native RDKit fingerprints. In addition, 2D Mordred descriptors are available through the import of the Mordred library.39 Finally, two types of fragment descriptors are implemented natively via Chython functions – circular fragments CircuS (first introduced in ref. 31) and linear fragments ChyLine (a new implementation). The brief information on these descriptors and details of their implementation are discussed in this section. The Python objects that calculate each type of descriptors are coded as scikit-learn transformers and realize the standard  and
and  functions, enabling easy integration of those into pipelines and unifying the input and output when used in the end user's code.
functions, enabling easy integration of those into pipelines and unifying the input and output when used in the end user's code.
Calculation of molecular fingerprints is implemented in the  class. The initialization of the object requires the indication of the fingerprint type (
class. The initialization of the object requires the indication of the fingerprint type ( argument). The possible values for this argument are
argument). The possible values for this argument are  for Morgan fingerprints with or without the features,
for Morgan fingerprints with or without the features, 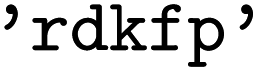 for RDKit fingerprints of any topology,
for RDKit fingerprints of any topology,  for RDKit layered fingerprints, and
for RDKit layered fingerprints, and  for the corresponding types.
for the corresponding types.  argument determines the length of the bit vector the object will calculate.
argument determines the length of the bit vector the object will calculate.  indicates the maximum length/radius for Morgan and RDKit fingerprints and does not affect other types. Additional parameters (e.g., calculation of Morgan fingerprints with chemical features) could be passed via the
indicates the maximum length/radius for Morgan and RDKit fingerprints and does not affect other types. Additional parameters (e.g., calculation of Morgan fingerprints with chemical features) could be passed via the  argument.
argument.
CircuS (Ccrcular substructures) fragments account for fragments of circular topology, i.e., atoms and their environments within a certain radius. The user must indicate the desired lower and upper limits for the size of substructures, as the topological radius. A size of 0 means only the atom itself, a size of 1 – an atom and all atoms directly connected to it, and so on. The algorithm will run through all atoms in the molecule (or CGR) and enumerate all possible substructures. Due to the way the substructure extraction is implemented in the Chython library, all bonds between selected atoms will be present, which may be slightly counterintuitive and differs from the way Morgan fingerprints are calculated in RDKit (see an example above, Fig. 1). A new functionality is implemented via the argument  : if set to
: if set to  , the algorithm will instead go through all bonds in the molecules, extracting augmented substructures centered on those bonds (example shown above). An older implementation of the CircuS calculator31 calculating fragment counts via a substructure search is also available as the
, the algorithm will instead go through all bonds in the molecules, extracting augmented substructures centered on those bonds (example shown above). An older implementation of the CircuS calculator31 calculating fragment counts via a substructure search is also available as the  class. However, that version is extremely slow and is not recommended for use.
class. However, that version is extremely slow and is not recommended for use.
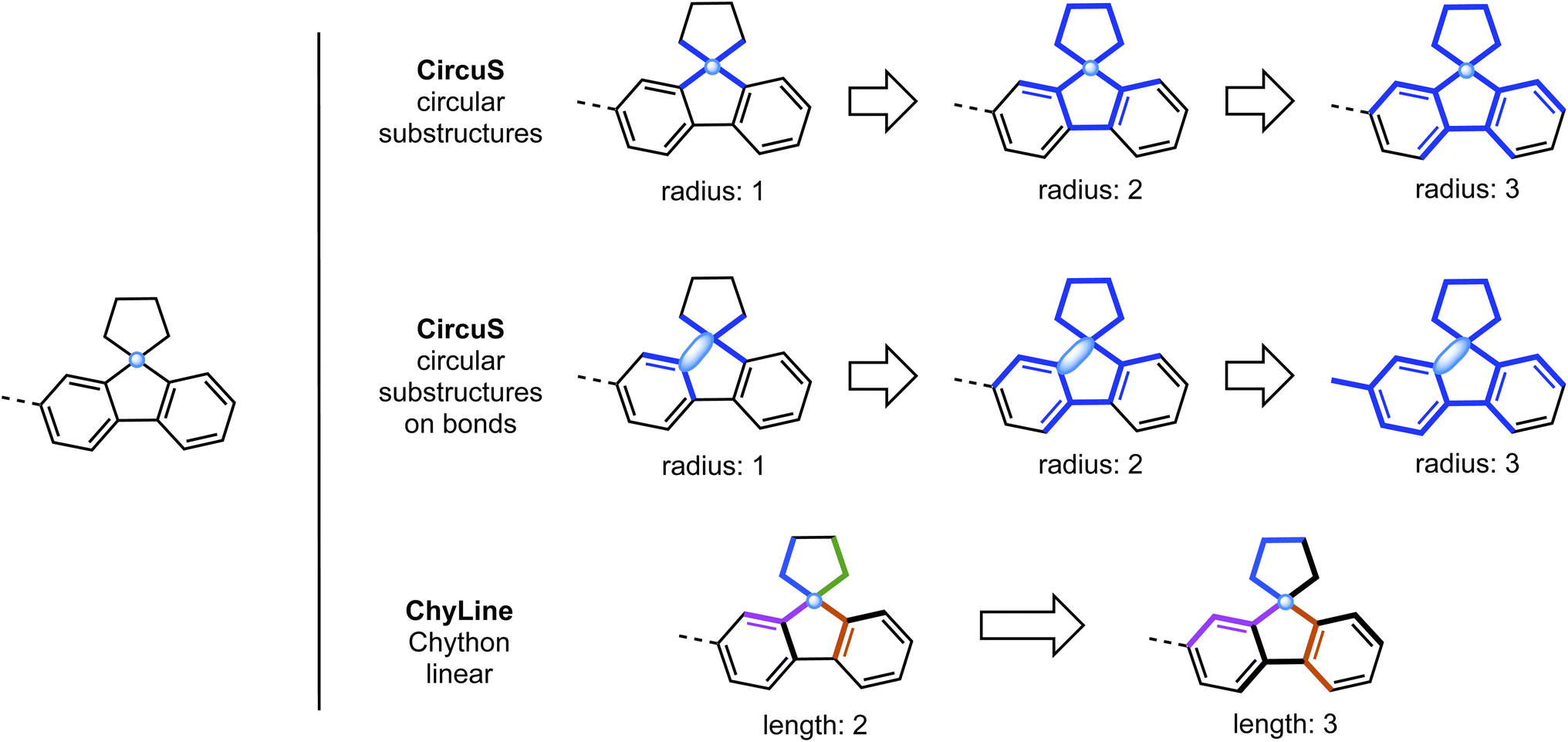 | ||
| Fig. 1 Comparison of fragments generated by CircuS (top and middle) and ChyLine (bottom) fragments, starting from the same center atom (indicated by the light blue dot). Bold blue lines in CircuS examples indicate the substructure of the specified radius. In ChyLine examples, the bold lines indicate the substructure of the given radius from where all linear fragments starting at the center atom would be taken. Different colors indicate different linear fragments of the same length. | ||
ChyLine (Chython Linear) fragments are linear fragments calculated using native Chython functions, specifically, from the 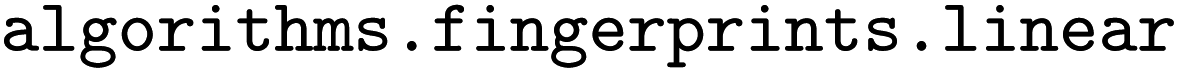 module. Their implementation is similar to CircuS and requires specifying the lower and upper limits for the lengths of fragments. The algorithm gathers all linear subgraphs in the molecule of the specified lengths (see examples in Fig. 1). Unlike the native RDKit fingerprints, ChyLine gathers the information on the frequencies of each linear substructure out of the box. Moreover, it can calculate the fragments of CGRs in addition to single molecules, similar to CircuS.
module. Their implementation is similar to CircuS and requires specifying the lower and upper limits for the lengths of fragments. The algorithm gathers all linear subgraphs in the molecule of the specified lengths (see examples in Fig. 1). Unlike the native RDKit fingerprints, ChyLine gathers the information on the frequencies of each linear substructure out of the box. Moreover, it can calculate the fragments of CGRs in addition to single molecules, similar to CircuS.
Descriptor calculators implement a  function that returns a Pandas DataFrame with the descriptor values for any data set after fitting. The column names in fragment calculators are the SMILES representations of the corresponding substructures. Note that in these cases, only the fragments that were initially recorded during fitting will be present in the resulting table and all new fragments that may be present in new molecules will be ignored. This is done in order to avoid feature number mismatch during ML model training and application. The fragment SMILES may also be accessed via the
function that returns a Pandas DataFrame with the descriptor values for any data set after fitting. The column names in fragment calculators are the SMILES representations of the corresponding substructures. Note that in these cases, only the fragments that were initially recorded during fitting will be present in the resulting table and all new fragments that may be present in new molecules will be ignored. This is done in order to avoid feature number mismatch during ML model training and application. The fragment SMILES may also be accessed via the  function.
function.
The platform also provides physico–chemical parameters for 152 solvents that can be used as descriptors. The data were extracted from the literature46 as tabular values, and include the empirical measurements of solvent acidity (SA), basicity (SB), dipolarity (SdP), and polarizability (SP). The  class implemented as a scikit-learn transformer takes an array of strings corresponding to the solvent names and outputs a table with the four abovementioned values. The available solvent names are provided in the variable
class implemented as a scikit-learn transformer takes an array of strings corresponding to the solvent names and outputs a table with the four abovementioned values. The available solvent names are provided in the variable  .
.
Finally, a utility class for calculating concatenated descriptors (for either different structures, or different descriptor types) is also introduced as  . Rather than an array of molecules, it takes a DataFrame with several molecule columns, and the
. Rather than an array of molecules, it takes a DataFrame with several molecule columns, and the  parameter specifies which calculator would be used for each column as a list of pairings ”column name” – ”descriptor calculator” (similar to the Pipeline implementation in scikit-learn). The idea behind this implementation is to allow seamless calculation of descriptors for mixtures or reactions via concatenation of features. The
parameter specifies which calculator would be used for each column as a list of pairings ”column name” – ”descriptor calculator” (similar to the Pipeline implementation in scikit-learn). The idea behind this implementation is to allow seamless calculation of descriptors for mixtures or reactions via concatenation of features. The  class facilitates such concatenation, allowing to specify which descriptors need to be used for each species, and returning a unified table with columns labeled according to their respective components. Similarly, solvent descriptors or numerical parameters may also be passed into a
class facilitates such concatenation, allowing to specify which descriptors need to be used for each species, and returning a unified table with columns labeled according to their respective components. Similarly, solvent descriptors or numerical parameters may also be passed into a  .
.
DOPtools also allows to calculate fragments of various topologies for reactions represented as CGRs out of the box. The ChyLine and CircuS calculators presented here internally transform a mapped reaction into a CGR during the calculations; thus no additional transformation steps are required. The fragments that contain dynamic bonds or atoms will be annotated in the CGR SMILES format introduced previously.47 For example, a single bond formation is noted as [.>–], while a bond changing order from single to double is represented as [–>=]. Note that, since RDKit or Mordred do not support CGR structures, the calculation of fingerprints or physico–chemical parameters for CGRs is not implemented.
Some examples of the descriptor calculation functions for both molecules and reactions (in both concatenation and CGR formats) are shown in Fig. 2. These examples are taken from the tutorials available in the library's GitHub repository,37 where the source code and data are deposited.
 | ||
| Fig. 2 Examples of code and output of fragment calculation functions of DOPtools. (A) Calculation of CircuS fragments; (B) calculation of ChyLine fragments. Both examples use a photoswitch data set.45 (C) Calculation of concatenated fragments for catalyst enantioselectivity modeling, including structural descriptors of substituents and solvent descriptors (data from ref. 31 are used). (D) Calculation of reaction fragments via condensed graph of reaction representation. Fragmentors can handle fully mapped reactions and transform them into CGR internally. Dynamic bonds are represented in fragments using the CGR SMILES notation.11 | ||
 submodule.
DOPtools provides a Python implementation of the ColorAtom method40 for CircuS and ChyLine fragments. ColorAtom allows to interpret the model predictions through atomic contributions. In this approach, the weights of all fragments are calculated as a function of prediction change. For regression, this is the partial derivative of the prediction over this fragment's occurrence number. For classification, it is inversely proportional to the required change in the descriptor value that would change the prediction (thus, the smaller the change in the descriptor value that causes the change in the predicted class, the more important the fragment is). Atoms involved in all fragments accumulate their weights as the score, which is then visualized by assigning colors to positive and negative contributions. This helps in drawing conclusions about which modifications to the structure may be beneficial for further improvement of the studied property. By default, regression models are visualized using a divergent color scale (the atoms contributing to higher values are colored in magenta, and atoms contributing to lower values are colored in green), and classification models use a single-color scale, with the intensity of the color normalized across the molecule, so that the relative contribution can be easily visualized (Fig. 3). Contributions across several molecules may be compared by scaling the colors to the maximum values between them. This is the case for models that take several molecular structures via ComplexFragmentor. If reactions are provided directly as structures, the user must specify whether they are visualized as an ensemble of reactants or products (see the example in the tutorials on the GitHub repository).
submodule.
DOPtools provides a Python implementation of the ColorAtom method40 for CircuS and ChyLine fragments. ColorAtom allows to interpret the model predictions through atomic contributions. In this approach, the weights of all fragments are calculated as a function of prediction change. For regression, this is the partial derivative of the prediction over this fragment's occurrence number. For classification, it is inversely proportional to the required change in the descriptor value that would change the prediction (thus, the smaller the change in the descriptor value that causes the change in the predicted class, the more important the fragment is). Atoms involved in all fragments accumulate their weights as the score, which is then visualized by assigning colors to positive and negative contributions. This helps in drawing conclusions about which modifications to the structure may be beneficial for further improvement of the studied property. By default, regression models are visualized using a divergent color scale (the atoms contributing to higher values are colored in magenta, and atoms contributing to lower values are colored in green), and classification models use a single-color scale, with the intensity of the color normalized across the molecule, so that the relative contribution can be easily visualized (Fig. 3). Contributions across several molecules may be compared by scaling the colors to the maximum values between them. This is the case for models that take several molecular structures via ComplexFragmentor. If reactions are provided directly as structures, the user must specify whether they are visualized as an ensemble of reactants or products (see the example in the tutorials on the GitHub repository).
 | ||
| Fig. 3 Model interpretation by ColorAtom. Top – a regression model with the standard color scheme (SVM regression model using CircuS fragments built on the photoswitch data set45). Relative atom contributions to the predictions are indicated by colors: green indicates that the atom's presence leads to a higher (more positive) property value and magenta – to a lower (more negative) property value. Middle – the same model, but with a custom color scheme and a colorbar to indicate the scale of atomic contributions. Bottom – a classification model with the standard color scheme (RF classification using ChyLine fragments built on blood–brain barrier penetration data48). | ||
After defining the object, the user needs to specify the pipeline (as a scikit-learn object) that is used for prediction, by calling the  function. The function assumes that the first object in the pipeline is the fragment calculator. Afterwards, the user can use the object to calculate the contribution and output them numerically with the
function. The function assumes that the first object in the pipeline is the fragment calculator. Afterwards, the user can use the object to calculate the contribution and output them numerically with the  function, or visualize them directly with the
function, or visualize them directly with the  function. The visualization is produced in SVG and HTML formats which can be directly visualized in Jupyter notebooks using Chython to depict the structure.
function. The visualization is produced in SVG and HTML formats which can be directly visualized in Jupyter notebooks using Chython to depict the structure.
2.2
 and
and  modules
modules
The 

 submodule contains functions for performing the tasks of descriptor enumeration and model hyperparameter optimization.
submodule contains functions for performing the tasks of descriptor enumeration and model hyperparameter optimization. 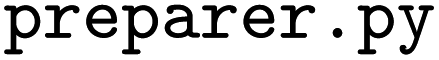 calculates and outputs the descriptor table files;
calculates and outputs the descriptor table files;  runs the algorithm for hyperparameter optimization on the chosen descriptor type. The submodule contains the corresponding CLI scripts
runs the algorithm for hyperparameter optimization on the chosen descriptor type. The submodule contains the corresponding CLI scripts  and
and  , as well as special scripts
, as well as special scripts  to rebuild a specified model from the parameters indicated in the result table and
to rebuild a specified model from the parameters indicated in the result table and  which includes utility functions for plotting the cross-validation (CV) results. The separation of the main function from CLI is done so that the batch descriptor calculation and model optimization could be performed separately in a Jupyter Notebook or embedded into custom scripts, while CLI provides the backend for server applications.
which includes utility functions for plotting the cross-validation (CV) results. The separation of the main function from CLI is done so that the batch descriptor calculation and model optimization could be performed separately in a Jupyter Notebook or embedded into custom scripts, while CLI provides the backend for server applications.
 accepts a structure table (csv or Excel format) to calculate a variety of descriptors. All options and their descriptions are shown in Table 2. The script can output descriptors in a CSV or a SVM format and allows for saving the trained descriptor calculator objects for each type. The SVM format is a representation of a sparse matrix where each line contains only non-zero elements along with their indices, making it highly suitable for fragments and fingerprints. The property is recorded in the first column in both formats. If the initial data table contains several property columns (i.e., the
accepts a structure table (csv or Excel format) to calculate a variety of descriptors. All options and their descriptions are shown in Table 2. The script can output descriptors in a CSV or a SVM format and allows for saving the trained descriptor calculator objects for each type. The SVM format is a representation of a sparse matrix where each line contains only non-zero elements along with their indices, making it highly suitable for fragments and fingerprints. The property is recorded in the first column in both formats. If the initial data table contains several property columns (i.e., the  argument is followed by more than one column name), separate files will be produced for each property. The script can sort the files by descriptor type, if a benchmark of each type is needed, or output them all in the same folder if the goal is to select the best descriptor type. The user may also concatenate the descriptors for several structure columns and/or solvents into one descriptor file (please note that only descriptors of one specific type at a time will be calculated for all columns, so combinatorial concatenation is not available at the moment). The descriptors that involve size parameters allow all possible choices to be indicated simultaneously in one run. The resulting file names follow the format of
argument is followed by more than one column name), separate files will be produced for each property. The script can sort the files by descriptor type, if a benchmark of each type is needed, or output them all in the same folder if the goal is to select the best descriptor type. The user may also concatenate the descriptors for several structure columns and/or solvents into one descriptor file (please note that only descriptors of one specific type at a time will be calculated for all columns, so combinatorial concatenation is not available at the moment). The descriptors that involve size parameters allow all possible choices to be indicated simultaneously in one run. The resulting file names follow the format of  . The basic set of descriptor parameters to calculate all fingerprints and fragments is given in the repository (
. The basic set of descriptor parameters to calculate all fingerprints and fragments is given in the repository ( ) and can be applied via the option
) and can be applied via the option  .
.
 script corresponding to the various molecular descriptors and their parameters
script corresponding to the various molecular descriptors and their parameters
| General | CLI option | Description |
|---|---|---|
a For all FP, the size of the bit vector can be parameterized as 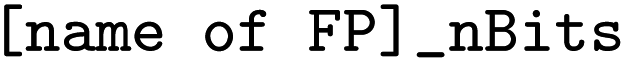 (an example is given for Morgan FP) (an example is given for Morgan FP)
|
||
| Input file |
|
Input file, requires csv or Excel format |
| Output directory |

|
Output directory |
| Output format |

|
The format for saving the descriptor files |
| Output option |

|
A toggle to save each descriptor type into a separate folder |
| Input structures |
|
The name of the column where the SMILES are stored |

|
Additional columns with SMILES, the descriptors for which will be calculated and concatenated together | |
| Solvents |

|
Column that contains the solvent names |
| Model target |

|
The column containing the modeled property (in numerical format) |

|
Alternative column names, for the cases where the column names in the input file contain spaces or the names are overly long | |
| Options |
|
Any number over 0 launches the calculation of descriptors in parallel |

|
Save the fragmentors in pickle format | |
| Descriptor type | CLI option | Description |
|---|---|---|
| Morgana |

|
Toggles for calculation of Morgan and Morgan feature FP, respectively |

|
Sets the size of the bit vector (default 1024) | |

|
Indicates the radius of Morgan FP | |
| RDKit FPa |

|
Toggles for calculation of RDKit FP, including linear and layered ones |

|
Indicates the maximum length of RDKit FP. | |
| Avalona |

|
Toggle for calculation of Avalon FP |
| Atom Pairsa |

|
Toggle for calculation of atom pair FP |
| Torsiona |
|
Toggle for calculation of torsion FP |
| CircuS |

|
Toggle for calculation of CircuS fragments |

|
Indicates the minimum and maximum radii for CircuS fragments. For each combination, separate files will be output | |
| ChyLine |

|
Toggle for calculation of ChyLine fragments |

|
Indicates the minimum and maximum lengths for ChyLine fragments. For each combination, separate files will be output | |
| Mordred |

|
Toggle for calculation of Mordred 2D |
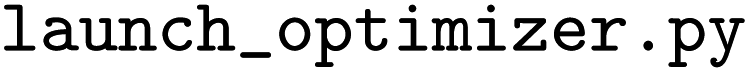 initiates the optimization of model hyperparameters using the descriptor files generated by the previous script. The optimization is powered by the Optuna library, which implements the Tree-structured Parzen Estimator (TPE).49 Descriptor spaces as well as the algorithm hyperparameters that are given in the
initiates the optimization of model hyperparameters using the descriptor files generated by the previous script. The optimization is powered by the Optuna library, which implements the Tree-structured Parzen Estimator (TPE).49 Descriptor spaces as well as the algorithm hyperparameters that are given in the  file are all subject to the optimization (the latter are given in Table 3). Currently, the script allows to choose from Support Vector Machines (
file are all subject to the optimization (the latter are given in Table 3). Currently, the script allows to choose from Support Vector Machines (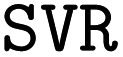 and
and  classes in scikit-learn), Random Forest (
classes in scikit-learn), Random Forest ( and
and  classes in scikit-learn) and XGBoost (
classes in scikit-learn) and XGBoost ( and
and  classes in the XGBoost library) as methods for both classification and regression, although other methods may be added by the user as they see fit. The main arguments are the input and output folders (
classes in the XGBoost library) as methods for both classification and regression, although other methods may be added by the user as they see fit. The main arguments are the input and output folders (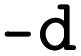 and
and 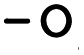 , respectively). The
, respectively). The  argument defines the ML algorithm that will be used. Model's performance is evaluated on the test set predictions in CV, and its parameters are given by the options
argument defines the ML algorithm that will be used. Model's performance is evaluated on the test set predictions in CV, and its parameters are given by the options  for the number of folds K in K-fold CV and
for the number of folds K in K-fold CV and  for repeated CV. All cross-validation during optimization is performed in a random manner; there are currently no options for stratification or a predetermined train-test split. The script will launch the optimization in parallel if the parameter
for repeated CV. All cross-validation during optimization is performed in a random manner; there are currently no options for stratification or a predetermined train-test split. The script will launch the optimization in parallel if the parameter 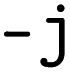 is used to indicate the number of CPU cores.
is used to indicate the number of CPU cores.  defines the time in seconds that is given to each process to finish, otherwise the process will be terminated (to prevent processes from getting stuck). Finally, an early stopping criterion is implemented to stop the optimization if a specific number of best models (
defines the time in seconds that is given to each process to finish, otherwise the process will be terminated (to prevent processes from getting stuck). Finally, an early stopping criterion is implemented to stop the optimization if a specific number of best models (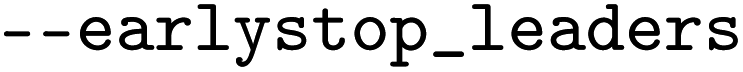 ) do not change for a certain number of steps (
) do not change for a certain number of steps ( ).
).
 script, as they are in the original libraries (Scikit-learn and XGBoost). These parameters and their possible values are given in the
script, as they are in the original libraries (Scikit-learn and XGBoost). These parameters and their possible values are given in the  file of the
file of the  module
module
| Method | Implementation | Hyperparameters |
|---|---|---|
| SVM | SVR, SVC |

|
| Random Forest | RandomForestRegressor, RandomForestClassifier |

|
| XGBoost | XGBRegressor, XGBClassifier |

|
After the optimization, the output folder will contain a folder for each successfully finished trial with CV prediction results for each repeat ( file) and the overall statistics (
file) and the overall statistics ( file). Also, two files with the scores and hyperparameters will be recorded, one with all trials (
file). Also, two files with the scores and hyperparameters will be recorded, one with all trials ( ) and one with the top 50, sorted by score (
) and one with the top 50, sorted by score ( ). The reported scores are RMSE, MAE and R2 for regression, and ROC AUC, accuracy, balanced accuracy and F1 score for classification.
). The reported scores are RMSE, MAE and R2 for regression, and ROC AUC, accuracy, balanced accuracy and F1 score for classification.
 allows to rebuild a pipeline containing the descriptor calculator, preprocessing and model using the hyperparameters obtained after optimization, and saves it as a scikit-learn object. The arguments for the script are the descriptor folder containing the descriptor calculator object in the
allows to rebuild a pipeline containing the descriptor calculator, preprocessing and model using the hyperparameters obtained after optimization, and saves it as a scikit-learn object. The arguments for the script are the descriptor folder containing the descriptor calculator object in the  format (
format ( ), the folder where the
), the folder where the  file is located for the models to be reproduced (
file is located for the models to be reproduced ( ), the number of the trial to reproduce (
), the number of the trial to reproduce ( ) and the output folder (
) and the output folder (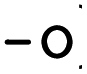 ).
).
The  script outputs the figure with the cross-validation results for a specific trial. The arguments are the input folder where the
script outputs the figure with the cross-validation results for a specific trial. The arguments are the input folder where the  file is located (
file is located ( ) and the output file name (
) and the output file name ( ). The optional parameters include the title of the plot (
). The optional parameters include the title of the plot ( ), the toggle for the output of the statistical scores (
), the toggle for the output of the statistical scores ( ), and the toggle for the error bar (
), and the toggle for the error bar ( ). The error bar is set to the standard deviation of the predictions across the repeats of the CV, so it would have no effect if a non-repeated CV was used during optimization. The script will use the property name from the
). The error bar is set to the standard deviation of the predictions across the repeats of the CV, so it would have no effect if a non-repeated CV was used during optimization. The script will use the property name from the  file for the axis names. The format of the
file for the axis names. The format of the  file and the examples of the regression and classification plots are shown in Fig. 4.
file and the examples of the regression and classification plots are shown in Fig. 4.
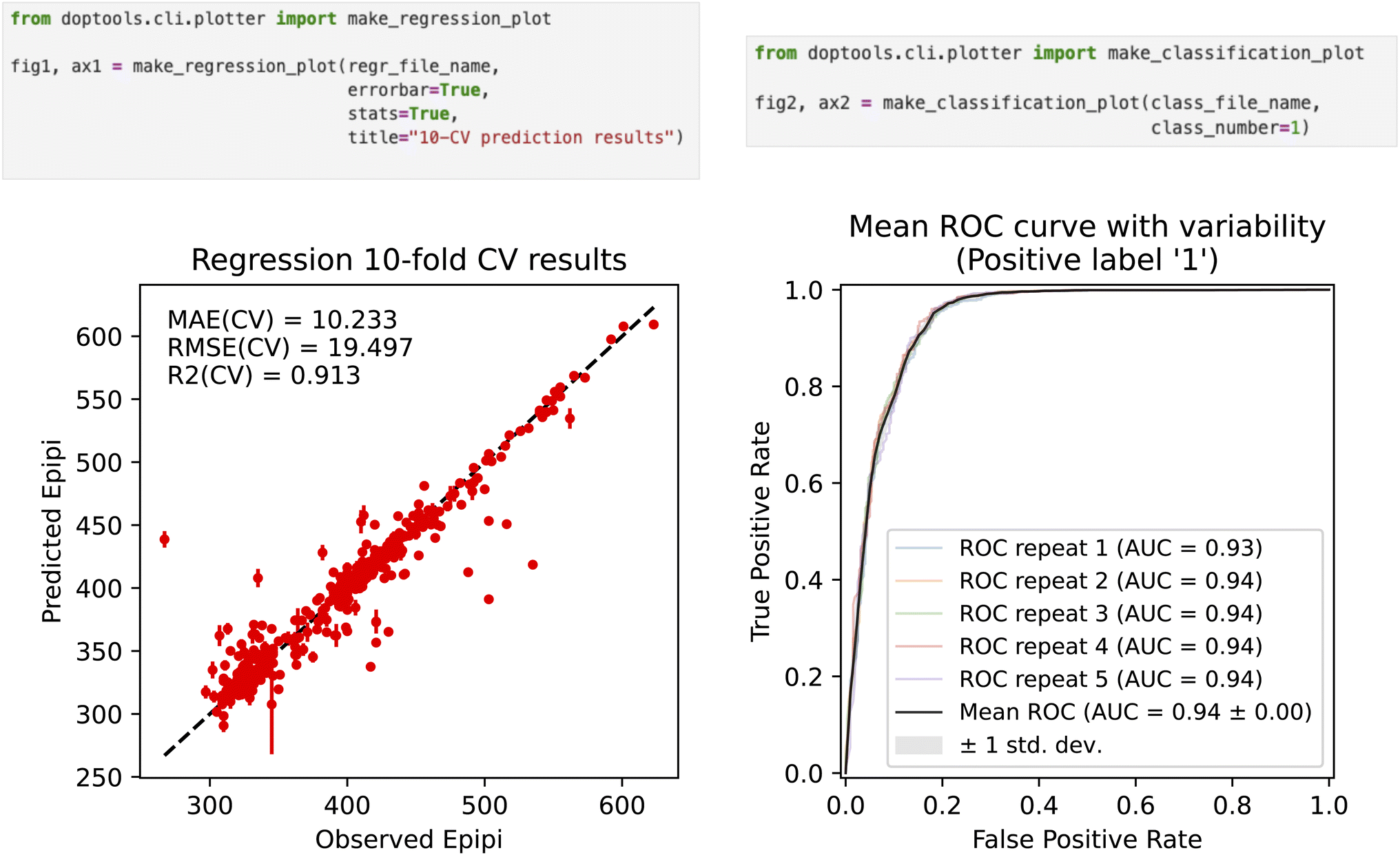 | ||
Fig. 4 Regression (on the left) and classification (on the right) plots produced by the  script from the optimization results files, using the code snippets shown on top. The example plot is for an SVM regression model using CircuS fragments built on the photoswitch data set45 to predict the maximum absorption wavelength. The classification model is built on a blood–brain barrier penetration data set48 using Avalon fingerprints. Both plots are for illustration purposes only. script from the optimization results files, using the code snippets shown on top. The example plot is for an SVM regression model using CircuS fragments built on the photoswitch data set45 to predict the maximum absorption wavelength. The classification model is built on a blood–brain barrier penetration data set48 using Avalon fingerprints. Both plots are for illustration purposes only. | ||
3 Discussion
DOPtools provides two ways for users to interact with the developed classes and functions. First, the classes for the calculation of structural descriptors, solvent descriptors, and ColorAtom implementation (i.e., module) can be freely integrated into custom scripts or Jupyter notebooks. As the calculators are written in a way that follows the scikit-learn notations, they can be seamlessly merged with existing pipelines using this library. In this case, the calculation of descriptors, data processing, and outputs can be fully customized, although the knowledge of Python coding would be required. Second, the CLI scripts from the
module) can be freely integrated into custom scripts or Jupyter notebooks. As the calculators are written in a way that follows the scikit-learn notations, they can be seamlessly merged with existing pipelines using this library. In this case, the calculation of descriptors, data processing, and outputs can be fully customized, although the knowledge of Python coding would be required. Second, the CLI scripts from the  and
and  modules can be used in the command line without any previous programming experience, as they only require the library to be installed and the input files to follow specific formats.
modules can be used in the command line without any previous programming experience, as they only require the library to be installed and the input files to follow specific formats.
Future developments of the library concern expanding the functions that are commonly required in QSPR modeling and chemical data analysis. One of the key areas to improve is related to memory management. In the current implementation, both  and
and  read the whole data set as the default behavior. While this is not an issue for smaller data sets and for certain types of descriptors, it may lead to overconsumption of memory and long calculation time for large data sets (over 10
read the whole data set as the default behavior. While this is not an issue for smaller data sets and for certain types of descriptors, it may lead to overconsumption of memory and long calculation time for large data sets (over 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 data points). Partial fitting in descriptor calculators, as well as data processing in batches, could resolve such issues; however, these features are not part of the current implementation and must be handled by the end user.
000 data points). Partial fitting in descriptor calculators, as well as data processing in batches, could resolve such issues; however, these features are not part of the current implementation and must be handled by the end user.
Another functionality that could prove useful in QSPR modeling, especially for the prediction of external compounds and virtual screening, is the implementation of applicability domain (AD)50 estimation. AD estimation is an important step in virtual screening and evaluation of modeling results. Currently, there are many ways to estimate the AD, including some methods that are specific for certain descriptor types51 (e.g., fragment control is unique for molecular fragments). However, at this time, DOPtools doesn't have any inherent implementation of these methods, and the end user must implement these themselves if needed.
4 Conclusions
DOPtools is a Python library that facilitates the calculation of different types of descriptors for molecules and reactions, including tabulated physico–chemical parameters for solvents, as well as ML model optimization, building and validation. The descriptor calculation functions are implemented following the API of the widely used scikit-learn library, to adhere to the best practices in the machine learning community. The library provides unique capabilities for calculating reaction fragments through the Condensed Graph of Reaction approach, facilitating modeling of reactions of mixed classes. The command-line interface allows to easily automatize the descriptor calculation and model hyperparameter optimization, enabling its use in server applications. Additional tools such as ColorAtom are available for model prediction interpretations and result plotting. While a limited number of ML algorithms are available out of the box, the modularity of the library and the general simplicity of the Python language facilitate the extension of its functions by the end user. The library is freely available on GitHub or PyPI, and tutorials for its basic functions from descriptor calculations to QSPR modeling and prediction are provided as supplements to this manuscript.Data availability
All data, including data sets used for illustration purposes, as well as the source code, reported in this manuscript are freely available in the DOPtools GitHub repository found at https://github.com/POSidorov/DOPtools. The library with all the code, tutorials and examples described in the manuscript, is also available in the Zenodo repository at DOI: 10.5281/zenodo.15007477.Author contributions
PS is the main developer and the main author of the manuscript. SB participated in the design of main functionalities and in testing the developed scripts. PG participated in the implementation of several utilities of the optimizer scripts, as well as code formatting and testing. TG developed the functions for calculating linear and circular fragments (Chython library). All authors participated in the discussion of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
PS, PG and SB acknowledge the financial support from the Institute for Chemical Reaction Design and Discovery (ICReDD), which was established by the World Premier International Research Initiative (WPI), MEXT, Japan, as well as the JSPS KAKENHI grant 23H03807. PS and PG also acknowledge the support from the List Sustainable Digital Transformation Catalyst Collaboration Research Platform offered by Hokkaido University. TG acknowledges the support from the subsidy allocated to Kazan Federal University for the state assignment in the sphere of scientific activities FZSM-2024-0002.Notes and references
- A. Cherkasov, E. N. Muratov, D. Fourches, A. Varnek, I. I. Baskin, M. Cronin, J. Dearden, P. Gramatica, Y. C. Martin, R. Todeschini, V. Consonni, V. E. Kuzmin, R. Cramer, R. Benigni, C. Yang, J. Rathman, L. Terfloth, J. Gasteiger, A. Richard and A. Tropsha, J. Med. Chem., 2014, 57, 4977–5010 CrossRef CAS PubMed.
- Cadence Molecular Sciences, OEChem TK, 2023 Search PubMed.
- CGC Inc., Molecular Operating Environment (MOE), 2011 Search PubMed.
- ChemAxon, Chemical Structure Representation Toolkit, 2023 Search PubMed.
- C. Steinbeck, Y. Han, S. Kuhn, O. Horlacher, E. Luttmann and E. Willighagen, J. Chem. Inf. Comput. Sci., 2003, 493–500 CrossRef CAS PubMed.
- G. Landrum, RDKit: Open-source cheminformatics, 2006 Search PubMed.
- N. M. O’boyle, M. Banck, C. A. James, C. Morley, T. Vandermeersch and G. R. Hutchison, Open Babel: An open chemical toolbox, 2011 Search PubMed.
- C. W. Yap, J. Comput. Chem., 2011, 32, 1466–1474 CrossRef CAS PubMed.
- A. Varnek, D. Fourches, D. Horvath, O. Klimchuk, C. Gaudin, P. Vayer, V. Solov’ev, F. Hoonakker, I. V. Tetko and G. Marcou, Curr. Comput.-Aided Drug Des., 2008, 4, 191 CrossRef CAS.
- J. Aires-de Sousa, Mol. Inf., 2024, 43, e202300190 CrossRef CAS PubMed.
- R. I. Nugmanov, R. N. Mukhametgaleev, T. Akhmetshin, T. R. Gimadiev, V. A. Afonina, T. I. Madzhidov and A. Varnek, J. Chem. Inf. Model., 2019, 59, 2516–2521 CrossRef CAS PubMed.
- R. Nugmanov, N. Dyubankova, A. Gedich and J. K. Wegner, J. Chem. Inf. Model., 2022, 62, 3307–3315 CrossRef CAS PubMed.
- J. Sieg, C. W. Feldmann, J. Hemmerich, C. Stork, F. Sandfort, P. Eiden and M. Mathea, ChemRxiv, 2024, preprint, DOI:10.26434/chemrxiv-2024-kd11b.
- E. J. Bjerrum, R. A. Bachorz, A. Bitton, O.-h. Choung, Y. Chen, C. Esposito, S. Viet Ha and A. Poehlmann, ChemRxiv, 2023, preprint, DOI:10.26434/chemrxiv-2023-fzqwd.
- M. Karim and R. M. Rahman, J. Software Eng. Appl., 2013, 6, 196–206 CrossRef.
- S. Sarumathi, N. Shanthi, S. Vidhya and P. Ranjetha, Int. J. Comput. Sci. Inf. Eng., 2015, 9, 473–480 Search PubMed.
- S. Beisken, T. Meinl, B. Wiswedel, L. F. de Figueiredo, M. Berthold and C. Steinbeck, BMC Bioinf., 2013, 14, 257 CrossRef PubMed.
- P. Ambure, A. K. Halder, H. G. Díaz and M. N. D. S. Cordeiro, J. Chem. Inf. Model., 2019, 59, 2538–2544 CrossRef CAS PubMed.
- D. Horvath, J. B. Brown, G. Marcou and A. Varnek, Challenges, 2014, 5, 450–472 CrossRef.
- S. L. Dixon, J. Duan, E. Smith, C. D. Von Bargen, W. Sherman and M. P. Repasky, Future Med. Chem., 2016, 8, 1825–1839 CrossRef CAS PubMed.
- M. Feurer, A. Klein, K. Eggensperger, J. Springenberg, M. Blum and F. Hutter, Adv. Neural Inf. Process. Syst., 2015, 28, 2962–2970 Search PubMed.
- M. Feurer, K. Eggensperger and S. Falkner, et al. , J. Mach. Learn. Res., 2022, 23, 11936–11996 Search PubMed.
- F. Pedregosa, G. Varoquaux, A. Gramfort, V. Michel, B. Thirion, O. Grisel, M. Blondel, P. Prettenhofer, R. Weiss, V. Dubourg, J. Vanderplas, A. Passos, D. Cournapeau, M. Brucher, M. Perrot and É. Duchesnay, J. Mach. Learn. Res., 2011, 12, 2825–2830 Search PubMed.
- W. McKinney, Python for High Performance and Scientific Computing, 2011, 14, 1–9 Search PubMed.
- T. Akiba, S. Sano, T. Yanase, T. Ohta and M. Koyama, Proceedings of the 25th ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, New York, NY, USA, 2019, pp. 2623–2631 Search PubMed.
- D. Dalmau and J. V. Alegre-Requena, Wiley Interdiscip. Rev. Comput. Mol. Sci., 2024, 14, e1733 CrossRef CAS.
- H. W. van den Maagdenberg, M. Šícho, D. A. Araripe, S. Luukkonen, L. Schoenmaker, M. Jespers, O. J. M. Béquignon, M. G. González, R. L. van den Broek, A. Bernatavicius, J. G. C. van Hasselt, P. H. van der Graaf and G. J. P. van Westen, J. Cheminf., 2024, 16, 128 Search PubMed.
- L. Mervin, A. Voronov, M. Kabeshov and O. Engkvist, ChemRxiv, 2024, preprint, DOI:10.26434/chemrxiv-2024-2rlk7-v2.
- J. Lanini, G. Santarossa, F. Sirockin, R. Lewis, N. Fechner, H. Misztela, S. Lewis, K. Maziarz, M. Stanley, M. Segler, N. Stiefl and N. Schneider, J. Chem. Inf. Model., 2023, 63, 4497–4504 Search PubMed.
- F. Hoonakker, N. Lachiche, A. Varnek and A. Wagner, Int. J. Artif. Intell. Tool., 2011, 20, 253–270 Search PubMed.
- N. Tsuji, P. Sidorov, C. Zhu, Y. Nagata, T. Gimadiev, A. Varnek and B. List, Angew. Chem., Int. Ed., 2023, 62, e202218659 CrossRef CAS PubMed.
- T. R. Gimadiev, T. I. Madzhidov, R. I. Nugmanov, I. I. Baskin, I. S. Antipin and A. Varnek, J. Comput.-Aided Mol. Des., 2018, 32, 401–414 Search PubMed.
- D. Horvath, G. Marcou, A. Varnek, S. Kayastha, A. de la Vega de León and J. Bajorath, J. Chem. Inf. Model., 2016, 56, 1631–1640 CAS.
- H. Drucker, C. J. C. Burges, L. Kaufman, A. J. Smola and V. Vapnik, Adv. Neural Inf. Process. Syst., 1997, 155–161 Search PubMed.
- T. Chen and C. Guestrin, Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, New York, NY, USA, 2016, pp. 785–794 Search PubMed.
- L. Breiman, Mach. Learn., 2001, 45, 5–32 Search PubMed.
- DOPtools GitHub repository, https://github.com/POSidorov/DOPtools, Accessed: 2024-12-12.
- J. Bergstra, D. Yamins and D. Cox, Proceedings of the Python in Science Conference, 2013, pp. 13–19 Search PubMed.
- H. Moriwaki, Y.-S. Tian, N. Kawashita and T. Takagi, J. Cheminf., 2018, 10, 1–14 Search PubMed.
- G. Marcou, D. Horvath, V. SolovEv, A. Arrault, P. Vayer and A. Varnek, Mol. Inf., 2012, 31, 639–642 CAS.
- D. Rogers and M. Hahn, J. Chem. Inf. Model., 2010, 50, 742–754 CAS.
- P. Gedeck, B. Rohde and C. Bartels, J. Chem. Inf. Model., 2006, 46, 1924–1936 CAS.
- R. E. Carhart, D. H. Smith and R. Venkataraghavan, J. Chem. Inf. Comput. Sci., 1985, 25, 64–73 CAS.
- R. Nilakantan, N. Bauman, J. S. Dixon and R. Venkataraghavan, J. Chem. Inf. Comput. Sci., 1987, 27, 82–85 Search PubMed.
- R. R. Griffiths, J. L. Greenfield, A. R. Thawani, A. R. Jamasb, H. B. Moss, A. Bourached, P. Jones, W. McCorkindale, A. A. Aldrick, M. J. Fuchter and A. A. Lee, Chem. Sci., 2022, 13, 13541–13551 Search PubMed.
- J. Catalán, J. Phys. Chem. B, 2009, 113, 5951–5960 Search PubMed.
- W. Bort, I. I. Baskin, T. Gimadiev, A. Mukanov, R. Nugmanov, P. Sidorov, G. Marcou, D. Horvath, O. Klimchuk, T. Madzhidov and A. Varnek, Sci. Rep., 2021, 11, 3178 Search PubMed.
- D. Roy, V. K. Hinge and A. Kovalenko, ACS Omega, 2019, 4, 16774–16780 Search PubMed.
- J. Bergstra, R. Bardenet, Y. Bengio and B. Kégl, Adv. Neural Inf. Process. Syst., 2011, 2546–2554 Search PubMed.
- T. I. Netzeva, A. P. Worth, T. Aldenberg, R. Benigni, T. D. Mark, P. Gramatica, J. S. Jaworska, S. Kahn, G. Klopman, A. Carol, G. Myatt, N. Nikolova-jeliazkova, G. Y. Patlewicz and R. Perkins, Altern. Lab. Anim., 2005, 2, 155–173 Search PubMed.
- A. Rakhimbekova, T. I. Madzhidov, R. I. Nugmanov, T. R. Gimadiev, I. I. Baskin and A. Varnek, Int. J. Mol. Sci., 2020, 21(15), 5542 Search PubMed.
| This journal is © The Royal Society of Chemistry 2025 |