
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Machine learning-driven optimization of the output force in photo-actuated organic crystals†
Kazuki
Ishizaki
 a,
Toru
Asahi
a and
Takuya
Taniguchi
*b
a,
Toru
Asahi
a and
Takuya
Taniguchi
*b
aDepartment of Advanced Science and Engineering, Graduate School of Advanced Science and Engineering, Waseda University, 3-4-1 Okubo, Shinjuku-ku, Tokyo 169-8555, Japan
bCenter for Data Science, Waseda University, 1-6-1 Nishiwaseda, Shinjuku-ku, Tokyo 169-8050, Japan. E-mail: takuya.taniguchi@aoni.waseda.jp
First published on 20th March 2025
Abstract
Photo-actuated organic crystals that can be remotely controlled by light are gaining attention as next-generation actuator materials. In the practical application of actuator materials, the mode of deformation and the output force are important properties. Since the output force depends on the crystal properties and experimental conditions, it is necessary to explore the optimal conditions from a vast parameter space. In this study, we employed two types of machine learning for molecular design and experimental optimization to maximize the blocking force. Machine learning in molecular design led to the creation of a material pool of salicylideneamine derivatives. Bayesian optimization was used for efficient sampling from the material pool for force measurements in the real world, achieving a maximum blocking force of 37.0 mN. This method was at least 73 times more efficient than the grid search approach.
Introduction
Actuator materials convert external stimuli into mechanical motion and are utilized in various fields, supporting our daily lives. For example, shape memory alloys can generate high output forces from heat, making them useful in the automotive components and robot drives.1 Electrically driven polymers are expected to be employed as artificial muscles and small medical devices due to their material flexibility.2 Forces in the nN–μN range can be utilized in the microelectromechanical systems.3 Actuator materials play important roles in a wide range of applications, depending on the forces they generate.Among actuator materials, photomechanical crystals deform upon light irradiation, making them attractive as lightweight and remotely controllable actuators. Previous studies have primarily reported on the free deformation of these crystals, revealing various deformation behaviors.4–10 When deformation occurs to an object, it can exert force and perform work.11–15 The maximum force is generated when the free deformation is perfectly blocked, which is defined as the blocking force, a key performance indicator for actuators. However, blocking force measurements for photomechanical crystals are rare due to the small size of the crystals and difficulty in constructing the experimental setup. Previously, we developed an experimental system that allowed us to directly measure the blocking force of photomechanical crystals.10,16
Controlling the blocking force of photomechanical crystals is crucial for their practical applications. While generating small forces can be readily achieved by adjusting the light intensity, increasing the maximum force remains challenging as it depends on multiple factors including crystal properties, crystal sizes, and experimental conditions. The relationship between these parameters and the generated force is not yet fully understood. Expanding the achievable maximum force could significantly broaden the potential applications of photomechanical crystals. Therefore, a comprehensive investigation of crystal properties and experimental parameters is essential. However, conducting a systematic grid search is impractical due to extensive time and resources required for the experiments. Machine learning approaches have recently proven effective in designing molecular materials and predicting crystal properties17–23 and offer a promising alternative. When predicting physical properties, striking the right balance between predictive performance and model interpretability is crucial, with the choice of model depending on the specific objectives. While Bayesian optimization is effective in optimizing the experimental conditions across various fields,24–27 its application to photomechanical crystals remains unexplored.
In this study, we employed two types of machine learning in the molecular design and experimental optimization of photomechanical crystals, successfully maximizing the blocking force (Fig. 1). First, the least absolute shrinkage and selection operator (LASSO) regression was used for molecular design of photomechanical crystals with different Young's moduli. Crystals of several compounds were prepared based on the molecular design, creating a material pool with varying Young's moduli and crystal sizes. Then, we experimentally measured the blocking force using Bayesian optimization for efficient sampling from the material pool, aiming to maximize the output force. These findings broaden the applicability of photomechanical molecular crystals and help optimize other properties of functional crystals.
 | ||
| Fig. 1 Workflow of this research. Machine learning (ML) was used to find the relationship between Young's modulus and functional groups and to find the optimal experimental condition. The human task involves the preparation of crystal samples and experimental assays to test the suggested conditions in the real world. | ||
Materials and methods
Dataset preparation
We performed a Google Scholar search using the queries “Organic crystals”, “Young's modulus”, and “Molecular crystals” “Young's modulus.” At the time of data curation, we identified 118 papers reporting at least one Young's modulus value, primarily obtained by nanoindentation. In many cases, multiple directional measurements were presented (e.g., along different crystallographic axes); therefore, we extracted each relevant data point. We also included bending test data and other measurements when available. As a result, a dataset containing 393 data was obtained. All extracted values are listed in the ESI data† and each source is identified by its DOI (Digital Object Identifier). To ensure data reliability, we cross-checked part of the dataset against a previously published dataset.28LASSO regression
The manually collected dataset has molecular information represented as simplified molecular input line entry system (SMILES) and the reported Young's modulus SMILES was then encoded as a numerical representation of the number of specific functional groups using the RDKit library. Each molecule was converted into an 85-dimensional vector with non-negative integers. After removing variables with high correlation among explanatory variables, we performed LASSO regression implemented by the scikit-learn library. In the model training, was used as the loss function. Here, n is the number of samples, y represents the actual measured value, ŷ is the predicted value, w represents the coefficients of the explanatory variables, and α is a hyperparameter. The α value was optimized to minimize
was used as the loss function. Here, n is the number of samples, y represents the actual measured value, ŷ is the predicted value, w represents the coefficients of the explanatory variables, and α is a hyperparameter. The α value was optimized to minimize 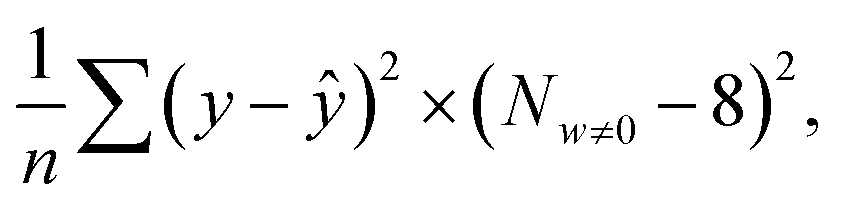 where Nw≠0 is the number of non-zero coefficients. This metric is the product of the mean square error and the constraint for variable selection. The dataset was split into training and test sets in an 8
where Nw≠0 is the number of non-zero coefficients. This metric is the product of the mean square error and the constraint for variable selection. The dataset was split into training and test sets in an 8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 ratio. The hyperparameters were tuned using 5-fold cross-validation on the training set.
:2 ratio. The hyperparameters were tuned using 5-fold cross-validation on the training set.
Material preparation
3,5-Di-tert-butyl-2-hydroxybenzaldehyde (2.5 mmol) and selected reactant (2.5 mmol) were dissolved in 2-propanol (15 mL) and the solution was heated at 150 °C under pressure for a sufficient period of time (1–3 hours depending on reactants) using a microwave apparatus (Monowave 300, AntonPaar) according to the literature.28 The resulting solution was evaporated and the obtained powder was recrystallized in methanol, ethanol, 1-propanol, or 2-propanol, yielding plate-like crystals.UV-visible (UV-vis) absorption measurement
The UV-vis diffuse reflectance spectra of the crystalline powder were measured using either a Lambda 650 spectrometer (PerkinElmer) or a V-650 spectrometer (JASCO) at 20 °C. The powder was mounted on a glass plate and covered with a quartz glass plate. Measurements were performed before and after photoirradiation (365 nm, 10 mW cm−2) on the quartz glass slide using a UV-LED (8332A, CCS Inc.). Measurements were performed between 250–800 nm at 1 nm resolution with a scan speed of 200 nm min−1.Crystal structure determination
Single-crystal X-ray diffraction data were obtained at 20 °C using a Rigaku R-AXIS RAPID diffractometer equipped with a monochromatic Cu-Kα radiation source (λ = 1.54187 Å). The crystal structures were solved using a direct method with SHELXT29 and refined with SHELXL30 on F2 using the full-matrix least-squares method. Calculations were performed using ShelXle and Olex2 graphical interfaces.31,32Nanoindentation and bending test
Young's modulus along the thickness direction was measured by nanoindentation (Nano Indenter G200, TOYO Corporation) and loading on the largest face. The loading rate was 0.03 mN s−1 and the hold time at the peak load (1 mN) was 10 s. The samples were fixed with glue on the glass plate. Five measurements at different positions were performed for each crystal. A Berkovich indenter was employed with a triangular pyramid structure made of diamond. The obtained P–h curves were analysed using the standard Oliver–Pharr method to determine Young's modulus. Young's modulus along the longitudinal direction was measured using a bending test with a universal testing machine (RTG-1210, A&D). The bending test was repeated on two or three different crystals of each compound to calculate the average value.Bayesian optimization of force measurement
Bayesian optimization was implemented using the bayes_opt library. The black box function was surrogated by Gaussian process regression. We used the upper confidence bound (UCB) as the acquisition function:| UCB(x) = μ(x) + κσ(x) |
We set the value range for each parameter. The measured Young's modulus was used as an identifier for the crystals and we used the values from the bending tests. Crystal sizes were defined within 0–3000 μm length, 200–2000 μm width, and 50–500 μm thickness. Irradiation utilized UV light at 365 nm, with an experimental setup capable of producing an intensity of 2080 mW cm−2 at 100% output. We conducted experiments with four additional light intensity conditions besides 365 nm since changing light intensity is easier than changing crystals. The four conditions were selected from 10, 25, 50, 75, and 100%, excluding the percentage closest to the Bayesian-suggested value. The explanatory variables were standardized and iterative regression was performed.
We first measured 10 data from 5 light conditions of 2 crystals. The blocking force was measured with a universal testing machine (RTG-1210, A&D). The jig approached a sample and stopped when the initial load was confirmed. Then, the crystal was irradiated with UV light at 365 nm (8332A, CCS Inc.) for 5 min at 20 °C. As we aimed to maximize the blocking force rather than investigate the cause of deformation, the sample was irradiated from top and bottom, adopting the larger force value. In the sampling by Bayesian optimization, the crystal closest to the suggested crystal sizes was manually selected. When crystals of the suggested size were unavailable, we either selected alternative crystals of compound (S)-4 or (S)-5 to update the surrogate function. Since adjusting the light intensity was relatively simple, four additional light intensities were tested outside of the suggested conditions.
Results and discussion
Molecular design by LASSO regression
We focused on salicylideneamine molecules, which are known to function as photomechanical crystals.10,16 Salicylideneamine molecules can be synthesized in a single step, and different compounds can be obtained by changing an amine reactant (Fig. 2a). For molecular design, we investigated the relationship between molecular substructures and Young's modulus. Young's modulus is the most fundamental mechanical property and it influences the blocking force. Here, we aimed to establish a material pool of salicylideneamine crystals with various Young's moduli and crystal sizes.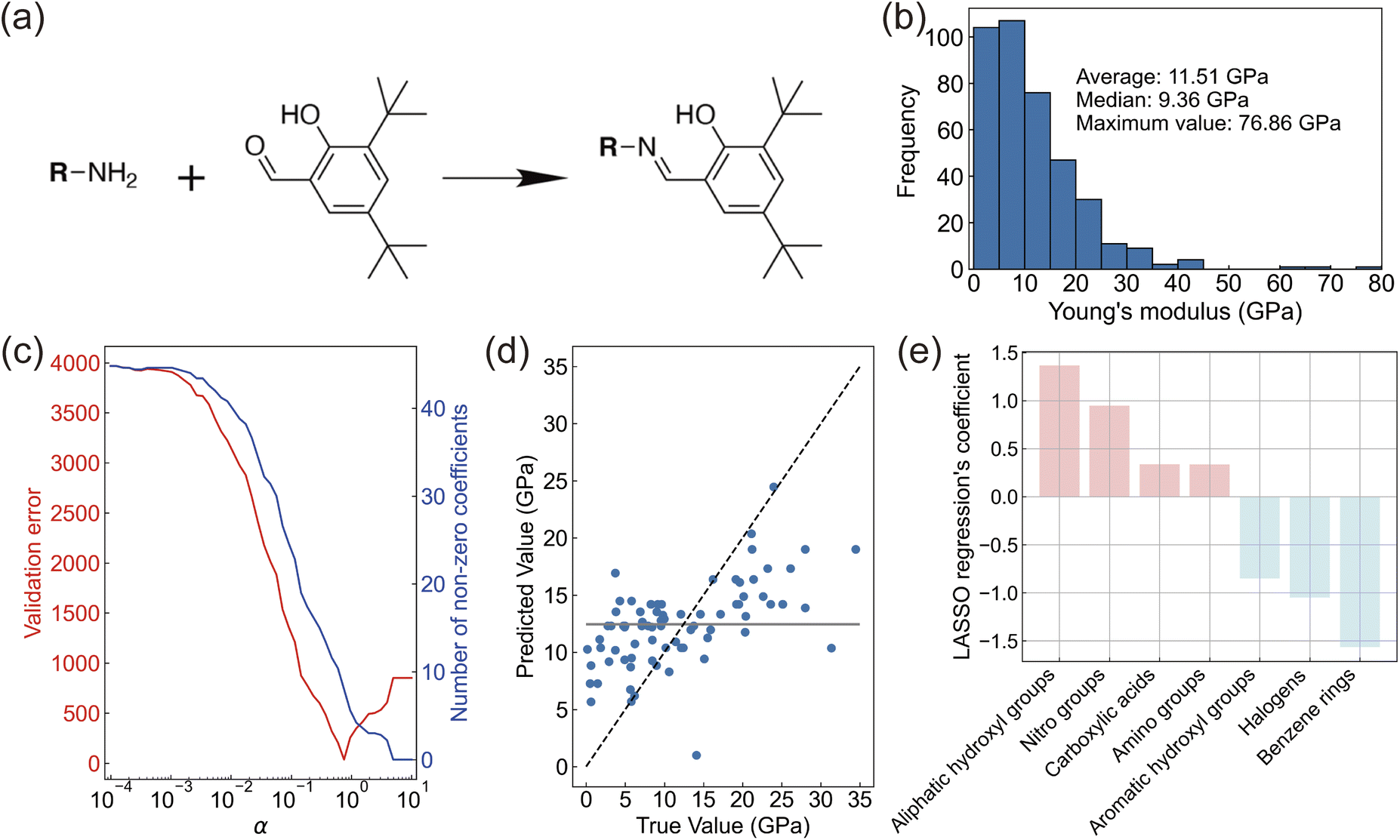 | ||
| Fig. 2 Molecular design aided by machine learning. (a) Target chemical reaction for obtaining photo-actuating molecular crystals. (b) Distribution of Young's modulus in the prepared dataset. (c) Hyperparameter optimization and the number of non-zero coefficients in the LASSO regression. (d) True-predicted plot of the optimized LASSO model. The grey line represents the mean model. (e) Regression coefficients whose variables were retained by the LASSO regression. | ||
In the prepared dataset containing 393 data points for machine learning, Young's modulus values distributed with a maximum value of 76.86 GPa, an average of 11.51 GPa, and a median of 9.36 GPa (Fig. 2b and ESI data†). We encoded the molecules to 85 dimensional vectors by the number of substructures implemented in the RDKit library. There are many ways to vectorize molecular structures, but we chose this molecular representation because it provides direct insights into the molecular structure of salicylideneamine reactants. After removing variables with high correlation among explanatory variables, 18 and 27 substructures showed positive and negative correlation with Young's modulus, respectively (ESI Fig. 1†). Substructures that form hydrogen bonds show a high positive correlation with Young's modulus. This is consistent with previous findings.33 Substructures with strong negative correlations should form relatively weak intermolecular interactions in crystals.
Our study focuses on regression analysis to gain insights into molecular design rather than merely improving predictive accuracy. Thus, enhancing interpretability aligns better with our analytical objectives. For this reason, we applied LASSO regression, a linear model that also enables variable selection. After hyperparameter optimization (Fig. 2c), we evaluated the accuracy of LASSO regression. The mean absolute error (MAE) for the training data was 6.0 GPa, while the MAE for the test data was 5.5 GPa (Fig. 2d). This metric is better than the reference metric of the mean model (MAE = 6.9 GPa), which assumes that there is no relationship between the input structure and output. Although basing the analysis solely on functional groups may not encapsulate all relevant factors (e.g., those modelled by graph neural networks), we believe this method is more suitable for identifying reactant compounds in organic synthesis. Furthermore, our dataset comprises experimentally measured values of Young's modulus, which naturally include measurement errors stemming from various factors, such as the crystal surface flatness and the specific conditions used for nanoindentation. As a result, it is not surprising that our prediction errors exceed those typically observed when working with purely computational databases such as the Materials Project or QM9. Nevertheless, we uncovered the relationship between molecular structure and Young's modulus and established design guidelines for salicylideneamine molecules.
The LASSO model retained seven variables with positive and negative coefficients (Fig. 2e). Even with slight changes in α, the LASSO regression coefficients remained almost stable, and the selected functional groups did not change. This demonstrates the robustness of variable selection (ESI Fig. 2†). Four variables were extracted as substructures that are likely to increase Young's modulus, while three variables were extracted as substructures that are likely to decrease Young's modulus. As mentioned earlier, substructures with positive coefficients tend to form hydrogen bonds in crystals, which is consistent with the increase in Young's modulus. Substructures with negative coefficients should be related with the flexibility of the crystal. Crystals containing benzene rings often form layered structures with weak interlayer interactions. Many elastically bendable crystals have this arrangement, which is consistent with a lower Young's modulus.34,35 Crystals forming halogen–halogen interactions are known for their plastic deformation,36 which is included in the training dataset. It is thought that the halogen substructure remained in the variable selection of LASSO regression because crystals that undergo plastic deformation have a small Young's modulus. The counterintuitive finding is regarding aromatic OH. In the dataset, methyl gallate has the highest number of aromatic OH groups, with three OH groups forming hydrogen bonds in the crystal. However, its Young's modulus is reportedly 9.38 GPa,37 which does not reach the average value. The existence of such data likely explains why aromatic OH showed a negative coefficient.
Based on the relationship obtained from LASSO regression, we chose commercially available reactants and synthesized salicylideneamine molecules (Fig. 3). We used reactants with nitro and carboxylic acid groups for likely positive fragments. We did not select aliphatic hydroxyl groups because the reactant already has an aromatic ring. We also excluded amino groups to prevent side reactions since salicylideneamine are synthesized by dehydration reactions between amino and carbonyl groups. For likely negative fragments, we used reactants with benzene rings and halogens. Since the reactant already has a phenyl ring, reactants with a naphthyl ring were used to verify the induction effect of the benzene fragment. Considering these to be sufficient, we did not use reactants with aromatic OH.
 | ||
| Fig. 3 Molecular structures synthesized in this work. | ||
Characterization of crystal structures and properties
All compounds were crystallized in a plate-like shape with varying crystal sizes. This allowed us to define the longest direction as length and the thinnest direction as thickness. The widest face appears perpendicular to the thickness. Eight out of ten crystals had common features according to X-ray crystallographic analysis (ESI Table 1†). Relatively strong molecular layers were stacked in a specific direction via interlayer van der Waals forces (Fig. 4). This is because the interlayer interactions are formed between tert-butyl groups between two molecular layers. There are two types of stacking directions: crystals with molecular layers stacked in the thickness direction and crystals stacked at an angle nearly perpendicular to the thickness. Among these, crystals with naphthyl groups all have molecular layers stacked in the thickness direction (Fig. 4d–f). For molecules with halogens, all crystals formed similar molecular arrangements except those with the –F group (Fig. 4h–j). Two crystals of compounds 2 and 7 did not show molecular layer stacking. The crystal of compound 2 forms hydrogen-bonded dimers due to the –COOH group but has a different molecular packing with the crystal of compound 1, which also forms hydrogen-bonded dimers. The crystal of compound 7 has a significantly different molecular packing compared to other halogenated crystals and does not show molecular layer stacking. | ||
| Fig. 4 Crystal structures of salicylideneamine compounds. (a–j) Crystals of compounds 1–10 in order. The top bold line represents the crystal surface of the largest face in the plate-shaped crystal. The highlighted region represents the interlayer van der Waals interactions. The longitudinal direction is indicated by an arrow or a circle with a cross mark. Hydrogen atoms and disordered tert-butyl substituents were omitted for clarity. | ||
In salicylideneamine crystals, the dihedral angle between the salicyl plane and the adjacent phenyl (or naphthyl) plane is an indicator of photoisomerization. Most of the synthesized crystals exhibited dihedral angles ranging from 20–50° (Table 1). An established empirical rule indicates that salicylideneamine crystals with dihedral angles exceeding 30° show higher propensity for photoisomerization.38 UV-vis absorption spectra of the crystalline powders showed absorption bands between 300–600 nm prior to UV irradiation, followed by enhanced absorption in the 400–600 nm range due to enol–keto photoisomerization (ESI Fig. 3†). Compounds 2 and 7 containing planar structures with dihedral angles of 8.48° and 21.24°, respectively, displayed minimal absorption changes.
| Compound | Dihedral angle (°) | Longitudinal axis | Largest face | E indent (GPa) | E bending (GPa) |
|---|---|---|---|---|---|
| a The average value of two independent molecules with slightly different conformations. b The crystal of compound 6 was not measured due to its small size. | |||||
| 1 | 36.21 | b-Axis | (001) | 7.32 (0.74) | 0.60 (0.25) |
| 2 | 8.48 | a-Axis | (010) | 8.54 (0.19) | 1.92 (0.33) |
| 3 | 51.58 | a-Axis | (001) | 6.04 (0.32) | 0.68 (0.17) |
| (S)-4 | 46.61a | b-Axis | (001) | 3.73 (0.07) | 1.83 (1.11) |
| (S)-5 | 36.99 | a-Axis | (001) | 4.75 (0.16) | 1.25 (1.81) |
| 6 | 46.84 | b-Axis | (001) | 3.13 (0.52) | —b |
| 7 | 21.24 | c-Axis | (100) | 12.88 (0.87) | 1.83 (0.37) |
| 8 | 28.54 | a-Axis | (001) | 6.20 (0.53) | 0.66 (0.53) |
| 9 | 27.51 | a-Axis | (001) | 7.58 (0.12) | 2.29 (1.62) |
| 10 | 24.88 | a-Axis | (001) | 1.99 (0.24) | 3.55 (2.84) |
It is important to identify Young's modulus, which is a parameter of force. Young's modulus was measured from different directions to characterize the anisotropy. First, nanoindentation was performed on the wide face of all plate-shaped crystals, measuring the Young's modulus in the thickness direction (Table 1 and ESI Fig. 4†). Nanoindentation is the most widespread and reliable method to measure Young's modulus of the molecular crystal by indenting the crystal surface up to hundreds of nm. Young's modulus of crystalline compounds 1–3 was larger than that of compounds 4–6 (Fig. 5a). This result is consistent with the prediction by LASSO because most of the dataset used for LASSO regression consisted of nanoindentation results and these crystals are thought to reflect the intermolecular interactions included in that dataset. However, halogenated crystals (compounds 7–10) did not align with predictions. Crystals of compounds 7–9 had relatively large Young's moduli. This is because the dataset includes halogenated crystals that form halogen bonds with relatively small Young's moduli, while halogenated crystals in this study do not form halogen bonds. For example, hexachlorobenzene, which is known for plastic deformation,36 has a relatively small Young's modulus in the dataset and contributes to a negative relationship in the regression because it has six halogen fragments.
 | ||
| Fig. 5 Young's modulus measured by nano-indentation and the bending test. (a) Nanoindentation results. The highlighted region agreed with the LASSO trend, while the halogenated crystals did not. (b) The relationship of Young's modulus determined by nanoindentation and the bending test. The crystal of compound 6 was not measured by the bending test due to its small size. | ||
Next, cantilever bending tests were conducted to measure Young's modulus in the longitudinal direction because it is difficult to measure the modulus of this direction by nanoindentation. Since Young's modulus measured by the bending test is a small fraction in the dataset, measurement results may differ from the LASSO trends. Crystals with larger Young's moduli in the thickness direction tended to have smaller Young's moduli in the length direction (Fig. 5b). This can be attributed to the anisotropy of organic crystals. A large Young's modulus in the thickness direction indicates strong intermolecular interactions in that direction. When strong interactions form in one direction, weaker interactions such as CH–π interactions and van der Waals forces become the main intermolecular interactions in other directions given that similar molecules are used in all crystals, explaining the smaller Young's modulus. The standard deviation of the bending test is larger than that of nanoindentation because the bending tests calculate Young's modulus from macroscopic deformation against load.
Optimization of the blocking force
Having established a material pool with varying Young's moduli and crystal sizes, we performed force measurements by sampling from this material pool using Bayesian optimization (Fig. 6a). The measured Young's modulus was used as an identifier for the crystals using values obtained from bending tests. Crystals of compound 7 were excluded from the entries as the modulus overlapped with that of compound 4, and crystals of compound 6 were also excluded due to their small size. This resulted in eight crystal entries. The ranges of crystal sizes and light intensities are described in the Materials and methods section. Once a crystal sample was selected, blocking force was monitored under light irradiation from above or below the crystal. | ||
| Fig. 6 Bayesian optimization of the blocking force of photo-actuation organic crystals. (a) Measurement process of the blocking force. (b) Time series of blocking force when the maximum values were updated during the experiments, with the arrow showing the value used for Bayesian optimization. (c) Progression of blocking force through the experiments using Bayesian optimization. (d) The frequency of the compound measured during the optimization experiments. (e) The relationship between the crystal volume and blocking force. | ||
In Bayesian optimization, the acquisition function used for sampling is crucial, and we employed upper confidence bound (UCB). Through pre-simulation, we set the hyperparameter of UCB to be κ = 5 for the optimal balance between exploration and exploitation (ESI Fig. 5†). Initially, we measured 10 points (five light conditions of two crystals), with the maximum value among these being Fmax = 9.8 mN (Fig. 6b). Here, Fmax is defined as the maximum blocking force minus the initial load. Then, we performed crystal sampling using Bayesian optimization and measured Fmax iteratively. Fmax increased progressively from 9.8 mN to 16.0 mN, then to 26.8 mN, 34.4 mN, and finally to 37.0 mN over the course of 100 experiments (Fig. 6c and ESI Table 2†). The decision to terminate the optimization process after 110 experiments was based on the convergence behaviour of Fmax. The final three updates of Fmax showed progressively smaller increments: from 26.8 mN to 34.4 mN (an increase of 7.6 mN), and then to 37.0 mN (an increase of only 2.6 mN). This diminishing rate of improvement, coupled with the fact that the last three maximum values were achieved using crystals of compound 3 under similar conditions, strongly suggests that the optimization was approaching the global maximum within our parameter space. The crystals of compound 3 were selected the most in the optimization process (Fig. 6d). While continuing the experiments might yield marginally higher forces, the decreasing magnitude of improvements indicated that we had likely identified the region of optimal conditions.
This study compares the force values achieved with those from previous research. The force of photomechanical crystals has been previously evaluated by the displacement of objects with known weights. The force achieved by photomechanical crystals is typically in the μN–mN range, with a maximum of around 10 mN.11–16 While our initial data also showed this value, we achieved a greater force than previously known by actively seeking to maximize it. On the other hand, the force density, which is the force divided by the crystal volume, reached a maximum of 1.7 × 108 N m−3, which is comparable to existing photomechanical crystals (107–109). Force density cancels out size effects, indicating that the light absorption properties and intermolecular interactions of our crystals are similar to those of other photomechanical crystals. Conversely, the significance of this study lies in demonstrating that larger forces can be achieved by sampling experimental conditions. The success of Bayesian optimization is evidenced by its ability to identify optimal combinations of crystal properties and sizes, rather than simply selecting the largest available crystals (Fig. 6e). If larger crystal volume automatically corresponded to greater force generation, a simple maximization of crystal sizes would suffice. However, the optimization process converged on specific combinations of crystal properties and dimensions, indicating that there is an optimal balance between crystal characteristics and size for maximizing the blocking force.
It is also important to consider the efficiency of sampling through Bayesian optimization. The material pool used for this force measurement included eight types of crystals. Although the ranges for length, width, and thickness differ, a grid search would require a total of 8000 crystal samplings if we sampled 10 discrete points along each size dimension. Assuming that the maximum value obtained through Bayesian optimization is the global maximum of the search space and the light conditions are the same, this method reduced the number of experiments to 110 out of 8000. Consequently, we have improved the experimental efficiency by at least 73 times, depending on the discretization. If we take finer intervals in the grid search, the total number of experiments increases exponentially, making the efficiency of Bayesian optimization stand out.
Finally, we discuss the mechanism behind the enhanced force generation. The superior performance of compound 3 can be attributed to its structural features: the large dihedral angle between the salicyl and phenyl planes (51.58°) enables efficient photoisomerization, while the nitro group enhances photoresponsivity. The molecular layers stacked along the thickness direction facilitate anisotropic thermal expansion.39 When exposed to light, compound 3 exhibits enol–keto photoisomerization and thermal effects. In thin crystals, photoisomerization causes bending away from the light source, while thermal expansion dominates in thick crystals.39 The maximum force generation observed during top illumination suggests significant thermal expansion in the thickness direction. The crystal's negative thermal expansion coefficient along the length direction near room temperature likely contributes to the overall mechanical response. The force generation increased with crystal size in regions of sufficient light intensity (ESI Fig. 6†).
Conclusions
We increased the maximum force of photomechanical crystals to a level higher than previously reported, aided by two machine learning models. LASSO regression revealed that certain substructures likely have positive and negative correlations with Young's modulus. Substructures that tend to form hydrogen bonds were selected as likely positive correlators, while benzene rings and halogens were identified as likely negative correlators. This enabled us to select appropriate substructures of salicylideneamine derivatives, and the measured Young's moduli of crystals without halogens was consistent with the LASSO trend. Subsequently, we sampled crystals for force measurements using Bayesian optimization. The active sampling achieved a blocking force of 37.0 mN in 110 measurements. Assuming this value represents the global maximum force in the parametric space, the exploration efficiency is at least 73 times higher compared to the grid search. This study demonstrates the effectiveness of combining machine learning approaches: LASSO regression for molecular design and Bayesian optimization for efficient sampling in the development of high-performance photomechanical crystals. Our findings establish a new benchmark for photomechanical crystal force output and provide a generalizable framework for material optimization that could be extended to other molecular crystal systems.Data availability
Crystallographic data for compounds 1, 2, 3, (S)-5, 6, 7, 8, 9 and 10 have been deposited at the Cambridge Crystallographic Data Centre under 2391778 (for 1), 2391779 (for 2), 2391780 (for 3), 2391781 (for (S)-5), 2391782 (for 6), 2391783 (for 7), 2391784 (for 8), 2391785 (for 9) and 2391786 (for 10). The data supporting this article have been included as part of the ESI.† The dataset used for LASSO regression was extracted from previously reported studies, which are referenced in the ESI data.† The data analysis scripts and the dataset of this article are available at https://github.com/kazuki143/Machine-Learning-Driven-Optimization-of-Output-Force-in-Photo--Actuated-Organic-Crystals. The code and dataset are also archived in Zenodo with the following URL: https://doi.org/10.5281/zenodo.15048712.Author contributions
K. I.: data curation, formal analysis, investigation, methodology, software, visualization, funding acquisition, writing – original draft, and writing – review & editing; T. A.: resources and supervision; T. T.: conceptualization, investigation, methodology, software, funding acquisition, project administration, supervision, writing – original draft, and writing – review & editing. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was financially supported by JSPS Grant-in-Aid (19K23638, 20H04677, 22K14747, 24K17748), the Waseda University Grant for Special Research Projects (2019C-646, 2020C-530, 2021C-404, 2022C-313, 2023C-292, 2023R-050, 2024C-297), JST ACT-X (JPMJAX23DD), Yashima Environment Technology Foundation, JST SPRING (JPMJSP2128), Waseda Research Institute for Science and Engineering, Grant-in-Aid for Young Scientists (Early Bird), academist Prize Phase III Monthly Supported Projects, and FY2023 and FY2024 Waseda University – ENEOS FS Research Grant. K. I. also thanks the members of Prof. Taro Hitosugi's laboratory for fruitful discussions on Bayesian optimization.References
- J. M. Jani, M. Leary, A. Subic and M. A. Gibson, Mater. Des., 2014, 56, 1078–1113 CrossRef.
- Biomedical Applications of Electroactive Polymer Actuators, ed. F. Carpi and E. Smela, John Wiley & Sons, 2009 Search PubMed.
- J. Wang, D. Yao, R. Wang, Z. Gao, M. Liu, X. Ye and X. Li, Extreme Mech. Lett., 2024, 72, 102228 CrossRef.
- P. Naumov, S. C. Sahoo, B. A. Zakharov and E. V. Boldyreva, Angew. Chem., 2013, 125, 10174–10179 CrossRef.
- T. Kim, L. Zhu, L. J. Mueller and C. J. Bardeen, J. Am. Chem. Soc., 2014, 136, 6617–6625 CrossRef CAS PubMed.
- T. Seki, K. Sakurada, M. Muromoto and H. Ito, Chem. Sci., 2015, 6, 1491–1497 RSC.
- D. Kitagawa, R. Tanaka and S. Kobatake, CrystEngComm, 2016, 18, 7236–7240 RSC.
- L. Zhu, R. O. Al-Kaysi and C. J. Bardeen, Angew. Chem., Int. Ed., 2016, 55, 7073–7076 CrossRef CAS PubMed.
- D. Kitagawa, H. Tsujioka, F. Tong, X. Dong, C. J. Bardeen and S. Kobatake, J. Am. Chem. Soc., 2018, 140, 4208–4212 CrossRef CAS PubMed.
- T. Taniguchi, K. Ishizaki, D. Takagi, K. Nishimura, H. Shigemune, M. Kuramochi, Y. C. Sasaki, H. Koshima and T. Asahi, Commun. Chem., 2022, 5, 4 CrossRef CAS PubMed.
- M. Morimoto and M. Irie, J. Am. Chem. Soc., 2010, 132, 14172–14178 CrossRef CAS PubMed.
- H. Koshima, R. Matsuo, M. Matsudomi, Y. Uemura and M. Shiro, Cryst. Growth Des., 2013, 13, 4330–4337 CrossRef CAS.
- J. Mahmoud Halabi, E. Ahmed, S. Sofela and P. Naumov, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2020604118 CrossRef PubMed.
- I. Tahir, E. Ahmed, D. P. Karothu, F. Fsehaye, J. Mahmoud Halabi and P. Naumov, J. Am. Chem. Soc., 2024, 146, 30174–30182 CAS.
- J. Lin, J. Zhou, L. Li, I. Tahir, S. Wu, P. Naumov and J. Gong, Nat. Commun., 2024, 15, 3633 CrossRef CAS PubMed.
- K. Ishizaki, R. Sugimoto, Y. Hagiwara, H. Koshima, T. Taniguchi and T. Asahi, CrystEngComm, 2021, 23, 5839–5847 CAS.
- S. Wengert, G. Csányi, K. Reuter and J. T. Margraf, Chem. Sci., 2021, 12, 4536–4546 CAS.
- J. Mahmoud Halabi, M. B. Al-Handawi, R. Ceballos and P. Naumov, J. Am. Chem. Soc., 2023, 145, 12173–12180 CAS.
- T. Taniguchi, M. Hosokawa and T. Asahi, ACS Omega, 2023, 8, 39481–39489 CAS.
- D. Takagi, K. Ishizaki, T. Asahi and T. Taniguchi, Digital Discovery, 2023, 2, 1126–1133 RSC.
- T. Taniguchi, CrystEngComm, 2024, 26, 631–638 RSC.
- T. Taniguchi, Faraday Discuss., 2025, 256, 139–155 RSC.
- P. W. Butler, R. Hafizi and G. M. Day, J. Phys. Chem. A, 2024, 128, 945–957 CrossRef CAS PubMed.
- D. M. Packwood and T. Hitosugi, Appl. Phys. Express, 2017, 10, 065502 CrossRef.
- B. Lei, T. Q. Kirk, A. Bhattacharya, D. Pati, X. Qian, R. Arroyave and B. K. Mallick, npj Comput. Mater., 2021, 7, 194 CrossRef.
- R. J. Hickman, M. Aldeghi, F. Häse and A. Aspuru-Guzik, Digital Discovery, 2022, 1, 732–744 RSC.
- S. Kobayashi, R. Shimizu, Y. Ando and T. Hitosugi, ACS Mater. Lett., 2023, 5, 2711–2717 CrossRef CAS.
- H. E. Smith, S. L. Cook and M. E. Warren Jr, J. Org. Chem., 1964, 29, 2265–2272 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A:Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C:Struct. Chem., 2015, 71, 3–8 Search PubMed.
- C. B. Hübschle, G. M. Sheldrick and B. Dittrich, J. Appl. Crystallogr., 2011, 44, 1281–1284 CrossRef PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- D. P. Karothu, J. Mahmoud Halabi, E. Ahmed, R. Ferreira, P. R. Spackman, M. A. Spackman and P. Naumov, Angew. Chem., Int. Ed., 2022, 61, e202113988 CrossRef CAS PubMed.
- S. Varughese, M. S. Kiran, U. Ramamurty and G. R. Desiraju, Chem.–Asian J., 2012, 7, 2118–2125 CrossRef CAS PubMed.
- S. Hayashi, Symmetry, 2020, 12, 2022 CrossRef CAS.
- M. K. Panda, S. Ghosh, N. Yasuda, T. Moriwaki, G. D. Mukherjee, C. M. Reddy and P. Naumov, Nat. Chem., 2015, 7, 65–72 CrossRef CAS PubMed.
- S. Chattoraj, L. Shi and C. C. Sun, CrystEngComm, 2010, 12, 2466–2472 CAS.
- H. Sugiyama and H. Uekusa, CrystEngComm, 2018, 20, 2144–2151 RSC.
- S. Hasebe, Y. Hagiwara, T. Ueno, T. Asahi and H. Koshima, Chem. Sci., 2024, 15, 1088–1097 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Details of the dataset, additional results and experimental results. CCDC 2391778–2391786. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dd00380b |
| This journal is © The Royal Society of Chemistry 2025 |