
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Learning on compressed molecular representations
Jan
Weinreich†
a and
Daniel
Probst†
 *b
*b
aUniversity of Vienna, Faculty of Physics, Kolingasse 14-16, Wien, Austria
bLTS2, Institute of Electrical and Micro Engineering, École polytechnique fédérale de Lausanne, Lausanne, Switzerland. E-mail: daniel.probst@epfl.ch
First published on 4th November 2024
Abstract
Last year, a preprint gained notoriety, proposing that a k-nearest neighbour classifier is able to outperform large-language models using compressed text as input and normalised compression distance (NCD) as a metric. In chemistry and biochemistry, molecules are often represented as strings, such as SMILES for small molecules or single-letter amino acid sequences for proteins. Here, we extend the previously introduced approach with support for regression and multitask classification and subsequently apply it to the prediction of molecular properties and protein–ligand binding affinities. We further propose converting numerical descriptors into string representations, enabling the integration of text input with domain-informed numerical descriptors. Finally, we show that the method can achieve performance competitive with chemical fingerprint- and GNN-based methodologies in general, and perform better than comparable methods on quantum chemistry and protein–ligand binding affinity prediction tasks.
1 Introduction
Machine learning methods to classify or predict the properties of molecules have become omnipresent tools in chemical and biological research. Classification tasks include categorising molecules into toxic and non-toxic, protein-binding and non-binding, or otherwise pharmacological active or inactive compounds. Meanwhile, regression tasks encompass predicting various physicochemical and pharmacological properties, such as solubility and lipophilicity, protein–ligand binding affinity, or even quantum chemical properties. With the rise of deep learning during the past decade, molecular classification and property prediction have increasingly been carried out by ever-larger models with mixed results, as in tasks such as pharmacokinetic property prediction, where data remains scarce, deep learning methods have yet to perform significantly better than ensemble methods.1 Across all machine learning approaches, the most utilised methods are fingerprint-, SMILES-, and graph-based approaches, where molecular feature vectors, text representations of molecules, and molecular graphs, respectively, are used as the input of the respective class of models (MLPs, transformers, and GNNs).2–4 Even though the text-based SMILES encoding of a molecule is often called a 1D representation (as opposed to the “2D” molecular graph and the 3D molecular structure), a SMILES string contains all information of its respective molecular graph, as it is constructed by traversing said graph using a depth-first search (DFS) algorithm.5 Furthermore, it also contains implicit and explicit information on the 3D structure of the molecule, as molecular structure is tied to molecular topology, and molecular chirality is often directly defined using the SMILES notation.Recently, a parameter-free text classification approach based on Gzip compression has been proposed, which has shown excellent performance compared to deep learning architectures, such as transformers, on text-classification benchmark data sets.6 The intuition guiding the method is to exploit the capability of lossless compressors, such as Gzip, to capture regularity using a statistical model that enables to assign shorter codes to high-probability sequences. It is then assumed that texts in the same category share similar regularity and are thus close in compression space under a normalised compression distance (NCD) metric.7 A k-nearest neighbour classifier is then used to classify text under the NCD metric. As the SMILES string encoding of molecular graphs was proved to be a well-performing molecular representation for applying natural language processing (NLP) methods, such as transformers or locality-sensitive hashing (LSH),2,8 we hypothesise that the methodology presented by Jiang et al.6 will also yield good results for chemical tasks, and a comparison to other commonly used methods is warranted.
Here, we report an implementation of the Gzip-based text representation method, initially introduced by Jiang et al.,6 targeted towards chemical machine learning problems. We present two algorithms denoted MolZip and MolZip-Vec, both capable of single- and multimodal molecular classification and regression, with MolZip-Vec also allowing for the incorporation of real-valued vectors to embed precomputed chemical values. We compare our implementation to various other methods that do not rely on pretraining, including molecular fingerprint-based approaches and graph neural networks (GNNs), on molecular classification and regression tasks that include a binding-affinity prediction problem which we cast as a multimodal task by including the molecular SMILES of the ligand and the amino acid sequence of the protein. We show that this conceptually simple and inexpensive method works not only for the classification and clustering of data in a natural language processing context but also on SMILES-encoded molecules without requiring time-consuming training on specialised hardware, such as GPUs. In addition, we extend the methodology to support most chemical machine learning tasks through an open-source Python library.
2 Results & discussion
We benchmark the proposed methodology on a subset of the MoleculeNet benchmark for molecular machine learning and compare it against a selection of non-pretrained baselines for fingerprint- and GNN-based methods that underlie most current machine learning methodologies used for chemical property prediction. Random forest (RF) and support vector machine (SVM) use binary extended-connectivity fingerprints, ECFP,9,10 as input, graph convolutional networks (GCN) and graph isomorphism network (GIN) use the molecular graph as input,11,12SchNet and Multi-View graph convolutional network (MGCN) take graphs as input and explicitly model quantum chemical interactions within molecules,13,14 and D-MPNN is a directed message-passing neural network that takes graphs and molecular descriptors as input.15 The current state of the art on the benchmark has been achieved by Molformer-XL,4 which was pretrained on 1.1 billion molecules for approximately 208 hours on 16 NVIDIA V100 GPUs and then fine-tuned for another 12 hours, has not been included in Tables 1 and 2 as we focus on non-pretrained methods. Furthermore, we extended MolZip towards predicting protein–ligand binding affinities and compared the approach with GNN-based methods, which have seen continuous use and advancements over the past years. For all our experiments, we have chosen Gzip as the compressor as it generally shows the best performance when compared to LZ4 and Snappy (Table 5). Finally, we ran additional experiments for k-nearest neighbour-based classification and regression using ECFP as a control.| Dataset | BBBP19 | ClinTox | HIV | SIDER |
|---|---|---|---|---|
| RF | 71.4 ± 0.0 |
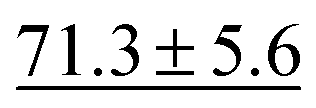
|
78.1 ± 0.6 | 68.4 ± 0.9 |
| SVM | 72.9 ± 0.0 |

|
79.2 ± 0.0 | 68.2 ± 1.3 |
| kNN |

|
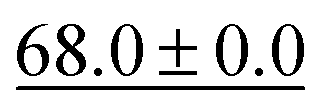
|
— | 61.3 ± 0.0 |
| GCN | 71.8 ± 0.9 |

|
74.0 ± 3.0 |

|
| GIN |

|

|
75.3 ± 1.9 |

|
| SchNet | 84.8 ± 2.2 |
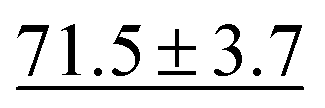
|

|
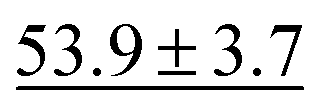
|
| MGCN | 85.0 ± 6.4 |

|
73.8 ± 1.6 |

|
| D-MPNN | 71.2 ± 3.8 |

|
75.0 ± 2.1 | 63.2 ± 2.3 |
| MolZip | 64.8 ± 0.0 |

|
68.8 ± 0.0 | 57.9 ± 0.0 |
| MolZip Aug | 65.9 ± 2.0 | 81.6 ± 1.5 | 71.2 ± 0.5 |

|
| MolZip Vec |

|
59.8 ± 0.0 |

|
58.1 ± 0.0 |
| Dataset | FreeSolv | ESOL | Lipo | QM8 |
|---|---|---|---|---|
| RF | 2.03 ± 0.22 |
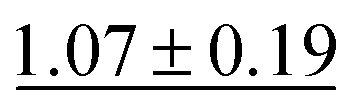
|
0.88 ± 0.04 |

|
| SVM | 3.14 ± 0.00 |

|
0.82 ± 0.00 |

|
| kNN |

|
0.87 ± 0.00 |

|
— |
| GCN | 2.87 ± 0.14 |

|
0.85 ± 0.08 |

|
| GIN | 2.76 ± 0.18 |

|
0.85 ± 0.07 |

|
| SchNet | 3.22 ± 0.76 |

|

|
0.020 ± 0.00 |
| MGCN |

|
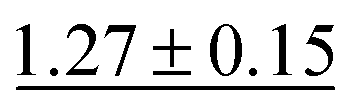
|

|
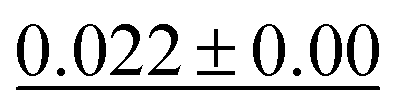
|
| D-MPNN | 2.18 ± 0.91 | 0.98 ± 0.26 | 0.65 ± 0.05 | 0.014 ± 0.00 |
| MolZip | 3.75 ± 0.00 | 1.33 ± 0.00 | 1.04 ± 0.00 | 0.028 ± 0.00 |
| MolZip Aug |
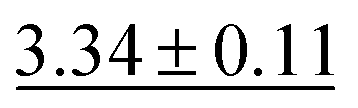
|
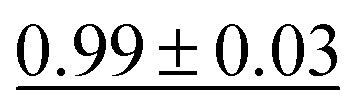
|
0.97 ± 0.02 | 0.026 ± 0.00 |
| MolZip Vec | 3.36 ± 0.00 | 1.16 ± 0.00 |

|

|
2.1 Classification
We follow the method proposed by Jiang et al.6 for the classification tasks and extend it with multiprocessing and nearest-neighbour weighing to support imbalanced data sets better. In addition, we implement a framework which provides serialisable text transformations on the input SMILES, including the translation into alternative string-based molecular representations (DeepSMILES and SELFIES) and SMILES-based augmentation, which augments a sample by concatenating a user-chosen number of different valid SMILES representations of a given molecule.16–18 For both MolZip and MolZip-Vec, we choose the parameter k = 5 for the k-nearest neighbour classification and assume that all data sets are imbalanced, therefore adjusting the kNN classification based on class weights that are calculated using the scikit-learn (v1.3.1) utility function .
.
Before benchmarking and comparing transformer-based methods, we evaluated the effect of translation and augmentation transformations. Table 4 compares the performance of SMILES, DeepSMILES, and SELFIES-encoded molecules with otherwise default parameters (k = 5, no augmentation) on various data sets. Based on these results, we decided to use SMILES encoding for our implementation, as it provides a balanced baseline across all evaluated data sets. However, the superior results of the SELFIES- and DeepSmiles-encodings on various data sets show that the encoding can indeed have a strong influence on the observed performance. Evaluating the effect of augmentation, which concatenates multiple variants of SMILES-encodings of the same molecule (e.g. starting the depth-first search, which constructs the SMILES, at a different atom), using the BBBP19 and BACE (classification) sets showed mixed results. While the performance of MolZip on the BACE (classification) data set that was later not used in further testing could have been pushed by approximately 10% (Fig. 2b), a lack of correlation of the positive effect on the validation and test set, as well as generally lower performance on the BBBP19 set (Fig. 2a), led us to report the non-augmented classification metrics as well. The same holds for MolZip-Vec, as presented in Fig. 2. Further investigation of the effect of the amount of augmentation (2-fold, 4-fold, 6-fold, and 11-fold, Table 6) showed the best overall performance for the 11-fold augmentation over the investigated classification and regression data sets, which led us to choose it as the default for MolZip Aug.
The results reported in Table 1 show that our compression-based methods reach competitive performance compared to fingerprint- and graph-based methods, while being conceptually exceptionally simple.
2.2 Molecular property prediction
We implemented regression functionality by taking the arithmetic mean of the k-nearest neighbours weighted by the similarity (=one minus distance) of their normalised compression distance NCD to the query (eqn (3)). For all regression tasks, we also choose k = 5. As for the classification tasks, we evaluated the SMILES-encoding against DeepSMILES and SELFIES and again chose SMILES over the two alternatives for benchmarking (Table 4). We further evaluated the effects of augmentation for regression tasks on the two data sets ESOL and BACE (regression). Interestingly, and unlike our evaluation of augmentation on classification tasks, augmentation on regression tasks has a general, and in some cases significant, positive effect on performance (Fig. 2c, d and Table 6): Augmenting each SMILES in the ESOL data set with an additional 19 SMILES, that represent the same molecule but differ in atom-order, would decrease the RMSE measured for MolZip by 28% from 1.510 to 1.097. As with the classification, we choose the 11-fold augmentation and report it in Table 2 as MolZip Aug.The results reported in Table 2 show that our compression-based methods reach competitive performance. The performance on the QM8 data set where the task is to predict quantum–chemical properties of a molecule is especially intriguing, as our method performs as well as MGCN, which includes quantum mechanical information.
2.3 Binding affinity prediction
In addition to molecular property prediction, we tested the ability of the compression-based approach to predict protein–ligand binding affinities—an essential metric for rational drug design, which aims to find a drug candidate, given structural information on a disease-associated protein.20 The protein–ligand binding affinity describes whether and how strong a ligand binds non-covalently to a protein, usually causing a conformational change of the protein and potentially leading to a therapeutic effect.21 The prediction of the binding affinity, given a potential ligand and a protein's structure or amino acid sequence, is therefore of interest to computational chemistry. Over the past years, geometric deep learning, specifically graph neural network-based approaches, have emerged as the most investigated methods to predict binding affinities, as they are capable of capturing topological and spatial features important to protein–ligand binding.22–24To tackle the challenge of protein–ligand binding affinity prediction using MolZip and MolZip-Vec, we implemented a data loader capable of loading and concatenating different modalities, namely SMILES and amino acid sequences, and pass it to a MolZip or MolZip-Vec regressor (Table 3). As we evaluated the method on the PCBbind data set,25 the following information was provided for each protein–ligand complex: (i) structural and compositional data for the ligand, (ii) structural and compositional data for amino acids that are part of the binding pocket of the protein, and (iii) structural and compositional data for the entire protein. From this data, we generated the following encodings: (1) for the ligand, a SMILES string, (2) for the binding pocket, a SMILES string and a one-letter amino acid string sequence, where amino acids that are not part of the binding pocket are replaced by an X, and (3) for the protein, a one-letter amino acid string sequence. These encodings provided us with four modalities (one molecule representation, two binding pocket representations, and one whole-protein representation) that can be combined arbitrarily through concatenation. Exploratory benchmark results for the combinations ligand (SMILES), binding pocket (SMILES), binding pocket (amino acid sequence), whole-protein (amino acid sequence), ligand (SMILES) + binding pocket (SMILES), ligand (SMILES) + binding pocket (amino acid sequence), and ligand (SMLIES) + whole-protein (amino acid sequence) can be found in Table 7. The combination ligand (SMILES) + binding pocket (amino acid sequence) provided the best results.
| Model | RMSE | MAE | R | |
|---|---|---|---|---|
| a With three-fold augmented SMILES. The average and standard deviation is taken from 5 runs with random SMILES permutations. | ||||
| GraphDTA | GCN | 1.735 ± 0.034 | 1.343 ± 0.037 | 0.613 ± 0.016 |
| GAT | 1.765 ± 0.026 | 1.354 ± 0.033 | 0.601 ± 0.016 | |
| GIN | 1.640 ± 0.044 | 1.261 ± 0.044 | 0.667 ± 0.018 | |
| GAT-GCN | 1.562 ± 0.022 | 1.191 ± 0.016 | 0.697 ± 0.008 | |
| GNN-based | SGCN | 1.583 ± 0.033 | 1.250 ± 0.036 | 0.686 ± 0.015 |
| GNN-DTI | 1.492 ± 0.025 | 1.192 ± 0.032 | 0.736 ± 0.021 | |
| D-MPNN | 1.493 ± 0.016 | 1.188 ± 0.009 | 0.729 ± 0.006 | |
| MAT | 1.457 ± 0.037 | 1.154 ± 0.037 | 0.747 ± 0.013 | |
| DimeNet | 1.453 ± 0.027 | 1.138 ± 0.026 | 0.752 ± 0.010 | |
| CMPNN | 1.408 ± 0.028 | 1.117 ± 0.031 | 0.765 ± 0.009 | |
| Compression-based | MolZip | 1.508 ± 0.000 | 1.190 ± 0.000 | 0.720 ± 0.000 |
| MolZip Aug | 1.422 ± 0.017 | 1.131 ± 0.014 | 0.757 ± 0.007 | |
| MolZip Vec | 1.675 ± 0.000 | 1.300 ± 0.000 | 0.648 ± 0.000 | |
Comparing our best results against baseline GraphDTA- and GNN-based methods, it becomes evident that MolZip performs exceptionally well. It does not only perform better than basic GNNs, including GCN, GAT, and GIN, that used atom features as node attributes for the molecular graph and the protein sequence as inputs,24 but also better than methods that include geometric information in the form of atom-wise protein–ligand interactions, such as GNN-DTI.26 The introduction of molecular descriptors with MolZip-Vec reduces the performance to that of GraphDTA methods, hinting at the importance of a relatively fuzzy representation of the ligand to a well-performing compression-based model.
2.4 Implications for chemical information retrieval
Compression-based representation of molecules may have implications beyond machine learning. As chemical databases such as ZINC or GDB contain billions of molecules, and even partially human-curated databases such as PubChem contain more than 100 million unique molecules, retrieving information based on chemical features is becoming increasingly important.27–29 Currently, most searches rely on graph topological similarity based on molecular fingerprints, precomputed stored chemical descriptors, or a combination of both.30 With the findings presented in this study, we have shown that the lossless compression-based combination of molecular structure and chemical descriptors, used as an input for MolZip-Vec, presents a low-memory alternative to established methods discussed by Warr et al.30 that allows for direct structure and property-based storage, similarity search and indexing.The ability to index and search molecules similar to a commonly used molecular fingerprint, ECFP (extended-connectivity fingerprint),32 is apparent when visually inspecting the TMAP plots in Fig. 1, where the high-dimensional ECFP and compression spaces of the BBBP data set19 are visualized by embedding a minimum spanning tree calculated in the original spaces in the Euclidean plane. TMAP (tree-MAP) is a data visualization method that represents large, high-dimensional data sets as two-dimensional trees, preserving local neighbourhoods with higher accuracy than other methods.31 They both show similar clusters of molecules capable of passing the blood–brain barrier.
 | ||
| Fig. 1 TMAP visualisation of the BBBP data set. Points represent molecules that are capable (blue) and incapable (orange) of penetrating the blood–brain barrier. Visual inspection of the plot confirms that MolZip encoding (b) is as capable as ECFP encoding (a) to identify both local and global similarities of molecules and cluster them accordingly.19,31 | ||
Finally, the adaptability of compression-based representations facilitates the handling of dynamic databases where new molecules are continually added, as it eliminates the need for retraining complex models or recomputing extensive descriptor sets. Implementing these techniques could significantly accelerate tasks such as virtual screening, lead optimization, and the identification of novel compounds with desired characteristics.
3 Methods
3.1 Implementation
We implemented the compression-based classification adapting the code presented in the original preprint and extended it with support for multiprocessing, class weights, multi-task classification, and regression.6 Inspired by the Normalised Compression Distance (NCD) from the original preprint, we define the distance between molecules x and y as | (1) |
This definition of NCD takes on values in the closed interval [0,1], providing a normalized measure of similarity between two strings based on their compressibility. It achieves its minimum value of 0 when the two strings are identical (x = y), as the compressed concatenation of identical strings does not add any new information beyond what is already in x or y. Conversely, the NCD approaches 1 when the strings are completely dissimilar and incompressible; in this case, x and y share no common patterns or redundancy for a compressor to exploit, so concatenating them does not reduce the overall compressed size. Thus, due to the properties of compression and the design of the NCD formula, the NCD is non-negative and does not exceed 1.
We are using UTF-8 encoding for text compression, consistent with the original method by Jiang et al.6
We changed the implementation of the k nearest-neighbor classifier by weighting the class counts Ci among the k nearest-neighbors using the formula
Cwi = CiWi(1 − ![[d with combining macron]](https://www.rsc.org/images/entities/i_char_0064_0304.gif) i) i) | (2) |
the mean distance (NCD) between the query point and the k nearest neighbors belonging to class i. The class weights were computed using the function  from the Python package scikit-learn. For the k nearest-neighbour regression, a simple distance weighted kNN regressor was implemented in the form of
from the Python package scikit-learn. For the k nearest-neighbour regression, a simple distance weighted kNN regressor was implemented in the form of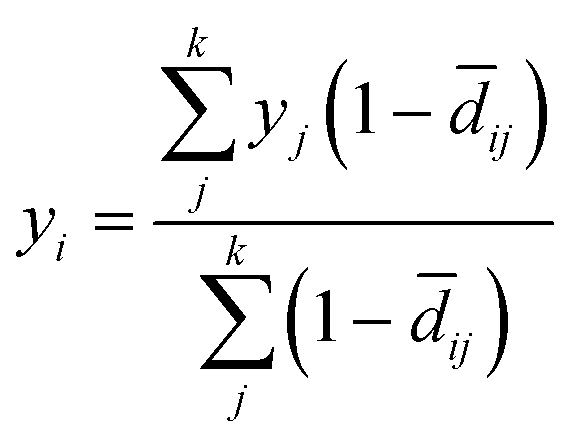 | (3) |
ij is the distance (NCD) between the query point i and the k nearest neighbours j, yj the values of the k nearest neighbours, and yi the predicted value.
Multiprocessing has been implemented using the Python standard library ( ).
).
3.2 MolZip-Vec
For MolZip-Vec, we combined SMILES strings with numerical descriptors of molecules commonly used in chemoinformatics. Specifically, we utilized a vector comprising 200 molecular descriptors from the RDKit cheminformatics library RDKit,33 which are typically used to augment graphs in molecular graph representation learning.15 A complete list of the 200 descriptors can be found in the documentation of the descriptastorus (v2.6.1) Python package. In order to combine and compress the numerical descriptors with the molecular string representation, the values are binned and subsequently translated into a set of non-ASCII Unicode characters. The three molecular string representations (SMILES, DeepSMILES, and SELFIES) used in this work only use ASCII characters, so collisions are avoided. Empirically, we found that 256 is a suitable number of bins. A special character prefixes negative values to represent positive and negative bins distinctly. Each string-based representation of the numerical vector is concatenated to the corresponding SMILES string, significantly improving the RMSE of several datasets listed in Table 2. In Fig. 3, we show that including numerical vectors increasingly improves the performance with growing training set size on the FreeSolv data set.34 Note that the computational cost for the prediction is slightly higher because of the increased string length.3.3 Benchmarking
The Moleculenet benchmarking results as well as the scaffold splitting method were taken from Wang et al.10 PDBbind benchmark results for GraphDTA and GNN-based methods were taken from Li et al.22All benchmarks with the exception of the runs involving k-optimisation were run on an Intel Core i7-13700K CPU with a total of 16 cores (8 performance and 8 efficiency cores) with a maximum power draw of 253 W. Together, all classification and regression benchmarks took 43 h 55 m to complete. All energy came from renewable sources (hydropower and solar energy).
4 Conclusion
By applying the proposed Gzip-based text classification method by Jiang et al.6 to multiple molecular classification tasks and extending it to regression problems, we verified its validity and utility beyond natural language processing tasks. While the claims by Jiang et al.6 in regards to performance comparisons with large language models were rather optimistic, the adapted methodology showed good performance compared to often-used fingerprint- and graph-based methods in a chemistry and biochemistry setting. Furthermore, it is in itself highly intriguing that a method based on differences in the length of Gzip compressed string representations of molecules can yield comparable or even superior performance compared to methods that were and continue to be in use for more than a decade. We have also shown that the methodology can be extended to multimodal binding affinity tasks, where SMILES strings and amino acid sequences are jointly compressed. On the PDBbind data set, our proposed method performs better than all GraphDTA- and most GNN-based methods, including those incorporating spatial information. We belief that this method represents a superior baseline for future developments compared to other approaches, as it is exceptionally easy to reproduce—a problem often encountered in highly parameterised methods. Additionally, we have demonstrated that integrating molecular SMILES strings with string-converted chemical descriptors can significantly enhance the accuracy compared to using SMILES input alone. Finally, we discuss how such a method could be of interest outside machine learning and support a new generation of chemical information retrieval in ultra-large databases. However, certain limitations and challenges still need to be addressed, including the relatively high time complexity of the kNN-based approach and the elucidation of the reasons for significant gaps in performance on specific data sets compared to the state-of-the-art.Data availability
The code for learning on compressed molecular representations as well as all scripts required to reproduce the results presented in the article can be found at https://github.com/daenuprobst/molzip with release https://doi.org/10.5281/zenodo.11643143 (https://zenodo.org/records/11643143).Conflicts of interest
There are no conflicts to declare.Appendix
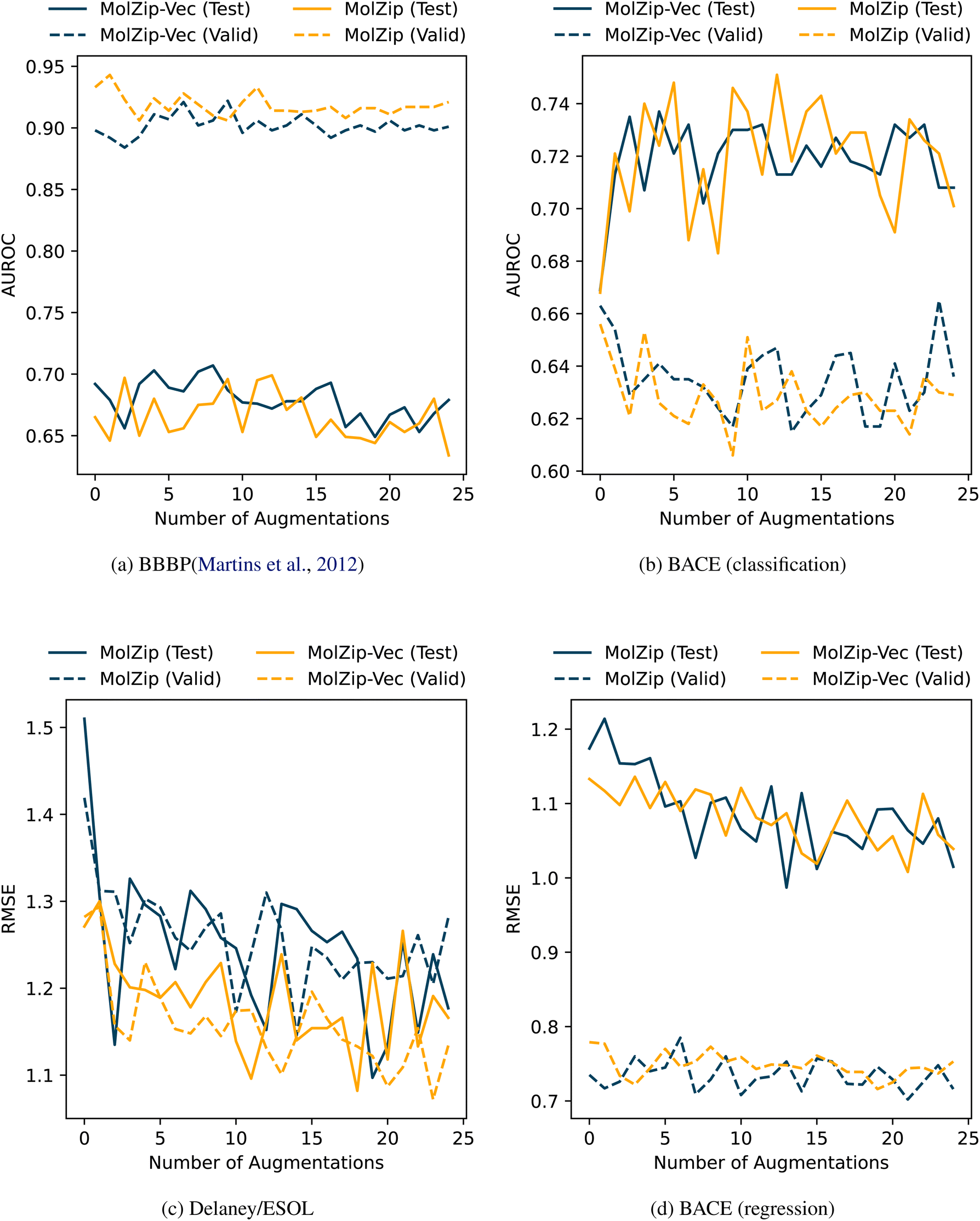 | ||
| Fig. 2 Influence of data augmentation (randomised SMILES) on validation and test results. In the four data sets BBBP (a), BACE classification (b), as well as Delaney/ESOL (c) and BACE regression (d), data augmentation did not yield consistent improvements. | ||
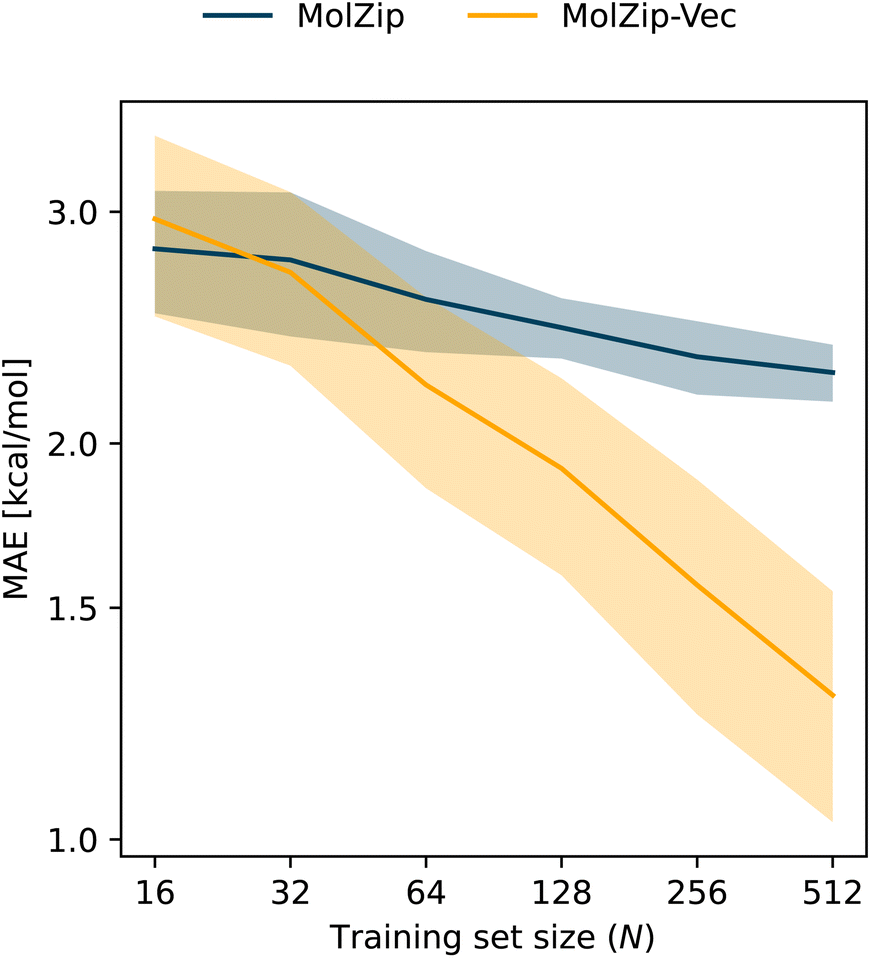 | ||
| Fig. 3 Comparing SMILES and a combination with molecular property vectors (SMILES + property vector). Learning curves i.e. mean absolute error (MAE) evaluated using 10-fold random splits of the FreeSolv34 database for solvation free energies. The x-axis shows the number of training examples N added at constant test set size. The curves show the average over the splits and the shadow the standard deviation. | ||
| Data set | Split | Metric | SMILES | DeepSMILES | SELFIES |
|---|---|---|---|---|---|
| BBBP19 | Scaffold | AUROC | 0.648 | 0.638 | 0.642 |
| ClinTox | Scaffold | AUROC | 0.914 | 0.920 | 0.693 |
| HIV | Scaffold | AUROC | 0.688 | 0.699 | 0.705 |
| SIDER | Scaffold | AUROC | 0.579 | 0.575 | 0.584 |
| FreeSolv | Scaffold | RMSE | 3.754 | 3.734 | 3.139 |
| ESOL | Scaffold | RMSE | 1.325 | 1.203 | 1.265 |
| LIPO | Scaffold | RMSE | 1.035 | 1.031 | 1.027 |
| QM8 | Scaffold | RMSE | 0.045 | 0.044 | 0.046 |
| Data set | Split | Metric | Gzip | LZ4 | Snappy |
|---|---|---|---|---|---|
| BBBP19 | Scaffold | AUROC | 0.648 | 0.691 | 0.641 |
| ClinTox | Scaffold | AUROC | 0.914 | 0.870 | 0.882 |
| HIV | Scaffold | AUROC | 0.688 | 0.687 | 0.667 |
| SIDER | Scaffold | AUROC | 0.579 | 0.584 | 0.594 |
| FreeSolv | Scaffold | RMSE | 3.754 | 5.586 | 5.343 |
| ESOL | Scaffold | RMSE | 1.325 | 1.460 | 1.736 |
| LIPO | Scaffold | RMSE | 1.035 | 0.979 | 1.072 |
| QM8 | Scaffold | RMSE | 0.045 | 0.047 | 0.050 |
| Data set | Split | Metric | None | +1 | +3 | +5 | +10 |
|---|---|---|---|---|---|---|---|
| BBBP19 | Scaffold | AUROC | 0.648 | 0.634 | 0.664 | 0.652 | 0.682 |
| ClinTox | Scaffold | AUROC | 0.914 | 0.865 | 0.824 | 0.810 | 0.802 |
| HIV | Scaffold | AUROC | 0.688 | 0.700 | 0.711 | 0.703 | 0.714 |
| SIDER | Scaffold | AUROC | 0.579 | 0.599 | 0.612 | 0.599 | 0.626 |
| FreeSolv | Scaffold | RMSE | 3.754 | 3.458 | 3.157 | 3.221 | 3.158 |
| ESOL | Scaffold | RMSE | 1.325 | 1.101 | 1.092 | 1.013 | 0.937 |
| LIPO | Scaffold | RMSE | 1.035 | 0.977 | 0.990 | 0.988 | 0.962 |
| QM8 | Scaffold | RMSE | 0.045 | 0.043 | 0.042 | 0.041 | 0.041 |
| Modalities | RMSE | MAE | R |
|---|---|---|---|
| Ligand (SMILES) | 1.776 | 1.416 | 0.591 |
| Pocket (SMILES) | 1.653 | 1.311 | 0.653 |
| Pocket (AA Seq.) | 1.665 | 1.303 | 0.644 |
| Protein (AA Seq.) | 1.885 | 1.512 | 0.525 |
| Ligand (SMILES) + pocket (SMILES) | 1.598 | 1.258 | 0.679 |
| Ligand (SMILES) + pocket (AA Seq.) | 1.504 | 1.187 | 0.721 |
| Ligand (SMILES) + protein (AA Seq.) | 1.688 | 1.307 | 0.633 |
References
- E. N. Muratov, J. Bajorath, R. P. Sheridan, I. V. Tetko, D. Filimonov, V. Poroikov, T. I. Oprea, I. I. Baskin, A. Varnek, A. Roitberg, O. Isayev, S. Curtalolo, D. Fourches, Y. Cohen, A. Aspuru-Guzik, D. A. Winkler, D. Agrafiotis, A. Cherkasov and A. Tropsha, QSAR without borders, Chem. Soc. Rev., 2020, 49(11), 3525–3564, 10.1039/D0CS00098A
.
- D. Probst and J.-L. Reymond, A probabilistic molecular fingerprint for big data settings, J. Cheminf., 2018, 10(1), 66, DOI:10.1186/s13321-018-0321-8
- P. Reiser, M. Neubert, A. Eberhard, L. Torresi, C. Zhou, C. Shao, H. Metni, C. van Hoesel, H. Schopmans, T. Sommer and P. Friederich, Graph neural networks for materials science and chemistry, Commun. Mater., 2022, 3(1), 1–18, DOI:10.1038/s43246-022-00315-6
- J. Ross, B. Belgodere, V. Chenthamarakshan, I. Padhi, Y. Mroueh and P. Das, Large-scale chemical language representations capture molecular structure and properties, Nat. Mach. Intell., 2022, 4(12), 1256–1264, DOI:10.1038/s42256-022-00580-7
- D. Weininger, SMILES, a chemical language and information system. 1. introduction to methodology and encoding rules, J. Chem. Inf. Comput. Sci., 1988, 28(1), 31–36, DOI:10.1021/ci00057a005
-
Z. Jiang, M. Yang, M. Tsirlin, R. Tang, Y. Dai, and J. Lin. “low-resource” text classification: A parameter-free classification method with compressors, in Findings of the Association for Computational Linguistics: ACL 2023, ed. A. Rogers, J. Boyd-Graber, and N. Okazaki, Association for Computational Linguistics, Toronto, Canada, 2023, pp. 6810–6828, DOI:10.18653/v1/2023.findings-acl.426
- M. Li, X. Chen, X. Li, B. Ma and P. M. B. Vitanyi, The similarity metric, IEEE Trans. Inf. Theory, 2004, 50(12), 3250–3264, DOI:10.1109/TIT.2004.838101
- P. Schwaller, T. Laino, T. Gaudin, P. Bolgar, C. A. Hunter, C. Bekas and A. A. Lee, Molecular Transformer: A Model for Uncertainty-Calibrated Chemical Reaction Prediction, ACS Cent. Sci., 2019, 5(9), 1572–1583, DOI:10.1021/acscentsci.9b00576
- D. Rogers and M. Hahn, Extended-connectivity fingerprints, J. Chem. Inf. Model., 2010, 50, 742–754, DOI:10.1021/ci100050t
- Y. Wang, J. Wang, Z. Cao and A. Barati Farimani, Molecular contrastive learning of representations via graph neural networks, Nat. Mach. Intell., 2022, 4(3), 279–287, DOI:10.1038/s42256-022-00447-x
- T. N. Kipf and M. Welling, Semi-supervised classification with graph convolutional networks, 2016, preprint, arXiv:1609.02907, DOI:10.48550/arXiv.1609.02907, http://arxiv.org/abs/1609.02907.
- K. Xu, W. Hu, J. Leskovec, and S. Jegelka, How powerful are graph neural networks?, arXiv, 2018, preprint, arXiv:1810.00826, DOI:10.48550/arXiv.1810.00826, http://arxiv.org/abs/1810.00826.
- K. T. Schütt, H. E. Sauceda, P.-J. Kindermans, A. Tkatchenko and K.-R. Müller, SchNet – a deep learning architecture for molecules and materials, J. Chem. Phys., 2018, 148(24), 241722, DOI:10.1063/1.5019779
- C. Lu, Q. Liu, C. Wang, Z. Huang, P. Lin and L. He, Molecular property prediction: A multilevel quantum interactions modeling perspective, Proc. AAAI Conf. Artif. Intell., 2019, 33(1), 1052–1060, DOI:10.1609/aaai.v33i01.33011052
- K. Yang, K. Swanson, W. Jin, C. Coley, P. Eiden, H. Gao, A. Guzman-Perez, T. Hopper, B. Kelley, M. Mathea, A. Palmer, V. Settels, T. Jaakkola, K. Jensen and R. Barzilay, Analyzing Learned Molecular Representations for Property Prediction, J. Chem. Inf. Model., 2019, 59(8), 3370–3388, DOI:10.1021/acs.jcim.9b00237
- E. Jannik Bjerrum, SMILES Enumeration as Data Augmentation for Neural Network Modeling of Molecules, arXiv, 2017, preprint, arXiv:1703.07076, DOI:10.48550/arXiv.1703.07076, http://arxiv.org/abs/1703.07076.
- N. O’Boyle and A. Dalke, DeepSMILES: An Adaptation of SMILES for Use in Machine-Learning of Chemical Structures, ChemRxiv, 2018, https://chemrxiv.org/engage/chemrxiv/article-details/60c73ed6567dfe7e5fec388d Search PubMed.
- M. Krenn, Q. Ai, S. Barthel, N. Carson, A. Frei, N. C. Frey, P. Friederich, T. Gaudin, A. A. Gayle, K. M. Jablonka, R. F. Lameiro, D. Lemm, A. Lo, S. M. Moosavi, J. M. Nápoles-Duarte, A. K. Nigam, R. Pollice, K. Rajan, U. Schatzschneider, P. Schwaller, M. Skreta, B. Smit, F. Strieth-Kalthoff, C. Sun, G. Tom, G. F. von Rudorff, A. Wang, A. D. White, A. Young, R. Yu and A. Aspuru-Guzik, SELFIES and the future of molecular string representations, Patterns, 2022, 3(10), 100588, DOI:10.1016/j.patter.2022.100588
- I. F. Martins, A. L. Teixeira, L. Pinheiro and A. O. Falcao, A Bayesian approach to in silico blood–brain barrier penetration modeling, J. Chem. Inf. Model., 2012, 52(6), 1686–1697, DOI:10.1021/ci300124c
- P. J. Gane and P. M. Dean, Recent advances in structure-based rational drug design, Curr. Opin. Struct. Biol., 2000, 10(4), 401–404, DOI:10.1016/S0959-440X(00)00105-6
- M. A. Williams, Protein-ligand interactions: fundamentals, Methods Mol. Biol., 2013, 1008, 3–34, DOI:10.1007/978-1-62703-398-5_1
-
S. Li, J. Zhou, T. Xu, L. Huang, F. Wang, H. Xiong, W. Huang, D. Dou, and H. Xiong. Structure-aware Interactive Graph Neural Networks for the Prediction of Protein-Ligand Binding Affinity. KDD ’21, Proceedings of the 27th ACM SIGKDD conference on knowledge discovery & data miningAssociation for Computing Machinery, New York, NY, USA, 2021, pp. 975–985, DOI:10.1145/3447548.3467311
- O. Méndez-Lucio, M. Ahmad, E. Antonio del Rio-Chanona and J. K. Wegner, A geometric deep learning approach to predict binding conformations of bioactive molecules, Nat. Mach. Intell., 2021, 3(12), 1033–1039, DOI:10.1038/s42256-021-00409-9
- T. Nguyen, H. Le, T. P. Quinn, T. Nguyen, T. Duy Le and S. Venkatesh, GraphDTA: predicting drug–target binding affinity with graph neural networks, Bioinformatics, 2021, 37(8), 1140–1147, DOI:10.1093/bioinformatics/btaa921
- Z. Liu, Y. Li, L. Han, J. Li, J. Liu, Z. Zhao, W. Nie, Y. Liu and R. Wang, PDB-wide collection of binding data: current status of the PDBbind database, Bioinformatics, 2015, 31(3), 405–412, DOI:10.1093/bioinformatics/btu626
- J. Lim, S. Ryu, K. Park, Y. Joong Choe, J. Ham and W. Y. Kim, Predicting Drug–Target Interaction Using a Novel Graph Neural Network with 3D Structure-Embedded Graph Representation, J. Chem. Inf. Model., 2019, 59(9), 3981–3988, DOI:10.1021/acs.jcim.9b00387
- J. J. Irwin and B. K. Shoichet, ZINC - A Free Database of Commercially Available Compounds for Virtual Screening, J. Chem. Inf. Model., 2005, 45(1), 177–182, DOI:10.1021/ci049714+
- R. Visini, M. Awale and J.-L. Reymond, Fragment Database FDB-17, J. Chem. Inf. Model., 2017, 57(4), 700–709, DOI:10.1021/acs.jcim.7b00020
- S. Kim, J. Chen, T. Cheng, A. Gindulyte, J. He, S. He, Q. Li, B. A. Shoemaker, P. A. Thiessen, B. Yu, L. Zaslavsky, J. Zhang and E. E. Bolton, PubChem 2019 update: improved access to chemical data, Nucleic Acids Res., 2019, 47(D1), D1102–D1109, DOI:10.1093/nar/gky1033
- W. A. Warr, M. C. Nicklaus, C. A. Nicolaou and M. Rarey, Exploration of Ultralarge Compound Collections for Drug Discovery, J. Chem. Inf. Model., 2022, 62(9), 2021–2034, DOI:10.1021/acs.jcim.2c00224
- D. Probst and J.-L. Reymond, Visualization of very large high-dimensional data sets as minimum spanning trees, J. Cheminf., 2020, 12(1) DOI:10.1186/s13321-020-0416-x
- D. Rogers and M. Hahn, Extended-Connectivity Fingerprints, J. Chem. Inf. Model., 2010, 50(5), 742–754, DOI:10.1021/ci100050t
- RDKit, Rdkit: Open-source cheminformatics, 2023, URL https://www.rdkit.org, accessed 28/9/2023.
- D. L. Mobley and J. P. Guthrie, FreeSolv: a database of experimental and calculated hydration free energies, with input files, J. Comput.-Aided Mol. Des., 2014, 711–720, DOI:10.1007/s10822-014-9747-x
Footnote |
| † Equal contribution. |
| This journal is © The Royal Society of Chemistry 2025 |