
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Isomerisation of phosphabutyne and a photochemical route to phosphabutadiyne (HC3P), a phosphorus analogue of cyanoacetylene†
Arun-Libertsen
Lawzer
 *a,
Elavenil
Ganesan
a,
Thomas
Custer
a,
Jean-Claude
Guillemin
b and
Robert
Kołos
a
*a,
Elavenil
Ganesan
a,
Thomas
Custer
a,
Jean-Claude
Guillemin
b and
Robert
Kołos
a
aInstitute of Physical Chemistry, Polish Academy of Sciences, Ul. Marcina Kasprzaka 44/52, 01-224, Warsaw, Poland. E-mail: alawzer@ichf.edu.pl; eganesan@ichf.edu.pl; tcuster@ichf.edu.pl; rkolos@ichf.edu.pl
bUniversity of Rennes, Ecole Nationale Superieure de Chimie de Rennes, CNRS-IRCR, 6226, F-35000, Rennes, France. E-mail: jean-claude.guillemin@ensc-rennes.fr
First published on 24th January 2025
Abstract
The photochemistry of phosphabut-1-yne, CH3CH2CP, was investigated by means of infrared spectroscopy assisted by theoretical (DFT) predictions. The UV-irradiated compound, isolated in a cryogenic argon matrix, undergoes isomerization and dissociation. Several isomers of phosphabutyne, in addition to phosphabutadiyne (HC3P), ethynylphosphinidene (HCCP), and phoshaethyne (HCP) are formed as the main photoproducts. Vibrational spectra of astrochemically relevant molecules HC3P and CH2CHCP (vinylphosphaethyne), have been detected and analyzed here for the first time.
Introduction
Phosphaalkynes and phosphaalkenes are reactive molecules which have been used as synthetic building blocks for more than a decade.1–4 Following the generation of phosphaethyne (H–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) P, the phosphorus analogue of hydrogen cyanide) by Gier,5 several other kinetically unstable phosphaalkynes (e.g. NC–CP, HCC–CP and H2C
P, the phosphorus analogue of hydrogen cyanide) by Gier,5 several other kinetically unstable phosphaalkynes (e.g. NC–CP, HCC–CP and H2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH–CP) were produced using flow pyrolysis and detected using microwave spectroscopy.6–9 Without appropriate, bulky stabilizing substituents, most of these molecules with phosphorus–carbon multiple bonds undergo oligomerization,10–13 making the smallest of them difficult to synthesize under normal laboratory conditions. tert-Butyl-substituted phosphaethyne (3,3′-dimethylphosphabutyne) was the first molecule with a phosphaalkyne unit synthesized.14,15 Given that cyanoacetylene16 and vinyl cyanide17 are commonly detected interstellar molecules, the spectroscopy of their P-bearing analogues phosphabutadiyne (HC3P) and vinylphosphaethyne (CH2CHCP) is also of interest to astrochemistry. To date, only a few phosphorus containing molecules have been detected in the interstellar medium: CP radical,18 HCP, PN, PO radical, CCP radical, and tentatively, NCCP.19,20 Phosphaalkenes and phosphaallenes, just like phosphaalkynes, are kinetically unstable unless stabilized with bulky, electron-rich substituents.21–25 Phosphaethene (CH2PH) has been characterized with microwave spectroscopy26,27 and two of its IR bands were tentatively identified.28 The simplest phosphaallene CH2CPH was proposed as an intermediate in base-catalyzed thermal rearrangements of alkynylphosphines to phosphaalkynes29 (Scheme 1(a)) and later characterized as a photoproduct of phosphapropyne rearrangement in an argon matrix.30
CH–CP) were produced using flow pyrolysis and detected using microwave spectroscopy.6–9 Without appropriate, bulky stabilizing substituents, most of these molecules with phosphorus–carbon multiple bonds undergo oligomerization,10–13 making the smallest of them difficult to synthesize under normal laboratory conditions. tert-Butyl-substituted phosphaethyne (3,3′-dimethylphosphabutyne) was the first molecule with a phosphaalkyne unit synthesized.14,15 Given that cyanoacetylene16 and vinyl cyanide17 are commonly detected interstellar molecules, the spectroscopy of their P-bearing analogues phosphabutadiyne (HC3P) and vinylphosphaethyne (CH2CHCP) is also of interest to astrochemistry. To date, only a few phosphorus containing molecules have been detected in the interstellar medium: CP radical,18 HCP, PN, PO radical, CCP radical, and tentatively, NCCP.19,20 Phosphaalkenes and phosphaallenes, just like phosphaalkynes, are kinetically unstable unless stabilized with bulky, electron-rich substituents.21–25 Phosphaethene (CH2PH) has been characterized with microwave spectroscopy26,27 and two of its IR bands were tentatively identified.28 The simplest phosphaallene CH2CPH was proposed as an intermediate in base-catalyzed thermal rearrangements of alkynylphosphines to phosphaalkynes29 (Scheme 1(a)) and later characterized as a photoproduct of phosphapropyne rearrangement in an argon matrix.30

The cryogenic technique of rare-gas matrix isolation provides the opportunity to characterize reactive molecules often classified as intermediates.31 In solid argon, UV-photolyzed phosphapropyne undergoes rearrangement and dehydrogenation to form molecules such as phosphaallene and triplet ethynylphosphinidene (HCCP)30 (Scheme 1(b)). Phosphinidenes are phosphorus analogues of nitrenes and our knowledge of these intermediate species is growing slowly but still limited to just a few compounds.32 In this context, it was of interest to explore the photochemistry of phosphabut-1-yne, CH3–CH2–CP (hereafter phosphabutyne or 1) as a way to obtain HC3P, CH2CHCP, open-shell organophosphorus species, and other phosphorus-containing unsaturated molecules of potential interest for spectroscopy of reactive intermediates and astrochemistry. Particularly important was the goal to investigate the completely unexplored infrared spectroscopy of HC3P and CH2CHCP. Thus far, only the microwave, i.e. purely rotational transitions of these two molecules have been reported.
The characterisation of the molecular vibrations of such exotic phosphorus-bearing molecules is of interest to the rapidly growing field of IR astrospectroscopy.33
Results and discussion
Photochemistry and isomerization of phosphabutyne
Irradiation of phosphabutyne (1) at 254 nm induces isomerization (hydrogen and/or methyl group migration) and dissociation. We characterized the species indicated in Scheme 2. Product ratios could be varied by using additional precursors and radiation sources, allowing us to distinguish products and their corresponding IR spectra. Most of the IR bands of the species (Scheme 2) formed through the UV-photolysis of 1 have been identified (ESI,† S10). The presence of 1-propynylphosphine 3, propadienylphosphine 5, and propargylphosphine 6 was confirmed with IR absorption spectra of the authentic substances (obtained through preparative synthesis) isolated in argon matrices. Spectra measured during the photolysis of 1, combined with theoretical (DFT) predictions of band frequencies and intensities, made it possible to follow the time evolution of the relative concentrations of photoproducts (Fig. 1).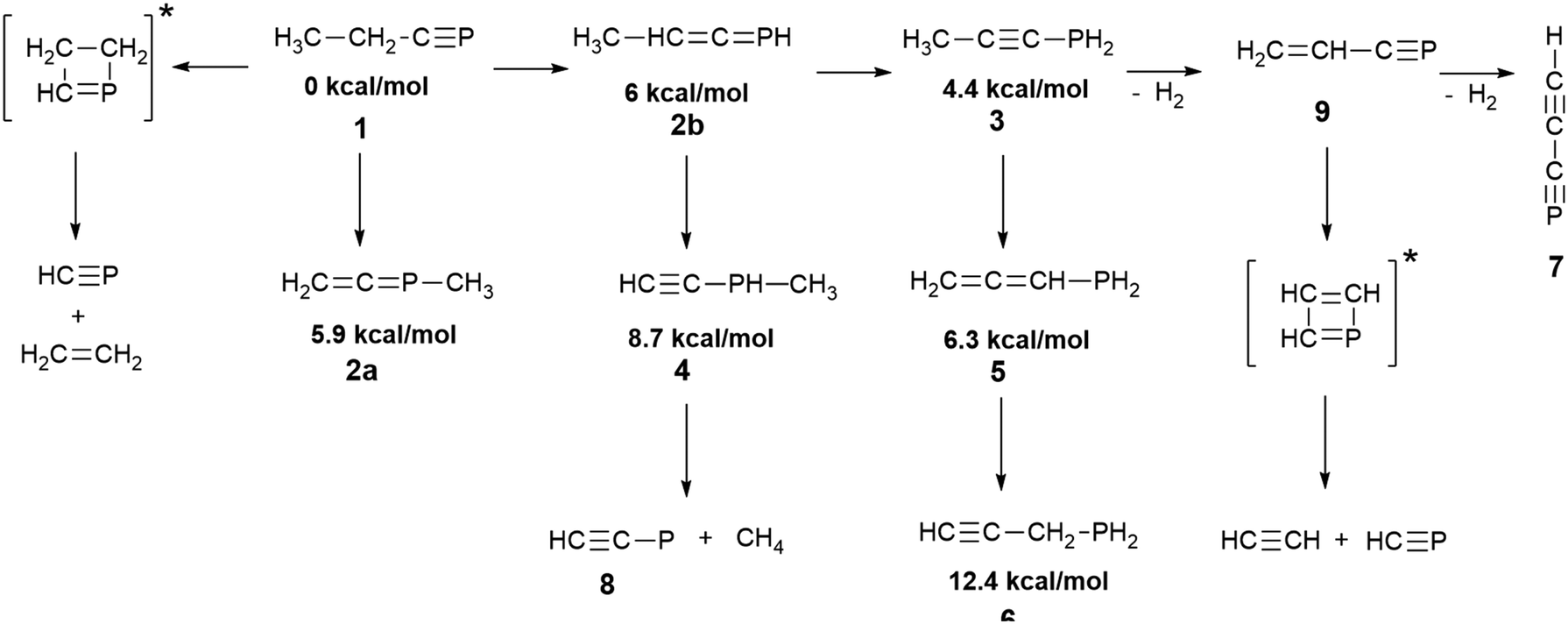 | ||
| Scheme 2 Products formed in the 254 nm photolysis of phosphabutyne in solid argon. The placement of the arrows, while compatible with Fig. 1 and 7, is provisional. DFT-derived relative electronic energies of the isomers of C3H5P species are provided. | ||
 | ||
| Fig. 1 Isomerization (1–6) and dehydrogenation (7,9) products traced over the course of 254 nm photolysis of phosphabutyne (1) in solid Ar, scaled with respect to the initial concentration of the precursor, taken as 100. Integrated intensities measured for the bands centered at 1551.2 cm−1 (1), 858 cm−1 (2a), 809 cm−1 (2b), 2216.9 cm−1 (3), 629 cm−1 (4), 831 cm−1 (5), 635 cm−1 (6), 1524.3 cm−1 (7), and 924 cm−1 (9) were used in conjunction with the B3LYP/aug-cc-pVTZ-derived absolute IR band strengths of 38.7 km mol−1 (1), 44.1 km mol−1 (2a), 20.2 km mol−1 (2b), 34.2 km mol−1 (3), 40.7 km mol−1 (4), 52.6 km mol−1 (5), 20.0 km mol−1 (6), 35.7 km mol−1 (7), and 44.8 km mol−1 (9). | ||
Subsections below describe the details pertaining to identification of the individual molecules. DFT-predicted IR spectral parameters for species 1, 2a, 2b, 4, and 6, as well as the listing of bands observed for Ar matrix-isolated authentic compounds 3, 5, and 6 are provided as ESI.†
Phosphaallenes CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e001.gif) CP–CH3 (2a) and CH3–CHCPH (2b)
CP–CH3 (2a) and CH3–CHCPH (2b)
Substituted phosphaallenes such as 1-methyl-1-phosphaallene, 2a and 3-methyl-1-phosphaallene, 2b are the products of methyl group and hydrogen atom migration in 1, respectively. Their appearance among the photoproduced isomers is analogous to that of phosphaallene (CH2CPH) in the course of phosphapropyne (CH3CP) photolysis.30
The P–H bending, P–H stretching, and CC stretching modes of 2b appear at 884.3 cm−1, 2257.6 cm−1 and 1753.5 (broad) cm−1, respectively, close to the values reported for the unsubstituted phosphaallene30 (888.4 cm−1, 2264.4 cm−1 and 1732.9 cm−1). The bands due to the allenic C–H in-plane and out-of-plane bendings of 2b appear at 1262.1 cm−1 and 808.7 cm−1, respectively. Methyl unit bending bands around 1400 cm−1 are the weakest and difficult to assign due to the overlapping features of other products. Synthesis of the species 2a has been reported and some of its IR bands were measured at 77 K.34 The CC stretching bands of these phosphaallenes conveniently fall within an uncongested region allowing for reliable identification. The band reported for solid 2a at 77 K34 had a maximum at 1715 cm−1, while in the argon matrix we detect two peaks: 1715.8 cm−1 and 1737.4 cm−1, which probably reflect separate microenvironments (matrix “sites”). The CH2 wagging mode of the allenic unit in solid 2a was reported at 869 cm−1; in argon matrix it appears at 858.2 cm−1. Optical densities of the IR absorption features assigned to 2a are mutually correlated, as are those assigned to 2b and the corresponding frequencies agree well with the DFT predictions (Fig. 2 and ESI,† S2.3, S3.3). The time evolution curves (Fig. 1) indicate that both species are formed in the early stages of the photolysis. The P-methylated molecule 2a seems to be more photostable than 2b. Within our proposed reaction pathway (Scheme 2), 2b is a key intermediate species for consecutive photochemical transformations.
 | ||
| Fig. 2 Identification of 1-methyl-1-phosphaallene 2a and 3-methyl-1-phosphaallene 2b among the phosphabutyne 1 photolysis products in solid argon. Traces (a)–(c) are difference spectra (after-minus-before photolysis) showing the net effects of, respectively, 76 h, 3 h and 1.5 h of Hg-lamp irradiation; traces (d) and (e) are B3LYP/aug-cc-pVTZ predictions (frequency scaled by 0.96) for 2b and 2a, respectively. Numbers give the band wavenumbers assigned to 2a (italicized) and 2b. | ||
Propynylphosphine CH3–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e002.gif) C–PH2 (3)
C–PH2 (3)
According to DFT predictions, propynylphosphine (3) is the most thermodynamically stable structural modification among the isomers of phosphabutyne, and it is reasonable to expect a photoinduced transformation from 1 to 3, analogous to the rearrangement of CH3–CP to HCC–PH2.30 Here, 3 appeared as the main product at the early stages of the photolysis, but decayed almost completely upon prolonged irradiation (Fig. 1 and 3). Its identification was straightforward, given the perfect match with the spectrum of pure 3 in solid argon (Fig. 3). The species is likely formed via a two-step 1,3-hydrogen migration, passing through 2b. Hg-lamp photolysis of the matrix isolated authentic sample of 3 produces a mixture of photoproducts very similar to that obtained from 1 (ESI,† S4.4).
 | ||
| Fig. 3 Identification of propynylphosphine 3 among the phosphabutyne 1 photolysis products in solid argon. Traces (a) and (b) are difference spectra (after-minus-before photolysis) showing the net effects of, respectively, 76 h and 3 h of Hg-lamp irradiation (concentration of 3 peaks at the beginning photolysis); trace (c) represents the spectrum of pure 3 isolated in solid Ar. Numbers give the band wavenumbers assigned to 3. | ||
Ethynylmethylphosphine HCC–PH–CH3 (4)
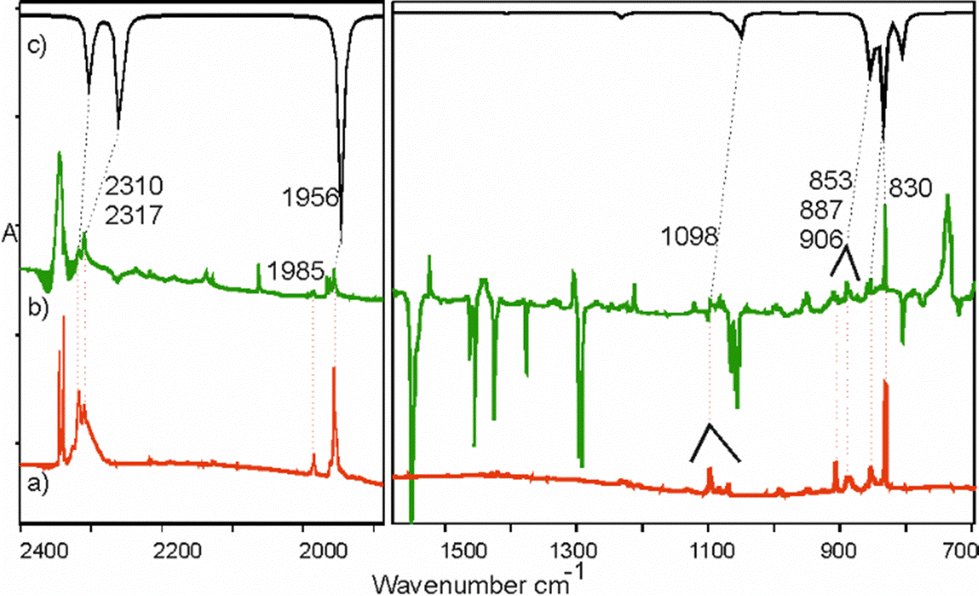
Species 4 is presumably formed from 2a through 1,3-hydrogen migration and/or from 2b through 1,3-methyl group migration towards the phosphorus center. IR bands of this molecule appear mostly as multiplets and are located in congested spectral regions. The C–H stretching vibration of the acetylenic unit of 4 overlaps the C–H stretchings of 6 and 7 (ESI,† S5.3b). A good candidate for the respective C–H bending band emerges prominently at 629.1 cm−1, in good agreement with the DFT prediction (Fig. 4 and S5.3a, ESI†). The PH stretching of 4 reported34 at 2260 cm−1 may correspond to a weak band observed at 2237.2 cm−1. The C–H stretching band at 3320.2 cm−1, a candidate for the main IR feature of 4, is observable at initial stages of the photolysis. Later on, however, it becomes overlapped by the much stronger C–H stretching band of HC3P (7) at 3322.7 cm−1 (ESI,† S5.3b). Considering the uncertainties, our identification of 4 (Fig. 4) is only tentative.
 | ||
| Fig. 4 Identification of ethynylmethylphosphine (4) among the phosphabutyne (1) photolysis products in solid argon. Topmost trace shows the B3LYP/aug-cc-pVTZ prediction for the IR spectrum of 4 (frequency scaled by 0.96). The other five traces are difference spectra (after-minus-before photolysis) illustrating (bottom to top) the net effect of 76 h, 46 h, 12 h, 6 h and 1.5 h of Hg-lamp irradiation. Numbers give the band wavenumbers assigned to 4 (spectral congestion makes the asterisked assignments less certain). | ||
Propadienylphosphine CH2CCH–PH2 (5)
Species (5) appears as one of two major products of the photolysis (along with HC3P). Accordingly, of all the observed C3H5P isomers, this species appears to be the least susceptible to any decomposition induced by 254 nm radiation. Species 5 may be formed via a 1,3-hydrogen migration from species 3 (and perhaps also from 2b). This conjecture was verified in a separate experiment (ESI,† S6.4) involving pure 3 isolated in solid Ar: isomer 5 emerged as an Hg-lamp photolysis product, its presence unambiguously confirmed by comparison to the spectrum of an authentic, matrix-isolated sample of 5 (Fig. 5).
 | ||
| Fig. 5 Identification of propadienylphosphine (5) among the phosphabutyne (1) photolysis products in solid argon. Trace (a) represents the spectrum of an authentic (synthesized) sample of 5 isolated in solid Ar. Trace (b) is the difference spectrum (after-minus-before photolysis) showing the net effects of 76 h of Hg-lamp irradiation; traces (c) illustrate B3LYP/aug-cc-pVTZ predictions (frequency scaled by 0.96) for 3. Numbers give the band wavenumbers assigned to 5. | ||
Species 5 may adopt conformations differing by the rotation angle of the PH2 unit around the C–P bond (Scheme 3). Gauche conformers (a pair of enantiomeric species) are 0.34 kcal mol−1 lower in energy than anti (ESI,† S6.1). Considering the band broadening and matrix-site effects induced by the solid environment, small spectral differences between gauche and anti species predicted by the DFT calculations (ESI,† S6.2) as well as their limited accuracy, we are unable to distinguish the rotamers in the present spectra. Our separate experiment with Hg-lamp irradiation of matrix-isolated 5 shows the compound to be a photochemical precursor for propargylphosphine, 6. Species 6 formed out of 5 along with other photodehydrogenation products: HCP, HCCP, HC3P, and acetylene (ESI,† S6.5).
 | ||
| Scheme 3 Rotamers of 5. | ||
Propargylphosphine HCC–CH2–PH2 (6)
As species 6 lacks any conjugation of π-electrons, with its acetylenic and phosphine moieties separated by a methylene unit, one can expect it to be relatively stable towards mid-UV irradiation.
Over the course of photolysis of 1, the C–H stretching and C–H bending bands of 6 overlap with those of 7 (HC3P) but the analysis of spectra corresponding to different irradiation times allowed us to separate the spectral features of the two species The authentic spectrum of matrix-isolated 6 was on hand, as the compound was always present as contaminant in the sample of 5, probably formed as coproduct during the synthesis (Fig. 6). In an Ar matrix, we observe most of the intense IR transitions which match well with the previously reported gas phase spectrum.35,36
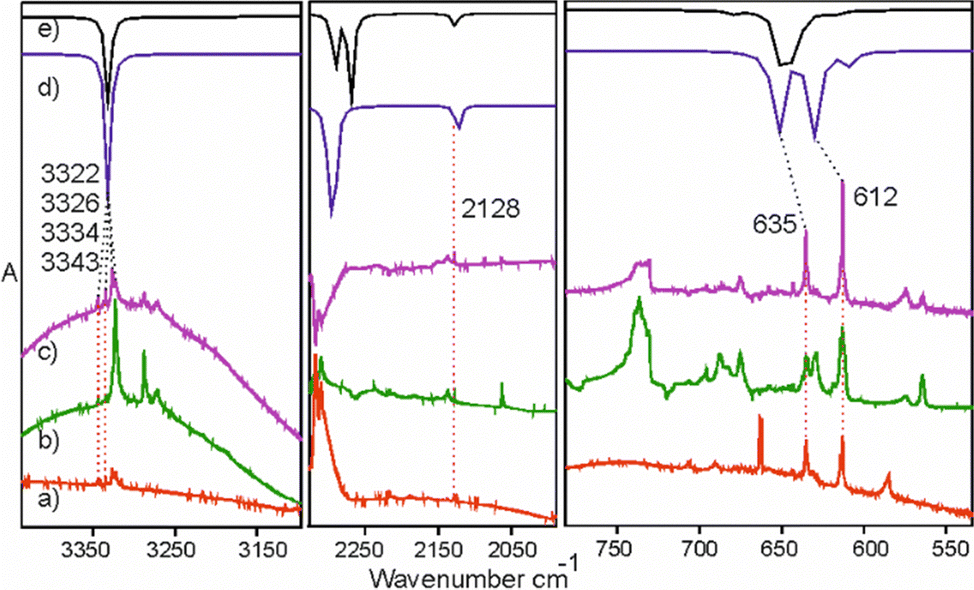 | ||
| Fig. 6 Identification of 6 among the photolysis products in solid Ar. Trace (a) is the spectrum of an authentic sample of 5 in Ar with 6 as an inseparable admixture. Trace (b) is the difference spectrum (after-minus-before photolysis) showing the net effect of 76 h of Hg-lamp irradiation of 1. Trace (c) is the difference spectrum showing the net effect of 24 h of Hg-lamp irradiation of 5. Traces (d) and (e) are B3LYP/aug-cc-pVTZ predictions (frequency scaled by 0.96) for the anti and gauche rotamers of 6, respectively. Wavenumbers of the bands assigned to 6 are provided. | ||
 | ||
| Fig. 7 Evolution of concentrations over the course of 254 nm photolysis of phosphabutyne 1 in solid argon, scaled with respect to the initial concentration of the precursor, taken as 100. Integrated intensities measured for the bands centered at 1524.3 cm−1 (7), 564 cm−1 (8), 924 cm−1 (9), 3271.7 (C2H2), 1304.2 (CH4), 675.5 (HCP) and 950.2 (C2H4) were used in conjunction with the B3LYP/aug-cc-pVTZ-derived absolute IR band strengths of 35.7 km mol−1 (7), 73.0 km mol−1 (8), 44.8 km mol−1 (9), 97 km mol−1 (C2H4), 35 km mol−1 (CH4), 15.7 km mol−1 (HCP) and 90 km mol−1 (C2H2). | ||
Analysis of spectra corresponding to different irradiation times allowed us to separate the spectral features of the two species.
Species 6 possesses two stable conformers, gauche and anti (Scheme 4 and ESI,† S7). According to our DFT prediction, the gauche species is just 0.31 kcal mol−1 lower in energy than anti. Calculations indicate the expected frequency difference between the two perpendicular acetylenic CH bending modes: 23 cm−1 for the anti and 8 cm−1 for the gauche rotamer (ESI,† S7.2). Experimentally, the respective IR bands are separated by 23 cm−1, indicating the dominance of the anti-form. Microwave spectroscopy37 also points to the latter as the preferred form.
 | ||
| Scheme 4 Rotamers of 6. | ||
Photodissociation
Based on previous experiments30 with matrix-isolated phosphapropyne, photodehydrogenation is expected to occur along with isomerisation upon irradiation of phosphabutyne. However, bands characteristic of acetylene (3271.2 cm−1), ethylene (950.2 cm−1 and 955.3cm−1), and HCP (675 cm−1, 682.2 cm−1 and 687 cm−1) indicate the presence of a C–C bond cleaving channel. These species likely form either through retro [2+2] cycloaddition38 from the isomers (Scheme 5 (a)) and the dehydrogenated products of 1 (Scheme 5 (b)) or through a concerted C–C dissociation during H-migration as C2H2/HCP and C2H4/HCP complexes in matrix cages. The related C2H2/HCN and C2H4/HCN adducts,39,40 in which HCN acts as a H-bond donor to the π-cloud of an unsaturated hydrocarbon, have been observed in low-temperature matrices. | ||
| Scheme 5 Proposed mechanism for the formation of HCP, C2H4, and C2H2 from the isomers of 1 (a) and the C3H3P isomer (b). | ||
For the complexes formed by HCP with acetylene and ethylene, our calculations reveal weak hydrogen bonding (binding energies below 2.0 kcal mol−1) depicted in Fig. 8: either HCP or a hydrocarbon molecule act as a H-bond donor. The vibrational modes most strongly affected by complexation will be those involved in this interaction. Examining shifts in the C–H stretching and bending of acetylene and HCP as well as the CH2 waging mode of ethylene compared with the uncomplexed species should indicate the nature of the complex produced in these experiments. A strong blue shift (about 25–40 cm−1) is predicted for the C–H bending frequency of HCP in complexes featuring HCP as the hydrogen bond donor (b and c in Fig. 8 and ESI,† S9). However, compared to the CH bending frequency of uncomplexed HCP (674.03 cm−1 gas phase,5,41 and 672.9 cm−1 in solid argon42), we observe three bands (675.1 cm−1, 682.2 cm−1 and 687.2 cm−1) with shifts of less than 15 cm−1. These shifts suggest the presence of some weaker complexes rather than b and c. Complex formation, whether into a, b, or c, splits the bending mode into out-of-plane and in-plane components, similar to what has recently been reported for the CH bending vibration of an HCP/HCl complex.42 The shift of out-of-plane CH bending of the C2H2/HCl complex is much less (2.8 cm−1) than the shift of in-plane CH bending (21.4 cm−1).42 In our measurements, we assume that the in-plane bending bands of complexed HCP are those found at 687.2 cm−1 (14.3 cm−1 shift from uncomplexed molecule) and 682.2 cm−1 (9.3 cm−1 shift) for C2H2/HCP and C2H4/HCP, respectively (inset, Fig. 9). The assumption is based on the behaviour of these two peaks over the course of photolysis of 1: the band at 682.2 cm−1 grows at earlier times than the one at 687.2 cm−1 (inset, Fig. 9), and Scheme 2 indicates that C2H4 is expected before the appearance of C2H2. The band at 675.1 cm−1 (shifted by +2.2 cm−1 with respect to uncomplexed HCP) is presumably due to the out-of-plane bending mode of C2H2/HCP. The above picture is consistent with the presence of C2H2/HCP complex a, where the discussed HCP bending frequency shift amounts to 7.9 cm−1 (B3LYP) or 12.5 cm−1 (MP2) for the in-plane mode and only 1.3 cm−1 (B3LYP) or 2.0 cm−1 (MP2) for the out-of-plane mode.
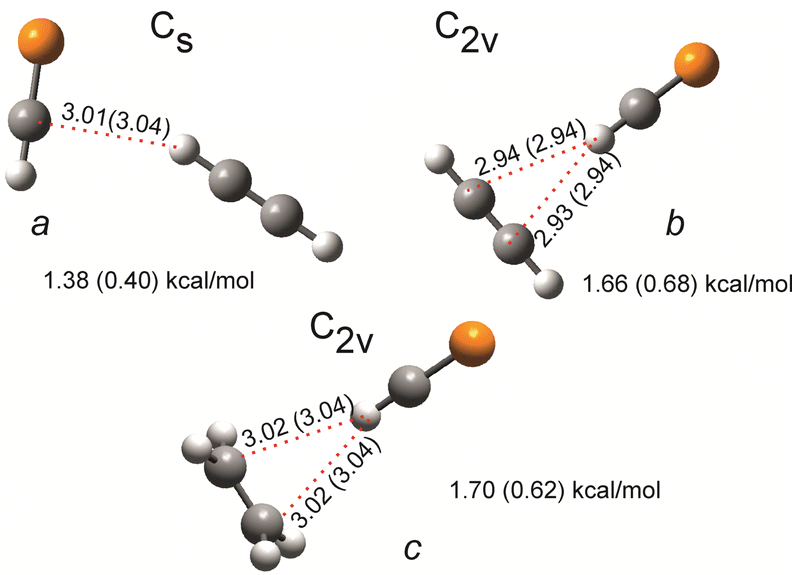 | ||
| Fig. 8 Complexes of HCP with acetylene and ethylene, as optimized at the MP2/aug-cc-pVTZ and B3LYP/aug-cc-pVTZ (in brackets) level of theory. The BSSE corrected interaction energies and bond length of the weak hydrogen bonds (in Angstrom) are indicated. The point groups symbols of each complex are given. | ||
 | ||
| Fig. 9 Identification of the dissociation products observed upon UV photolysis of phosphabutyne (1) in solid argon. Traces (a) and (b) are different spectra (after-minus-before photolysis) illustrating the net effect of 3h and 76 h of Hg-lamp irradiation. Topmost trace shows the B3LYP/aug-cc-pVTZ prediction (frequency scaled by 0.96) for the IR spectrum of HC3P (7). | ||
The complexation-induced shift observed for the CH stretching band of C2H2 also points to the presence of complex a in which HCP acts as an H-bond acceptor. The band shifts by −17.7 cm−1 from the frequency of 3288.9 cm−1 reported for the uncomplexed molecule43 isolated in solid Ar, while the corresponding shift predicted for a is −14.4 cm−1 (B3LYP) or −15.1 cm−1 (MP2). For complex b (where HCP acts as an hydrogen bond donor), this shift would, as calculated, be almost doubled. Unfortunately, the CH bending band of C2H2 is obscured by a strong feature at 736.6 cm−1 caused by the substrate window.
For the C2H4/HCP complex, the CH2 wagging mode of ethylene is observed with shift of +2.1 cm−1 and +7.3 cm−1 (the splitting is most likely due to matrix sites) with respect to the frequencies of 948 cm−1 reported44 for pure ethylene in solid Ar. The agreement of this shift with B3LYP and MP2 predictions for c (both methods indicate a value around +5 cm−1) is not conclusive, considering the shift in the HCP bending region. Nevertheless, the small observed blue shift in the CH2 wagging mode of ethylene suggests that HCP is bound to the π-cloud of the ethylene molecule in another orientation than in c. To understand the true nature of the C2H4/HCP complex, theoretical studies of complexes in argon cages and separate experiments involving solid argon doped with the mixtures of the pure substances are necessary.
The presence of HCCP (8) was confirmed based on the reported IR frequencies in an argon matrix.30 Its formation necessitates the elimination of CH4, likely from the P-methyl alkyne 4 (photoproduced methane is indeed observed here, through its band at 1304.6 cm−1). Growth curves for the two species (Fig. 7) are consistent with the assumption of their common origin, considering inevitable inaccuracies in the computed absolute IR intensities. No bands attributable to any other phosphinidene (a compound bearing a formally single C–P bond) were detected.
Three stretching fundamental vibrational frequencies, one bending fundamental, and one bending overtone of HC3P (7) are identified (Table 1 and Fig. 9 and 10). Anharmonic calculations confirm the high strength of the detected C–H bending overtone band (ESI,† S11). While the exact dehydrogenation pathways leading from 1 to 7 remain to be elucidated, the high and constantly growing concentration of HC3P suggests that the immediate precursor for 7 swiftly reaches a steady state and that 7 does not undergo any significant secondary photolysis.
| Mode | Vibrational frequency in cm−1 (absolute IR intensity in km mol−1) | |||
|---|---|---|---|---|
| Theorya (CCSD(T))/(cc-pVQZ) harmonic | Theoryb (B3LYP/aug-cc-pVTZ) harmonic | Theoryc (VPT2//B3LYP/aug-cc-pVTZ) anharmonic | Ar matrix | |
| a Harmonic approximation, not scaled (ref. 45). b Scaling factor 0.96 (this work). c This work. | ||||
| C–H str. (ν1) | 3453 | 3324.7(102) | 3337.7(113.6) | 3322.7 |
| C–C str. (ν2) | 2111 | 2076.0(8.7) | 2119.5(12.1) | 2062.8 |
| C–P str. (ν3) | 1544 | 1519.8(35.8) | 1563.1(91.2) | 1524.3 |
| HCCCP str. (ν4) | 687 | 680.4(0.2) | 708.7(0.8) | — |
| C–H bend. (ν5) | 621 | 622.2(80.4) | 635.0(513.8) | 611.2 |
| 611.8 | ||||
| 614, 613.1 | ||||
| CCP bend. (ν6) | 479 | 496.5(0.7) | 514.0(5.4) | — |
| CCP bend. (ν7) | 192 | 196.6(7.9) | 205.4(152.1) | — |
| Overtone (2ν5) | — | — | 1276.6 (93.8) | 1212.2 |
| 1255.5 (46.9) | ||||
 | ||
| Fig. 10 Difference spectra depicting the identification of HC3P (7) among the products generated by far-UV photolyses (160–190 nm, 6-hour irradiations) of Ar-matrix isolated compounds 1 (bottommost, brown trace), 3 (red), and 5 (blue). Top: B3LYP/aug-cc-pVTZ prediction for the IR spectrum of 7 (frequency scaled by 0.96). | ||
A series of additional experiments were conducted photolyzing Ar-matrix isolated compounds 1, 3, and 5 with far-UV (160–190 nm) radiation from a microwave-driven Xe lamp (Fig. 10). All of these far-UV photolyses produced 7 as the major product. The only C3H3P species detected here is vinylphosphaethyne (CH2CHCP, 9). It was difficult to identify following Hg-lamp and far-UV irradiation due to the weakness of its bands. Unambiguous assignment was only possible by performing a secondary photolysis using 308 nm light following photolysis at 254 nm (Fig. 11 and ESI,† S8). The identified IR frequencies are: 923.8 cm−1 (CH2 wagging), 967.4 (CH2 and CH twisting), 1529.1 cm−1 (C–P stretching), and 1614.0 cm−1 (C–C stretching). Molecule 9 is one probable precursor for 7.
 | ||
| Fig. 11 Bottom: Difference spectrum (after-minus-before photolysis) showing, for phosphabutyne 1 in solid Ar, the net effect of an additional 24 h irradiation at 308 nm after 6 days of 254 nm photolysis. Top: B3LYP/aug-cc-pVTZ prediction for the IR spectrum of 9 (frequency scaled by 0.96). Asterisked assignment is tentative. | ||
Conclusions
Photolysis of Ar matrix-isolated phosphabutyne, CH3–CH2–CP, with 254 nm radiation produces a variety of C3H5P isomers as well as photodissociation products. Isomers observed include CH2CP–CH3, CH3–CHCPH, HCC-PH–CH3 (tentative), HCC–CH2–PH2, CH3–CC–PH2, and CH2CCH–PH2. Formation of HCP, ethylene, and acetylene indicates that some of the isomers undergo a retro-cycloaddition reaction. Photodissociation consists mainly of the elimination of methane (leading to HCCP) and in dehydrogenation resulting in the formation of phosphabutadiyne (H–CC–CP) and vinylphosphaethyne (CH2CH–CP). These experiments gave access, for the first time, to the infrared spectra of HC3P, CH2CHCP, and CH3–CHCP. Five vibrational frequencies, including a bending overtone, of phosphabutadiyne (HC3P), the phosphorus analog of the astrochemically significant cyanoacetylene, were determined. A simplified reaction scheme was proposed, the verification of which, as well as a full description of the photochemical transformations of phosphabutyne, require dedicated quantum chemical studies.
Materials and methods
Diethylene glycol dibutyl ether (diglyme), tetraethylene glycol dimethyl ether (tetraglyme), lithium aluminum hydride, aluminum chloride, 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU), n-butyl lithium (n-BuLi) (2.5 M in hexanes) and ethyl bromide (CH3CH2Br) were purchased from the Aldrich Company and used without further purification.Synthesis
Phosphabutyne 1 was prepared in a three-step reaction following reported procedures (Scheme 6).46,47 Briefly, (1,1,1-trichloromethyl) phosphonic acid, diisopropyl ester was reacted with n-BuLi in the presence of LiCl before the addition of ethyl bromide to give (1,1-dichloropropyl)phosphonic acid, diisopropyl ester.47 In a chemoselective reduction with AlHCl2 in a high boiling solvent, diglyme, the 1,1-dichloropropylphosphine was formed, isolated at low pressure with gentle heating of the reaction mixture and condensed in a cold trap.48 The synthesis of phosphabutyne by bis-dehydrochlorination of the phosphine was performed at low temperature in diglyme, using the strong Lewis base 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU). For the two last steps, the product was isolated from the crude reaction mixture in a vacuum line by selective trapping at 183 K and 77 K, respectively. | ||
| Scheme 6 Synthesis of phosphabutyne 1. | ||
1-Propynylphosphine 3 and 1,2-propadienylphosphine 5 were synthesized as previously reported.48
Matrix isolation experiments
Precursor compounds were sublimed into an evacuated stainless-steel manifold and diluted with argon at approx. 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1000 ratio using standard manometric techniques. The gas mixture was condensed on a cold (ca. 10 K) cesium iodide window mounted to a closed-cycle helium cryostat (Advanced Research Systems DE-202SE refrigerator, ultimate temp. of 6 K) and exposed to radiation of a low-pressure Hg-lamp, dominated by resonance emission at 254 nm. A microwave-driven xenon lamp (Opthos instrument Co.) was used as a VUV continuum radiation source (150–190) nm. Photolysis progress was monitored using a Bruker Vertex 70 Fourier transform spectrometer featuring liquid nitrogen cooled MCT detector operating at its maximal resolution of 0.16 cm−1.
:1000 ratio using standard manometric techniques. The gas mixture was condensed on a cold (ca. 10 K) cesium iodide window mounted to a closed-cycle helium cryostat (Advanced Research Systems DE-202SE refrigerator, ultimate temp. of 6 K) and exposed to radiation of a low-pressure Hg-lamp, dominated by resonance emission at 254 nm. A microwave-driven xenon lamp (Opthos instrument Co.) was used as a VUV continuum radiation source (150–190) nm. Photolysis progress was monitored using a Bruker Vertex 70 Fourier transform spectrometer featuring liquid nitrogen cooled MCT detector operating at its maximal resolution of 0.16 cm−1.
Computational methods
Equilibrium molecular structures and the corresponding harmonic vibrational frequencies of molecular vibrations (as given by analytical second derivatives of the total energy, with respect to nuclear positions) were predicted at the DFT level employing the B3LYP functional49 and aug-cc-pVTZ basis set.50,51 The derived vibrational frequencies were scaled by a factor of 0.96 to take partial account of anharmonicity, incomplete inclusion of electron correlation effects, and deficiencies in the applied basis set. Zero-point energy correction was applied to the obtained electronic energy values. Default convergence criteria were used. For non-covalent complexes, the equilibrium geometry and IR frequencies were obtained from both Møller–Plesset perturbation theory of second order (MP2)52 and density functional theory (DFT) method with the B3LYP hybrid functional. The binding energies of the complexes were corrected for the basis set superposition errors (BSSE) using the counterpoise (CP) scheme of Boys and Bernardi.53 All computations were performed using the Gaussian 16 software suite.54Data availability
The authors confirm that the data supporting the results of this research article are available in the article and its ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the financial support from the PHC Polonium project no. BPN/BFR/2021/1/00028/U/00001. J. C. G. thanks for the support from “Programme National Physique et Chimie du Milieu Interstellaire” (PCMI) of CNRS/INSU with INC/INP co-funded by CEA and CNES.Notes and references
- M. Regitz, Chem. Rev., 1990, 90, 191–213 CrossRef CAS.
- J. F. Nixon, Chem. Soc. Rev., 1995, 24, 319–328 RSC.
- M. Regitz, J. Heterocycl. Chem., 1994, 31, 663–677 CrossRef CAS.
- K. Nakajima, S. Takata, K. Sakata and Y. Nishibayashi, Angew. Chem., Int. Ed., 2015, 54, 7597–7601 CrossRef CAS PubMed.
- T. E. Gier, J. Am. Chem. Soc., 1961, 83, 1769–1770 CrossRef CAS.
- J. C. T. R. Burckett St. Laurent, T. A. Cooper, H. W. Kroto, J. F. Nixon, O. Ohashi and K. Ohno, J. Mol. Struct., 1982, 78, 215–220 CrossRef.
- H. W. Kroto, J. F. Nixon and K. Ohno, J. Mol. Spectrosc., 1981, 90, 512–516 CrossRef CAS.
- K. Ohno, H. W. Kroto and J. F. Nixon, J. Mol. Spectrosc., 1981, 90, 507–511 CrossRef CAS.
- L. Bizzocchi, S. Thorwirth, F. Lewen and G. Winnewisser, J. Mol. Spectrosc., 2001, 205, 110–116 CrossRef CAS PubMed.
- U. Bergstraesser, Sci. Synth., 2004, 18, 427–444 Search PubMed.
- E. A. Cohen, G. A. McRae, H. Goldwhite, S. Di Stefano and R. A. Beaudet, Inorg. Chem., 1987, 26, 4000 CrossRef CAS.
- J. P. Albrand, S. P. Anderson, H. Goldwhite and L. Huff, Inorg. Chem., 1975, 14, 570–572 CrossRef CAS.
- W. J. Marshall, B. M. Fish, M. F. Schiffhauer, F. Davidson and C. N. McEwen, Organometallics, 2009, 28, 2410–2416 CrossRef.
- G. Becker, G. Gresser and W. Whl, Z. Naturforsch., B, 1981, 36, 16 CrossRef.
- M. Regitz, Chem. Rev., 1990, 90, 191–213 CrossRef CAS.
- C. M. Walmsley, G. Winnewisser and F. Toelle, Astron. Astrophys., 1980, 81, 245–250 CAS.
- H. E. Matthews and T. J. Sears, Astrophys. J., 1983, 272, 149–153 CrossRef CAS.
- M. Guélin, J. Cernicharo, G. Paubert and B. E. Turner, Astron. Astrophys., 1990, 230, L9–11 Search PubMed.
- M. Agúndez, J. Cernicharo and M. Guélin, Astron. Astrophys., 2014, 570, A45 CrossRef.
- M. Agúndez, J. Cernicharo and M. Guélin, Astrophys. J., 2007, 662, L91–L94 CrossRef.
- R. Appel, F. Knoch and V. Winkhaus, Chem. Ber., 1986, 119, 2466–2472 CrossRef CAS.
- G. Märkl, P. Kreitmeier, H. Nöth and K. Polbom, Angew. Chem., Int. Ed. Engl., 1990, 29, 927–929 CrossRef.
- M. Yoshifuji, K. Toyota, K. Shibayama and N. Inamoto, Tetrahedron Lett., 1984, 25, 1809–1812 CrossRef CAS.
- G. Märkl and S. Reitinger, Tetrahedron Lett., 1988, 29, 463–466 CrossRef.
- G. Märkl and U. Herold, Tetrahedron Lett., 1988, 29, 2935–2938 CrossRef.
- H. W. Kroto, J. F. Nixon and K. Ohno, J. Mol. Spectrosc., 1981, 90, 367–373 CrossRef CAS.
- L. Margules, J. Demaison, P. B. Sreeja and J.-C. Guillemin, J. Mol. Struct., 2006, 238, 234–240 CAS.
- B. Pellerin, P. Guenot and J.-M. Denis, Tetrahedron Lett., 1987, 28, 5811–5814 CrossRef CAS.
- J.-C. Guillemin, T. Janati and J.-M. Denis, J. Chem. Soc., Chem. Commun., 1992, 415–416 RSC.
- A.-L. Lawzer, T. Custer, J.-C. Guillemin and R. Kołos, Angew. Chem., Int. Ed., 2021, 60, 6400–6402 CrossRef CAS PubMed.
- R. A. Moss, Chem. Rev., 2013, 113(9), 6903–6904 CrossRef CAS PubMed.
- B. Lu and X. Zeng, Chem. – Eur. J., 2024, e202303283 CrossRef CAS PubMed.
- J.-P. Maillard, Philos. Trans. R. Soc., A, 2024, 382, 20230070 CrossRef PubMed.
- J.-C. Guillemin, T. Janati, J.-M. Denis, P. Guenot and P. Savignac, Tetrahedron Lett., 1994, 35, 245–248 CrossRef CAS.
- R. H. Shay, B. N. Diel, D. M. Schubert and A. D. Norman, Inorg. Chem., 1988, 27, 2378–2382 CrossRef CAS.
- J.-C. Guillemin and K. Malagu, Organometallics, 1999, 18, 5259–5263 CrossRef CAS.
- J. Demaison, J.-C. Guillemin and H. Mollendal, Inorg. Chem., 2001, 40, 3719–3724 CrossRef CAS PubMed.
- F. Mathey and P. Le Floch, J. Org. Chem., 2004, 69, 5100–5103 CrossRef CAS PubMed.
- A. Toumi, I. Couturier-Tamburelli, T. Chiavassa and N. Pietri, J. Phys. Chem. A, 2014, 118, 2453–2462 CrossRef CAS PubMed.
- A. Toumi, N. Pietri and I. Couturier-Tamburelli, Phys. Chem. Chem. Phys., 2015, 17, 30352–30363 RSC.
- M. Jung, B. P. Winnewisser and M. Winnewisser, J. Mol. Spectrosc., 1997, 413, 31–48 Search PubMed.
- J. Jiang, B. Lu, B. Zhu, G. Rauhut and X. Zeng, J. Phys. Chem. Lett., 2023, 14, 4327–4333 CrossRef CAS PubMed.
- K. Sundararajan and N. Ramanathan, J. Mol. Spectrosc., 2009, 920, 369–376 CAS.
- E. Rytter and D. M. Gruen, Spectrochim. Acta, Part A, 1979, 35A, 199–207 CrossRef CAS.
- L. Bizzocchi, C. D. Esposti and P. Botschwina, Chem. Phys. Lett., 2000, 411–417 CrossRef CAS.
- J.-C. Guillemin, T. Janati and J.-M. Denis, J. Org. Chem., 2001, 66, 7864–7868 CrossRef CAS PubMed.
- C. Grandin, E. About-Jaudet, N. Collignon, J.-M. Denis and P. Savignac, Heteroat. Chem., 1992, 3, 337–343 CrossRef CAS.
- J.-C. Guillemin, P. Savignac and J.-M. Denis, Inorg. Chem., 1991, 30, 2170–2173 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 7, 5648–5652 CrossRef.
- D. E. Woon and T. H. Dunning Jr., J. Chem. Phys., 1993, 98, 1358–1371 CrossRef CAS.
- K. A. Peterson, J. Chem. Phys., 2003, 119, 11099–11112 CrossRef CAS.
- C. Møller and M. S. Plesset, Phys. Rev., 1934, 46, 618–622 CrossRef.
- S. F. Boys and F. Bernardi, Mol. Phys., 1970, 19, 553–566 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision B.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cp04182h |
| This journal is © the Owner Societies 2025 |