
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePolyamidoxime (PAO) granules for solar-enhanced uranium extraction from seawater†
Xue
Zhang
,
Qianhong
Gao
and
Dadong
Shao
 *
*
School of Environmental and Biological Engineering, Nanjing University of Science and Technology, Nanjing 210094, PR China. E-mail: shaodadong@126.com
First published on 24th November 2023
Abstract
Extracting uranium (U(VI)) from seawater can effectively solve the shortage of uranium resources on land. The key to U(VI) extraction from seawater is preparing high performance U(VI) extraction materials. Although PAO based materials present outstanding performance in U(VI) extraction, their experimental adsorption capabilities are greatly restricted by its aggregation. To inhibit PAO aggregation, we used KMnO4 as a regulator to interfere with PAO aggregation in aqueous solutions, and the physicochemical properties of the obtained KMnO4@PAO were well characterized. Importantly, KMnO4@PAO also presents excellent photo-thermal conversion properties in various aqueous solutions, which are beneficial for U(VI) extraction. The adsorption properties of KMnO4@PAO for U(VI) under different solution conditions (pH, adsorption time, U(VI) ion concentration, and temperature) were studied by batch adsorption experiments. The results reveal that KMnO4@PAO presents excellent adsorption properties for U(VI), and its efficient photo-thermal conversion properties can increase solution temperature and U(VI) adsorption capability.
Environmental significanceUranium is the fundamental material of nuclear energy and is a representative actinide element of radioactive wastewater management. The immobilization and enrichment of uranium from aqueous solutions is an important for nuclear energy. |
Introduction
Nuclear energy is a vital part of the solution to global climate change especially under the current “two-carbon” target.1,2 Uranium is the primary material of nuclear energy, and terrestrial uranium ore is limited and will be exhausted in a few decades. The search for new uranium source is critical for the future of nuclear energy. The ∼4.5 billion tons of U(VI) in seawater is a thousand times that in terrestrial ore.3–6 Meanwhile, ∼27![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 tons of U(VI) is also replenished by rivers yearly.7,8 How to extract U(VI) from seawater has become a research hotspot, and many schemes, including chemical precipitation, evaporation, ion exchange, and membrane separation, have been reported for U(VI) extraction from seawater.9–14 Adsorption is widely regarded as one of the promising strategies for this target owing to its merits, such as simple operation and environmentally friendly process.15–20 After decades of research, people found that the material is one core of this task, directly determining the efficiency and comprehensive cost of U(VI) extraction. Developing materials with excellent comprehensive and economy properties is significant for U(VI) extraction.
000 tons of U(VI) is also replenished by rivers yearly.7,8 How to extract U(VI) from seawater has become a research hotspot, and many schemes, including chemical precipitation, evaporation, ion exchange, and membrane separation, have been reported for U(VI) extraction from seawater.9–14 Adsorption is widely regarded as one of the promising strategies for this target owing to its merits, such as simple operation and environmentally friendly process.15–20 After decades of research, people found that the material is one core of this task, directly determining the efficiency and comprehensive cost of U(VI) extraction. Developing materials with excellent comprehensive and economy properties is significant for U(VI) extraction.
Amidoxime (AO, –C(NH2)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–OH) contains both an oxime group and amino group, and the oxygen atom in the oxime group and the nitrogen atom in the amino group both contain free lone electron pairs. Therefore, it is easy for AO to form stable complexes with U(VI) ions,21–23 and it has become an indispensable part of various U(VI) extraction materials. The excellent stability of PAO based materials has been tested in seawater.17,23–26 Polyacrylonitrile (PAN) is widely used as the precursor for PAO because the core –C
N–OH) contains both an oxime group and amino group, and the oxygen atom in the oxime group and the nitrogen atom in the amino group both contain free lone electron pairs. Therefore, it is easy for AO to form stable complexes with U(VI) ions,21–23 and it has become an indispensable part of various U(VI) extraction materials. The excellent stability of PAO based materials has been tested in seawater.17,23–26 Polyacrylonitrile (PAN) is widely used as the precursor for PAO because the core –C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N in PAN can be easily converted into AO. Many methods are used to convert PAN into PAO, NH2OH amidoximation is the most classic method.27–31 NH2OH amidoximation technique includes homogeneous and heterogeneous methods, which were react in organic solvents31–33 and in aqueous solutions,28,34 respectively. Organic solvents used in the homogeneous method result in environmental pollution and high cost, which dramatically reduces the comprehensive competitiveness of PAO in U(VI) extraction. Meanwhile, PAN quickly settles down in aqueous solution, and most –CN in PAN cannot react with NH2OH in the heterogeneous method.
N in PAN can be easily converted into AO. Many methods are used to convert PAN into PAO, NH2OH amidoximation is the most classic method.27–31 NH2OH amidoximation technique includes homogeneous and heterogeneous methods, which were react in organic solvents31–33 and in aqueous solutions,28,34 respectively. Organic solvents used in the homogeneous method result in environmental pollution and high cost, which dramatically reduces the comprehensive competitiveness of PAO in U(VI) extraction. Meanwhile, PAN quickly settles down in aqueous solution, and most –CN in PAN cannot react with NH2OH in the heterogeneous method.
To inhibit PAN aggregation in aqueous solutions during the NH2OH amidoximation process, a new strategy is developed in this work by using KMnO4 as a regulator. The added KMnO4 can effectively inhibit PAN agglomeration, regulate PAN morphology, and endow PAO with photo-thermal properties. The adsorption and photo-thermal conversion properties of KMnO4@PAO in a variety of aqueous solutions, including ultrapure water, tap water, river water, and simulated seawater, have been systematically studied. This method can reduce the cost of PAO-based materials, in line with the concept of green development.
Results and discussion
Characterization
Fig. 1A shows the synthesis procedure and U(VI) adsorption of KMnO4@PAO. PAN as a macromolecule will undergo intramolecular agglomeration in aqueous solution and settle down as a block before reacting with NH2OH. Ultimately, it limits the application of PAO based materials in U(VI) recovery performance. In this work, we successfully suppressed the agglomeration of PAO in aqueous solution by using KMnO4 as a regulator, and the obtained KMnO4@PAO is uniform in aqueous solution (Fig. 1B). Particle size distributions of PAO and KMnO4@PAO were also studied by DLS measurement. Compared with PAO, the size of KMnO4@PAO is significantly decreased, and the diameters of PAO and KMnO4@PAO were concentrated at ∼1100 nm (Fig. 1C) and ∼400 nm, respectively. In a word, the PAO particle size can be easily adjusted by using KMnO4.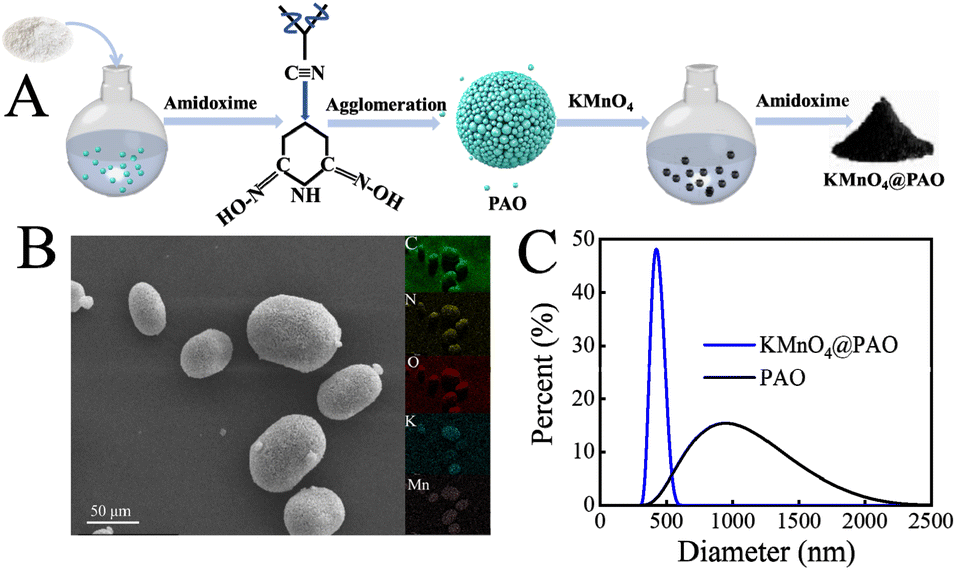 | ||
| Fig. 1 Diagram of KMnO4@PAO synthesis procedures (A); SEM images – EDS maps of KMnO4@PAO (B); size distributions of PAO and KMnO4@PAO (C). | ||
SEM was used to investigate the effect of KMnO4 and PAO amounts on the surface morphology of KMnO4@PAO. PAO amount did not shown significant impact on PAN surface topologies when the KMnO4 dosage was fixed at 0.20 g (Fig. 2A and B), whereas the agglomeration of PAO apparently increases with increasing KMnO4 amount when the PAO dosage was fixed at 2.0 g (Fig. 2C and D). When KMnO4 is introduced into the suspensions of PAN and NH2OH, the redox reaction between NH2OH and KMnO4 and the conversion of –CN to –C(NH2)N–OH coexist together, which restricts and promotes each other in aqueous solutions. The reaction is dominated by solution compositions. At low PAN KMnO4 content, the primary reaction in the solution is the amidoximation of PAN and NH2OH. The present KMnO4 can inhibit the generation of unsaturated amines, and finally obtain uniformly dispersed PAO particles. At high KMnO4 content, the dominating reaction in the solution is the reduction reaction of KMnO4 and NH2OH. Few –CN was reduced by NH2OH, and the unreacted –CN bond to each other to form unsaturated amine, and settle down in aqueous solution. It means that the morphology and size of KMnO4@PAO can be easily adjusted by controlling KMnO4 amounts. The KMnO4@PAO prepared with 2.0 g PAO and 0.20 g KMnO4 shows ideal morphology and size, and is selected for subsequent experiments.
 | ||
| Fig. 2 SEM images of KMnO4@PAO with different KMnO4 and PAO amounts. 0.20 g KMnO4 series: 1.0 g PAO (A); 2.0 g PAO (B). 2.0 g PAN series: 1.0 g KMnO4 (C); 1.5 g KMnO4 (D). The details of KMnO4@PAO preparation conditions are shown in Table S1.† | ||
The FT-IR technique was used to study the effects of KMnO4 on the surface functional groups and thermal stability of KMnO4@PAO. As shown in Fig. 3A, the weak FT-IR band at ∼2240 cm−1 is related to stretching vibration of residual –CN groups in PAO and KMnO4@PAO. The peaks at ∼1651 and ∼932 cm−1 can be respectively assigned to –CN and N–O vibrations, which reveals the high conversion of PAN to PAO.17,23 The FT-IR spectrum results show that the introduced KMnO4 cannot significantly affect the conversion of PAN to PAO during the amidoximation process.35
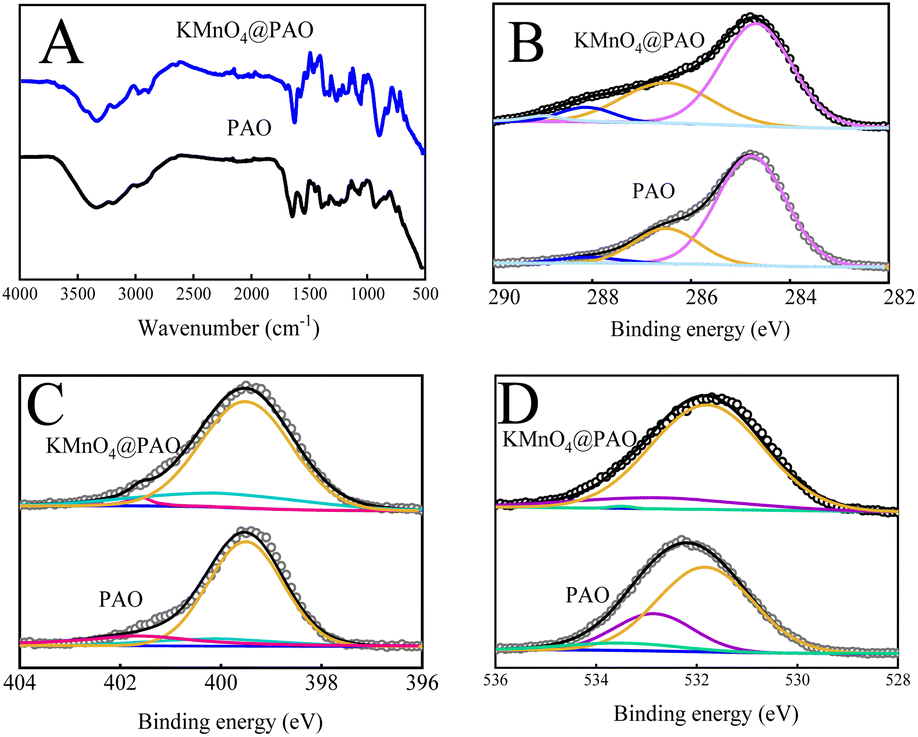 | ||
| Fig. 3 FT-IR (A), C 1s (B), N 1s (C), and O 1s (D) spectra of KMnO4@PAO and PAO. | ||
The functional groups on PAO and KMnO4@PAO surfaces are verified by XPS spectroscopy. The XPS peaks of O 1s, N 1s, and C 1s were located at ∼532 eV, ∼400 eV, and ∼285 eV, respectively.36 The XPS C 1s15,36 spectra of KMnO4@PAO and PAO can be deconvoluted into four peaks at 284.8 ± 0.1 eV, 286.5 ± 0.1 eV, 288.1 ± 0.1 eV and 288.9 ± 0.1 eV, which can be assigned to –CN, C–OH, –CO, and –COOH, respectively. The proportion of –CN in KMnO4 is significantly lower than that of PAO, indicating the higher conversion of PAN in KMnO4@PAO (Fig. 3B, Table S2†). PAO-based materials present excellent stability,23–26 The content of –COOH increases after adding KMnO4 during the conversion of PAN to PAO. It is speculated that KMnO4 works as an oxidant in the solution, and MnO2 is its product. MnO2 can catalyze the transformation of –CN into –C(NH2)N–OH, and promote the hydrolysis of –CN to –COOH in the presence of NaOH.18
The XPS N 1s6,16 spectra of KMnO4@PAO and PAO (Fig. 3C, Table S3†) can be deconvoluted into three species at 399.5 ± 0.1 eV, 400.0 ± 0.1 eV, and 401.6 ± 0.1 eV, which were attributed to N–H, H2N–CNOH, and –N+, respectively. As shown in Table S3,† the content of H2N–CNOH in KMnO4@PAO is significantly higher than that in PAO. The results of XPS C 1s and N 1s spectra reveal that more PAN was converted into PAO by KMnO4@PAO. The XPS O 1s6,23 spectra of KMnO4@PAO and PAO could be deconvoluted into three peaks of –COOH at 581.8 ± 0.1 eV, CO at 532.8 ± 0.1 eV, and –OH at 533.5 ± 0.1 eV (Fig. 3D, Table S4†). The identical XPS results of PAO and KMnO4@PAO indicate that their chemical bonding environments are similar. The introduced KMnO4 has no significant effect on the chemical environments of PAO.
U(VI) adsorption capability
Solution pH influences adsorption behavior by affecting the existing forms of U(VI) in solution and the surface charge of the adsorbent. Fig. 4A exhibits the adsorption capacities and zeta potentials of KMnO4@PAO at different pH values.23,30,31 The adsorption capacity gradually increases with increasing pH to 5, and then gradually decreased with further increasing pH. The KMnO4@PAO surface is positively charged due to protonation at pH < 5, and U(VI) is predominantly present as positively charged free UO22+. The strong electrostatic repulsion between KMnO4@PAO and UO22+ results in low adsorption capacity. With increasing solution pH, the protonation level of the KMnO4@PAO surface gradually decreases, which benefits U(VI) adsorption. At pH > 5.0, negatively charged U(VI) species (such as UO2(OH)3−) appear, and the KMnO4@PAO surface becomes negatively charged. Again, the electrostatic repulsion results in low adsorption capacity.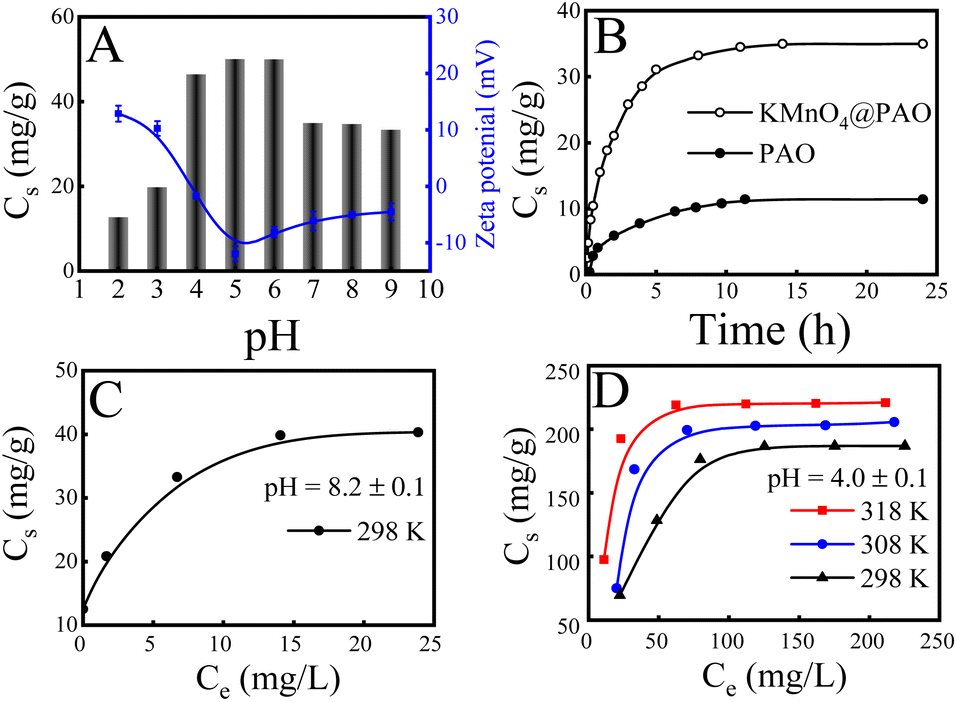 | ||
| Fig. 4 Effect of pH on zeta potential of KMnO4@PAO for U(VI) (A); effect of pH (A) and contact time on adsorption capability of KMnO4@PAO for U(VI) (B); adsorption isotherms of U(VI) on KMnO4@PAO and on PAO at pH = 8.2 ± 0.1 (C) and at pH = 4.0 ± 0.1 (D). m/V = 0.4 g L−1, I = 0.1 mol per L NaCl. (A) T = 298 ± 1 K, contact time: 24 h, C[U(VI)]initial = 20 mg L−1, pH = 8.2 ± 0.1. (B) T = 298 ± 1 K, C[U(VI)]initial = 20 mg L−1, pH = 8.2 ± 0.1. (C) T = 298 K ± 1 K, contact time: 24 h, pH = 8.2 ± 0.1. (D) Contact time: 24 h, pH = 4.0 ± 0.1. | ||
The effect of contact time on the extraction of U(VI) by KMnO4@PAO is also studied, and the result is shown in Fig. 4B. Due to the abundant adsorption sites, the adsorption of U(VI) on KMnO4@PAO rapidly increases with increasing contact time in the initial 5 h. The adsorption sites are gradually exhausted with the progress of the reaction, and the adsorption reaction slowly reaches an equilibrium. To deeply understand the adsorption process, pseudo first order and pseudo second order kinetic models4,6 are used to analyse kinetic adsorption data, and the relevant results are shown in Fig. S1 and Table S5.† According to the correlation coefficients (R2), the pseudo second order model is more suitable to describe the adsorption of U(VI) on the KMnO4@PAO surface, indicating that the enrichment of U(VI) on the KMnO4@PAO surface is a chemisorption process. From the pseudo second order model fitting results, the maximum adsorption capacities of U(VI) on KMnO4@PAO were 38.4 mg g−1 at pH 8.2 and 298 K, about 3 times that of PAO (13.1 mg g−1) under the same conditions. Results show that KMnO4 not only inhibits PAO agglomeration but also improves PAO adsorption capability. It further enhances the competitiveness of PAO as a candidate for U(VI) extraction from seawater.
The maximum adsorption capacity of KMnO4@PAO for U(VI) was explored by the adsorption isotherm technique at pH 8.2 and 298 K (Fig. 4C), and the adsorption data were analysed by commonly used Langmuir and Freundlich models (Fig. S2A†). According to the R2 values in Table S6,† the Langmuir model was more suitable to describe the adsorption process than the Freundlich model, demonstrating that adsorbed U(VI) was chemisorbed and formed a single layer on the KMnO4@PAO surface.26–28 After the addition of KMnO4, the adsorption capacity is significantly increased. This means that except PAO, the manganese related compounds in KMnO4@PAO can also recover U(VI), which can significantly solidify U(VI) in acidic solutions.37,38 To further expand the application scope of KMnO4@PAO, we explored the adsorption of KMnO4@PAO at pH 4. Experimental results show that KMnO4@PAO has good curing ability for U(VI), which benefits the application of PAO-based materials in wastewater management. To evaluate the maximum adsorption capacity (Cs,max) and the related thermodynamic parameters of KMnO4@PAO for U(VI) in acidic solutions, the adsorption isotherms of U(VI) on KMnO4@PAO were obtained at pH 4.0 and 298–318 K (Fig. 4D). The adsorption capacity of KMnO4@PAO for U(VI) gradually increases with increasing C[U(VI)]initial and solution temperature. The Cs,max value of KMnO4@PAO is 233 mg g−1 at 298 K and pH 4, which is competitive to hot materials (Table 1).
The experimental data were also fitted by the commonly used Langmuir and Freundlich models (Fig. S2B†), and the obtained fitting parameters are shown in Table S7.† The R2 values indicate that the Langmuir model again better describes the adsorption process of U(VI) on KMnO4@PAO in acidic solutions than the Freundlich model. Determining thermodynamic parameters (standard enthalpy change ΔH°, standard Gibbs free energy change ΔG°, and entropy change ΔS°) is very important for the effective evaluation of adsorption properties.36 The thermodynamic parameters for U(VI) adsorption on KMnO4@PAO are calculated and shown in Table S8.† The positive ΔH° value, positive ΔS° value, and negative ΔG° value indicate that U(VI) adsorption on thr KMnO4@PAO surface is a spontaneous and endothermic process. The positive ΔH° value reveals that increasing reaction temperature is conducive to the adsorption reaction, which is consistent with macroscopic experimental results.
Repeatability and stability are essential indices to evaluate the availability of the adsorbent. To screen suitable eluants for KMnO4@PAO, the effect of eluant type (0.1 mol L−1) on U(VI) desorption from the KMnO4@PAO surface was investigated. As shown in Fig. S3,† Na2CO3 shows a particularly prominent elution effect on U(VI) among the studied eluants (NaCO3, NaHCO3, NaOH, HCl, and HNO3).25 Na2CO3 is often used to elute U(VI), and the elution rates can reach 80–100%.5,11,13,19 So Na2CO3 is selected as the eluant in the cyclic experiment. U(VI) elution increases to ∼100% on increasing C[Na2CO3] to 0.5 mol L−1, and then the high level is retained (Fig. 5A). So, 0.5 mol per L Na2CO3 is selected for the following elution process. As shown in Fig. 5B, the adsorption capability KMnO4@PAO for U(VI) only decreases ∼6.1% even after 6 cycles.
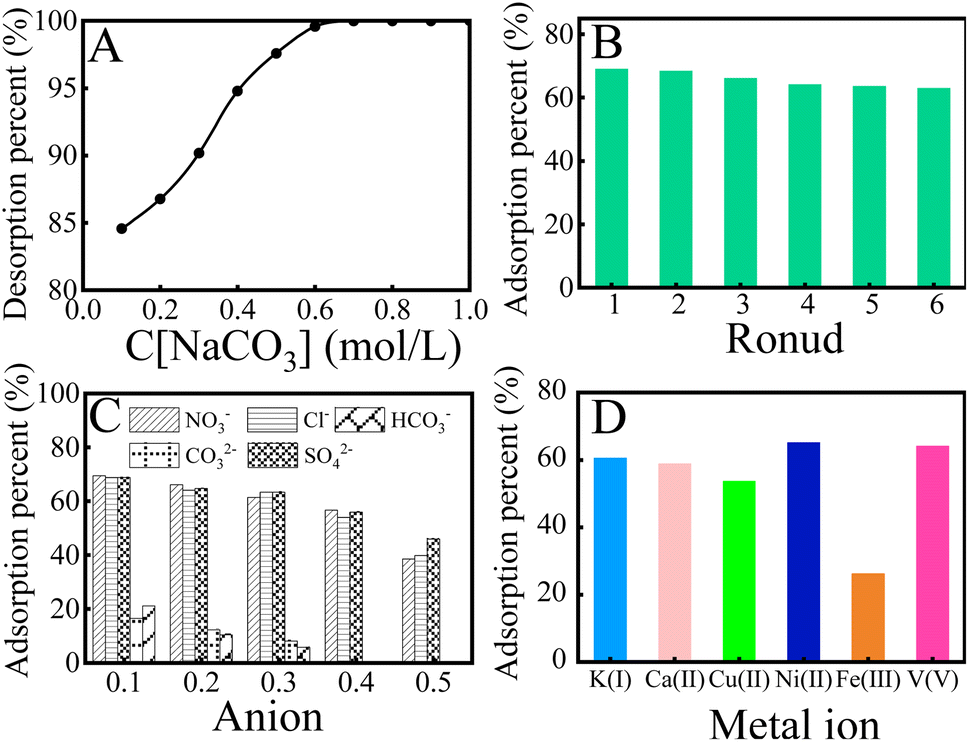 | ||
| Fig. 5 Effect of Na2CO3 concentration on the desorption of U(VI) from KMnO4@PAO (A); recycling application of KMnO4@PAO in U(VI) extraction (B); effect of anions (C) and cations (D) on the adsorption capability of KMnO4@PAO for U(VI). T = 298 ± 1 K, contact time: 24 h, C[U(VI)]initial = 20 mg L−1, m/V = 0.40 g L−1, pH = 8.2 ± 0.1. (A), (B) and (D): I = 0.1 mol per L NaCl. | ||
Seawater is a very complex ecosystem, and all known elements and compounds on earth can be found in seawater. These elements and compounds affect the recovery of U(VI) from seawater. The influence of major cations and anions in seawater on U(VI) recovery from seawater was investigated. As shown in Fig. 5C, the effect of Cl−, NO3−, and SO42− concentrations on U(VI) recovery from seawater can be neglected, whereas the recovery of U(VI) by KMnO4@PAO gradually decreased with increasing HCO3−/CO32− concentration. It is well known that Ca(II)/Mg(II)–U(VI)–CO3 species are the main existing forms of U(VI) in seawater, and their dissociation is usually the determining step of U(VI) recovery.5,11 Luckily, CO32−/HCO3− concentration is stable and below 0.1 mol L−1 in natural seawater.38 Since abundant competitive cations co-existed in natural seawater, the selective adsorption capacity of KMnO4@PAO for U(VI) cannot be ignored.13 As depicted in Fig. 5D, the U(VI) adsorption on the KMnO4@PAO surface is much higher than that of other coexisting metals. In this work, KMnO4 was used to regulate the PAO morphology. And the formed Mn related compounds have an excellent recovery effect on U(VI).37–39 On this basis, KMnO4@PAO shows high recovery capability for U(VI) against competitive cations.
Based on the excellent adsorption capability of KMnO4@PAO for U(VI), the application of KMnO4@PAO in extraction of U(VI) from real seawater was studied. Natural seawater was used to prepare U(VI) containing solutions with different initial concentrations. As shown in Table 2, KMnO4@PAO can quantitatively take up ∼50% of mg L−1 levels U(VI) from seawater. Experimental results further confirm the high efficiency of KMnO4@PAO in extraction of U(VI) from seawater.
| C[U(VI)] (mg L−1) | Adsorption (%) | |
|---|---|---|
| Initial | Final | |
| 10 | 5.01 | 49.9 |
| 8 | 3.25 | 47.5 |
| 5 | 2.21 | 27.9 |
| 3 | 1.55 | 14.5 |
KMnO4 may undergo photolysis and reduction reactions in aqueous solution. The products of KMnO4 are depending on solution conditions when it is used as an oxidizing agent. Whatever KMnO4 decomposed or reduced during the reaction, the resulted products are finally combined into black adsorbent powder (KMnO4@PAO). Usually black objects are a heat absorber, and can effectively absorb visible light. The adsorption of U(VI) on PAO is a endothermic reaction, and the increased temperature can enhance the adsorption process. The final product black KMnO4@PAO can effectively absorb sunlight and increase its temperature. This benefits the adsorption of U(VI) on KMnO4@PAO surface.26,40,41
In summary, KMnO4@PAO can work as a light conversion material that can absorb a wide range of sunlight, and the increased solution temperature further improves U(VI) recovery efficiency. As shown in Fig. 6A, after irradiation for 2 h at 0.20 W cm−2, the surface temperature of KMnO4@PAO increased to ∼30 °C, indicating the high photo-thermal conversion properties of KMnO4@PAO. The photo-thermal conversion properties of KMnO4@PAO (0.40 g L−1) in simulated seawater were also studied. It is incredible that just after irradiation for 2 h at 0.20 W cm−2, the temperature of simulated seawater was increased to nearly ∼34 °C (Fig. 6B). KMnO4@PAO material can well perform the task of photo-thermal conversion, and significantly increases solution temperature. The effect of water sources (simulated seawater, ultrapure water, tap water, and river water) and light sources (simulated sunlight and infrared light) on the recovery efficiency of U(VI) by KMnO4@PAO was also evaluated. According to Fig. 6C, the recovery capabilities of KMnO4@PAO for U(VI) were improved under sunlight and near-infrared light irradiation. Meanwhile, the adsorption rate of U(VI) on KMnO4@PAO under external light irradiation is much faster than that under dark conditions, and adsorption equilibrium is achieved in 2 h (Fig. 6D).
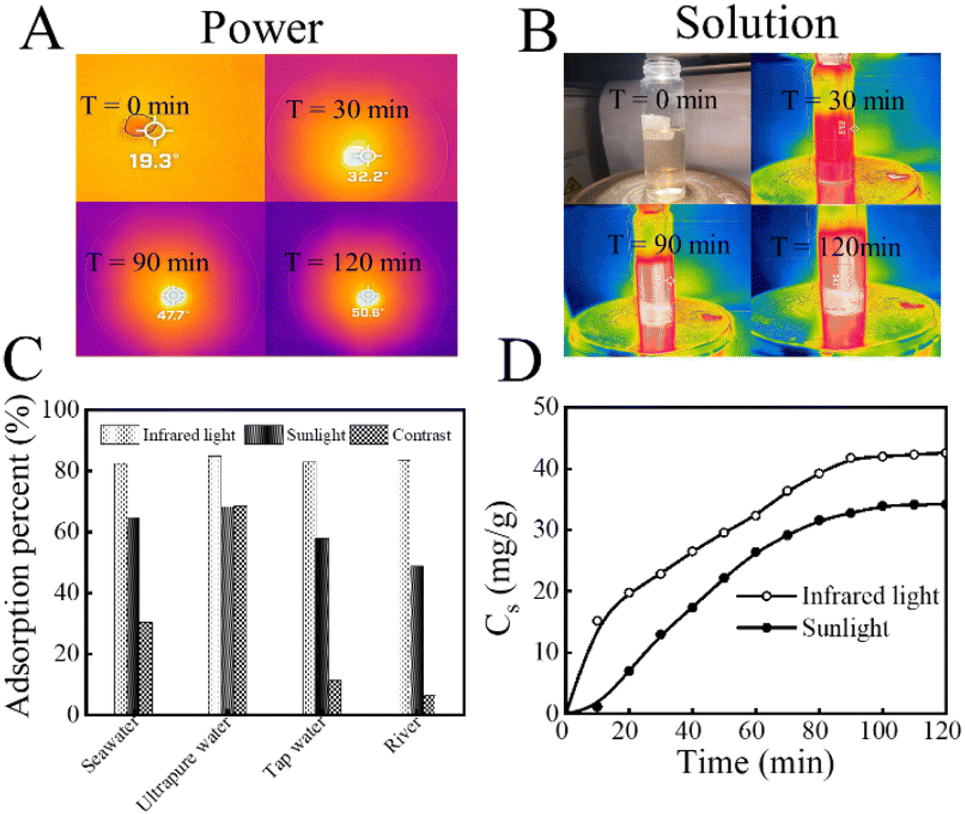 | ||
| Fig. 6 Infrared images of KMnO4@PAO powder (A) and 0.40 g per L KMnO4@PAO suspension (B) under simulated sunlight irradiation; adsorption capability (C) and kinetics of KMnO4@PAO for U(VI) under light irradiation (D). Light intensity: 0.20 W cm−2. T = 298 K ± 1 K, contact time: 24 h, C[U(VI)]initial = 20 mg L−1, m/V = 0.40 g L−1, pH = 8.2 ± 0.1, I = 0.1 mol per L NaCl. | ||
To further explore the influence of light and solution sources, the samples after the adsorption experiment were collected for FT-IR testing (Fig. 7). Experimental results show that the FT-IR spectra of KMnO4@PAO under different water sources and light sources are similar, which reveal the highly stable performance of KMnO4@PAO.
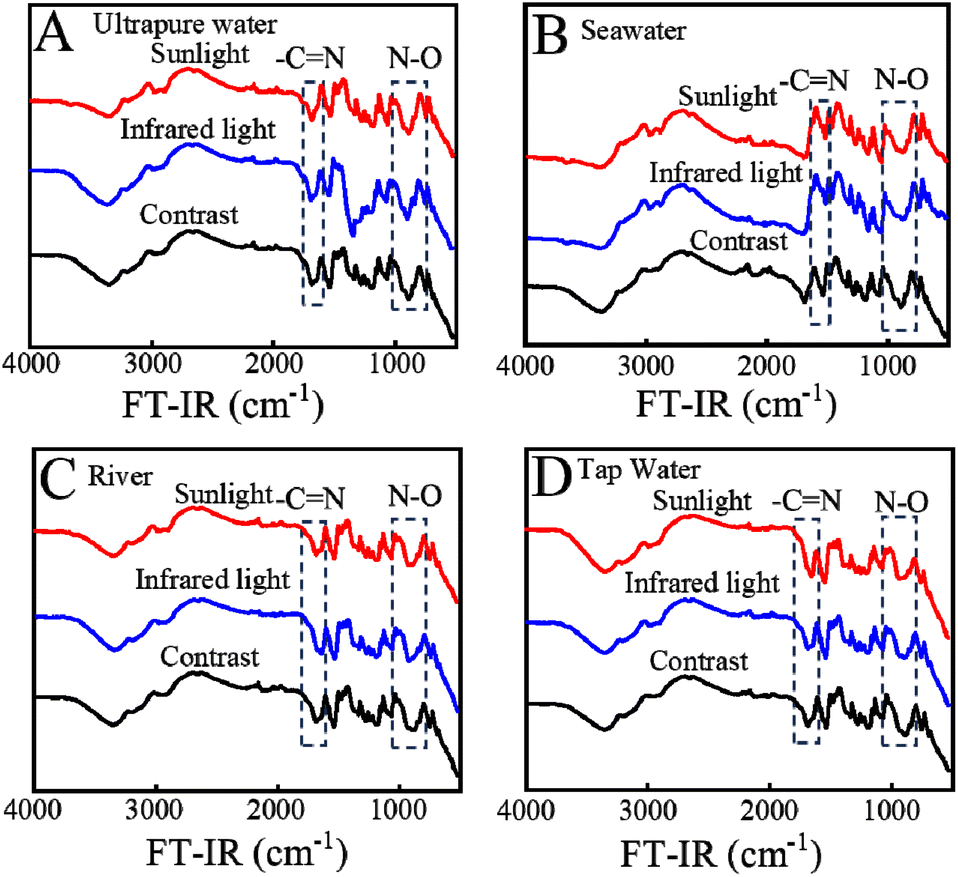 | ||
| Fig. 7 FT-IR spectra of U(VI) adsorbed by KMnO4@PAO under different illumination and solution conditions. | ||
The used KMnO4@PAO was collected to characterize morphology changes. As shown in Fig. 8A–D, KMnO4@PAO can well maintain its morphology after U(VI) adsorption. EDS-mapping shows that U(VI) was uniformly adsorbed on the KMnO4@PAO surface. The surface functional groups of PAO and KMnO4@PAO after U(VI) adsorption were characterized by XPS spectroscopy (Fig. 8E). The adsorbed U(VI) was evidenced by the new peak at 530.80 ± 0.1 eV (Fig. 8F) related to OUO,23 and the new peaks at ∼390 eV (Fig. 8G) were ascribed to to XPS U 4f.23 This reveals the high adsorption capabilities of PAO and KMnO4 for U(VI). The XPS Mn 2p spectrum of KMnO4@PAO can be deconvoluted into Mn 2p3/2 and Mn 2p1/2 peaks35,37–39 at 641.78 and 653.47 eV, respectively (Fig. 8H). This confirms the successful introduction of KMnO4 into PAO.
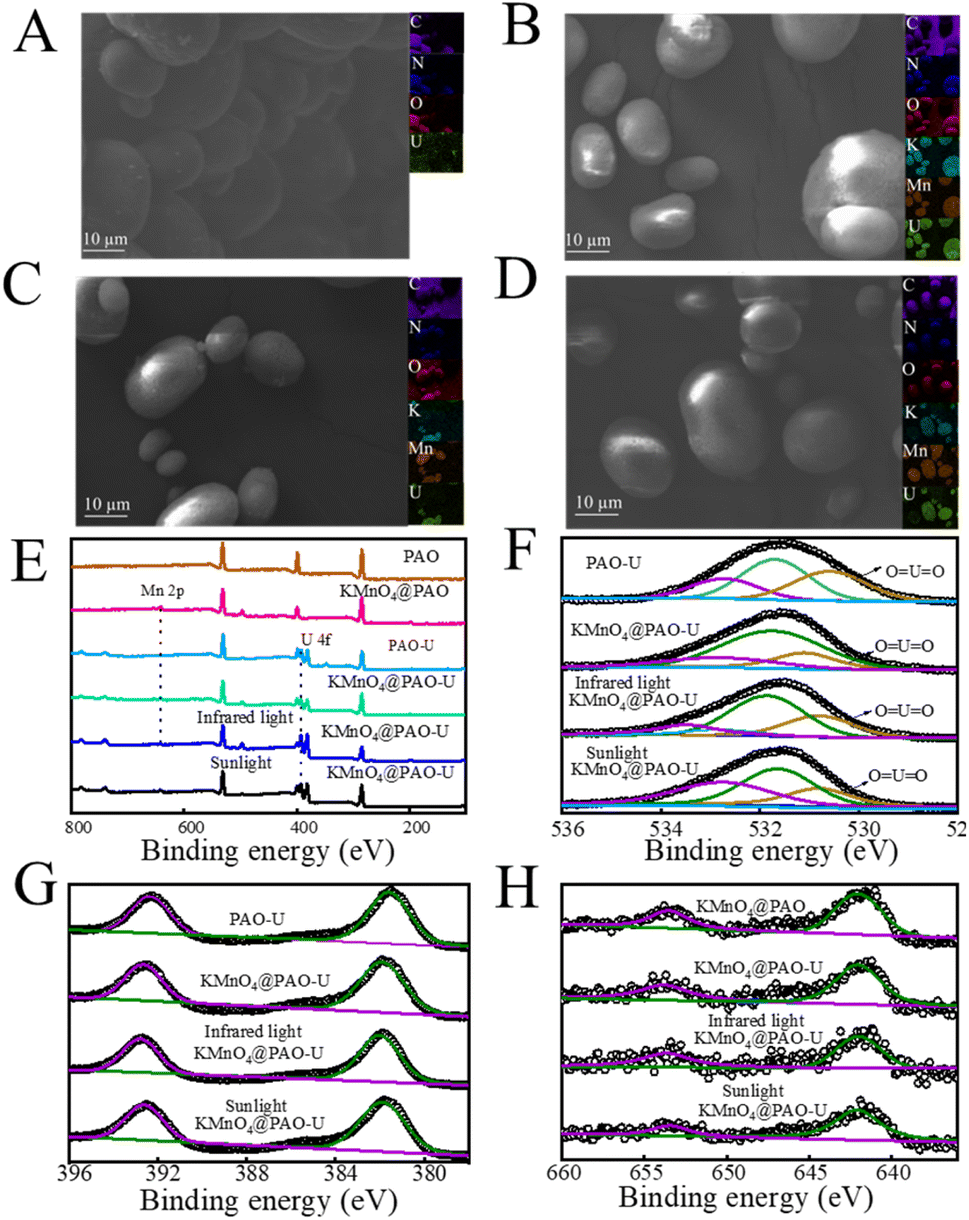 | ||
| Fig. 8 SEM images of PAO-U (A), KMnO4@PAO-U (B), infrared light KMnO4@PAO-U (C), and sunlight KMnO4@PAO-U (D). XPS survey spectra (E), O 1s (F), U 4f (G), and Mn 2p (H) of PAO-U and KMnO4@PAO. | ||
The introduced KMnO4 during the PAN amidoximation process can significantly decrease PAO size from 1100 nm to 400 nm (KMnO4@PAO), and solve the problem of PAO agglomeration. The introduced KMnO4 also endows KMnO4@PAO with photo-thermal conversion properties, and significantly increases the temperature of simulated seawater by ∼10 °C just by adding 0.4 g per L KMnO4@PAO under light irradiation conditions (2 h, 0.20 W cm−2). Given the excellent photo-thermal conversion efficiency of KMnO4@PAO, it is expected to be able to overcome seasonal and regional temperature differences and maintain high U(VI) recovery efficiency from seawater. The superb adsorption capability for U(VI), repeatability, and stability of KMnO4@PAO further highlight its application in extraction of U(VI) from seawater.
Conclusions
Experimental section
000 rpm for 10 min and filtered by using a 0.22 μm membrane filter. The final U(VI) concentrations (mg L−1 and μg L−1 levels) in supernatants were measured on an Optima 2100 DV inductively coupled plasma (ICP) atomic emission spectroscopy system (PerkinElmer) and on an ICP mass spectroscopy (ICP-MS, Thermo Scientific X-Series II) system, respectively.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge financial support from the National Natural Science Foundation of China (21976089, 22176098, and 22206081).Notes and references
- B. K. Sovacool, P. Schmid, A. Stirling, G. Walter and G. M. Kerron, Nat. Energy, 2020, 5, 928–935 CrossRef.
- A. Singh, S. Lal, N. Kumar, R. Yadav and S. Kumari, Environ. Sci. Pollut. Res., 2023, 30, 46185–46203 CrossRef CAS PubMed.
- L. Zhu, C. Zhang, F. Qin, F. Ma, C. Bi, R. Zhu, L. Liu, J. W. Bai, H. Dong and T. Satoh, J. Mol. Liq., 2022, 368, 120741 CrossRef CAS.
- J. Yu, H. Zhang, Q. Liu, J. Zhu, J. Yu, G. Sun, R. Li and J. Wang, J. Hazard. Mater., 2022, 440, 129735 CrossRef CAS.
- H. B. Pan, C. M. Wai, L. J. Kuo, G. Gill, G. Tian, L. Rao, S. Das, R. T. Mayes and C. J. Janke, ChemistrySelect, 2017, 2, 3769–3774 CrossRef CAS.
- J. Zhu, Y. Luo, J. Wang, J. Yu, Q. Liu, J. Liu, R. Chen, P. Liu and J. Wang, J. Mol. Liq., 2022, 368, 120744 CrossRef CAS.
- A. S. Ivanov, C. J. Leggett, B. F. Parker, Z. Zhang, J. Arnold, S. Dai, C. W. Abney, V. S. Bryantsev and L. Rao, Nat. Commun., 2017, 8, 1560 CrossRef PubMed.
- I. Tokarev and E. Yakovlev, Water, 2021, 13, 3514 CrossRef CAS.
- P. Ilaiyaraja, A. K. S. Deb, K. Sivasubramanian, D. Ponraju and B. Venkatraman, J. Hazard. Mater., 2013, 250, 155–166 CrossRef PubMed.
- C. W. Abney, R. T. Mayes, T. Saito and S. Dai, Chem. Rev., 2017, 117, 13935–14013 CrossRef CAS PubMed.
- H. B. Pan, L. J. Kuo, C. M. Wai, N. Miyamoto, R. Joshi, J. R. Wood, J. E. Strivens, C. J. Janke, Y. Oyola, S. Das, R. T. Mayes and G. A. Gill, Ind. Eng. Chem. Res., 2016, 55, 4313–4320 CrossRef CAS.
- T. Liu, X. Zhang, H. Wang, M. Chen, Y. Yuan, R. Zhang, Z. Xie, Y. Liu, H. Zhang and N. Wang, Chem. Eng. J., 2021, 412, 128700 CrossRef CAS.
- H. B. Pan, L. J. Kuo, C. M. Wai, N. Miyamoto, R. Joshi, J. R. Wood, J. E. Strivens, C. J. Janke, Y. Oyola, S. Das, R. T. Mayes and G. A. Gill, Ind. Eng. Chem. Res., 2016, 55, 4313–4320 CrossRef CAS.
- Z. Wang, Q. Meng, R. Ma, Z. Wang, Y. Yang, H. Sha, X. Ma, X. Ruan, X. Zou, Y. Yuan and G. Zhu, Chem, 2020, 6, 1683–1691 CAS.
- Y. Wang, Z. Lin, Q. Liu, J. Zhu, J. Liu, J. Yu, R. Chen, P. Liu and J. Wang, Chem. Eng. J., 2021, 425, 131538 CrossRef CAS.
- C. Bi, C. Zhang, W. Xu, F. Ma, L. Zhu, R. Zhu, Q. Qi, L. Liu, J. Bai and H. Dong, Desalination, 2023, 545, 116169 CrossRef CAS.
- Y. Wang, Z. Lin, J. Yu, J. Zhu, J. Liu, Q. Liu, R. Chen, P. Liu and J. Wang, J. Environ. Chem. Eng., 2023, 11, 109277 CrossRef CAS.
- H. B. Pan, C. M. Wai, L. J. Kuo, G. A. Gill, J. S. Wang, R. Joshi and C. J. Janke, Dalton Trans., 2020, 49, 2803–2810 RSC.
- H. B. Pan, L. J. Kuo, J. Wood, J. Strivens, G. A. Gill, C. J. Janke and C. M. Wai, RSC Adv., 2015, 5, 100715–100721 RSC.
- Q. Chen, X. Xue, Y. Liu, A. Guo, K. Chen, J. Yin, F. Yu, H. Zhu and X. Guo, J. Hazard. Mater., 2022, 438, 129524 CrossRef CAS PubMed.
- X. Guo, Y. Wang, X. Li, P. Huai and G. Wu, Mol. Phys., 2015, 113, 1327–1336 CrossRef CAS.
- J. Kim, C. Tsouris, R. T. Mayes, Y. Oyola, T. Saito, C. J. Janke, S. Dai, E. Schneider and D. Sachde, Sep. Sci. Technol., 2013, 48, 367–387 CrossRef CAS.
- D. Zhang, L. Liu, B. Zhao, X. Wang, H. Pang and S. Yu, Environ. Pollut., 2023, 317, 120826 CrossRef CAS PubMed.
- G. A. Gill, L. J. Kuo, C. J. Janke, J. Park, R. T. Jeters, G. T. Bonheyo, H. B. Pan, C. Wai, T. Khangaonkar, L. Bianucci, J. R. Wood, G. W. Marvin, S. Peterson, G. Abrecht, R. T. Mayes, C. Tsouris, Y. Oyola, J. E. Strivens, N. J. Schlafer, R. S. Addleman, W. Chouyyok, S. Das, J. Kim, K. Buesseler, C. Breier and E. D. Alessandro, Ind. Eng. Chem. Res., 2016, 55, 4264–4277 CrossRef CAS.
- L. J. Kuo, H. B. Pan, C. M. Wai, M. F. Byers, E. Schneider, J. E. Strivens, C. J. Janke, S. Das, R. T. Mayes, J. R. Wood, N. Schlafer and G. A. Gill, Ind. Eng. Chem. Res., 2017, 56, 11603–11611 CrossRef CAS.
- L. J. Kuo, G. A. Gill, C. Tsouris, L. Rao, H. B. Pan, C. M. Wai, C. J. Janke, J. E. Strivens, J. R. Wood, N. Schlafer and E. K. D. Alessandro, ChemistrySelect, 2018, 3, 843–848 CrossRef CAS.
- Z. Mi, D. Zhang, J. Wang, S. Bi, J. Liu, X. Gao, D. Zhang, Y. Jiang, Z. Li, Y. Zhu and Z. Liu, New J. Chem., 2022, 46, 6296–6306 RSC.
- C. C. Wan, R. Yu, L. Meng, D. Wang, T. Duan and L. Li, Desalination, 2020, 486, 114447 CrossRef.
- S. Shi, R. Wu, S. Meng, G. Xiao, C. Ma, G. Yang and N. Wang, J. Hazard. Mater., 2022, 436, 128983 CrossRef CAS PubMed.
- S. Lu, L. Chen, M. F. Hamza, C. He, X. Wang, Y. Wei and E. Guibal, Chem. Eng. J., 2019, 368, 459–473 CrossRef CAS.
- T. Liu, R. Zhang, M. Chen, Yi. Liu, Z. Xie, S. Tang, Y. Yuan and N. Wang, Adv. Funct. Mater., 2022, 32, 211104 Search PubMed.
- Y. Yue, B. Wang, X. Xu, D. Cai, L. Zhang, S. Hu, J. Xiao, T. Yang, D. Wang and H. Wu, Mater. Today Chem., 2022, 26, 101090 CrossRef CAS.
- C. Han, Y. Yue, X. Xu, D. Cai, Z. Liu, S. Chen, L. Luo, J. Xiao and D. Wang, New J. Chem., 2020, 44, 19445–19449 RSC.
- H. Heshmati, H. G. Gilani, M. T. Mostaedi and A. Haidary, J. Dispersion Sci. Technol., 2014, 35, 501–509 CrossRef CAS.
- S. Wang, G. Yuan, J. Yang, J. Bai, G. Wang and J. Yan, ChemSusChem, 2022, 15, 1–13 Search PubMed.
- D. Shao, G. Hou, F. Chi, X. Lu and X. Ren, RSC Adv., 2021, 11, 1909–1915 RSC.
- X. Li, J. Huang, Z. Shi, Y. Xie, Z. Xu, J. Long, G. Song, Y. Wang, G. Zhang, X. Luo, P. Zhang, S. Zha and H. Li, J. Environ. Manage., 2023, 342, 118088 CrossRef CAS PubMed.
- X. Zhang, L. Zhang, Y. Liu, M. Li, X. Wu, T. Jiang, C. Chen and Y. Peng, Environ. Pollut., 2020, 262, 114841 Search PubMed.
- H. Zhou, J. Yu, S. Liu, L. Wang and P. Li, Mater. Lett., 2023, 333, 133652 CrossRef CAS.
- Z. Guo, Y. Liu, Y. Zhang, X. Sun, F. Li, T. Bu, Q. Wang and L. Wang, Biomater. Sci., 2020, 8, 4266 RSC.
- M. Wen, X. Liu, N. Yu, P. Qiu, D. K. Macharia, M. Li, H. Zhang, Z. Chen and W. Lian, J. Colloid Interface Sci., 2022, 626, 77–88 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3va00179b |
| This journal is © The Royal Society of Chemistry 2024 |