
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn artificial cathode-electrolyte interphase enabling one-step sulfur transition in polyethylene oxide-based solid-state lithium–sulfur batteries†
Leicheng
Zhang‡
a,
Tianshuai
Wang‡
a,
Junjie
Chen‡
a,
Maochun
Wu
 *c and
Tianshou
Zhao
*ab
*c and
Tianshou
Zhao
*ab
aDepartment of Mechanical and Aerospace Engineering, The Hong Kong University of Science and Technology, Clear Water Bay, Kowloon, Hong Kong SAR, China. E-mail: zhaots@sustech.edu.cn
bDepartment of Mechanical and Energy Engineering, Southern University of Science and Technology, Shenzhen, 518055, China
cDepartment of Mechanical Engineering, The Hong Kong Polytechnic University, Hung Hom, Kowloon, Hong Kong SAR, China. E-mail: maochun.wu@polyu.edu.hk
First published on 15th August 2024
Abstract
All-solid-state lithium–sulfur (Li–S) batteries using polyethylene oxide (PEO)-based electrolytes hold the advantages of high theoretical energy density, cost-effectiveness, and high safety. However, the drawback of polysulfide dissolution in PEO results in a short battery lifespan. Here, we propose to construct an artificial cathode-electrolyte interphase (CEI) on the S cathode, which converts the S speciation pathway to a one-step solid transition, significantly mitigating the polysulfide migration in PEO. Surface analyses and theoretical calculations reveal the composition of the CEI and its effect on the reaction mechanism. As a result, the all-solid-state Li–S cell with the artificial CEI is able to deliver 873 mA h g−1 at 100 mA g−1 and maintain 739 mA h g−1 after 50 cycles, whereas the cell using the pristine S cathode retains only 364 mA h g−1. More remarkably, the artificial CEI enables the cell to achieve a high capacity retention rate of 83.1% at 300 mA g−1 over 200 cycles, demonstrating that our strategy of CEI manipulation effectively enhances the cycling reversibility of PEO-based solid-state Li–S batteries.
1. Introduction
The global electric vehicle and energy storage markets have been growing exponentially over the past few years, which has simultaneously boosted the vast consumption of power and energy-storage batteries.1–3 So far, lithium-ion batteries (LIBs) composed of graphite anodes and transition-metal oxide cathodes (e.g., LiFePO4 and LiCoO2) have dominated the battery market.4,5 However, with years of research and development by both industry and academia, the energy densities of LIBs have been progressively pushed to the theoretical limits, which cannot further meet the ever-growing demand for longer-lasting batteries with higher energy densities. Hence, lithium–sulfur (Li–S) batteries have received wide research attention because of the high capacity, non-toxicity, and low cost of S.6–9Nevertheless, Li–S batteries face several critical challenges, which hamper their industrial application. First, S undergoes a solid–liquid–solid multi-step transition to the final discharge product Li2S in conventional ether electrolytes. The intermediate polysulfides generated during the transition can easily dissolve in the ether electrolytes and shuttle between the Li anode and S cathode, triggering a series of side reactions, which result in a low coulombic efficiency and quick capacity fade.10,11 Moreover, the highly reductive Li metal may spontaneously react with the electrolyte solvents and Li salts, lowering the coulombic efficiency. Even worse, the possible formation of Li dendrites will lead to short-circuiting and bring about serious safety concerns.12–14
To date, numerous strategies have been proposed to tackle the above issues. Among them, the use of solid-state electrolytes (SSEs) in Li–S batteries is believed to simultaneously resolve both the shuttle effect and Li dendrite formation due to their dense structure, high modulus, and superior thermal stability.15–17 Solid-state Li–S batteries can be categorized based on the type of SSE used, which determines whether the S undergoes a one- or multi-step speciation pathway.17 With high ionic conductivity and excellent mechanical strength, inorganic SSEs such as oxides and sulfides have been widely studied in Li–S batteries.18–23 For instance, S-coated reduced graphene oxide (rGO@S) was homogeneously mixed with Li10GeP2S12 and conductive carbon, which ensured the percolation of the conductive pathway throughout the cathode.21 S underwent a one-step solid–solid conversion to the final discharge product without generating high-order polysulfides. Compared to sulfides, oxide SSEs usually show much better chemical stability and air/moisture tolerance. For example, a garnet SSE was fabricated with a bilayer structure, in which the dense layer against the Li metal inhibited dendrite formation, while the porous layer served as a mechanical support and host for S.20 It should be noted that the poor interfacial contact between electrolytes and S remains a critical challenge in solid-state Li–S batteries using oxide SSEs. One facile approach is to add a small amount of liquid electrolyte to facilitate ion conduction, which, however, results in the generation of soluble polysulfides, as evidenced by multiple voltage plateaus. Although the dense oxide SSEs can completely block the polysulfide migration to the Li anode, the use of liquid electrolytes still brings about the risks of leakage and combustion, thus compromising the advantages of solid-state batteries.17
By contrast, organic polymer electrolytes stand out as rational alternatives to inorganic ones due to their inherent good elasticity and flexibility, which result in intimate interfacial contact with electrodes.24–26 Polyethylene oxide (PEO)-based solid polymer electrolytes are primarily investigated in solid-state Li–S batteries owing to their superior ionic conductivity, feasible processibility, and low cost.27–33 More importantly, the repeated oxyethylene (EO) units show strong interaction with Li+, which endows the PEO with excellent solvation capability towards Li salts.17,26 Nonetheless, high-order polysulfides can also be generated and dissolved in PEO like in liquid ether electrolytes, resulting in a severe shuttle effect and loss of active materials.27,31 Recently, it has been reported that a compact cathode-electrolyte interphase (CEI) derived from advanced electrolytes could enable the direct one-step conversion of S,14,34–39 inspiring us to manipulate the S conversion chemistry and bypass the polysulfide dissolution in PEO.
In this contribution, we develop an artificial CEI on a S cathode, which enables a one-step solid–solid S transition in a PEO-based all-solid-state Li–S battery (ASSLSB). Experimental and calculational results show that by pre-cycling the S cathode in a concentrated liquid electrolyte, a compact LiF-rich CEI can be formed due to the prior decomposition of Li+-anion contact ion pairs and aggregates. The CEI isolates the active material from direct contact with PEO and prevents the cohesion of polysulfides, which radically suppresses their dissolution in PEO (Fig. 1). As a result, the PEO-based Li–S cell with the artificial CEI delivers a high capacity of 873 mA h g−1 initially and maintains 739 mA h g−1 after 50 cycles, while the pristine cell retains only 41.7% of its original capacity. More remarkably, the artificial CEI enables the all-solid-state Li–S cell to achieve a high capacity retention rate of 83.1% after 200 cycles at 300 mA g−1.
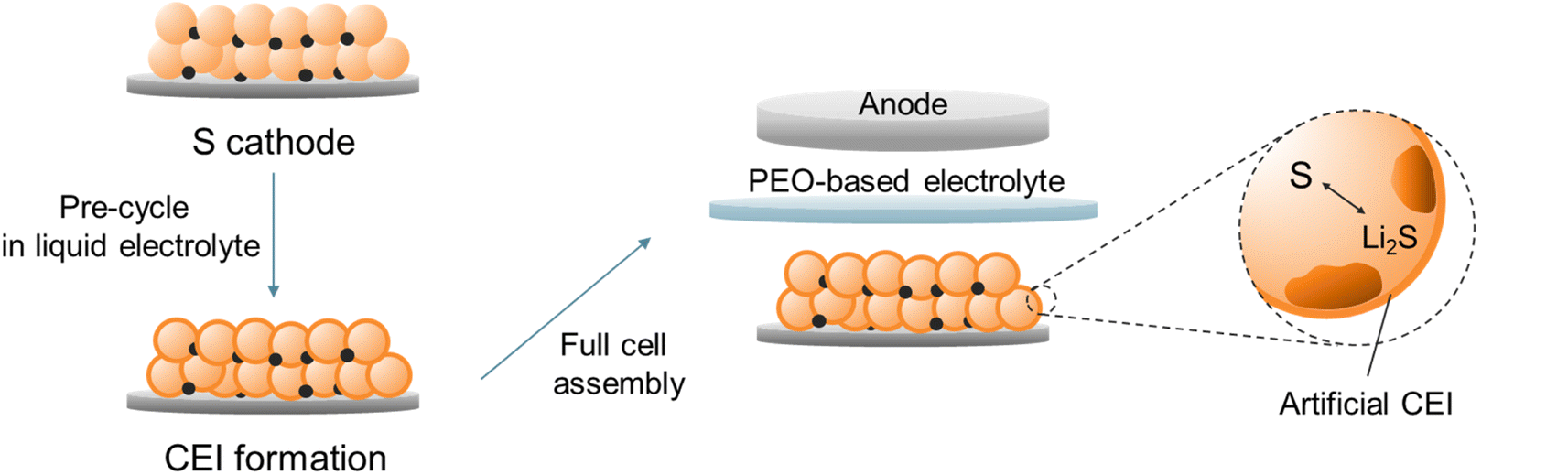 | ||
| Fig. 1 Schematic illustration of the artificial CEI formation on the S cathode and reassembly of the PEO-based all-solid-state Li–S battery. | ||
2. Results and discussion
2.1. Electrochemical characterization of S cathodes in different liquid electrolytes
The artificial CEI was formed by simply pre-cycling the S cathodes in sacrificial cells using Li metal and concentrated 12 M lithium bis(fluoro sulphonyl)imide (LiFSI) in 1,2-dimethoxyethane (DME). The electrochemical behavior of the S cathode using conventional diluted 1 M lithium bis(trifluoromethane sulphonyl)imide (LiTFSI) in DME/1,3-dioxolane (DOL) was also characterized for comparison. As shown in Fig. 2a, S undergoes a typical “solid–liquid–solid” transition in the diluted ether electrolyte with the formation of highly soluble Li2Sn (n = 4, 6, 8) as intermediates.10 When operated in the concentrated electrolyte, the first discharge plateau corresponding to the polysulfide generation is significantly suppressed and gradually diminished after several cycles (Fig. 2b), which is attributed to the limited amount of free DME solvents that can hardly further solvate the polysulfides.37 The subsequent long-sloped plateau is ascribed to the direct conversion of S to the final Li2S, while the descending trend can be explained by the increasing internal resistance.17 In a dilute electrolyte, sulfur undergoes a transformation into polysulfides, and the dissolution of polysulfides in the electrolyte results in a yellow coloration, as shown in the digital photo in Fig. 2a. In contrast, in a concentrated electrolyte (Fig. 2b), the dissolution of polysulfides is inhibited, and sulfur directly undergoes a one-step conversion into Li2S. Therefore, the color of the electrolyte remains unchanged. As shown in Fig. 2c, the cell using diluted electrolyte displays two distinct reduction peaks in the cyclic voltammetry (CV) curve, which are ascribed to the two-step transition of S. By contrast, the first reduction peak is largely suppressed when the concentrated electrolyte is used (Fig. 2d). The galvanostatic intermittent titration technique (GITT) was performed to unveil the reaction mechanism of S in different electrolytes.40 As shown in Fig. 2e, the open-circuit voltage (OCV) during discharge in diluted electrolyte exhibits two-plateau regions, wherein the voltage polarization of the liquid phase transition (Li2S8 to Li2S4) is much lower than that of the final solid phase transition (Li2S2 to Li2S). In contrast, the discharge OCV with the concentrated electrolyte only shows one plateau of ∼2.0 V, and the voltage polarization remains almost constant over the entire process, indicating a single-phase transition (Fig. 2f). These results demonstrate the distinct conversion pathways of S dictated by the different electrolyte compositions.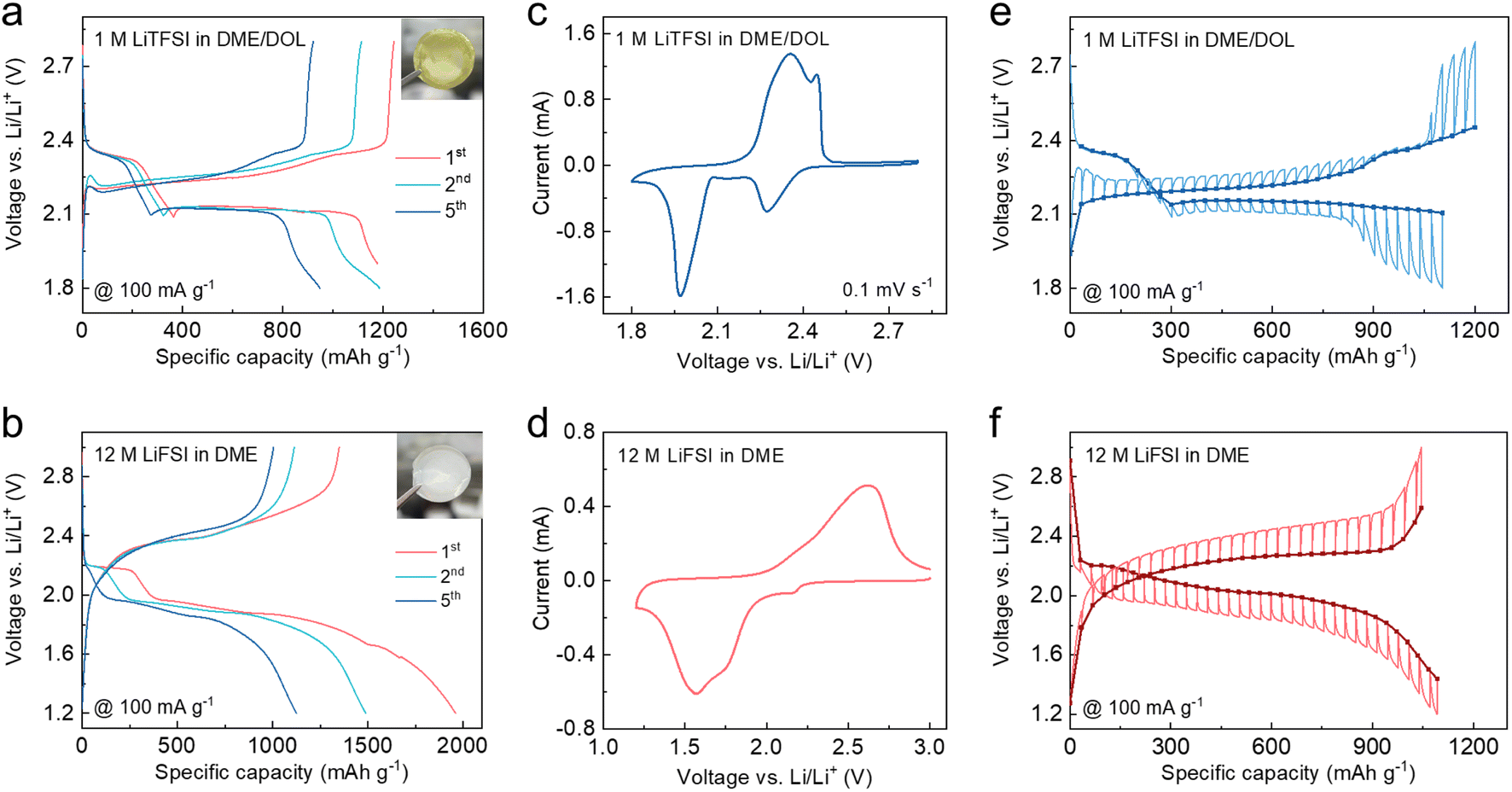 | ||
| Fig. 2 (a and b) Galvanostatic voltage profiles ((insets): digital photos of separators retrieved from cycled cells), (c and d) CV curves at a scan rate of 0.1 mV s−1, and (e and f) GITT curves of Li–S cells using (a, c and e) 1 M LiTFSI in DME/DOL and (b, d and f) 12 M LiFSI in DME. | ||
2.2. CEI characterization of cycled S cathodes
To reveal the mechanism underlying the distinct reaction pathways, X-ray photoelectron spectroscopy (XPS) with Ar+ sputtering was performed on the cycled S cathodes to investigate the composition of the CEI formed in different electrolytes. As depicted in Fig. 3a and S1,† the intensity of C 1s (representative of organic compounds) decreases along with the etching depth, while that of Li 1s and F 1s (representative of inorganic compounds) increases in both diluted and concentrated electrolytes, which well matches the two-sublayer SEI architecture as established in previous studies.41–43 Notably, the organic components of the CEI formed in the concentrated electrolyte are much less than those in its diluted counterpart, which can be attributed to the suppressed solvent decomposition as a result of limited free DME molecules.37,44 In contrast, the content of LiF in the CEI formed in the concentrated electrolyte is much higher than that in the diluted electrolyte throughout the etching depth, indicating that the decomposition of FSI− anions mainly contributes to the CEI formation in the concentrated electrolyte (Fig. 3b). The dense LiF-rich CEI layer is believed to isolate the active S from the solvation of solvent molecules, thereby enabling the direct transition of S to the final discharge product after several activation cycles.14,34 Time-of-flight secondary ion mass spectroscopy (ToF-SIMS) also provided the ESI† CEI, as shown in Fig. S2.† For the concentrated electrolyte, the content of C2HO− (representing the organic components) is less than that of the diluted electrolyte; the LiF2− content (representing the inorganic components) is higher than that of the diluted electrolyte, and the content increases with the depth. This shows that the CEI generated in the concentrated electrolyte has lower organic components and higher inorganic components than that in the diluted electrolyte, which is consistent with the results from XPS tests.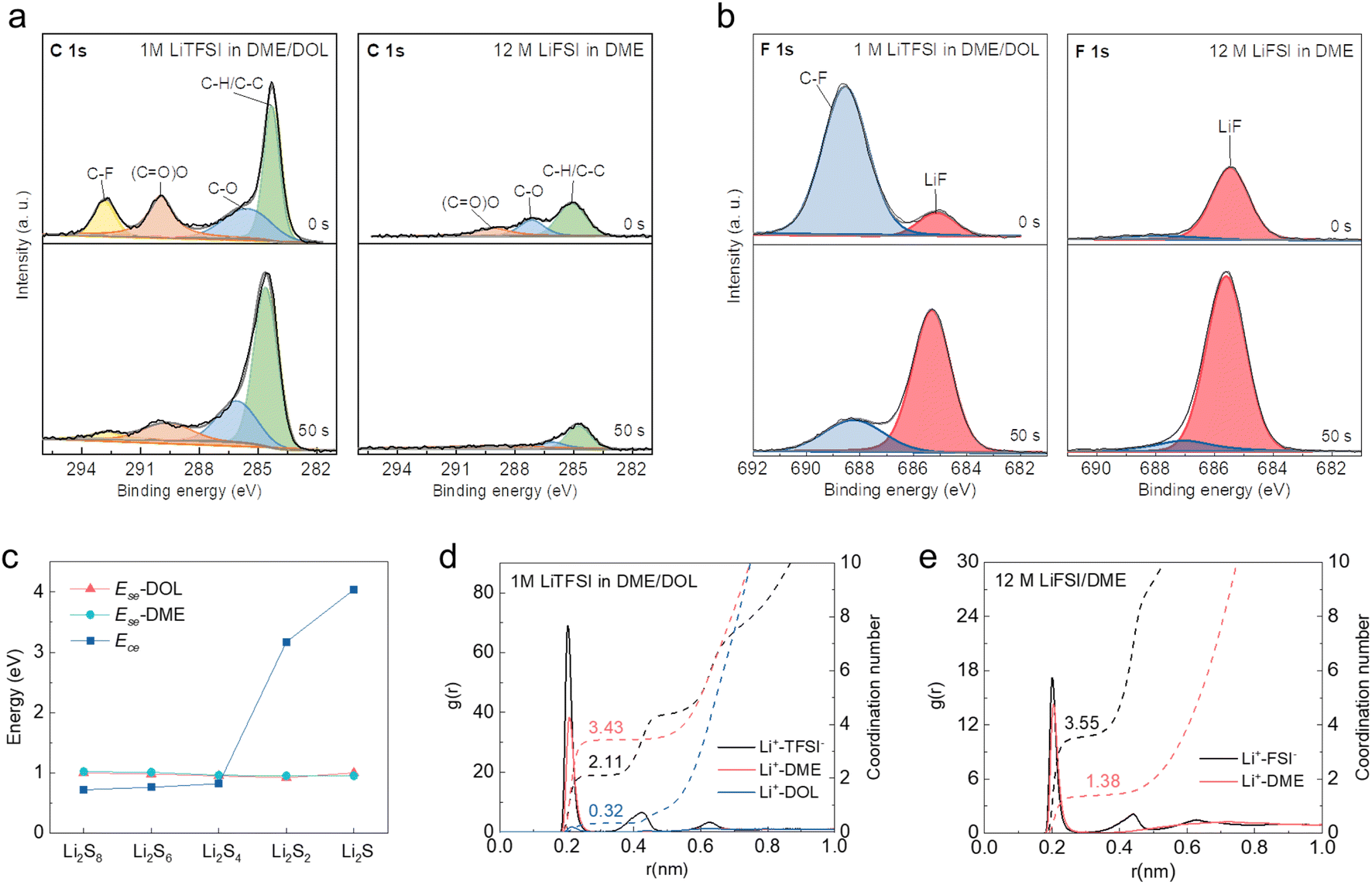 | ||
| Fig. 3 High resolution (a) C 1s and (b) F 1s spectra of S cathodes cycled in 1 M LiTFSI in DME/DOL and 12 M LiFSI in DME at the etching times of 0 and 50 s. (c) Ece of polysulfides and their Ese with DME and DOL. Coordination number distribution of (d) 1 M LiTFSI in DME/DOL and (e) 12 M LiFSI in DME. | ||
To gain a deeper understanding of the S reaction mechanism in two different electrolytes, theoretical calculations were performed on the cohesive energies (Ece) and solvation energies (Ese) of polysulfides in DME and DOL solvents. Their competitive solvation potential (ΔP = Ece − Ese) dictates whether the polysulfides can be dissolved in the electrolyte solvents. As shown in Fig. 3c, the cohesive energies of high-order Li2Sn (n ≥ 4) are lower than their solvation energies in DME and DOL with only single Li–O interaction (Fig. S3†), and the Ese increases with the number of solvent molecules (Fig. S4†), which indicates the soluble properties of high-order polysulfides. In contrast, Li2S and Li2S2 show an intrinsic solid nature based on the calculation results. The different solubility of polysulfides results in the solid–liquid–solid transition of S in the diluted electrolyte with sufficient free solvent molecules. However, the dramatic reduction of free solvents can suppress polysulfide dissolution. Molecule dynamics simulations based on GROMACS were further performed. For the system of 1 M LiTFSI in DME/DOL, the primary solvation shell of Li+ is around 0.2 nm, which is composed of Li+-TFSI−, Li+-DME, and Li+-DOL pairs (Fig. 3d). Coordination number distribution functions reveal that there are 0.32 DOL and 3.43 DME molecules in the primary solvation shell (PSS) of Li+, with many free DME and DOL molecules remaining in the electrolyte. In sharp contrast, there are only 1.43 DME molecules in the PSS of Li+ in 12 M LiFSI in DME, which is insufficient compared to the number of Li+ ions (Fig. 3e). The solvent molecules are effectively trapped in the salt solvation sheaths at such a high concentration, leaving few free DME molecules available to further solvate polysulfides. Moreover, the solvation structure is converted from the solvated-separated ion pairs in diluted electrolytes to contact ion pairs and aggregates in the concentrated electrolyte system, resulting in the prior decomposition of FSI− anions to form a LiF-rich CEI, which is consistent with the XPS results.
2.3. Electrochemical characterization of the S@CEI cathode in a PEO-based ASSLSB
To explore whether the CEI formed in the concentrated electrolyte can preserve the functionality of converting the S reaction pathway in solid-state cells, the pre-cycled S cathodes with the CEI (S@CEI) were retrieved and paired with PEO-based composite electrolytes and Li metal in coin cells. The composite polymer electrolyte comprises a polyvinylidene difluoride (PVDF) scaffold filled with PEO and garnet nanoparticles (PPG, Fig. S5†).45 Meanwhile, solid-state cells using a pristine S cathode with the PEO binder (S/PEO) and a S cathode pre-cycled in 1 M LiTFSI in DME/DOL were also prepared for comparison. All cell tests were conducted at 60 °C to facilitate ion conduction in solid-state cells. Firstly, we conducted EIS tests to characterize the influence of the interface impendence from the CEI layer (as shown in Fig. S6†). It can be observed that by forming an artificial CEI on the surface of the S cathode before assembling the PEO-based all-solid-state battery, the interface resistance is significantly reduced compared to the battery without the CEI layer. This indicates that the pre-formed CEI effectively forms an ion pathway, facilitating good contact between the cathode and the PEO electrolyte.As shown in Fig. 4a, the discharge voltage profile of the Li | PPG | S/PEO cell exhibits two plateaus similar to those in the diluted electrolyte. This is because the EO units of PEO possess strong solvation ability towards Li+ salts, which results in the dissolution of polysulfides in the PEO and the subsequent two-step transition.27,31 The solid-state cell using the S cathode pre-cycled in the diluted electrolyte also exhibits similar two-plateau discharge behavior but with an enlarged overpotential (Fig. S7†), which indicates that the CEI formed in the conventional electrolyte cannot change the reaction pathway of S in the solid-state cell. In sharp contrast, the voltage profile of the cell using the S@CEI cathode shows one long-sloped plateau identical to that of the cell using concentrated electrolyte (Fig. 4b), suggesting a direct transition between S and Li2S. The reduction peak at ∼2.4 V of the CV curve corresponds to the polysulfide generation in the cell using the S/PEO cathode (Fig. 4c), while no peak above 2.2 V is observed in the S@CEI cell (Fig. 4d), which demonstrates that the LiF-rich CEI thoroughly blocks the dissolution of polysulfides in the PEO. As shown in Fig. S8a and b,† the changes in voltage polarization in the GITT curves of both kinds of cells present analogical trends to those in their liquid counterparts, which again confirms the distinct conversion mechanisms of S attributed to the CEI manipulation strategy.
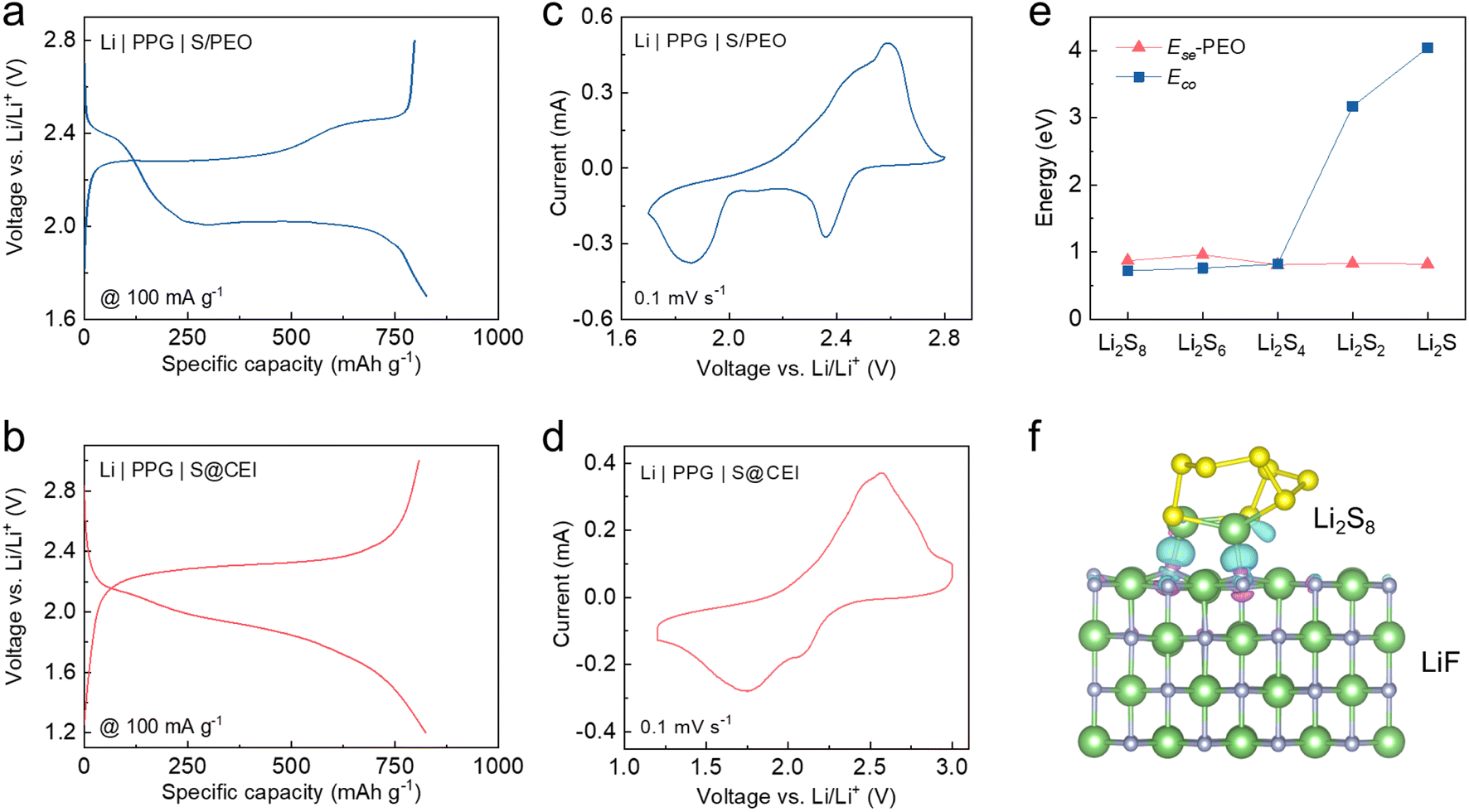 | ||
| Fig. 4 (a and b) Galvanostatic voltage profiles and (c and d) CV curves at a scan rate of 0.1 mV s−1 of (a and c) Li | PPG | S/PEO cell and (b and d) Li | PPG | S@CEI cell at 60 °C. (e) Ece of polysulfides and their Ese with PEO. (f) Charge density difference between LiF and Li2S8. The light blue and purple sections represent the electron accumulation and loss parts, respectively. | ||
The Ece and Ese of polysulfides in the PEO system were also calculated. As shown in Fig. 4e and S9,† the stronger Li2Sn (n ≥ 4)–PEO binding drives the dissolution of high-order polysulfides in PEO like in the diluted ether electrolytes, which explains the similar two-plateau discharge behavior of the S/PEO cathode. For the S@CEI cathode, the compact LiF-rich CEI can isolate the S from direct contact with PEO, which prevents the polysulfide solvation. Additionally, the Ece of Li2Sn (n ≥ 4) is lower than their adsorption energies on the (001) surface of LiF (Fig. S10†), and the charge density difference of LiF–Li2S8 in Fig. 4f shows that F can bond with Li+. These results suggest that the LiF surface can suppress the aggregation of high-order polysulfides in the adsorbed state due to the negative ΔP value, which contributes to the direct solid–solid transition of S to Li2S/Li2S2.
2.4. Electrochemical performance of the Li | PPG | S@CEI cell
To demonstrate the possibility for practical application, cycling stability tests were conducted on the coin cells using the S@CEI cathode paired with PPG and Li metal. As shown in Fig. 5a, the cell achieves a high initial reversible capacity of 873 mA h g−1 and maintains 739 mA h g−1 after 50 cycles, corresponding to a retention rate of 84.7%. In contrast, the cell using the S/PEO cathode only maintains 41.7% of its original capacity after 50 cycles, which can be attributed to the severe irreversible polysulfide migration in the PEO.27 The voltage profiles of the S@CEI cell in Fig. 5b show identical long-sloped plateaus over 50 cycles, indicating the good stability of the CEI, which sustains the direct conversion of S without polysulfide migration. Fig. 5c and d show the discharge capacities and voltage profiles of the Li | PPG | S@CEI cell at varying current densities. The battery can deliver capacities of 870, 682, 345, and 247 mA h g−1 at 0.1, 0.2, 0.5, and 1.0 A g−1, respectively. When the current turns back to 0.2 A g−1, 625 mA h g−1 is recovered, suggesting excellent rate capability. Fig. 5e shows the long-term cycling test of the Li | PPG | S@CEI cell at a current density of 0.3 A g−1. Even after 200 cycles, the capacity only drops from 679 to 564 mA h g−1, corresponding to a retention rate of 83.1%, which suggests that the artificial CEI effectively improves the cycling stability of the S cathode in the PEO-based polymer electrolyte. Moreover, as shown in Fig. S13 and Table S1,† within PEO-based Li–S batteries reported in the literature, the artificial CEI contributes outstanding performance in terms of cycle number, retention, mass loading of sulfur, and specific capacity.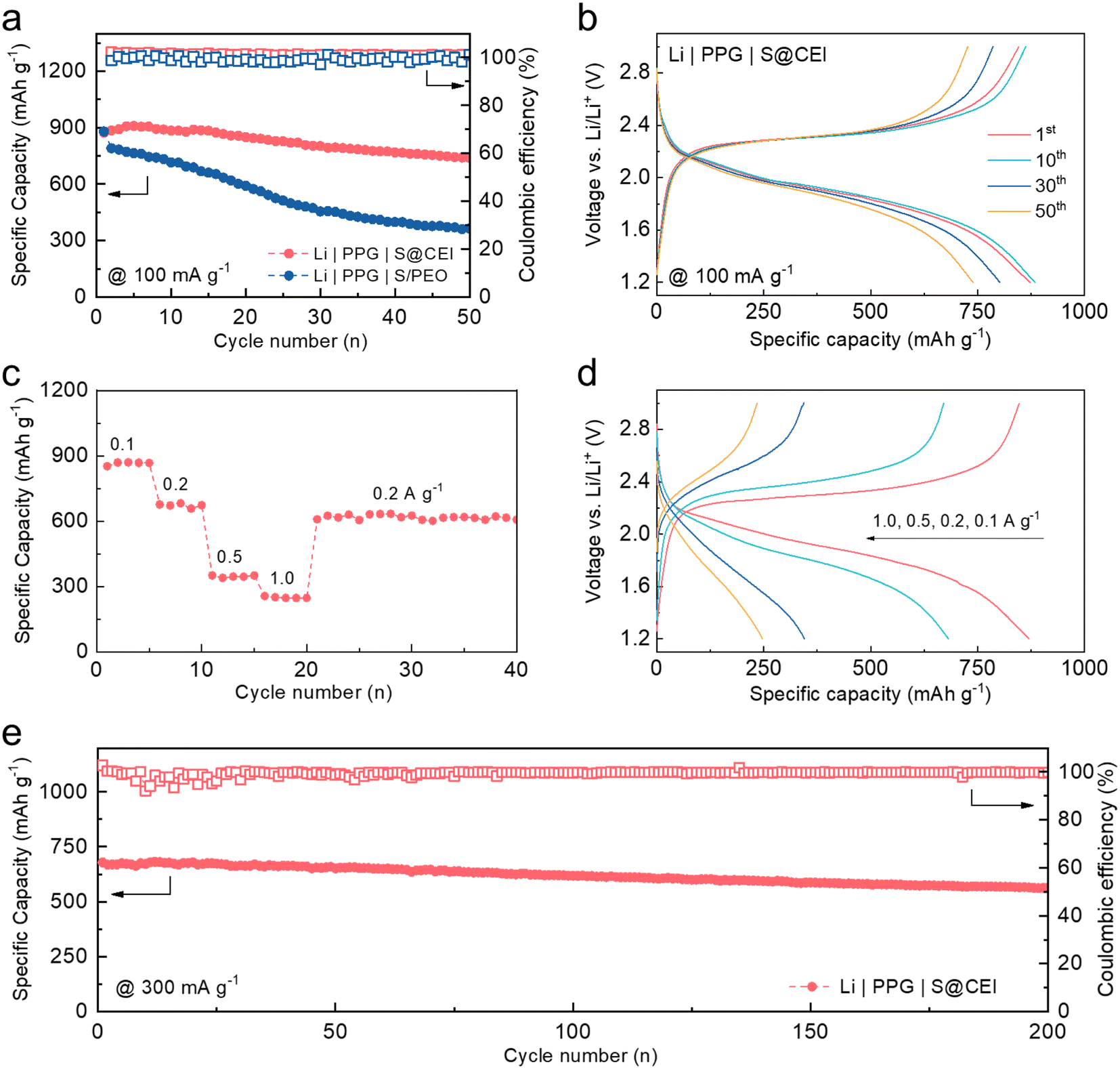 | ||
| Fig. 5 (a) Discharge capacities of Li | PPG | S@CEI and Li | PPG | S/PEO cells at 100 mA g−1 for 50 cycles. (b) Representative voltage profiles of the Li | PPG | S@CEI cell. (c) Discharge capacities and (d) voltage profiles of the Li | PPG | S@CEI cell at varying current densities. (e) Long-term cycling performance of the Li | PPG | S@CEI cell at 300 mA g−1. All cell tests were conducted at 60 °C. | ||
3. Conclusion
In summary, a facile CEI manipulation strategy is developed for solid-state Li–S batteries by pre-cycling a S cathode in a concentrated LiFSI in DME electrolyte. XPS and theoretical calculation results reveal that the LiF-rich CEI can suppress the aggregation of high-order polysulfides and prevent their dissolution in PEO, thus enabling the one-step transition of S to the final discharge product. Consequently, the Li | PPG | S@CEI cell achieves a high initial reversible capacity of 873 mA h g−1 and maintains 739 mA h g−1 after 50 cycles. More remarkably, the cell with the artificial CEI achieves a high capacity retention rate of 83.1% over 200 cycles, indicating excellent stability of the artificial CEI. Our strategy of CEI manipulation significantly contributes to the development of long-term cycling PEO-based ASSLSBs.4. Experimental section
4.1. Preparation of the artificial CEI on the S cathode
The S powder and mesoporous carbon (Aladdin, China) were mixed in a mass ratio of 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 and kept at 155 °C for 12 h in a Teflon-sealed autoclave filled with Ar gas. Then S/mesoporous C, polyvinylidene fluoride (PVDF) binder, and Super P were mixed in a mass ratio of 8:1:1 in N-methyl-2-pyrrolidone (NMP), which was then cast on carbon-coated aluminum foil to obtain the S cathode. The electrode was dried at 80 °C under vacuum for two days to remove the solvent completely, and the S loading was measured to be around 0.8 mg cm−2.
:3 and kept at 155 °C for 12 h in a Teflon-sealed autoclave filled with Ar gas. Then S/mesoporous C, polyvinylidene fluoride (PVDF) binder, and Super P were mixed in a mass ratio of 8:1:1 in N-methyl-2-pyrrolidone (NMP), which was then cast on carbon-coated aluminum foil to obtain the S cathode. The electrode was dried at 80 °C under vacuum for two days to remove the solvent completely, and the S loading was measured to be around 0.8 mg cm−2.
To form the artificial CEI, sacrificial cells were assembled using Li foil, Celgard 2400 separators, and S cathodes with the addition of 12 M lithium bis(fluoro sulphonyl)imide (LiFSI) in 1,2-dimethoxyethane (DME), or 1 M lithium bis(trifluoromethane sulphonyl)imide (LiTFSI) in DME/1,3-dioxolane (DOL) (v/v, 1:1) in an Ar-filled glove box. The “M” herein refers to the mole of salts dissolved in a liter of solvent (not the electrolyte solution). After galvanostatic discharge and charge for 7 cycles within the voltage window of 1.2–3 V (in 12 M LiFSI in DME) or 1.8–2.8 V (in 1 M LiTFSI in DME/DOL), the S@CEI cathodes were fully charged and retrieved from the disassembled cells, which were then thoroughly rinsed with DME solvents and vacuum dried at 45 °C overnight.
4.2. Preparation of PPG solid electrolyte
The PPG was prepared following a method reported by our group previously.45 The polyvinylidene difluoride (PVDF) fiber network was fabricated by electrospinning. 200 mg PEO (Mw = 600000), 100 mg LiTFSI, and 40 mg LLZTO nanoparticles (200–400 nm) were uniformly mixed in 5 mL acetonitrile by ultrasonication and then stirred at 60 °C overnight. Then, the PEO/LiTFSI/garnet solution was infiltrated into the PVDF network placed on a polytetrafluoroethylene (PTFE) plate, followed by evaporation at room temperature and then under vacuum at 60 °C for 48 h to remove the solvent completely. Finally, the free-standing PPG was peeled off the PTFE plate for cell assembly.
4.3. Electrochemical measurements
To prepare the S cathode with the PEO binder (S/PEO), S@mesoporous C, Super P, PEO, and LiTFSI were mixed in a mass ratio of 8:1:0.75:0.25 in acetonitrile, which was then cast on carbon-coated aluminum foil, followed by evaporation at room temperature and then at 65 °C under vacuum overnight. To assemble the all-solid-state Li–S cells, the S@CEI or S/PEO cathodes were paired with PPG and Li foil in CR2032 coin cells in an Ar-filled glove box. The cycling tests were conducted at 60 °C within the voltage window of 1.2–3.0 V (Li | PPG | S@CEI) or 1.7–2.8 V (Li | PPG | S/PEO) using a Neware CT-4008W testing system.
CV tests were performed on an electrochemical workstation (Biologic, SP 200) with a scan rate of 0.1 mV s−1. The GITT was also conducted on the electrochemical workstation by discharging/charging the cell at 100 mA g−1 for 20 min and then holding it for 2 h repeatedly until it reached its lower/upper cut-off voltage.
4.4. Calculation methods
To obtain the two kinds of polysulfides, including molecular phases as Li2Sn (n = 1, 2, 4, 6, 8) clusters and the solid phase of Li2S2, structure prediction was performed using crystal structure analysis by particle swarm optimization.46 For each polysulfide configuration, more than 10000 structures were generated and computed, while the structures with the lowest energy were extracted. Here, clusters were defined as monomers and dimers of the corresponding molecular phases and could be expressed as (Li2Sn)x (n = 1, 2, 4, 6, 8; x = 1, 2) (Fig. S11†). We have adopted this method to investigate the aggregated polysulfide phases and achieved convincing results.47
The formation energies (EFor) of polysulfides (Fig. S12†) were calculated as follows:
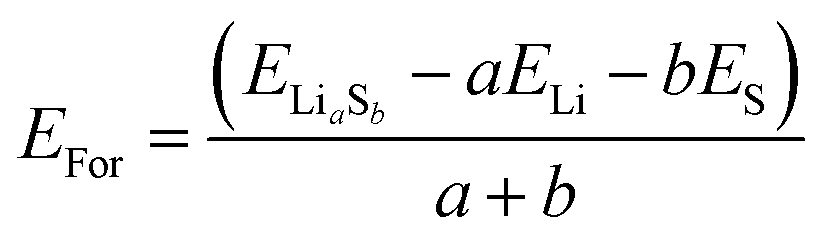 | (1) |
The cohesive energies of polysulfides were calculated using:
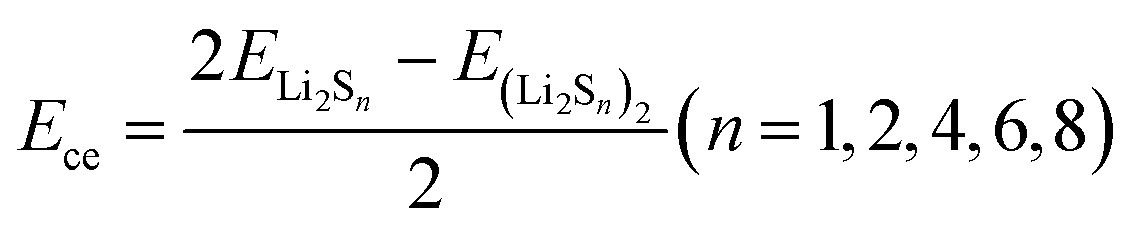 | (2) |
| Ese=(EElec + ELi2Sn) − EElec–Li2Sn (n = 1, 2, 4, 6, 8) | (3) |
Material dynamics (MD) simulations were performed using the GROMACS 2018.8 simulation package with amber force fields.48,49 We generated the atomic charges of TFSI−, FSI−, DME, and DOL by the restrained electrostatic potential fitting procedure (RESP) through Multiwfn software.50 The equilibrium and production simulations were calculated in the NPT ensemble at a constant pressure (1.01325 bar) and temperature (298.15 K) in a cubic box with periodic boundary conditions in all xyz Cartesian directions. For equilibrium processes, the temperature was maintained by V-rescale coupling with a time constant of 0.2 ps. The Berendsen barostat was adopted for controlling the pressure with a coupling constant of 0.5 ps. Equilibrium simulation ran for 2 ns. For production simulation, the V-rescale coupling and Parrinello–Rahman barostat were used to control the temperature and pressure, respectively. Production simulation ran for 10 ns (2 fs per step and simulating 5 × 106 steps) to obtain the equilibrium Li+ coordination shell structure at room temperature for electrolytes. Electrostatic interactions were treated using the Particle-Mesh-Ewald (PME) method.51 The coordination number of molecules of type i in the first solvation shell surrounding a single molecule of type j was calculated as:
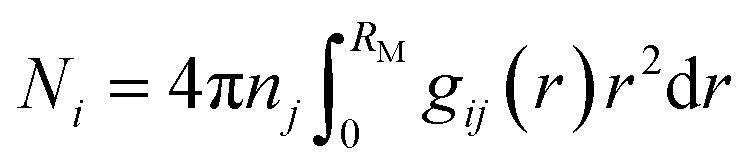 | (4) |
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work described in this paper was supported by a grant from the Research Grants Council of the Hong Kong Special Administrative Region, China (Project No. R6005-20) and Guangdong Major Project of Basic and Applied Basic Research (2023B0303000002).References
- M. A. Rajaeifar, P. Ghadimi, M. Raugei, Y. Wu and O. Heidrich, Resour., Conserv. Recycl., 2022, 180, 106144 CrossRef.
- J. Archsmith, E. Muehlegger and D. S. Rapson, Environ. Energy Pol. Econ., 2022, 3, 71–110 Search PubMed.
- N. D. Popovich, D. Rajagopal, E. Tasar and A. Phadke, Nat. Energy, 2021, 6, 1017–1025 CrossRef.
- Y. Yang, E. G. Okonkwo, G. Huang, S. Xu, W. Sun and Y. He, Energy Storage Mater., 2021, 36, 186–212 CrossRef.
- M. Weiss, R. Ruess, J. Kasnatscheew, Y. Levartovsky, N. R. Levy, P. Minnmann, L. Stolz, T. Waldmann, M. Wohlfahrt-Mehrens, D. Aurbach, M. Winter, Y. Ein-Eli and J. Janek, Adv. Energy Mater., 2021, 11, 2101126 CrossRef CAS.
- L. Zhou, D. L. Danilov, R. A. Eichel and P. H. L. Notten, Adv. Energy Mater., 2021, 11, 2001304 CrossRef CAS.
- Y. Zhang, X. Zhang, S. R. P. Silva, B. Ding, P. Zhang and G. Shao, Advanced Science, 2022, 9, 1–34 Search PubMed.
- C. Zhao, G. L. Xu, Z. Yu, L. Zhang, I. Hwang, Y. X. Mo, Y. Ren, L. Cheng, C. J. Sun, Y. Ren, X. Zuo, J. T. Li, S. G. Sun, K. Amine and T. Zhao, Nat. Nanotechnol., 2021, 16, 166–173 CrossRef CAS PubMed.
- L. Zhang, C. Zhao, Q. Jian, M. Wu and T. Zhao, J. Power Sources, 2021, 506, 230174 CrossRef CAS.
- F. Y. Fan, W. C. Carter and Y. M. Chiang, Adv. Mater., 2015, 27, 5203–5209 CrossRef CAS.
- H. Wang, Y. Shao, H. Pan, X. Feng, Y. Chen, Y. S. Liu, E. D. Walter, M. H. Engelhard, K. S. Han, T. Deng, G. Ren, D. Lu, X. Lu, W. Xu, C. Wang, J. Feng, K. T. Mueller, J. Guo, K. R. Zavadil and J. G. Zhang, Nano Energy, 2020, 76, 105041 CrossRef CAS.
- H. Hong, N. A. R. Che Mohamad, K. Chae, F. Marques Mota and D. H. Kim, J. Mater. Chem. A, 2021, 9, 10012–10038 RSC.
- Z. Yu, H. Wang, X. Kong, W. Huang, Y. Tsao, D. G. Mackanic, K. Wang, X. Wang, W. Huang, S. Choudhury, Y. Zheng, C. V. Amanchukwu, S. T. Hung, Y. Ma, E. G. Lomeli, J. Qin, Y. Cui and Z. Bao, Nat. Energy, 2020, 5, 526–533 CrossRef CAS.
- C. Zhao, G. L. Xu, T. Zhao and K. Amine, Angew. Chem., Int. Ed., 2020, 59, 17634–17640 CrossRef CAS PubMed.
- S. Li, W. Zhang, J. Zheng, M. Lv, H. Song and L. Du, Adv. Energy Mater., 2021, 11, 1–24 Search PubMed.
- W. Huang, J. Wang, M. R. Braun, Z. Zhang, Y. Li, D. T. Boyle, P. C. McIntyre and Y. Cui, Matter, 2019, 1, 1232–1245 CrossRef.
- H. Pan, Z. Cheng, P. He and H. Zhou, Energy Fuel., 2020, 34, 11942–11961 CrossRef CAS.
- S. A. Pervez, B. P. Vinayan, M. A. Cambaz, G. Melinte, T. Diemant, T. Braun, G. Karkera, R. J. Behm and M. Fichtner, J. Mater. Chem. A, 2020, 8, 16451–16462 RSC.
- Y. Lu, X. Huang, Z. Song, K. Rui, Q. Wang, S. Gu, J. Yang, T. Xiu, M. E. Badding and Z. Wen, Energy Storage Mater., 2018, 15, 282–290 CrossRef.
- K. Fu, Y. Gong, G. T. Hitz, D. W. McOwen, Y. Li, S. Xu, Y. Wen, L. Zhang, C. Wang, G. Pastel, J. Dai, B. Liu, H. Xie, Y. Yao, E. D. Wachsman and L. Hu, Energy Environ. Sci., 2017, 10, 1568–1575 RSC.
- X. Yao, N. Huang, F. Han, Q. Zhang, H. Wan, J. P. Mwizerwa, C. Wang and X. Xu, Adv. Energy Mater., 2017, 7, 1–9 Search PubMed.
- Y. Hao, S. Wang, F. Xu, Y. Liu, N. Feng, P. He and H. Zhou, ACS Appl. Mater. Interfaces, 2017, 9, 3375–33739 Search PubMed.
- R. Xu, Z. Wu, S. Zhang, X. Wang, Y. Xia, X. Xia, X. Huang and J. Tu, Chem.–Eur. J., 2017, 23, 13950–13956 CrossRef CAS.
- S. Tang, W. Guo and Y. Fu, Adv. Energy Mater., 2021, 11, 1–29 Search PubMed.
- X. Yang, K. R. Adair, X. Gao and X. Sun, Energy Environ. Sci., 2021, 14, 643–671 RSC.
- Z. Zou, Y. Li, Z. Lu, D. Wang, Y. Cui, B. Guo, Y. Li, X. Liang, J. Feng, H. Li, C. W. Nan, M. Armand, L. Chen, K. Xu and S. Shi, Chem. Rev., 2020, 120, 4169–4221 CrossRef CAS PubMed.
- R. Fang, H. Xu, B. Xu, X. Li, Y. Li and J. B. Goodenough, Adv. Funct. Mater., 2021, 31, 1–7 Search PubMed.
- H. Zhang, X. Judez, A. Santiago, M. Martinez-Ibañez, M. Á. Muñoz-Márquez, J. Carrasco, C. Li, G. G. Eshetu and M. Armand, Adv. Energy Mater., 2019, 9, 1–14 Search PubMed.
- G. G. Eshetu, X. Judez, C. Li, M. Martinez-Ibañez, I. Gracia, O. Bondarchuk, J. Carrasco, L. M. Rodriguez-Martinez, H. Zhang and M. Armand, J. Am. Chem. Soc., 2018, 140, 9921–9933 CrossRef CAS PubMed.
- L. Zhong, S. Wang, M. Xiao, W. Liu, D. Han, Z. Li, J. Qin, Y. Li, S. Zhang, S. Huang and Y. Meng, Energy Storage Mater., 2021, 41, 563–570 CrossRef.
- Y. X. Song, Y. Shi, J. Wan, S. Y. Lang, X. C. Hu, H. J. Yan, B. Liu, Y. G. Guo, R. Wen and L. J. Wan, Energy Environ. Sci., 2019, 12, 2496–2506 RSC.
- S. M. Liu, M. X. Chen, Y. Xie, D. H. Liu, J. F. Zheng, X. Xiong, H. Jiang, L. C. Wang, H. Luo and K. Han, Rare Met., 2023, 42, 2562–2576 CrossRef CAS.
- Z. C. Li, T. Y. Li, Y. R. Deng, W. H. Tang, X. D. Wang, J. L. Yang, Q. Liu, L. Zhang, Q. Wang and R. P. Liu, Rare Met., 2022, 41, 2834–2843 CrossRef CAS.
- G. L. Xu, H. Sun, C. Luo, L. Estevez, M. Zhuang, H. Gao, R. Amine, H. Wang, X. Zhang, C. J. Sun, Y. Liu, Y. Ren, S. M. Heald, C. Wang, Z. Chen and K. Amine, Adv. Energy Mater., 2019, 9, 1–14 Search PubMed.
- L. Zhang, C. Zhao, Y. Lin, M. Wu and T. Zhao, J. Mater. Chem. A, 2021, 9, 25426–25434 RSC.
- C. Zhao, A. Daali, I. Hwang, T. Li, X. Huang, D. Robertson, Z. Yang, S. Trask, W. Xu, C. C. Sun, G. G. Xu and K. Amine, Angew. Chem., Int. Ed., 2022, e202203466 CAS.
- J. Zheng, X. Fan, G. Ji, H. Wang, S. Hou, K. C. DeMella, S. R. Raghavan, J. Wang, K. Xu and C. Wang, Nano Energy, 2018, 50, 431–440 CrossRef CAS.
- J. Zheng, G. Ji, X. Fan, J. Chen, Q. Li, H. Wang, Y. Yang, K. C. DeMella, S. R. Raghavan and C. Wang, Adv. Energy Mater., 2019, 9, 1–9 Search PubMed.
- H. Duan, L. Li, X. Fu, Y. Deng and G. Chen, Chem. Eng. J., 2022, 450, 138208 CrossRef CAS.
- Y. Lu, X. Li, J. Liang, L. Hu, Y. Zhu and Y. Qian, Nanoscale, 2016, 8, 17616–17622 RSC.
- J. Chen, X. Fan, Q. Li, H. Yang, M. R. Khoshi, Y. Xu, S. Hwang, L. Chen, X. Ji, C. Yang, H. He, C. Wang, E. Garfunkel, D. Su, O. Borodin and C. Wang, Nat. Energy, 2020, 5, 386–397 CrossRef CAS.
- J. Zheng, J. Tian, D. Wu, M. Gu, W. Xu, C. Wang, F. Gao, M. H. Engelhard, J. G. Zhang, J. Liu and J. Xiao, Nano Lett., 2014, 14, 2345–2352 CrossRef CAS.
- E. Peled and S. Menkin, J. Electrochem. Soc., 2017, 164, A1703–A1719 CrossRef CAS.
- T. Li, X. Q. Zhang, P. Shi and Q. Zhang, Joule, 2019, 3, 1–15 CrossRef.
- Y. Lin, M. Wu, J. Sun, L. Zhang, Q. Jian and T. Zhao, Adv. Energy Mater., 2021, 2101612, 1–9 Search PubMed.
- Y. Wang, J. Lv, L. Zhu and Y. Ma, Comput. Phys. Commun., 2012, 183, 2063–2070 CrossRef CAS.
- J. Xiao, G. Zhou, H. Chen, X. Feng, D. Legut, Y. Fan, T. Wang, Y. Cui and Q. Zhang, Nano Lett., 2019, 19, 7487–7493 CrossRef CAS PubMed.
- H. J. C. Berendsen, J. P. M. Postma, W. F. Van Gunsteren, A. Dinola and J. R. Haak, J. Chem. Phys., 1984, 81, 3684–3690 CrossRef CAS.
- C. I. Bayly, K. M. Merz, D. M. Ferguson, W. D. Cornell, T. Fox, J. W. Caldwell, P. A. Kollman, P. Cieplak, I. R. Gould and D. C. Spellmeyer, J. Am. Chem. Soc., 1995, 117, 5179–5197 CrossRef.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS.
- T. Darden, D. York and L. Pedersen, J. Chem. Phys., 1993, 98, 10089–10092 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta02413c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |