
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Study of degradation mechanisms in aqueous-processed Ni-rich cathodes for enhanced sustainability of batteries†
Heyin
Chen
 *a,
Agnes-Matilda
Mattsson
a,
Laura
King
a,
Haidong
Liu
a,
Ida
Nielsen
a,
Tove
Ericson
a,
Alexei
Preobrajenski
b,
William R.
Brant
*a and
Maria
Hahlin
*ac
*a,
Agnes-Matilda
Mattsson
a,
Laura
King
a,
Haidong
Liu
a,
Ida
Nielsen
a,
Tove
Ericson
a,
Alexei
Preobrajenski
b,
William R.
Brant
*a and
Maria
Hahlin
*ac
aDepartment of Chemistry –Ångström Laboratory, Uppsala University, Box 538, 751 20 Uppsala, Sweden. E-mail: heyin.chen@kemi.uu.se
bMAX IV Laboratory, Lund University, Box 118, 221 00 Lund, Sweden
cDepartment of Physics and Astronomy, Uppsala University, Box 516, 75120 Uppsala, Sweden
First published on 27th August 2024
Abstract
Traditionally, Ni-rich-layered oxide cathodes for lithium-ion batteries are produced utilizing N-methyl-2-pyrrolidone (NMP)-processed casting. However, to avoid using the reprotoxic solvent NMP, aqueous processing becomes one of the options. In this study, H2O-processed LiNi0.8Mn0.1Co0.1O2 (NMC811) electrodes have been prepared to compare with the NMP-processed counterparts to investigate the degradation mechanism. The thick cathode–electrolyte interphase (CEI), NiO-like phase formation, and the growth of electrochemically inactive NMC particles after long-term cycling lead to capacity decay. In addition, phosphoric acid (H3PO4) was utilized to lower the pH value during the water-processed electrode preparation, to avoid corrosion of the aluminium current collector. The use of H3PO4 enhanced the capacity retention of NMC811 electrodes, likely owing to the formation of a LiF-rich CEI layer in the initial cycle(s) and the alleviated formation of electrochemically inactive NMC particles. Additionally, reaction inhomogeneity is present in H3PO4-modified electrodes, which is attributed to various Li-ion reinsertion resistances throughout the porous electrode during long-term cycling. Although the performance of the water-processed NMC811 electrode is not reaching the level of NMP-processed electrodes, this study provides key insights into the involved degradation mechanisms and demonstrates a viable pathway for the development of sustainable battery manufacturing processes.
Introduction
Recently, Ni-rich transition-metal oxides (LiNixMnyCozO2 Ni-rich NMC, x ≥ 0.7) have gained interest in the lithium-ion battery (LIB) field mainly due to their high specific capacities (up to 220 mA h g−1) and lower cost.1,2 The development of LiNi0.8Co0.1Mn0.1O2 (NMC811) and other Ni-rich layered oxides has allowed them to be utilized as cathode materials in electric vehicle LIBs.3 Traditionally, NMC811 electrodes are produced using N-methyl-2-pyrrolidone (NMP) solvent, which is both toxic and expensive.4 To pursue a more sustainable and greener production of NMC811 electrodes, water-based processing has been studied. However, this comes with challenges.5–7 For example, during water processing, lithium carbonate residuals on the particle surface can be removed.8 However, it has been reported that Ni-rich NMC materials can react with water through Li+/H+ exchange and further generate LiOH,9 this in turn increases the pH and further leads to corrosion of the Al current collector during slurry coating.10 In addition, during water-based processing, Li-ions at near-surface regions are reported to leach out into the aqueous medium and the vacancy can be filled by transition metals, with possible formation of NiO rock-salt phase in near-surface regions.11 All of these processes are detrimental to the battery's performance. Therefore, the development of mitigation strategies is crucial for the successful implementation of water-based processing.A few solutions have previously been considered, where NMC surface modification is a major strategy, for example, (1) coating with Al2O3, ZrO2, and TiO2 on the particle surface with atomic layer deposition (ALD) technique,12 (2) coating with cobalt boride, polyaniline, and Li3PO4 during the synthesis,13–15 and (3) using phosphoric acid to obtain a phosphate coating on the surface.10,16,17 Compared to expensive ALD coating and time-consuming synthesis coating, in situ coating with phosphoric acid during slurry casting is easy and thus cheap to achieve since it only includes the addition of the acid during the mixing of the NMC materials with slurry binder and H2O. It has also been shown that phosphoric acid can be used as a pH controller to avoid Al current collector corrosion.11,18 This is an important property as the corrosion product Al(OH)4− may react with electrolyte leading to co-solvent, ethylene carbonate, decomposition, and the corrosion product may also increase the charge transfer resistance at the Al/NMC electrode interface, which leads to capacity decay in the initial cycles.19 It is claimed that a phosphate coating can also improve the electrochemical cycling performance by stabilizing the NMC 523 electrode/electrolyte interphase and forming Li3P as a good ionic conductor component in the cathode electrolyte interphase (CEI).10 Also, the addition of H3PO4 acid in the slurry seems to stabilize the electrode/electrolyte interface.11 However, not much work has been done on NMC811 electrodes processed in water, and particularly it is not clear how the H3PO4-modified surface influences the CEI formation. In addition, the structural degradation mechanisms of water-processed electrodes during long-term cycling need to be investigated if it is to be implemented on a larger scale.
In this work, a systematic study was performed on aqueous-processed NMC811 electrodes, specifically the electrochemical performance, CEI layer formation, phase transitions, and charge transfer resistance. Water-processed electrodes (H2ONMC), H3PO4 modified electrodes (HPONMC) containing a small amount of phosphoric acid (0.5 wt% of total electrode material mass), and as a reference the NMP-processed electrodes (NMPNMC) were prepared. Generally, the results show that H2O-processed electrodes exhibit a thicker CEI layer leading to a inhibited H2 → H3 phase transition during long-term cycling, as determined by photoelectron spectroscopy (PES) and operando X-ray diffraction (XRD) measurements, respectively. Compared to the H2ONMC electrode, the HPONMC electrode had an improved cycling performance with a stable charge transfer resistance, which may be due to the reduction of an electrochemically inactive phase forming after extended cycling. Even though H3PO4 modification during electrode preparation cannot fully protect the NMC811 surface from reacting with H2O during water processing, it still inspires the development of a potential coating material/method for a more protective layer.
Experimental section
Electrode preparation
The cathode slurries were prepared by mixing active material NMC811 (Northvolt), conductive carbon black (Super C65T, Imerys) and polyvinylidene fluoride (PVDF, KYNAR) or sodium carboxymethyl cellulose (Na-CMC, Dow Wolff Cellulosics) binder in a mass ratio of 90%![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5%:5% using N-methyl-2-pyrrolidene (NMP) or deionized water as solvent, respectively. Another type of cathode slurry was prepared by adding 0.5 wt% phosphoric acid (H3PO4, Sigma-Aldrich, purity 99.99%) into the aqueous slurry mentioned above, and the mass ratio of the active material was reduced to 89.5%. The mixture was ball milled for 1 h at 25 Hz using an MM 400 mixer milling (Retsch). The resulting slurry was cast on carbon-coated Al foil (SHL) using a doctor blade, pre-dried at 70 °C for 5 h, and dried at 120 °C for 12 h in vacuum. The NMC811 electrodes were punched into a 13 mm disc with active material (AM) mass loading 2–3 mg cm−2 (coating thickness 16 μm) and 3.5–4 mg cm−2 (coating thickness 25 μm).
:5%:5% using N-methyl-2-pyrrolidene (NMP) or deionized water as solvent, respectively. Another type of cathode slurry was prepared by adding 0.5 wt% phosphoric acid (H3PO4, Sigma-Aldrich, purity 99.99%) into the aqueous slurry mentioned above, and the mass ratio of the active material was reduced to 89.5%. The mixture was ball milled for 1 h at 25 Hz using an MM 400 mixer milling (Retsch). The resulting slurry was cast on carbon-coated Al foil (SHL) using a doctor blade, pre-dried at 70 °C for 5 h, and dried at 120 °C for 12 h in vacuum. The NMC811 electrodes were punched into a 13 mm disc with active material (AM) mass loading 2–3 mg cm−2 (coating thickness 16 μm) and 3.5–4 mg cm−2 (coating thickness 25 μm).
Cell preparation and electrochemical measurements
NMC811 electrodes (AM mass loading 2–3 mg cm−2) were combined with 15 mm Li chip (China Energy Lithium Co., Ltd) as the negative electrode which were separated by Celgard 2325 in batteries. 1 mol L−1 LiPF6 in a mixture of ethylene carbonate/diethylene carbonate (EC/DEC 1:1 by volume, Solvionic, 99.9% purity) was utilized as the electrolyte. Single-layer pouch cells were assembled in an Ar-filled glovebox. The electrochemical cycling performance was evaluated galvanostatically between 3.0 V to 4.3 V vs. Li+/Li by using an Arbin analysis system. Following 5 formation cycles at a 0.1C rate, the batteries underwent 100 cycles at room temperature with a 0.5C rate (1C = 200 mA h g−1). Electrochemical impedance spectroscopy (EIS) measurements were carried out in the frequency range of 100 kHz to 10 mHz and with a 10 mV amplitude perturbation by a VMP instrument (Bio-Logic) at an open circuit voltage (OCV) state before electrochemical cycling, after 5 formation cycles, and every 20 cycles at 0.5C. Before each EIS measurement, the battery rested at OCV for one hour.
Ex situ photoelectron spectroscopy (PES) and X-ray absorption spectroscopy (XAS) analysis
For both PES and XAS measurements, NMPNMC, H2ONMC, and HPONMC electrodes were measured as pristine, after the initial cycle, and after 105 cycles. All of the pouch cells were kept fully discharged and disassembled in an Ar-filled glovebox. The cycled cathode electrodes were extracted from the pouch cells and rinsed with DMC drop-wise for a few seconds to remove electrolyte residue. The electrodes were mounted on Omicron sample plates using conductive adhesive PELCO tabs (Ted Pella, Inc) after drying under Ar and transferred to the FlexPES beamline endstation through an air-tight sample transfer vessel filled with Ar. Soft X-ray PES and XAS measurements were carried out at the FlexPES beamline at the MAX IV Laboratory, Lund.20 In addition, the disassembled electrodes were measured with Al Kα source (1486.7 eV) using a Kratos instrument (AXIS Supra+). Samples were transferred from an Ar-filled glovebox to the instrument using a sample holder with a lid pumped to −80 kPa relative to atmospheric pressure. The samples were transferred within 5 minutes without exposure to air.To obtain chemical composition in the outermost layer of electrodes with comparable depth profiling, P 2p, C 1s, O 1s, and F 1s core level spectra were measured with photon energies 275 eV, 425 eV, 675 eV, and 820 eV, which gives similar kinetic energy of photoelectrons (∼150 eV) and a probing depth of ∼2 nm (probing depth being 3* estimated inelastic mean free path of electrons). The binding energy in all spectra was referenced to an adventitious hydrocarbon peak at 285 eV. For the O 1s and F 1s spectra binding energy calibration, the C 1s spectra were also obtained using a photon energy of 675 eV and 820 eV, respectively. To calibrate P 2p collected with photon energy 275 eV, P 2p and C 1s spectra were collected with a photon energy of 720 eV to be utilized as references. Acquired spectra were curve fitted with the Igor Pro 9 software with a combination of Gaussian and Lorentzian functions, following linear background subtraction. The full-width half-maximum is fixed to 0.2 eV in the Lorentzian function and variable in the Gaussian function with a range of 1–1.6 eV (except for the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C peak in carbon black set to 0.7 eV). For each collected C 1s, O 1s, and F 1s spectrum, the peak intensity was normalized to 1, except for the F 1s spectra measured from the pristine H2ONMC and HPONMC electrodes due to their zero intensity. For P 2p, the spectra collected from the pristine samples were normalized to the same background intensity, while the spectra from the cycled electrodes were normalized to a different background intensity. This adjustment was necessary because these two batches of data were collected from two different endstations at the synchrotron, and no direct comparison of the intensity between these sets of spectra should be made.
C peak in carbon black set to 0.7 eV). For each collected C 1s, O 1s, and F 1s spectrum, the peak intensity was normalized to 1, except for the F 1s spectra measured from the pristine H2ONMC and HPONMC electrodes due to their zero intensity. For P 2p, the spectra collected from the pristine samples were normalized to the same background intensity, while the spectra from the cycled electrodes were normalized to a different background intensity. This adjustment was necessary because these two batches of data were collected from two different endstations at the synchrotron, and no direct comparison of the intensity between these sets of spectra should be made.
XAS measurements were conducted for all electrodes and NiO, LiNiO2, Li2CO3, LiF, and LiOH references using drain current. Ni L-edge, O K-edge, and Li K-edge spectra were measured in total electron yield (TEY, depth information about 5 nm) mode.21,22 In addition, Ni L-edge fluorescence was detected using SDD (Rayspec) for the pristine electrodes. The obtained spectra were normalized by the I0 signal from a clean gold sensor in the beamline, and the background was further corrected to ensure constant pre-edge intensity. The presented Ni L-edge and O K-edge spectra were normalized to a point after the absorption edge.
Operando X-ray diffraction (XRD) measurements
To obtain a good signal-to-noise ratio, NMC811 electrodes (AM mass loading 3.5–4 mg cm−2) were assembled into pouch batteries. After 5 formation cycles and 55 cycles (0.1C for 5 cycles followed by 0.5C for 50 cycles), pouch cells were taken for operando XRD measurements. A dedicated cell holder, described previously23 was modified to hold six cells for the current study. The six mounted pouch cells were measured one after another continuously. One second exposure was needed to obtain one diffraction pattern and the time interval between patterns from the same sample is around 50 seconds. Galvanostatic cycling of the pouch cells was performed using a Bio-Logic VMP potentiostat at 0.2C (40 μA mg−1) within a voltage window of 3 V–4.3 V vs. Li+/Li. Diffraction data was collected using a Dectris Pilatus3 2 M CdTe detector with a photon energy of 35 keV (λ = 0.3542 Å). These measurements were performed at the DanMAX beamline at MAX IV Laboratory, Lund, Sweden.Quantitative analysis of d-spacing changes of the 003 reflection was obtained from peak fitting using a Voigt function performed in using the Fityk software.24 A linear background was applied to all data sets. For the majority of the data, a single peak was sufficient to fit the data. In instances where asymmetry was observed or where a second peak was identified, a second peak was included in the fitting. An example of the single and double-peak fitting is shown in Fig. S1 (ESI†). The full width at half maximum (FWHM) values of the main peak at the 003 reflection vary from 0.015 Å−1 to around 0.03 Å−1 depending on states of charge. The peak shape, a combination of Gaussian and Lorentzian functions, remains constant during peak fitting. In a double-peak fitting, the shapes of the two peaks were set to be identical. The shifts in fitted peak position can be used to roughly calculate changes in the c-lattice parameter through their corresponding d-spacings.
Inductively coupled plasma atomic emission spectrometry (ICP-OES)
Pouch cells were assembled in the same manner as mentioned above except for the separator usage. Instead of using Celgard separators, glass fibre separators were selected in order to capture more electrolyte during battery cycling. After 105 cycles, separator from each pouch cell was disassembled and centrifuged. The electrolyte captured by glass fibre was collected by centrifugation. The ICP-OES was calibrated with a known amount of quantified element concentration ranging from 0.001 ppm to 1 ppm. This calibration was conducted using a commercial multi-elements calibration standard (PerkinElmer). The obtained electrolytes were diluted by a factor of 200 using 5% HNO3 solution before running measurements.Results and discussion
Before presenting the data, it is important to highlight the labelling method used for the samples. The NMP-processed electrodes are named as NMPNMC, and two types of water-processed electrodes are written as H2ONMC (without H3PO4 acid modified), and HPONMC (with H3PO4 acid modified).Electrochemical cycling performance
The electrochemical behaviour of NMPNMC, H2ONMC and HPONMC electrodes are shown in Fig. 1, where (a) depicts the long-term galvanostatic cycling performance. H2ONMC electrodes have the worst capacity retention after 105 cycles with 71.8% of initial discharge capacity, followed by HPONMC electrodes with 77.3%, and NMPNMC electrode with 89.5%. As displayed in Fig. 1b, H2ONMC and HPONMC electrodes have less charge/discharge capacity already in the initial cycle compared to NMPNMC electrodes, which could be linked to Li-ions leaching out during water-processed slurry preparation as previously observed.11,25 In addition, a higher voltage polarization can be seen in water-processed electrodes, which can be linked to a NMC811 surface reaction with H2O during slurry preparation resulting in Li+/H+ exchange and formation of LiOH and Li2CO3 on the electrode surface.8,9,11,26 As supported by the XAS data below, the presence of a NiO-like rock-salt phase may be linked to the increase in surface resistance. In the 105th cycle, H2ONMC electrodes have increased overpotential (>0.5 V) compared to the 6th cycle, as shown in Fig. 1h. As shown by the PES data below, this overpotential increase with increased cycling index may be due to a thick CEI layer formation during long-term cycling, which likely increases the Li-ions diffusion resistance. | ||
| Fig. 1 Electrochemical performance of NMPNMC, H2ONMC, and HPONMC electrodes in half cells. (a) The discharge capacity of three electrodes with galvanostatic cycling at 0.1C for 5 cycles and 0.5C for 100 cycles within the voltage range of 3.0–4.3 V vs. Li+/Li, averaged results from 5 batteries, (b) charge–discharge profile of 1st cycle, (c) charge–discharge profile of 105th cycle, the dQ dV−1 curves for NMPNMC electrode (d and g), H2ONMC, (e and h) and HPONMC (f and i) at various cycles at 0.1C and 0.5C respectively. | ||
The differential capacity (dQ dV−1) profiles are illustrated with various cycle numbers of the three types of electrodes in Fig. 1(d–i). The initial cycle shows a similar overpotential as the charge–discharge profile for all three types of electrodes, and negligible polarization can be seen between cycles 2–5 at 0.1C in Fig. 1d–f. During charge, NMC811 particles undergo phase transitions from hexagonal 1 to monoclinic (H1 → M), monoclinic to hexagonal 2 (M → H2), and hexagonal 2 to hexagonal 3 (H2 → H3) phase. In the dQ dV− 1 plots these phase transitions coincide with the distinct peaks at ∼3.75 V, ∼4.0 V, and ∼4.2 V.1,27,28 As can be seen in Fig. 1g, the NMPNMC electrode has a maintained peak intensity at 0.5C for 100 cycles, indicating a good phase transition reversibility and structure stability after extended cycling. In contrast, for the H2ONMC electrodes, dramatic shifts of all the dQ dV−1 peaks and peak intensity decay for the H2–H3 phase transition can be seen during cycling at 0.5C in Fig. 1h. These peak shifts provide evidence of surface polarization due to high resistance which impacts the occurrence of the phase transitions. Many reasons can cause the peak intensity drop, and we propose that structural degradation in the near-surface regions leads to the incomplete phase transition. With the following XAS and XRD measurement results, the above-mentioned suggestions are confirmed to be reasonable. Compared to H2ONMC, HPONMC electrodes exhibit reduced surface polarization, shown in the dQ dV−1 peaks in Fig. 1i, however, it still displayed higher surface polarization than the NMPNMC electrode (Fig. 1g). The intensity drops observed in HPONMC electrodes are less severe compared to those in H2ONMC electrodes. These results indicate that phosphoric acid modification can improve structure reversibility in the near-surface regions of H2O-processed electrodes during battery cycling, although not yet reaching the performance level of NMP-processed electrodes.
Analysis of CEI layer formation with PES
Soft tunable X-ray ex situ PES measurements were utilized to investigate the chemical species on the electrodes surface. As shown in Fig. 2–6, C 1s, O 1s, P 2p, and F 1s spectra were collected on the three types of cathodes, pristine, after the initial cycle, and after 105 cycles. Curve fitting was performed on the core-level spectra, and peak assignment is shown in the individual core-level spectra. Specifically, in the C 1s spectra, the peak at 284.2–284.4 eV was assigned to CC originating from carbon black and the C–H peak at ∼285 eV assigned to adventitious carbon from air.26 The peak at ∼286 eV is assigned to ![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) H2–CF2 (from binder PVDF in NMP-processed electrodes) and C–O/C–OH species.26,29 CO and OC–O species give peak features at ∼288 eV and ∼289 eV, respectively.29 The peak at ∼290 eV binding energy is assigned to CH2–F2 (from binder PVDF in NMP-processed electrodes) and CO32−.5 The peak at ∼291 eV has contributions from CFx (x ≥ 2) species, which are likely side products from electrolyte degradation in the H2O-processed electrodes.30
H2–CF2 (from binder PVDF in NMP-processed electrodes) and C–O/C–OH species.26,29 CO and OC–O species give peak features at ∼288 eV and ∼289 eV, respectively.29 The peak at ∼290 eV binding energy is assigned to CH2–F2 (from binder PVDF in NMP-processed electrodes) and CO32−.5 The peak at ∼291 eV has contributions from CFx (x ≥ 2) species, which are likely side products from electrolyte degradation in the H2O-processed electrodes.30
 | ||
| Fig. 2 C 1s spectra collected on NMPNMC (a), H2ONMC (b), and HPONMC (c) electrodes as pristine (top) in the discharge state after the initial cycle (middle) and after 105 cycles (bottom) with photon energy 425 eV. | ||
 | ||
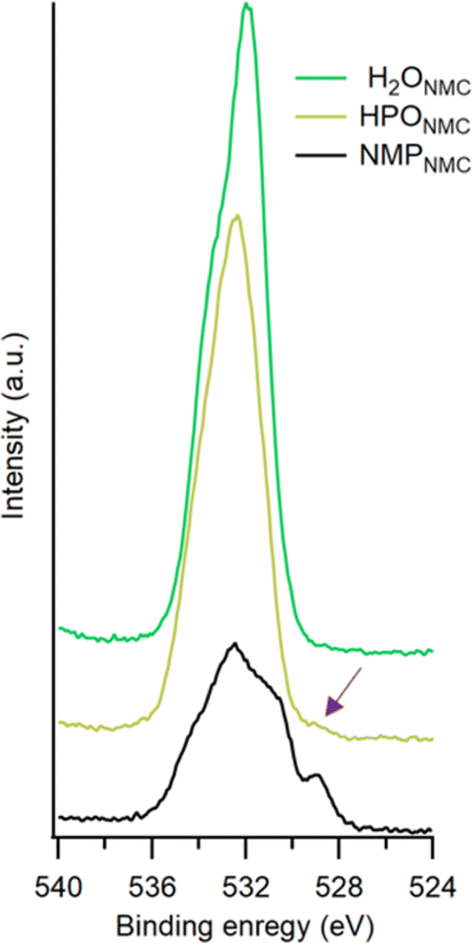
| Fig. 3 O 1s spectra collected on NMPNMC (a), H2ONMC (b), and HPONMC (c) electrodes as pristine (top), after 1 cycle (middle) and after 105 cycles (bottom) with photon energy 675 eV. | ||
 | ||
| Fig. 4 O 1s spectra measured on NMCNMP, H2ONMC and HPONMC electrodes after 105 cycles with photon energy 1486.7 eV. | ||
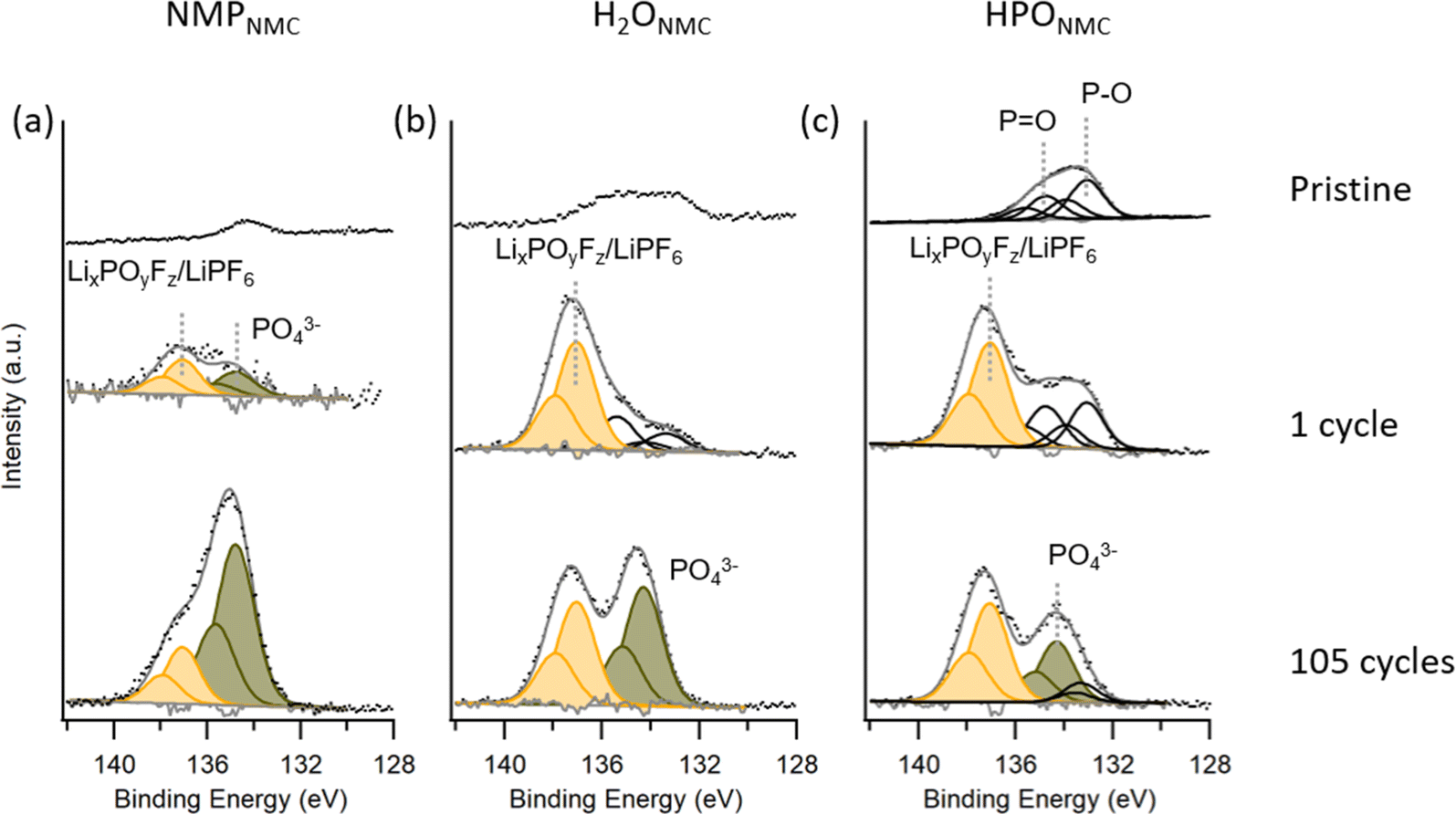 | ||
| Fig. 5 P 2p spectra collected on NMPNMC (a), H2ONMC (b), and HPONMC (c) electrodes as pristine (top), after 1 cycle (middle), and after 105 cycles (bottom) with photon energy 275 eV. | ||
 | ||
| Fig. 6 F 1s spectra collected on NMPNMC (a), H2ONMC (b), and HPONMC (c) electrodes as pristine (top), after 1 cycle (middle) and after 105 cycles (bottom) with photon energy 820 eV. | ||
In the soft X-ray C 1s PES spectra CO32− signals can be seen for the pristine H2ONMC and HPONMC electrodes, which most likely originates from reactions of CO2 with the surface.29 This result is in contrast to the pristine NMPNMC electrode, where no such species can be confirmed. It was previously determined in our research that the presence of water facilitates the formation of carboxylate species, and indeed an increase in the relative peak ratio of O–CO to CO suggests that this also could occur for the H2O-processed electrodes in this study.31 The Na-CMC binder in the pristine H2ONMC and HPONMC show peak intensity corresponding to C–O, CO, and O–CO species. The majority of adventitious hydrocarbon species were removed after 1 cycle for both H2ONMC and HPONMC electrodes, as seen from a relatively stronger CC peak. The relative ratios between other C 1s peaks indicate that both the H2ONMC and HPONMC electrodes are after 1 cycle rather similar to their pristine counterparts. However, upon longer cycling a thicker CEI layer is building up, mainly consisting of C–H, C–O, O–CO, CO32− containing compounds. Further, more electronegative C moieties were detectable among the electrolyte degradation products, e.g. CFx species, after 105 cycles. In NMPNMC electrodes, negligible changes can be seen between the pristine electrodes and after they were cycled once. After 105 cycles, O–CO species from electrolyte degradation forms in the CEI layer. Also, CH2–F2 and CO32− species together gave intensity contributions to the peak at 290 eV binding energy. In summary, more adventitious hydrocarbon and carbonate species accumulated on the pristine H2O-processed electrodes compared to NMP-processed electrodes. After 105 cycles, CO32− species were detected in the CEI layer in all three types of electrodes, and CFx (x ≥ 2) species were also found on the H2ONMC and HPONMC electrode surfaces.
From O 1s spectra in Fig. 3, the peak at lowest binding energy ∼529 eV is assigned to M−O, the structural oxygen in the NMC811 materials. The following peak at ∼531 eV (referred to as surface O) can have a number of origins; both M–OH and CO32− species generally show a peak at ∼531 eV in the O 1s spectra, and our previous research identifies the presence of them on the NMC811 powder surface during storage.31 Thus, M–OH and CO32− species may exist on the pristine electrodes, hence giving intensity contribution in O 1s spectra. Also, R–OLi species are known to show a peak at ∼531 eV in O 1s spectra, which may be generated after electrical cycling.32,33 As shown in Fig. 3, the peak assigned to M–OH, CO32−, and R–OLi species is labeled as surface O. CO and C–O give intensity at ∼532 eV and 533 eV respectively.29,34 After the initial cycle, in the NMPNMC electrode, the peak at ∼534.2 eV is assigned to P–O species, which is contributed by salt decomposition into phosphate compounds. While, after 105 cycles, the peak shifts to slightly higher binder energy (∼534.5 eV), which could be due to the intensity contribution from the LixPOyFz species. In pristine H2ONMC and HPONMC electrodes, the peaks at ∼534 eV are attributed to C–OH species from the Na-CMC binder.35,36 After electrochemical cycling, P–O species may give intensity contribution to the peak at ∼534 eV, together with C–OH species in water-processed electrodes. The peaks at the highest binding energy ∼535 eV are attributed to LixPOyFz, which is assigned to degradation products from the electrolyte. In the pristine H2ONMC and HPONMC electrodes and following 1 cycle, peak ratios of C–O to CO species in C 1s vary in O 1s spectra, which is due to contribution from the PO species at ∼533 eV, together with C–O species in O 1s. The presence of phosphorus species is supported by the P 2p spectra, shown in Fig. 5.
With the soft X-ray PES data, signal from structural oxygen (M–O species) is visible in all three types of electrodes, as pristine and after the initial cycle. However, after 105 cycles, as the CEI layer grows, M–O species are not detectable with the soft X-rays in any of the samples. However, with an Al Kα source, M−O contribution to the O 1s spectrum can be seen in the NMPNMC electrode (see Fig. 4). This result implies that a thicker CEI layer was formed in the H2O-processed electrodes than in the NMP-processed electrodes. In the HPONMC electrode, a minor peak intensity corresponding to M−O species is observable, suggesting that M–O buried by CEI remains detectable. This implies that after 105 cycles, the HPONMC electrode exhibits a slightly thinner CEI layer compared to the H2ONMC electrode (see the overlaid spectra and fitted spectra in Fig. S2 in ESI†). The interpretation of the thickness of the CEI, as obtained from the C 1s and O 1s spectra, supports each other, however, a small difference is present. Particularly, the C 1s spectra suggest a slightly thinner CEI, compared to what the O 1s spectra do, exemplified by the clear CC peak for all electrodes after 105 cycles, while at the same time, the M–O peak in the O 1s spectra is barely visible. This phenomenon most likely originates from some carbon black particles not being buried by the binder during electrode preparation. Thus, they were not impacted or covered by the resulting CEI layer on the cycled electrodes.
In P 2p spectra (Fig. 5), peaks at lower binding energy are assigned to phosphate and PO/P–O groups and peaks at higher binding energy are attributed to LixPOyFz and LiPF6 salt residue.29 Pristine HPONMC electrodes show PO/P–O species on the surface due to the H3PO4 acid modification, while the pristine NMPNMC and H2ONMC electrodes contain a smaller quantity of phosphorus compounds, which is assumed to dissolve into electrolyte while cycling, see Fig. S3 in ESI.† After 1 cycle, H2ONMC and HPONMC electrodes show a relatively higher contribution from LixPOyFz/LiPF6 compared to the NMPNMC electrode. This can be linked to the OH group from Na-CMC binder reacting with LiPF6 salt dissociation product PF5, which is reported by Passerini et al.29 As described in reaction scheme (I), the reaction generates POxFy-like products. In addition to that, PF5 reacts with H2O residue, and the product POF3 further generates LixPOyFz, as described in reaction scheme (II). In contrast, the formation of LixPOyFz species on the surface of the PVDF-based electrode is mainly due to the H2O residue, following the reaction scheme (II).
| LiPF6 ↔ LiF + PF5 |
| PF5 + R−OH → R−O−PF4 + HF | (Reaction scheme I) |
| PF5 + H2O → 2HF + POF3 |
| POF3 + ne− + nLi+ → LixPOyFz + LiF | (Reaction scheme II) |
After 105 cycles, major contributions from phosphate can be detected in the NMPNMC electrode, while both H2ONMC and HPONMC electrodes show similar phosphate and LixPOyFz/PF6− quantities. The phosphate-rich CEI layer is reported to stabilize the particle surface structure,37 and lithium phosphate has good ionic conductivity.15 Thus, a phosphate-rich CEI on the NMPNMC compared to the H2O-processed electrodes can be a contributing factor for the better cycling performance of NMPNMC electrodes.
As displayed in Fig. 6, for the pristine NMPNMC electrode, PVDF binder contributes to the peak at ∼688 eV and 689 eV, and no detectable F 1s signal can be observed in pristine H2ONMC and HPONMC electrodes. After the initial cycle, LiF (∼685 eV) and LixPOyFz (∼687 eV) peak intensity can be seen in F 1s spectra,29 which shows the presence of these species in the CEI layer in all three electrode types. Compared to H2ONMC electrodes, HPONMC electrodes have relatively more LiF product at the surface. LiF is formed in both electrodes according to the scheme (II). In addition to that, the larger amounts of LiF observed in the HPONMC could be due to a reaction between HF (side product from LiPF6 degradation reaction with H2O residue and the hydroxyl group) and pre-existing phosphorus species, probably LixH3−xPO4 species, resulting in the extra formation of LiF, according to reaction scheme (III). LiF is considered to give good ionic conduction in the CEI layer, and this result supports the smaller impedance observed for the HPONMC electrodes compared to the H2ONMC electrodes. Further details will be demonstrated in the following EIS results.
| Li3−xHxPO4 + HF → Li2−xHx+1PO4 + LiF | (Reaction scheme III) |
The thicker CEI layers after 105 cycles are seen for all electrodes and contain relatively more LixPOyFz compared to salt residues. This ratio increased from NMPNMC to HPO NMC, with the largest ratio for H2ONMC. This could indicate an increased reactivity between the salt and the NMC surface in the same order.
To summarize, from the PES data collected from various pristine and cycled electrodes we can draw the following conclusions: (1) pristine H2O-processed electrodes are prone to accumulate adventitious carbon species on the surface and also have carbonate species on the surface that react with CO2 during storage; (2) for the cycled H2O-processed electrodes, the CEI layer contains LiF, Li2CO3, and LixPOyFz after the initial cycles, and phosphate is gradually increasing during long-term cycling. (3) Compared to NMPNMC electrodes, both H2O-based electrodes have a thicker CEI layer; and (4) HPONMC electrodes have pre-existing phosphorus species on the surface, which react to form relatively more LiF in the CEI layer compared to the H2ONMC.
Surface investigation with XAS
Fig. 7 shows the Li K-edge TEY XAS spectra for references and the electrodes under investigation. Since the majority of Ni is trivalent in NMC811 materials, a LiNiO2 reference is utilized in this work to show the comparable feature given by Li+ surrounded by Ni3+ and O2− in NMC811 materials. LiNiO2 material mainly gives XAS features at ∼62 eV and 67 eV photon energy. LiF shows a peak at ∼62 eV and a peak at ∼70 eV and compared to LiNiO2, the LiF peaks are relatively sharper. The overall structure of the Li XAS from NMPNMC shows strong similarity with the LiNiO2 reference spectrum, while both the HPONMC and H2ONMC show a larger deviation. Compared to pristine NMPNMC electrodes, pristine H2ONMC and HPONMC electrodes have less peak intensity at ∼62 eV, which may be due to Li-ions leaching in the near-surface region during water processing. This finding is in the line with Li+/H+ exchange, as reported by Pritzl et al.8 The feature at ∼67 eV may be contributed from Li2CO3 and LiOH. The presence of carbonate species on the pristine H2O-processed NMC electrodes can be proved from the C 1s spectra in Fig. 2b and c. Apart from that, LiOH is expected on the electrode surface, and it gives peak intensity at ∼67 eV.38 During cycling, NMPNMC electrodes do not show obvious changes except for a more visible/sharper peak at 62 eV, implicating the formation of more LiF species in the CEI layer after 105 cycles. These results also fit with the relative changes observed for the F 1s spectra. However, for the cycled H2O-processed electrodes, strong LiF features can be seen already after 1 cycle, and the intensity of the characteristic LiF peak drops significantly after 105 cycles. From the PES data, a thickening CEI was observed with cycling, and the lower LiF peak in the XAS after 105 cycles is interpreted as the initially formed LiF becoming buried under the CEI layer during the following cycles. Still, there is intensity in the Li K-edge XAS and these may originate from CEI species e.g. phosphate, LixPOyFz, and residual LiPF6.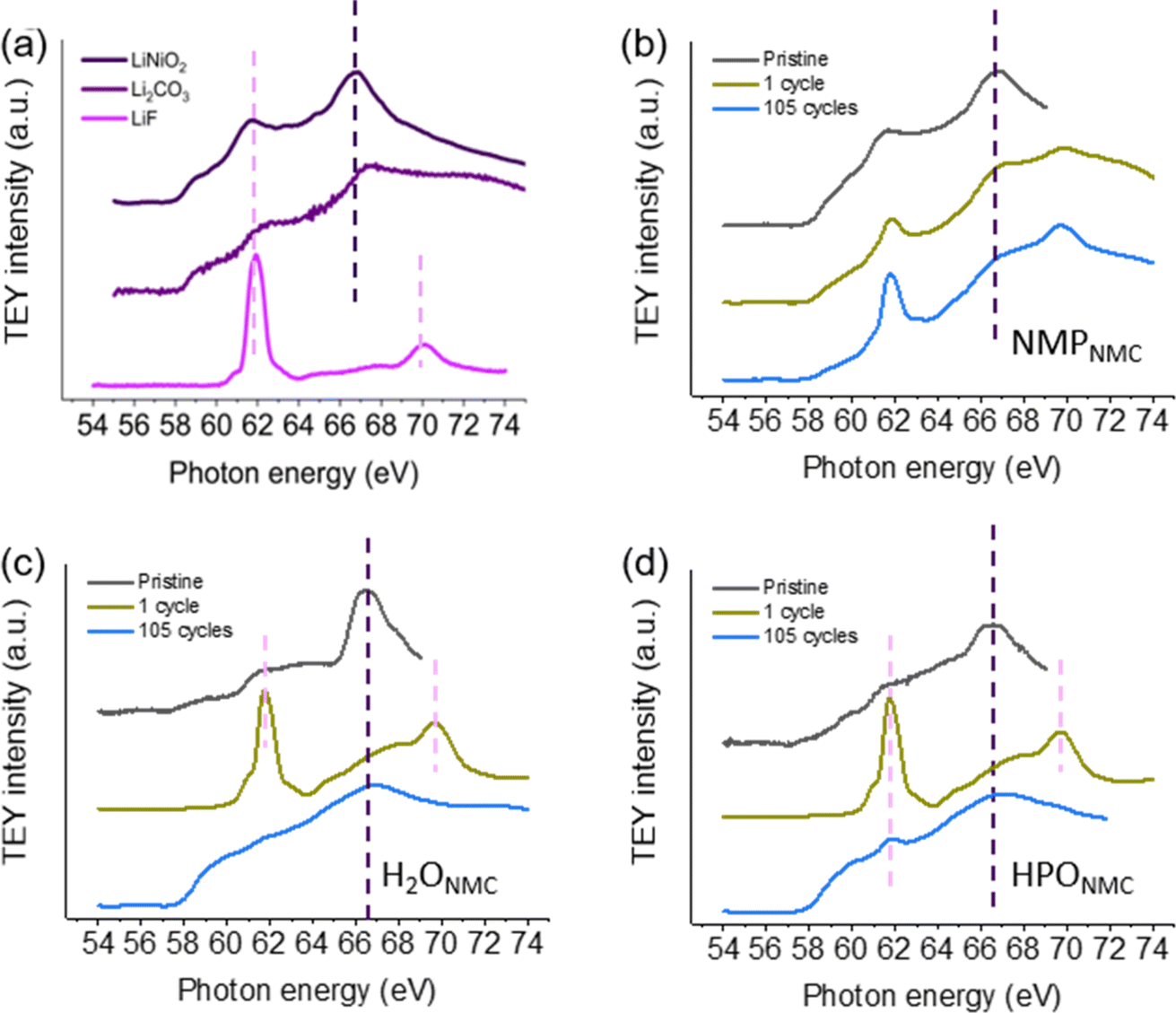 | ||
| Fig. 7 Li K-edge data in TEY mode for (a) reference materials including LiNiO2, Li2O3, and LiF. (b) NMPNMC electrodes, (c) H2ONMC electrodes, and (d) HPONMC electrodes as pristine, after the 1 cycle, and after 105 cycles. | ||
O K-edge XAS spectra measured for reference chemicals and the electrodes are shown in Fig. 8. The feature at ∼534 eV is attributed to π* orbitals in Li2CO3.22 LiNiO2 and NiO give contributions to the peak at ∼528 eV and 532 eV respectively, as can be seen in Fig. 8(a). In addition, LiNiO2 also has a feature at ∼534 eV from CO32− species, which is due to its air instability and formation of Li2CO3 on the surface during storage.39 Compared to the pristine NMPNMC electrode, H2ONMC, and HPONMC electrodes have relatively higher peak intensity at 532 eV compared to the peak at 528 eV, which indicates that a NiO-like rock-salt phase is formed on the surface of these H2O-processed electrodes.25 As reported by Bichon et al., in the aqueous medium, Li-ions at near-surface regions of NMC materials will leach. Following this, transition metals can fill the lithium site and the reduction of the Ni3+ ions to Ni2+ together with oxygen loss leads to a NiO-like rock-salt phase in this lithium-deficient near-surface region.11,39,40 Interestingly, after the initial cycle, all electrodes in this study have a slightly higher intensity ratio of O–Ni3+ feature to O–Ni2+, labelled with dashed lines in Fig. 8b–d. The proposed explanation is that after the initial discharge, not all the Ni3+ can be fully reduced back to Ni2+, which also matches the capacity loss in the initial discharge. The NMPNMC electrode, on the other hand, shows almost complete reversibility. After 105 cycles, the relative peak intensity ratio between O–Ni3+ to O–Ni2+ decreased also for the NMPNMC electrode, and this is likely due to the NiO-like phase formation in line with previously reported results.28 In the H2ONMC and HPONMC electrodes after 105 cycles, due to the surface sensitivity of TEY mode, the O K-edge spectra mainly show information from the CEI layer rather than the NMC811 active material. Thus, the presence of O–Ni3+ and O–Ni2+ peaks indicates some Ni-ions were trapped in the CEI possibly linked to HF attack.41 Also, from ICP-OES results, transition metal diffusion into electrolyte is identified (see Table S1, ESI†). A substantial amount of Li2CO3 species is produced after long-term cycling, which is confirmed by the peak intensity at ∼534 eV in Fig. 8c and d. This result matches the collected C 1s PES spectra as well as Li K-edge XAS spectra.
 | ||
| Fig. 8 O K-edge data in TEY mode for (a) reference materials including Li2O3, LiNiO2, and NiO. (b) NMPNMC electrodes, (c) H2ONMC electrodes, and (d) HPONMC electrodes as pristine, after the 1 cycle, and after 105 cycles. | ||
Ni L-edge XAS spectra were obtained for pristine NMPNMC, H2ONMC, and HPONMC electrodes, following the initial cycle, and after 105 cycles, as shown in Fig. 9a–c. Fig. 9(d) displays the spectra from the reference compounds NiO and LiNiO2, and the doublet peak near 853 eV and 855 eV labelled with ‘L3 low’ and ‘L3 high’ corresponds to the 2p3/2 → 3d transition under two different photon energies. The ratio of peak intensity of L3 high to L3 low gives qualitative information about the oxidation state of Ni, with a higher value meaning a higher amount of Ni3+.2,22,42 Thus, the L3 high to L3 low peak intensity ratios have been calculated for each spectrum, as shown in Fig. 9e, to investigate the evolution of the oxidation state of Ni at the near-surface region in each sample. As for the pristine electrodes, H2ONMC and HPONMC have a lower oxidation state on Ni compared to NMPNMC, which supports the NiO phase formation in the H2O-processed electrodes. After the initial cycle, the intensity ratio values increase in all three types of electrodes. Thus, the same conclusion can be drawn as from the O K-edge data that some Ni3+ ions were not reduced back to Ni2+ during the initial discharge. After 105 cycles, the peak intensity ratio drops for the NMPNMC electrode, indicating NiO formation during cycling. This data also matches the interpretation of the O K-edge XAS data presented earlier. The Ni L-edge data from H2ONMC and HPONMC electrodes after 105 cycles gives information on the CEI layer rather than the active materials, which also confirms the presence of Ni-ions in the CEI layer.
 | ||
| Fig. 9 Ni L-edge XAS spectra in TEY mode for: (a) NMPNMC, (b) H2ONMC, and (c) HPONMC electrodes as pristine, after the 1 cycle, and after 105 cycles (oscillations in the spectrum collected from the HPONMC electrode after 105 cycles are not real data points due to measurement artifacts), (d) reference materials including LiNiO2 and NiO; (e) relative TEY peak intensity ratio of Ni L3 high/L3 low with three different electrodes under various cycling conditions; (f) fluorescence measurements with three pristine electrodes. | ||
Operando XRD measurements following the phase transition
In order to study the impact of water processing on the structural phase transitions of NMC811 particles, another batch of electrodes were produced with a higher mass loading of active material (3.5–4 mg cm−2) aiming to obtain diffraction patterns with a good signal to noise ratio. The pre-cycled electrochemical data for the aged batteries is shown in Fig. S4 (ESI†). Waterfall plots of the diffraction pattern of the 003 reflection is displayed in Fig. S5 (ESI†). Fig. 10 shows the two-dimensional contour plots for the 003 reflection of fresh and aged batteries for cathode electrodes of NMPNMC, H2ONMC, and HPONMC at a current rate of C/5. The simulated (in CrystalDiffract) Q value of the 003 reflection for pristine NMC811 is shown with white dashed lines.43 Fresh cells behave very similarly to one another (see Fig. 10a–c) with the 003 reflection shifting towards a smaller Q value, corresponding to a gradual expansion of the interlayer spacing, until the voltage passes ∼4.0 V, which includes H1 → M and M → H2 phase transition. The H2 → H3 phase transition takes place at ∼4.2 V (see dQ dV−1 plot in Fig. 1), where the interlayer spacing quickly shrinks.1,44 After 55 cycles, the phase transition process in the NMPNMC electrode shows negligible changes relative to the fresh cell (Fig. 10a and d). In contrast, the H2 → H3 phase transition in the H2ONMC and HPONMC electrodes does not appear to go to completion after 55 cycles, as displayed in Fig. 10e and f. Consequently, the total volume change during the H2 → H3 transition is less due to reduced Li-ions extraction, which may stem from high internal resistance. This is evidenced by the larger voltage drop observed in the cycling profile shown in Fig. 10e and f. | ||
| Fig. 10 Operando synchrotron XRD characterization of 003 reflection of NMPNMC (a and d), NMC_ H2O (b and e), and HPONMC (c and f) electrodes after 5 formation cycles and 55 cycles along with the cycling profile at 0.5C. | ||
d-Spacing values for the 003 reflection were determined through peak fitting. In the case of single-peak fitting for the 003 reflection, one d-spacing value will be obtained, while with a double-peak fitting, two d-spacing values were obtained, corresponding to two different phases (see Fig. S1†). As depicted in Fig. 10a and d, a fitting using a single peak (and therefore with a single phase) was sufficient for all states of charge of the NMPNMC electrode. In comparison to the fresh NMPNMC electrode, the aged NMPNMC electrode shows negligible changes to the phase transition. This observation suggests that NMPNMC electrodes exhibit minimal resistance to Li-ions insertion and extraction from the bulk material. Unlike NMPNMC electrodes, a single peak was insufficient for fitting the 003 reflection for H2ONMC and HPONMC electrodes and subsequently a second peak (phase) was included in the fitting, respectively (see Fig. S6 and S7†). In the H2ONMC electrode, the second peak appears at ∼4.3 V, and the d-spacing values for this peak (shown in green in Fig. 10b and e) are fairly invariant. We interpret this phenomenon as some delithiated particles in the electrode experiencing a higher resistance during the H2 → H3 phase transition and becoming electrochemically inactive.
After aging, a more electrochemically inactive phase exists in the H2ONMC electrode compared to the fresh one, as the peak fitting shows in Fig. S4.† It is proposed that the presence of the NiO-like rock-salt phase in the fresh cell leads to the gradual growth of the inactive phase during cycling. In HPONMC electrodes, rather than peak asymmetry, the main reflection became significantly broadened during the H3 → H2 phase transition in discharge (see example in Fig. S7†). Two or more phases existed during discharge from 4.3 V to 4.0 V in the fresh cell (Fig. 10c) or 3.9 V in the aged cell (Fig. 10f) (see the green box in Fig. 10). Further, in both the fresh and aged electrodes, two or more phases exhibit expansion along the c-axis of the unit cell revealing some electrochemical activity, even though the rate of expansion varies. A possible explanation for this is that rather than particles becoming electrochemically inactive, different parts of the electrode are experiencing varying resistance to Li-ions reinsertion and react at different rates during discharge. Instead of referring to a sluggish phase with a lower unit cell change rate, they are distinguished by naming phase 1, phase 2, and phase 3. This difference in reaction kinetics is significant enough to be detected via XRD as shown in Fig. S7.† In the fresh and aged HPONMC electrodes at ∼4.1 V, the peak broadens significantly and cannot be fitted with two phases. This implies that the beam is diffracting from parts of the electrode that are at different states of charge, where some regions (phase 3) are more sluggish than others (see Fig. S7†). d-Spacing values from phase 1 and phase 2 are shown in blue and green color, respectively, in Fig. 10f. This data suggests that HPONMC electrodes experience significant reaction inhomogeneity. However, other techniques are needed to ascertain the spatial distribution of this inhomogeneity. For example, if it exists on a particle level or if it varies as a function of depth in the electrode. Compared to the aged H2ONMC electrode, the resistance in the aged HPONMC electrode is less pronounced since all the phases were electrochemically active, unlike the electrochemically inactive phase formation in the aged H2ONMC electrode.
Charge transfer resistance with EIS measurements
The EIS measurements were conducted before electrochemical cycling, after 5 cycles at a current rate of 0.1C, and every 20 cycles at a current rate of 0.5C, as illustrated in Fig. 11. NMPNMC electrodes exhibit the lowest charge transfer resistance, remaining constant during long-term cycling. In contrast, H2ONMC electrodes experience a dramatic increase in charge transfer resistance during long-term cycling, which is most likely from the growth of the electrochemically inactive phase. This observation is corroborated by the results of operando XRD measurements. With phosphoric acid modification on the surface, HPONMC electrodes maintain a relatively constant charge transfer resistance during long-term cycling, which is higher than that in NMPNMC electrodes. This result suggests that the increased charge transfer resistance in HPONMC electrodes may lead to phase inhomogeneity in the electrode during battery cycling, particularly during the Li-reinsertion process, as also observed in the operando XRD measurements. However, the charge transfer resistance in HPONMC electrodes is smaller than the ones in H2ONMC electrodes, where an electrochemically inactive phase was formed. | ||
| Fig. 11 EIS measurements of (a) NMPNMC, (b) H2ONMC, and (c) HPONMC electrodes after various cycles. | ||
Conclusions
In this work, we manage to understand the capacity degradation mechanisms of the H2O-based casting with Ni-rich cathode materials. A more reactive surface and a NiO-like rock-salt phase in the near-surface regions are obtained with NMC811 electrodes after water processing. During long-term cycling, compared to the NMPNMC electrodes, a thicker CEI layer and larger surface polarization are observed in H2O-processed electrodes. In addition, an electrochemically inactive phase and sluggish phase formation lead to capacity decay in H2ONMC and HPONMC electrodes, respectively. HPONMC electrodes provide enhanced capacity retention compared to H2ONMC electrodes, which is likely due to LiF-rich CEI, less charge transfer resistance, and the alleviation of the growth of the electrochemically inactive phase. Regardless, the high resistance during the H3 → H2 phase transition results in loss of capacity retention in HPONMC electrodes.Data availability
The data underlying this article will be shared on reasonable request to the corresponding author.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the Swedish Energy Agency (48678-1, P2020-90112, and P2022-00055), Swedish Research Council (2021-04987), and STandUP for energy for financial support. The financial support by EU Horizon 2020 innovation program under grant agreement number 875527 – HYDRA project is acknowledged by the authors. The authors gratefully acknowledge funding from Stiftelsen för Strategisk Forskning (SSF) within the Swedish National Graduate School in Neutron Scattering, SwedNess (GSn15–0008). We acknowledge MAX IV Laboratory for time on FlexPES Beamline (proposal #20221259, #20220527, and #20230127), and DanMAX beamline (proposal #20221292). Research conducted at MAX IV Laboratory, a Swedish national user facility, is supported by the Swedish Research council under contract 2018-07152, the Swedish Governmental Agency for Innovation Systems under contract 2018-04969, and Formas under contract 2019-02496. DanMAX is funded by the NUFI grant no. 4059-00009B. Authors would like to thank Alexander Generalov (MAX IV laboratory) for the help with the python scripts for the XAS data treatment. We gratefully acknowledge Yu-Chin Huang for the ICP measurements, and Qianhui Liu and Alexandra Ulander (Uppsala University) for the useful discussion and help during the beamtime.References
- T. Liu, et al., Rational design of mechanically robust Ni-rich cathode materials via concentration gradient strategy, Nat. Commun., 2021, 12, 6024 CrossRef CAS PubMed.
- B. Ying, et al., Monitoring the Formation of Nickel-Poor and Nickel-Rich Oxide Cathode Materials for Lithium-Ion Batteries with Synchrotron Radiation, Chem. Mater., 2023, 35, 1514–1526 CrossRef CAS PubMed.
- C. Xu, et al,. Bulk fatigue induced by surface reconstruction in layered Ni-rich cathodes for Li-ion batteries, Nat. Mater., 2021, 20, 84–92 CrossRef CAS PubMed.
- M. Wood, et al., Chemical stability and long-term cell performance of low-cobalt, Ni-Rich cathodes prepared by aqueous processing for high-energy Li-Ion batteries, Energy Storage Mater., 2020, 24, 188–197 CrossRef.
- F. Wu, et al., Enabling High-Stability of Aqueous-Processed Nickel-Rich Positive Electrodes in Lithium Metal Batteries, Small, 2022, 2203874 CrossRef CAS PubMed.
- S. Radloff, G. Carbonari, R. G. Scurtu, M. Hölzle and M. Wohlfahrt-Mehrens, Fluorine-free water-based Ni-rich positive electrodes and their performance in pouch- and 21700-type cells, J. Power Sources, 2023, 553, 232253 CrossRef CAS.
- S. Radloff, R.-G. Scurtu, M. Hölzle and M. Wohlfahrt-Mehrens, Applying Established Water-Based Binders to Aqueous Processing of LiNi0.83Co0.12Mn0.05O2 Positive Electrodes, J. Electrochem. Soc., 2021, 168, 100506 CrossRef CAS.
- D. Pritzl, et al., Editors' Choice—Washing of Nickel-Rich Cathode Materials for Lithium-Ion Batteries: Towards a Mechanistic Understanding, J. Electrochem. Soc., 2019, 166, A4056–A4066 CrossRef CAS.
- L. Hartmann, D. Pritzl, H. Beyer and H. A. Gasteiger, Evidence for Li+/H+ Exchange during Ambient Storage of Ni-Rich Cathode Active Materials, J. Electrochem. Soc., 2021, 168, 70507 CrossRef CAS.
- M. Bichon, D. Sotta, E. De Vito, W. Porcher and B. Lestriez, Performance and ageing behavior of water-processed LiNi0.5Mn0.3Co0.2O2/Graphite lithium-ion cells, J. Power Sources, 2021, 483, 229097 CrossRef CAS.
- M. Bichon, et al., Study of Immersion of LiNi0.5Mn0.3Co0.2O2 Material in Water for Aqueous Processing of Positive Electrode for Li-Ion Batteries, ACS Appl. Mater. Interfaces, 2019, 11, 18331–18341 CrossRef CAS PubMed.
- M. Lee, et al., Powder Coatings via Atomic Layer Deposition for Batteries: A Review, Chem. Mater., 2022, 34(8), 3639–3587 CrossRef.
- M. Yoon, et al., Reactive boride infusion stabilizes Ni-rich cathodes for lithium-ion batteries, Nat. Energy, 2021, 6, 362–371 CrossRef CAS.
- Y. Yoon, S. Shin and M. W. Shin, Fundamental Understanding of the Effect of a Polyaniline Coating Layer on Cation Mixing and Chemical States of LiNi0.9Co0.085Mn0.015O2 for Li-Ion Batteries, ACS Appl. Polym. Mater., 2023, 5(2), 1344–1353 CrossRef CAS.
- H. G. Song, J. Y. Kim, K. T. Kim and Y. J. Park, Enhanced electrochemical properties of Li(Ni0.4Co0.3Mn0.3)O2 cathode by surface modification using Li3PO4-based materials, J. Power Sources, 2011, 196, 6847–6855 CrossRef CAS.
- F. Wu, et al., Enabling High-Stability of Aqueous-Processed Nickel-Rich Positive Electrodes in Lithium Metal Batteries, Small, 2022, 18, 2203874 CrossRef CAS PubMed.
- N. Loeffler, et al., In Situ Coating of Li[Ni0.33Mn0.33Co0.33]O2Particles to Enable Aqueous Electrode Processing, ChemSusChem, 2016, 9, 1112–1117 CrossRef CAS PubMed.
- W. Bauer, F. A. Çetinel, M. Müller and U. Kaufmann, Effects of pH control by acid addition at the aqueous processing of cathodes for lithium ion batteries, Electrochim. Acta, 2019, 317, 112–119 CrossRef CAS.
- I. Doberdò, et al., Enabling aqueous binders for lithium battery cathodes – carbon coating of aluminum current collector, J. Power Sources, 2014, 248, 1000–1006 CrossRef.
- A. Preobrajenski, et al., FlexPES: a versatile soft X-ray beamline at MAX IV Laboratory, J. Synchrotron Radiat., 2023, 30, 831–840 CrossRef CAS PubMed.
- C. D. Quilty, et al., Elucidating Cathode Degradation Mechanisms in LiNi0.8Mn0.1Co0.1O2 (NMC811)/Graphite Cells Under Fast Charge Rates Using Operando Synchrotron Characterization, J. Electrochem. Soc., 2022, 169, 020545 CrossRef CAS.
- C. Tian, et al., Distinct Surface and Bulk Thermal Behaviors of LiNi0.6Mn0.2Co0.2O2 Cathode Materials as a Function of State of Charge, ACS Appl. Mater. Interfaces, 2020, 12, 11643–11656 CrossRef CAS PubMed.
- O. Gustafsson, A. Schökel and W. R. Brant, Design and Operation of an Operando Synchrotron Diffraction Cell Enabling Fast Cycling of Battery Materials, Batteries Supercaps, 2021, 4, 1599–1604 CrossRef CAS.
- M. Wojdyr, Fityk: a general-purpose peak fitting program, J. Appl. Crystallogr., 2010, 43, 1126–1128 CrossRef CAS.
- W. Lee, et al., Destabilization of the Surface Structure of Ni-Rich Layered Materials by Water-Washing Process, Energy Storage Mater., 2022, 44, 441–451 CrossRef.
- H. Liu, et al., Understanding the Roles of Tris(trimethylsilyl) Phosphite (TMSPi) in LiNi0.8Mn0.1Co0.1O2 (NMC811)/Silicon–Graphite (Si–Gr) Lithium-Ion Batteries, Adv. Mater. Interfaces, 2020, 7, 2000277 CrossRef CAS.
- L. Shaw and M. Ashuri, Coating – A potent method to enhance electrochemical performance of Li(NixMnyCoz)O2 cathodes for Li-ion batteries, Adv. Mater. Lett., 2019, 10, 369–380 CrossRef CAS.
- X. Ou, et al., Enabling high energy lithium metal batteries via single-crystal Ni-rich cathode material co-doping strategy, Nat. Commun., 2022, 13, 2319 CrossRef CAS PubMed.
- M. Hekmatfar, A. Kazzazi, G. G. Eshetu, I. Hasa and S. Passerini, Understanding the Electrode/Electrolyte Interface Layer on the Li-Rich Nickel Manganese Cobalt Layered Oxide Cathode by XPS, ACS Appl. Mater. Interfaces, 2019, 11, 43166–43179 CrossRef CAS PubMed.
- Z.-S. Wu, et al., Regulating Electrode/Electrolyte Interfacial Chemistry Enables 4.6 V Ultra-Stable Fast Charging of Commercial LiCoO2, Energy Environ. Sci., 2024, 17, 3021–3031 RSC.
- H. Chen, et al., Investigating Surface Reactivity of a Ni-Rich Cathode Material toward CO2, H2O, and O2 Using Ambient Pressure X-ray Photoelectron Spectroscopy, ACS Appl. Energy Mater., 2023, 6, 11458–11467 CrossRef CAS.
- J. H. Lim, Y. Myung, M. Yang and J. W. Lee, Facile Formation of a LiF-Carbon Layer as an Artificial Cathodic Electrolyte Interphase through Encapsulation of a Cathode with Carbon Monofluoride, ACS Appl. Mater. Interfaces, 2021, 13, 31741–31748 CrossRef CAS PubMed.
- H. Zhang, et al., Enhancing Thermal and High-Voltage Cycling Stability of Ni-Rich Layered Cathodes through a Ti-Doping-Induced Surface-Disordered Structure, ACS Appl. Energy Mater., 2022, 5, 12673–12681 CrossRef CAS.
- S. Park, et al., Replacing conventional battery electrolyte additives with dioxolone derivatives for high-energy-density lithium-ion batteries, Nat. Commun., 2021, 12, 1–12 CrossRef PubMed.
- V. S. A. Piriya, et al., Synergistic Role of Electrolyte and Binder for Enhanced Electrochemical Storage for Sodium-Ion Battery, ACS Omega, 2018, 3, 9945–9955 CrossRef CAS PubMed.
- J. a Lawton, et al., A Major Constituent of Brown Algae for, Science, 2011, 334, 75–79 CrossRef PubMed.
- C. Yang, et al., Phosphate-Rich Interface for a Highly Stable and Safe 4.6 V LiCoO2 Cathode, Adv. Mater., 2023, 35, 2210966 CrossRef CAS PubMed.
- U. Boesenberg, et al., Asymmetric pathways in the electrochemical conversion reaction of NiO as battery electrode with high storage capacity, Sci. Rep., 2014, 4, 1–9 Search PubMed.
- H. S. Liu, Z. R. Zhang, Z. L. Gong and Y. Yang, Origin of Deterioration for LiNiO[sub 2] Cathode Material during Storage in Air, Electrochem. Solid-State Lett., 2004, 7, A190 CrossRef CAS.
- S. Radloff, et al., Mitigating water-induced surface degradation in water-based Ni-rich Li-ion battery electrodes, J. Power Sources, 2023, 580, 233314 CrossRef CAS.
- Y. Chen, et al., Armoring LiNi1/3Co1/3Mn1/3O2 Cathode with Reliable Fluorinated Organic–Inorganic Hybrid Interphase Layer toward Durable High Rate Battery, Adv. Funct. Mater., 2020, 30, 1–12 Search PubMed.
- K. Kleiner, et al., Unraveling the Degradation Process of LiNi0.8Co0.15Al0.05O2 Electrodes in Commercial Lithium Ion Batteries by Electronic Structure Investigations, ACS Appl. Mater. Interfaces, 2015, 7, 19589–19600 CrossRef CAS PubMed.
- Generated Using CrystalDiffract®: A Powder Diffraction Program for Mac and Windows, CrystalMaker Software Ltd, Oxford, England, https://www.crystalmaker.com Search PubMed.
- H. H. Ryu, K. J. Park, C. S. Yoon and Y. K. Sun, Capacity fading of Ni-rich Li[NixCoyMn1−x−y]O2 (0.6 ≤ x ≤ 0.95) Cathodes for High-Energy-Density Lithium-Ion Batteries: Bulk or Surface Degradation?, Chem. Mater., 2018, 30, 1155–1163 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta03592e |
| This journal is © The Royal Society of Chemistry 2024 |