
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Conductive carbon embedded beneath cathode active material for longevity of solid-state batteries†
Young-Woon
Byeon‡
a,
Sizhuo
Yang‡
b,
Guang
Yang
 c,
Dong-Min
Kim
b,
Venkata Sai
Avvaru
a,
Tofunmi
Ogunfunmi
d,
Mary
Scott
bd,
Brett A.
Helms
ab,
Jeffrey
Urban
*b and
Haegyeom
Kim
*a
c,
Dong-Min
Kim
b,
Venkata Sai
Avvaru
a,
Tofunmi
Ogunfunmi
d,
Mary
Scott
bd,
Brett A.
Helms
ab,
Jeffrey
Urban
*b and
Haegyeom
Kim
*a
aMaterials Sciences Division, Lawrence Berkeley National Laboratory, Berkeley, CA, USA. E-mail: haegyumkim@lbl.gov
bMolecular Foundry Division, Lawrence Berkeley National Laboratory, Berkeley, CA, USA. E-mail: jjurban@lbl.gov
cChemical Sciences Division, Oak Ridge National Laboratory, Oak Ridge, TN, USA
dMaterials Science and Engineering, University of California, Berkeley, CA, USA
First published on 4th March 2024
Abstract
A composite structure was developed for use in all-solid-state batteries that consists of a conductive 3D reduced graphene oxide framework embedded beneath cathode active material particles. This unique structure offers significant advantages when combined with a sulfide solid electrolyte as the heterogeneous distribution of the conductive carbon in the composite cathode ensures good contact between the carbon and cathode particles for facile electron transfer while a direct contact between the carbon and sulfide solid electrolyte is avoided or minimized. This approach assists in preventing or reducing unwanted irreversible faradaic reactions. As a result, the newly developed composite of cathode particles decorated on a 3D reduced graphene oxide framework delivers higher specific capacity with improved cycling stability compared with a typical composite cathode consisting of a homogenous mixture of the cathode active material, carbon nanofibers, and sulfide solid electrolyte.
 Haegyeom Kim | Haegyeom Kim is a Career Staff Scientist at the Materials Sciences Division of Lawrence Berkeley National Laboratory (LBNL). He received his PhD in 2015 from Seoul National University and was a postdoctoral researcher at the LBNL until early 2019. His research interest lies in the materials design for energy storage and conversion materials based on the fundamental understanding of the synthesis process-structure-property relationship. He has published more than 95 peer-reviewed articles and 6 patents until now. He was selected as a Clarivate's ‘Highly Cited Researcher (HCR)’, and won several awards, including 2023 ACS Materials Au Rising Star, Young Scientist Award from the International Society for Solid-State Ionics, ECS Battery Division Postdoctoral Associate Research Award. |
1. Introduction
All-solid-state batteries (ASSBs) hold great promise, including improved safety and higher energy density, compared with conventional lithium-ion batteries (LIBs) as the non-flammable solid electrolyte (SE) can be combined with an energy-dense lithium-metal anode.1–3 Given these technical advantages, various types of SEs have been studied and developed, including sulfides, halides, and oxides as well as their polymer composites.4–7 Among these materials, sulfide-based SEs exhibit superior ionic conductivity of 10−3 to 10−4 S cm−1 at room temperature, which is even comparable to that of liquid organic electrolytes.4,8 However, sulfide-based SEs have a narrow electrochemical stability window9 and poor chemical/electrochemical compatibility with electrodes,3,10–14 which remain grand challenges.Argyrodite (Li6PS5X, X = Cl, Br) SEs possess both high ionic conductivity and relatively good interfacial compatibility against a lithium-metal anode compared with other sulfide SEs such as Li10GeP2S12 and Li3PS4 electrolytes.15,16 However, in the composite cathode, both the cathode active material (CAM)/argyrodite SE and carbon/argyrodite SE interfaces are not thermodynamically stable.10,17,18 Recent studies have demonstrated that inorganic coating layers such as LiNbO3 (LNO),19 Li2O–ZrO2 (LZO),20 and Li1.175Nb0.645Ti0.4O3 (LNTO)21 can suppress the degradation at the CAM/argyrodite SE interface. Whereas cation substitutions barely affect their intrinsic oxidative electrochemical stability when sulfur remains a major part of the chemical composition,22 partial substitution of oxygen with sulfur has been suggested to increase the oxidation limit.23,24 Coating of halide SEs with higher oxidative stability on the CAM also results in improved cycling stability as the formation of a detrimental interface between the CAM and argyrodite SE can be avoided, and the halide SE functions as a Li-conducting buffer layer in this composite cathode design.25
The use of an electronically conductive carbon additive in the composite cathode is necessary to provide facile electron transfers to the CAMs and create uniform current distributions. However, sulfide SEs, including argyrodites, are readily decomposed when they form an interface with the conductive carbon, and the decomposition is even accelerated when the composite cathode is charged to high voltage.26 Recent studies show that the morphology and quality (i.e., defects and functional groups) of carbon significantly affect the electrochemical performance of ASSBs.18,27–29 For example, Darren et al. showed that the low-surface-area carbon (vapor-grown carbon fiber) delivered a lower irreversible capacity in the composite cathode than high-surface-area carbon (carbon black).18 Park et al. demonstrated that the use of graphitized carbon with fewer functional groups in the composite cathode improved the reversible capacity and cycling stability.29
Although conductive carbon coating on the CAM surface is frequently used in conventional LIBs to improve the electronic conductivity of the cathode,30,31 this approach is not suitable for ASSBs using sulfide SEs because the coated carbon must contact the SEs, which would lead to detrimental decomposition reactions (Fig. 1a). Therefore, low-surface-area carbon nanofibers (CNFs) or carbon nanotubes (CNTs) are widely used in the composite cathode of ASSBs.17,18,20,32 These low-surface-area carbon additives can reduce the interfacial area between carbon and SEs.18 However, the homogeneously mixed CNFs with CAM and SE cannot prevent direct contact between the carbon additive and SEs, and we cannot control these interfaces (Fig. 1b). In a very recent study, Lee and colleagues developed zinc oxide (ZnO)-coated CNF to reduce the contact area between the carbon additive and SEs.17
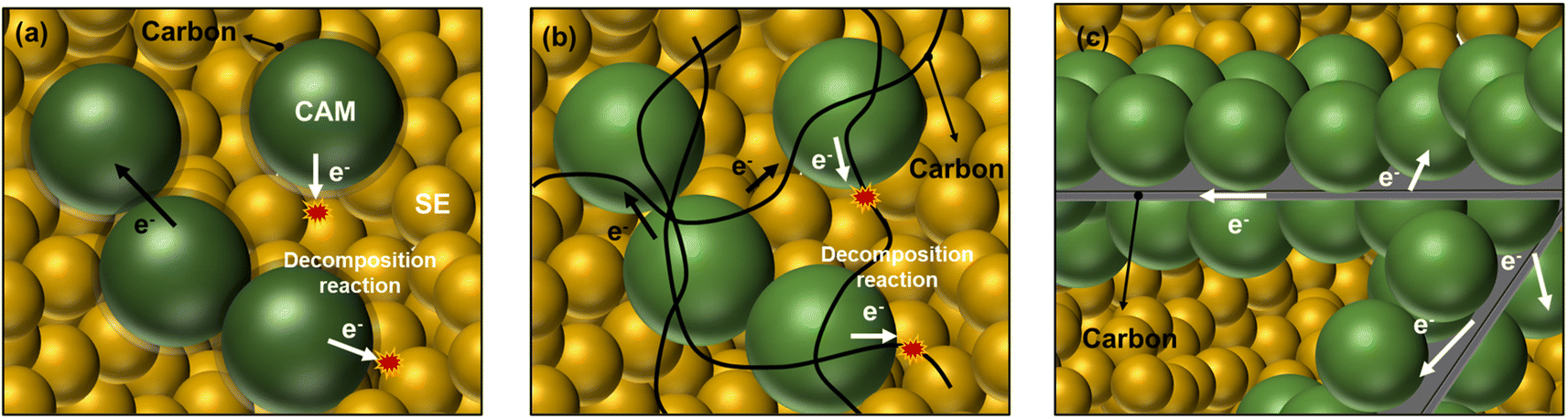 | ||
| Fig. 1 Schematics of carbon–CAM composite with SEs. (a) Carbon-coated CAM mixture with SEs. (b) Mixture of CAM, CNF, and SE. (c) Carbon embedded beneath CAM and its mixture with SE. | ||
In the current study, an unconventional CAM–carbon structure was designed where an electronically conductive carbon framework is embedded beneath CAM particles, as illustrated in Fig. 1c. In this unique structure, electron transfers to CAM particles can be facilitated through the carbon framework while the detrimental interface between the carbon additive and SE can be avoided or at least minimized when this CAM–carbon structure is mixed with a SE. Herein, we applied this strategy to LiNbO3 (LNO)-coated polycrystalline LiNi0.8Mn0.1Co0.1O2 (pc-NMC) and LNO-coated single-crystalline LiNi0.6Mn0.2Co0.2O2 (sc-NMC622), and these materials demonstrated improved cycling performance.
2. Experimental section
2.1. Materials synthesis
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) solution. Then, 1000 mg of pc-NMC811 (or sc-NMC622) particles were dispersed in that solution at a concentration of 50 mg mL−1. The dispersion was ultrasonically treated for 1 h and then stirred at 600 rpm at RT for 24 h. The products were obtained and washed with MeOH and acetone. LNO-coated pc-NMC811 and sc-NMC622 are purchased from MSE supplies LLC.
:1 ratio) in H2O and sonicated for 1 h. Then, 57% hydrogen iodide (HI) was added to the dispersion and stirred for 2 h at 90 °C. After cooling down to RT, the composite was washed with water and MeOH to obtain the desired RGO–carbon composite.
:1) solution. Then, 1000 mg of pc-NMC811 (or sc-NMC622) particles were dispersed in that solution at a concentration of 50 mg mL−1. The dispersion was ultrasonically treated for 1 h and then stirred at 600 rpm at RT for 24 h. The products were obtained and washed with MeOH and acetone. LNO-coated pc-NMC811 and sc-NMC622 are purchased from MSE supplies LLC.
:1 ratio) in H2O and sonicated for 1 h. Then, 57% hydrogen iodide (HI) was added to the dispersion and stirred for 2 h at 90 °C. After cooling down to RT, the composite was washed with water and MeOH to obtain the desired RGO–carbon composite.
2.2. Characterization
X-ray diffraction (XRD) analysis was conducted using a Rigaku Miniflex 600 equipped with a Cu Kα radiation source. The surface morphology and microstructure of the samples were examined using scanning electron microscopy (SEM; PHENOM PW-100-017 or FEI Quanta FEG-250). Energy-dispersive X-ray spectroscopy (EDS) elemental mappings were obtained using transmission electron microscopy (TEM; FEI TitanX 60-300). The Kα X-ray edges were used to construct a map of each element (C, N, O, Si, Mn, Co, Ni, Cu, and Nb) to avoid energy overlaps between certain elements in the samples.Thermogravimetric analysis (TGA) was performed with a TGA5500 (TA instruments) using an aluminum pan. The measurements were conducted at a heating rate of 5 °C min−1 from 40 °C to 650 °C after drying at 120 °C for 1 h before measurement to remove absorbed moisture on the sample surface. Fourier-transform infrared (FTIR) spectra were recorded on a Nicolet iS50 FT-IR system (Thermo Fisher, USA). Raman measurements were performed using a Renishaw Invia Raman spectrometer equipped with a laser of 514.5 nm excitation wavelength. Raman mapping measurements were conducted on a confocal Raman spectrometer (Horiba, Xplorer 405 nm edge laser, long working distance objective = 50×, grating with 2400 grooves per mm, numerical aperture (N.A.) = 0.42, local power < 500 μW). The laser spot diameter was estimated to be 1 μm. The scan region was set to 80 × 40 μm2, with a 1 μm2 per pixel step size. The integration time was set to 2 s for each point. Autofocus mode was applied at each XY point to ensure consistent focus on the sample surface. All the Raman mappings were analyzed using Horiba Labspec 6 imaging and spectroscopy software, and the K-means clustering algorithm was integrated into the Scikit-learn platform using a previously reported similar method.33,34 The SEM images of the composite cathodes were obtained from the cross-sectioned particles using a Scios Dual Beam focused-ion beam (FIB)/SEM (FEI/Thermo Fisher) at the QB3 facility at the University of California, Berkeley. The composite cathode pellets were first cross-sectioned using the Ga+ ion beam for material. The composite cathode pellets were milled with a beam current of 0.9–6.5 nA and then polished with a beam current of 93 pA to 0.46 nA. SEM/EDX analysis was conducted on the milled samples.
2.3. Electrochemical testing
The all-solid-state Li-metal batteries were constructed using a commercial battery cell enclosure system (model WCBR-SLD-S, Wellcos Inc.). Firstly, 50 mg of LPSCl powder (D50 ∼ 1 μm, Ampcera Inc.) was pressed using two stainless-steel rods at a pressure of 100 MPa to fabricate a thin LPSCl pellet. To fabricate the composite cathodes, the synthesized NMC/RGO composite (78 wt%) was homogeneously mixed with LPSCl powder (22 wt%) and then uniformly applied on the LPSCl solid-electrolyte layer. For the reference NMC/CNF composite, NMC and CNF were initially mixed to achieve homogeneity, followed by the subsequent addition of LPSCl powder to make the composite. The composite cathode and LPSCl SE layer were pressed under 250 MPa to secure contact between the composite cathode and SE layers. To add the anode layer, an In metal disk was first placed onto the other side of the LPSCl pellet, followed by placing a Li-metal disk onto the indium (In) disk (composition: Li0.22In0.78). In our experiments, we used 7.0–7.3 mg of CAM for each cell, which corresponds to the CAM loading density of 9.1–9.5 mg cm−2. The assembled ASSB cell was then pressed under 30 MPa during the galvanostatic cycling performed in a voltage window of 2.5–4.3 V (vs. Li/Li+). All electrochemical testing was conducted using a Maccor Model 2200 cycler at 50 °C.3. Results
3.1. Polycrystalline LiNi0.8Mn0.1Co0.1O2/reduced graphene oxide framework composite
Fig. 2 illustrates the fabrication process for the CAM-3D reduced graphene oxide (RGO) framework composite. First, a porous 3D RGO framework was prepared by dispersing acid-treated porous carbon and single-layer graphene oxide (GO) in H2O. The objective of the acid treatment of the porous carbon was to improve its dispersion ability in H2O, which aids the hybridization with single-layer GO. After that, the porous 3D RGO framework was reduced by HI aqueous solution. The porous 3D RGO framework exhibits graphitic features, as confirmed by XRD (see ESI Fig. S1a†). Typical D- and G-bands of RGO are confirmed at ∼1340 and ∼1580 cm−1, respectively, by Raman spectroscopy (ESI Fig. S1b†). The ID/IG ratio of the RGO framework is estimated to be 0.59. ESI Fig. S1c† presents SEM images of the 3D RGO framework where graphene flakes are attached on the porous carbon framework. In this structure, porous carbon is the framework and RGO is added to increase the electrical conductivity. After the reduction, the obtained 3D RGO framework was mixed with PEI-modified NMC in MeOH. To synthesize PEI-modified NMC particles, we used PVP and PEI polymers in which PVP was used to prevent NMC particle agglomeration and PEI was used to create a positively charged surface layer on NMC particles. The resulting mixture of PEI-modified NMC and 3D RGO framework was sonicated for 2 h and stirred overnight to produce the desired CAM–carbon composite. In this process, the PEI-coated NMC particles, which have positive surface charges attracts the 3D RGO framework with the negatively charged surface. The presence of the PEI coating on pc-NMC811 was confirmed by the N–H stretching and bending peaks in the IR analysis, as illustrated in ESI Fig. S2a† and the EDS mapping in ESI Fig. S2b.† We also confirm that the pc-NMC811 particles and their morphologies were not damaged by the PEI coating process, as shown in the SEM images in ESI Fig. S2c and d.†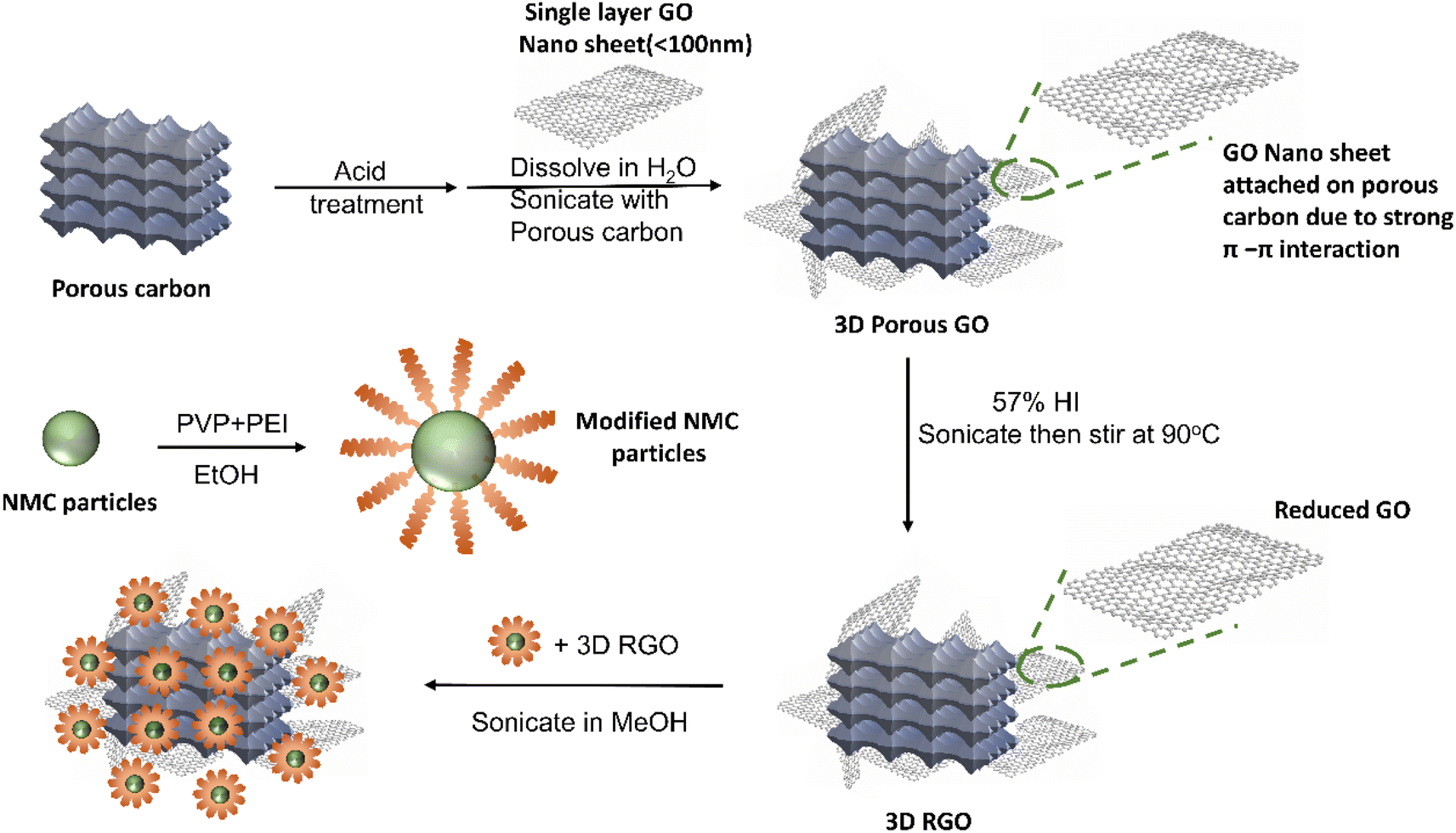 | ||
| Fig. 2 Schematic of the fabrication of the CAM–RGO framework composite. | ||
Following the synthesis process described above, we developed LiNbO3 (LNO)-coated polycrystalline LiNi0.8Mn0.1Co0.1O2/RGO composites (hereafter, pc-NMC811/RGO) with varied carbon contents. The target carbon contents were 0.5 wt%, 1.0 wt%, 2.0 wt%, and 4.0 wt%. The carbon contents in the pc-NMC811/RGO composites were estimated using TGA as shown in Fig. 3a. The weight loss from 100 °C to 650 °C of the pc-NMC811/RGO is attributable to the decomposition of any carbon in the composite, which includes RGO and porous carbon.35 Hereafter, we used the RGO content to refer to the carbon conent in the composite structure for simplicity. The RGO contents of the composites are 0.52 wt%, 1.6 wt%, 3.5 wt%, and 5.2 wt%, respectively. The RGO contents calculated from the TGA results are used in the following sections. Fig. 3b presents XRD patterns of pc-NMC811 and its RGO composites, without any noticeable impurity phases or structure changes of pc-NMC811 after the synthesis. Fig. 3c–f presents SEM images of the pc-NMC811/RGO composites with varied carbon contents. We found that the 3D RGO framework is well covered by pc-NMC811 cathode particles (secondary particle size: 5–20 μm), and only a limited surface of the RGO framework remained uncovered by pc-NMC811 when the RGO content is below 5.2 wt%, as confirmed by the SEM images. In contrast, 3D-RGO is not fully covered by pc-NMC811 particles when a relatively high RGO content of 5.2 wt% is used (Fig. 3f). To demonstrate that pc-NMC811 particles are selectively attached on the 3D-RGO framework in our synthesis process, we synthesized the control group where pc-NMC811 without PEI coating was mixed with 3D-RGO. In this control group, we found that pc-NMC811 particles are randomly distributed and that some of the pc-NMC811 particles are not attached on the 3D-RGO framework (ESI Fig. S3a†). In contrast, all the PEI-coated pc-NMC811 particles are covered on the 3D-RGO framework (ESI Fig. S3b†), which is clear evidence that the PEI coating on pc-NMC811 particles plays a key role in creating good contact between the pc-NMC811 particles and 3D-RGO framework.
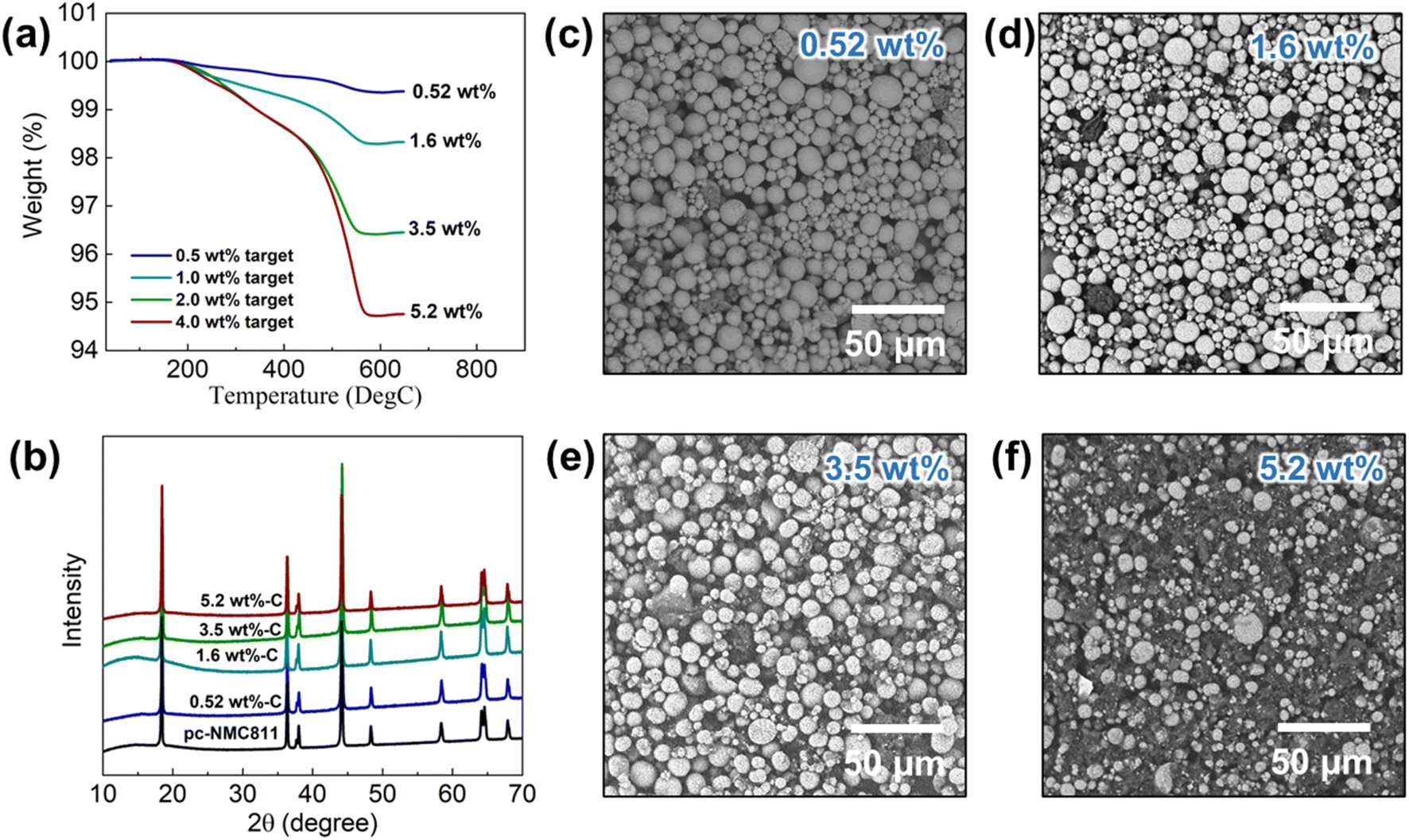 | ||
| Fig. 3 Materials characterization of pc-NMC811/RGO composites at varied RGO contents. (a) TGA results, (b) XRD patterns, and (c)–(f) SEM images of pc-NMC811/RGO composites. | ||
To evaluate our hypothesis that using conductive carbon embedded beneath cathode particles can reduce the interfacial area between the conductive carbon and SEs in the composite cathode, we mixed the pc-NMC811/RGO composite with LPSCl SE, pelletized the mixture, and then conducted Raman mapping as shown in Fig. 4a. All Raman mapping frames were analyzed using the K-means clustering algorithm, which has been proven effective in extracting heterogeneous chemical and structural information in several previous studies.36,37 The Raman peak intensities were normalized by the highest peak intensity from LPSCl or its decomposed products at 400–600 cm−1.38 The lower part of the mapping area consists solely of LPSCl SE. In Fig. 4a, the carbon-rich areas with a relatively high intensity of Raman peaks at 1200–1800 cm−1, which are typical Raman peaks from carbon (i.e., D-band at 1377 cm−1 and G-band at 1584 cm−1), are colored red.39,40 The average domain size of the carbon-rich area is 10–20 μm, which is consistent with the pc-NMC811 particle size. Since pc-NMC811 particles are decorated on the RGO framework, as confirmed by the SEM images in Fig. 3, we expect pc-NMC811 particles to be present in the carbon-rich areas. The green areas, showing a reduced intensity of the typical Raman carbon peaks, correspond to LPSCl-rich domains. Due to overlapping Raman peaks of pc-NMC811 and LPSCl, distinguishing pc-NMC811 particles from LPSCl in the composite cathode is not feasible.38,41 Nevertheless, our results confirm that the distribution of the carbon framework is highly heterogeneous in the composite cathode, which suggests a reduced contact area between the RGO framework and LPSCl SE. We expect that the contact area (if any) between RGO and LPSCl SE will still lead to unwanted side reactions because we did not control the functionalities of the carbon. The gray, brown, and orange colors in Fig. 4 indicate LPSCl only areas from the LPSCl SE. Each color has a slightly different peak shape at Raman shift below 700 cm−1, which is attributable to the distinct local structural environments. In contrast, Fig. 4b displays a composite cathode where CNFs were randomly mixed with LPSCl SE and pc-NMC811 particles using a mortar and pestle. In this control group, a homogeneous carbon-rich Raman response (red color) is observed throughout the mapping, indicating an even distribution of CNFs in the composite cathode. We expect that the homogeneous distribution of carbon in the composite cathode may facilitate uniform electron transfer throughout the composite cathode; however, it does not prevent detrimental contact between carbon and the LPSCl SE. These findings indicate that our composite cathode design, featuring conductive carbon embedded beneath the cathode active material, can effectively reduce the interfacial area between carbon and the LPSCl SE. We anticipate that carbon embedded beneath the cathode particles can still provide facile electron transport pathways to the cathode particles.
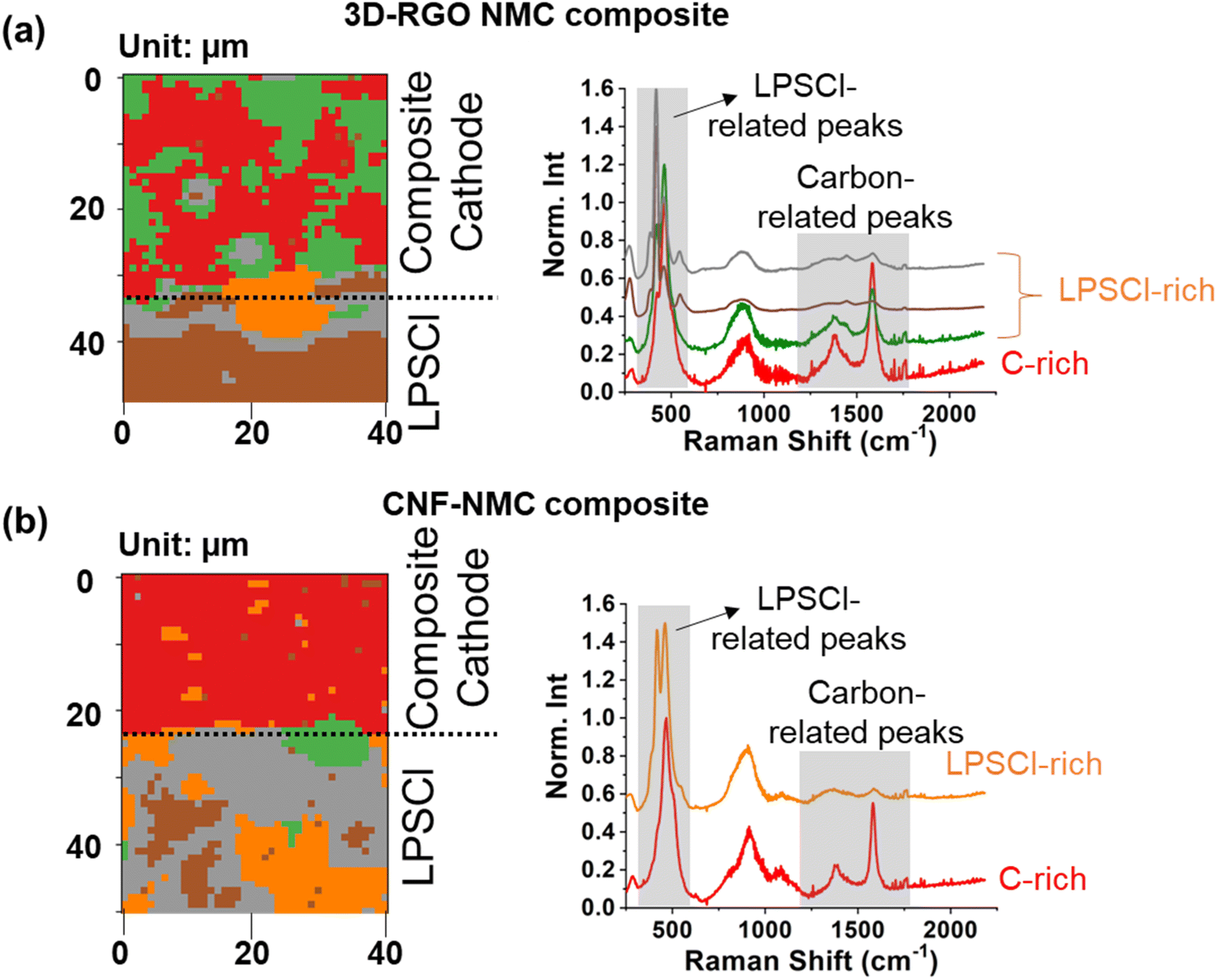 | ||
| Fig. 4 Raman mapping based on K-means clustering analysis for (a) composite cathode consisting of pc-NMC811/RGO and LPSCl SE and (b) composite cathode consisting of pc-NMC811, CNF, and LPSCl SE. The black dotted line indicates the location of the composite cathode|LPSCl interface. | ||
Fig. 5a presents charge–discharge profiles of the pc-NMC811/RGO composite cathodes with varied RGO contents, a CNF composite cathode, and a cathode without carbon additive. The composite cathodes were prepared by mixing RGO composites (78 wt%) and LPSCl SEs (22 wt%). We used Li–In (composition: Li0.22In0.78) alloy anodes, and the voltage was converted to vs. Li/Li+. All the solid-state battery cells were tested at 50 °C under 30 MPa. The composite cathode with CNF shows a relatively large charge capacity in the low-voltage regions below 3.5 V (vs. Li/Li+), whereas the control group without carbon additive shows no capacity in the low-voltage regions (indicated by the green box in Fig. 5a). These results confirm that the capacity obtained from the low-voltage regions <3.5 V (vs. Li/Li+) originates from the decomposition of LPSCl at LPSCl/carbon interfaces. Interestingly, unwanted side reactions at <3.5 V (vs. Li/Li+) are greatly suppressed in the RGO composite cathode, as indicated in the highlighted area in Fig. 5a. Fig. 5b summarizes the charge and discharge capacities in the first cycle. The charge and discharge capacities gradually increase as the RGO content increases from 0.52 wt% to 3.5 wt% from 163 to 232 mA h g−1 for charging and from 104 to 164 mA h g−1 for discharging at C/10 (1C = 200 mA g−1). The charge and discharge capacities decrease when the RGO content increases to 5.2 wt%. In addition, the composite cathode with 5.2 wt% RGO has lower coulombic efficiency (59.4%) than the composite cathodes with lower RGO contents (71.2% for 1.6 wt% RGO and 70.6% for 3.5 wt% RGO). Low coulombic efficiency in 5.2 wt% RGO sample could be related to the RGO framework surfaces which are not covered by pc-NMC811 particles that led to the decomposition of LPSCl at the interface during the charge process. The 3.5 wt% RGO and 1.6 wt% RGO composites exhibit improved reversible discharge capacities and coulombic efficiency compared with the composite cathode containing CNF (2 wt%), as shown in Fig. 5b. We also found that the 3.5 wt% RGO and 1.6 wt% RGO composites show lower polarization than the composite cathode containing CNF in Fig. 5a. The improved capacities and coulombic efficiencies can be attributed to the electronically conductive carbon being embedded beneath the pc-NMC811 particles and less carbon surface forming interfaces with LPSCl in the RGO composite system. Fig. 5c shows the cycling stability of the pc-NMC811/RGO composites and control groups. All the RGO composites deliver improved cycling stability compared with the composite cathode with CNF. More specifically, the 3.5 wt% RGO and 1.6 wt% RGO composites maintain 94.7 and 106 mA h g−1, respectively, after 15 cycles. The 0.52 wt% RGO composite exhibits lower discharge capacity in the first cycle than the other RGO composites but it retains the highest capacity of 114.6 mA h g−1 after 15 cycles and 70 mA h g−1 after 30 cycles. Nevertheless, capacity decay was still observed as cycling progressed in the RGO composites. ESI Fig. S4† confirms that some primary NMC811 particles were detached from the secondary particle. In these RGO composites, the detached particles could not be connected to electronically conductive carbon, and their surfaces were not protected by a LNO coating layer, which may have led to capacity degradation upon repeated charge–discharge cycling. Therefore, we extended our study to single-crystal NMC particles, as discussed in the next section.
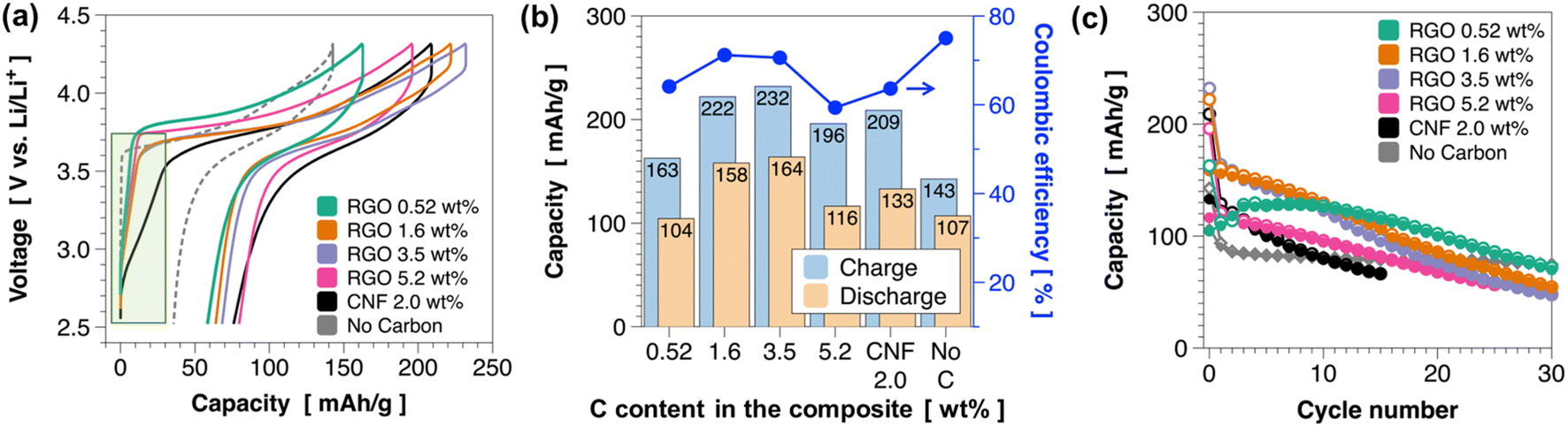 | ||
| Fig. 5 Electrochemical properties of pc-NMC811/RGO composites. (a) The first charge–discharge profiles of pc-NMC811/RGO composites with varied RGO contents. Composite cathodes with CNF and no carbon are used as control groups. (b) Initial charge and discharge capacities of pc-NMC811/RGO composites and control groups. The right y-axis indicates the coulombic efficiency at the first cycle. (c) Cycling stability of pc-NMC811/RGO composites and control groups. | ||
3.2. Single-crystalline LiNi0.6Mn0.2Co0.2O2/RGO framework composite
We used LNO-coated single-crystal NMC622 (sc-NMC622) cathode particles (particle size: 1–5 μm) to create the RGO composite to avoid the issue of particle detachment from the secondary particle agglomerates that occurred in pc-NMC811/RGO composites. The same synthesis procedures that were applied to the pc-NMC811/RGO composites were used for the synthesis of sc-NMC622/RGO composites. Fig. 6a presents the TGA results of the sc-NMC622/RGO composites. The target RGO contents in the synthesis process were 2 wt%, 4 wt%, and 6 wt%. The weight loss from 100 °C to 650 °C of the sc-NMC622/RGO in TGA is attributable to the decomposition of any carbon in the composite, which includes RGO and porous carbon.35 Hereafter, we used the RGO content to refer to the carbon conent in the composite structure for simplicity. The RGO contents were estimated to be 1.6 wt%, 2.0 wt%, and 3.1 wt%, respectively, in the composites. Hereafter, the estimated RGO contents will be used to refer to the samples. No changes in the XRD patterns of sc-NMC622 were observed after the RGO composite synthesis, as shown in Fig. 6b, indicating that the synthesis process does not affect the crystal structure of the sc-NMC622 particles and that decomposition of sc-NMC622 did not occur. Fig. 6c–e display SEM images of sc-NMC622/RGO composites at varied RGO contents. In the 1.6 wt% RGO sample, the RGO surface is fully decorated with sc-NMC622 particles, and it is difficult to observe the RGO surface in Fig. 6c. This structure can reduce the contact area between carbon framework and LPSCl SE in the composite cathode structure. However, we suspect that some sc-NMC622 particles may not be directly connected to the electronically conductive RGO framework due to a high sc-NMC622 loading on the RGO framework, which can increase the polarization in the electrochemical test. When the RGO content increased to 2.0 wt%, some of the RGO was detected under the sc-NMC622 particles in the SEM image (Fig. 6d). When the RGO content increased up to 3.1 wt%, more RGO surfaces were exposed and not covered by sc-NMC622 particles. These uncovered surfaces likely react with the LPSCl SE in the composite cathode.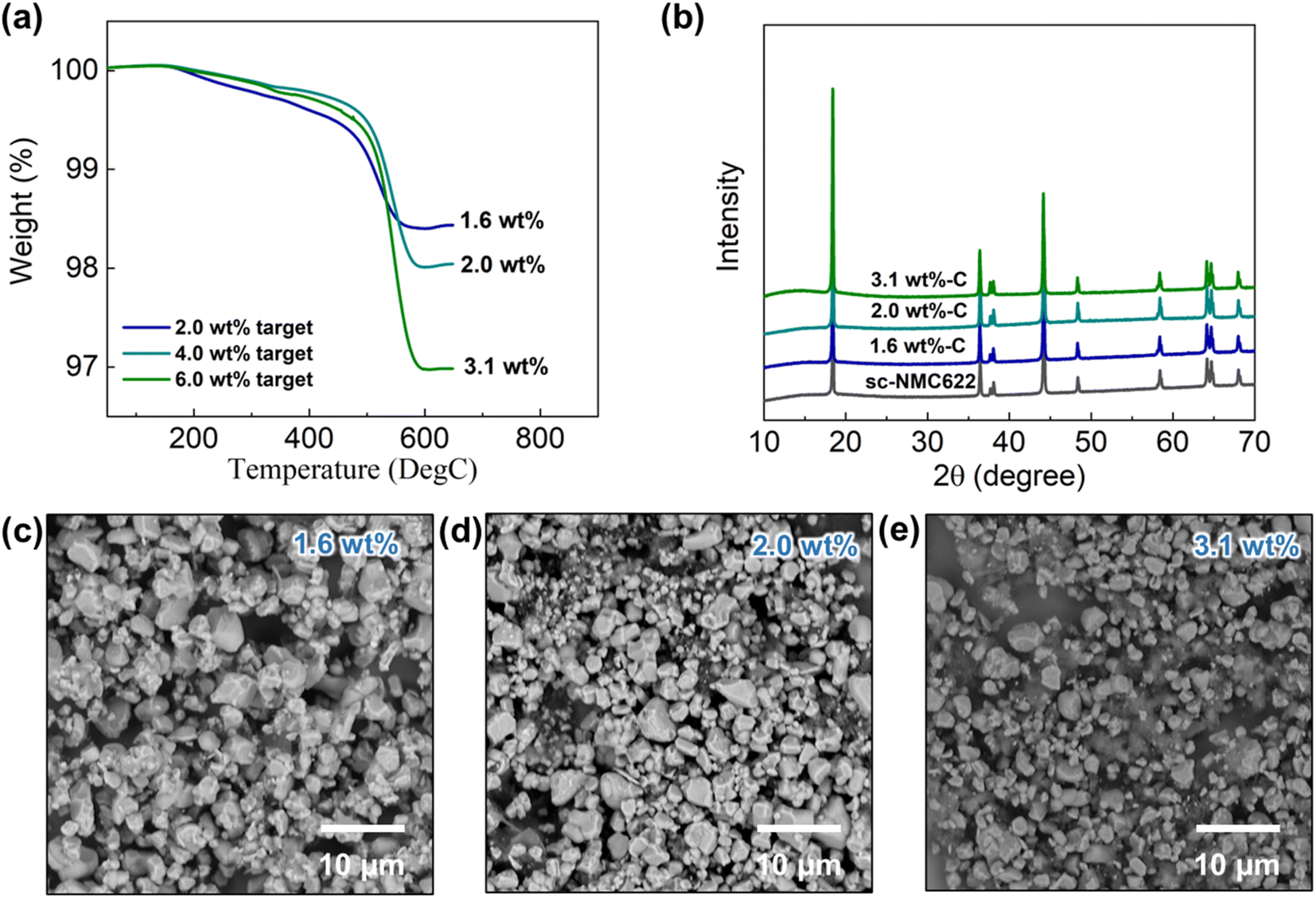 | ||
| Fig. 6 Materials characterization of sc-NMC622/RGO composites at varied RGO contents. (a) TGA results, (b) XRD patterns, and (c)–(e) SEM images of sc-NMC622/RGO composites. | ||
The three sc-NMC622/RGO composites were tested in ASSB cells using LPSCl as the SE and a Li–In (composition: Li0.22In0.78) alloy as the counter electrode. Electrochemical tests were conducted at 50 °C under 30 MPa. Fig. 7a presents charge–discharge profiles of the sc-NMC622/RGO composites with varied RGO contents of 1.6 wt%, 2.0 wt%, and 3.1 wt%. The first charge and discharge capacities are 138.6 mA h g−1 (charge) and 104.4 mA h g−1 (discharge), 183.5 mA h g−1 (charge) and 155.4 mA h g−1 (discharge), and 201.4 mA h g−1 (charge) and 171.0 mA h g−1 (discharge) for the 1.6 wt%, 2.0 wt%, and 3.1 wt% RGO composites, respectively, at C/20 (1C = 170 mA g−1). The first cycle coulombic efficiency of the 1.6 wt%, 2.0 wt%, and 3.1 wt% RGO composites are 75.3%, 84.7%, and 84.9%, respectively. The inset in Fig. 7a shows that the capacity obtained in a relatively low-voltage region (<3.5 V vs. Li/Li+) decreases when the RGO content decreases. This result indicates that the RGO composite with lower RGO content has a lower interfacial area between the RGO framework and LPSCl SE in the composite cathode. However, the RGO composite with low RGO content (1.6 wt%) exhibits a larger polarization than the RGO composites with higher RGO contents (2.0 wt% and 3.1 wt%). These results may indicate that some of the sc-NMC622 particles are not connected to the electronically conductive RGO framework in the 1.6 wt% RGO composite because of the low RGO content. Notably, much lower charge capacity at <3.5 V vs. Li/Li+ was observed in the sc-NMC622/RGO composites compared with the pc-NMC811/RGO composites. This is attributable to the sc-NMC622 particles being well connected to the RGO framework without breaking.
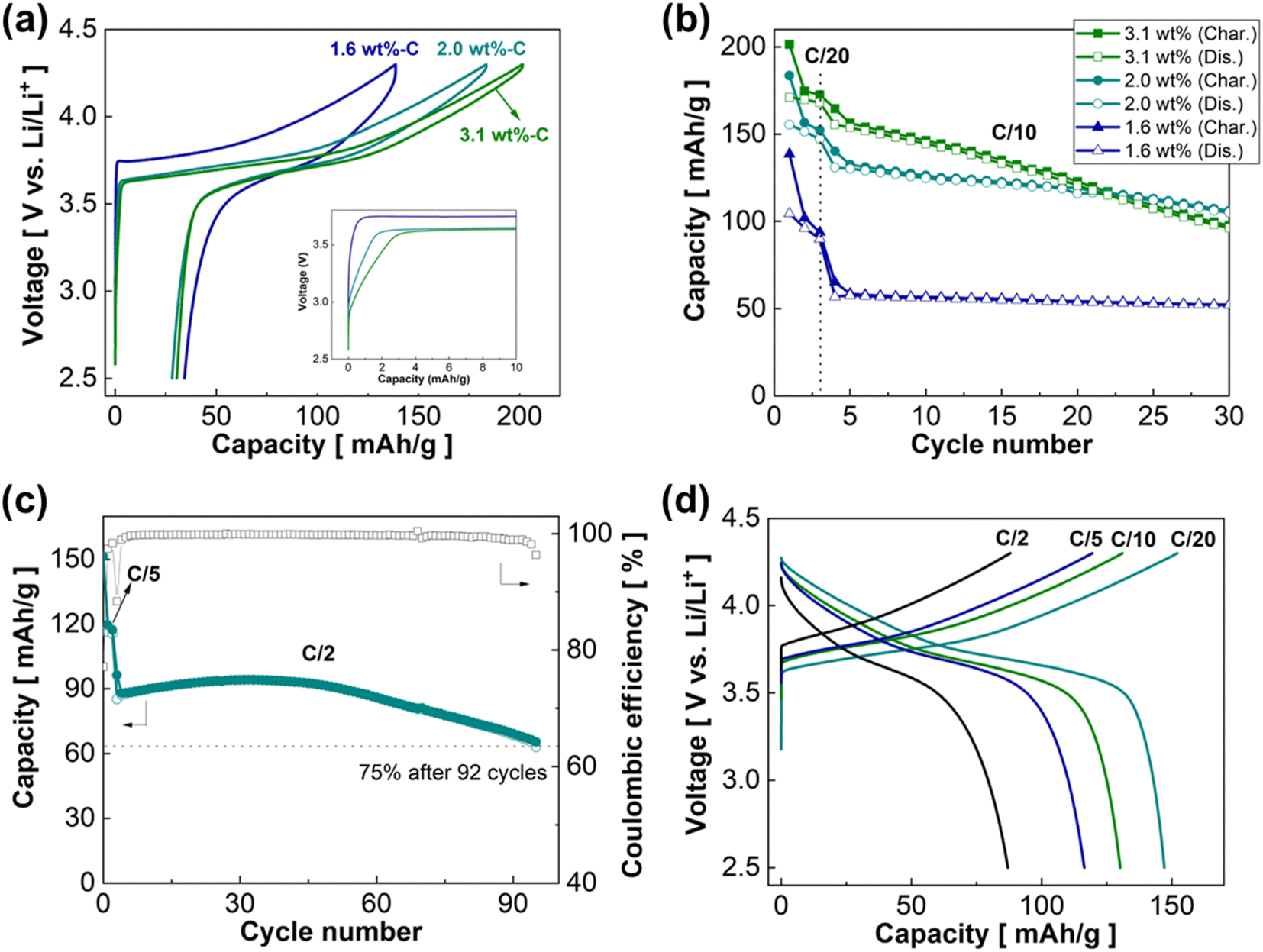 | ||
| Fig. 7 Electrochemical properties of sc-NMC622/RGO composites. (a) Charge–discharge profiles of sc-NMC622/RGO composites with varied RGO contents. Inset: magnified area of the initial charging process up to 10 mA h g−1. (b) Cycling stability of sc-NMC622/RGO composites. (c) Cycling stability of 2.0 wt% RGO composite at C/5 and C/10. (d) Rate capability of 2.0 wt% RGO composite. | ||
Fig. 7b shows the cycling stability of the sc-NMC622/RGO composites. The initial three cycles were performed at C/20, and the current rate increased to C/10 for the following cycles. After 30 cycles, the 3.1 wt% RGO composite maintains a capacity of 96.2 mA h g−1, corresponding to 62% retention at C/10. The 2.0 wt% RGO and 1.6 wt% RGO composites retain capacities of 105.0 and 52.0 mA h g−1, which correspond to capacity retention of 80% and 91.5%, respectively, after 30 cycles at C/10. We found a trade-off in the RGO content of the composite. A high RGO content delivers higher first discharge capacity but shows faster capacity decay over cycles than lower RGO content. Fig. 7c shows the cycling stability of the 2.0 wt% RGO composite at C/2 after the initial three cycles at C/5. The 2.0 wt% RGO composite maintains 75% (63.0 mA h g−1) of the capacity at C/2 after 92 cycles. The first-cycle coulombic efficiency is 77.1% and increases gradually upon cycling up to 99.7–99.96%, which might indicate the formation of passivating cathode-electrolyte interphase (CEI). After 60 cycles, the coulombic efficiency and capacity begins to decrease. Fig. 7d displays the charge–discharge profiles of the 2.0 wt% RGO composite at varied current rates of C/20, C/10, C/5, and C/2. The 2.0 wt% RGO composite delivers capacities of 147.1, 130.2, 116.4, and 87.0 mA g−1 at C/20, C/10, C/5, and C/2, respectively.
4. Discussion
In this work, we developed a new composite structure consisting of an electronically conductive RGO framework embedded beneath CAM particles using both polycrystalline (pc) and single-crystalline (sc) NMC particles. Although both the pc-NMC811/RGO and sc-NMC622/RGO composites delivered reasonably good electrochemical performance, the sc-NMC622/RGO composite exhibited improved cycling stability (Fig. 5 and 7). The different compositions of pc-NMC811 and sc-NMC622 may have affected the electrochemical performance. However, we suspect that the improved cycling stability in the sc-NMC622/RGO composite is mainly attributable to all the sc-NMC622 particles being well attached on the RGO framework and sc-NMC622 not breaking down during the mixing process with LPSCl SE particles. In the pc-NMC811/RGO composite, we observed that some pc-NMC811 particles were broken and that small primary particles were detached from the polycrystalline composite, as shown in ESI Fig. S4,† after mixing with LPSCl SE particles. Because the outer surface of the secondary particles was covered by a LNO coating layer, the primary particles were not protected by LNO after the primary particles were detached from the secondary particles. These unprotected surfaces led to unwanted side reactions with LPSCl SE particles both chemically and electrochemically. In addition, these detached primary particles could not be effectively connected to the RGO framework.Our RGO framework had negatively charged oxygen-containing functional groups at the surface, which were used to attract the positively charged PEI-coated NMC particles (either pc-NMC811 or sc-NMC622). These attractions created a unique structure where an electronically conductive RGO framework was embedded beneath NMC particles. However, it is known that the functional groups and defects in the carbon additive can form a functional-group-induced insulating layer at the SE interface.29 Although most of the surface of the RGO framework was covered by NMC particles, it is expected that some of the surface may form an interface with LPSCl. In this case, the functional groups at the surface of the RGO framework can accelerate decomposition of LPSCl.
5. Conclusion
In this study, we developed a conductive 3D RGO framework embedded beneath the CAM (pc-NMC811 and sc-NMC622). In the composite cathode for the solid-state battery, the newly developed composite of NMC particles decorated on the 3D RGO framework exhibited higher specific capacity and improved cycling stability compared with a typical composite cathode, where the CAM, CNF, and LPSCl SE are homogeneously mixed. In our composite cathode where the NMC/RGO composite and LPSCl SE were mixed, the heterogeneous distribution of the electronically conductive RGO framework, which preferably contacted with NMC particles, prevented the formation of a detrimental interface between RGO framework and LPSCl SE. This study demonstrates the importance of designing carbon–CAM composite structures where the electronically conductive carbon is embedded beneath the CAM to provide facilitated electron pathways while preventing direct contact between the carbon and SE in the composite cathode of solid-state batteries.Author contributions
Y. W. B.: investigation, data curation, visualization, writing-original draft. S. Y.: investigation, data curation, visualization, writing-original draft. G. Y.: investigation, data curation, visualization. D. M. K.: investigation, data curation. V. S. A.: investigation, data curation. T. O.: investigation, data curation. M. S.: supervision. B. H.: supervision. J. U.: funding acquisition, supervision. H. K.: conceptualization, funding acquisition, data curation, investigation, visualization, supervision, writing-original draft. All: writing-review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Assistant Secretary for Energy Efficiency and Renewable Energy, Vehicle Technologies Office, under the Advanced Battery Materials Research (BMR) Program, of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231. Work at the Molecular Foundry was supported by the Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231.References
- Y. Xiao, Y. Wang, S.-H. Bo, J. C. Kim, L. J. Miara and G. Ceder, Understanding interface stability in solid-state batteries, Nat. Rev. Mater., 2020, 5, 105–126 CrossRef CAS
.
- C. Yu, F. Zhao, J. Luo, L. Zhang and X. Sun, Recent development of lithium argyrodite solid-state electrolytes for solid-state batteries: synthesis, structure, stability and dynamics, Nano Energy, 2021, 83, 105858 CrossRef CAS
- A. Banerjee, X. Wang, C. Fang, E. A. Wu and Y. S. Meng, Interfaces and Interphases in All-Solid-State Batteries with Inorganic Solid Electrolytes, Chem. Rev., 2020, 120, 6878–6933 CrossRef CAS
- Q. Zhang, D. Cao, Y. Ma, A. Natan, P. Aurora and H. Zhu, Sulfide-Based Solid-State Electrolytes: Synthesis, Stability, and Potential for All-Solid-State Batteries, Adv. Mater., 2019, 31, 1901131 CrossRef CAS
- Y. Nikodimos, W.-N. Su and B. J. Hwang, Halide Solid-State Electrolytes: Stability and Application for High Voltage All-Solid-State Li Batteries, Adv. Energy Mater., 2023, 13, 2202854 CrossRef CAS
- K. J. Kim, M. Balaish, M. Wadaguchi, L. Kong and J. L. M. Rupp, Solid-State Li–Metal Batteries: Challenges and Horizons of Oxide and Sulfide Solid Electrolytes and Their Interfaces, Adv. Energy Mater., 2021, 11, 2002689 CrossRef CAS
- S. Li, S.-Q. Zhang, L. Shen, Q. Liu, J.-B. Ma, W. Lv, Y.-B. He and Q.-H. Yang, Progress and Perspective of Ceramic/Polymer Composite Solid Electrolytes for Lithium Batteries, Advanced Science, 2020, 7, 1903088 CrossRef CAS
- S. Chen, D. Xie, G. Liu, J. P. Mwizerwa, Q. Zhang, Y. Zhao, X. Xu and X. Yao, Sulfide solid electrolytes for all-solid-state lithium batteries: structure, conductivity, stability and application, Energy Storage Mater., 2018, 14, 58–74 CrossRef
- W. D. Richards, L. J. Miara, Y. Wang, J. C. Kim and G. Ceder, Interface Stability in Solid-State Batteries, Chem. Mater., 2016, 28, 266–273 CrossRef CAS
- S.-K. Jung, H. Gwon, S.-S. Lee, H. Kim, J. C. Lee, J. G. Chung, S. Y. Park, Y. Aihara and D. Im, Understanding the effects of chemical reactions at the cathode–electrolyte interface in sulfide based all-solid-state batteries, J. Mater. Chem. A, 2019, 7, 22967–22976 RSC
- J. Haruyama, K. Sodeyama and Y. Tateyama, Cation Mixing Properties toward Co Diffusion at the LiCoO2 Cathode/Sulfide Electrolyte Interface in a Solid-State Battery, ACS Appl. Mater. Interfaces, 2017, 9, 286–292 CrossRef CAS PubMed
- X. Wu, M. Mirolo, C. A. F. Vaz, P. Novák and M. El Kazzi, Reactivity and Potential Profile across the Electrochemical LiCoO2–Li3PS4 Interface Probed by Operando X-ray Photoelectron Spectroscopy, ACS Appl. Mater. Interfaces, 2021, 13, 42670–42681 CrossRef CAS PubMed
- A. Sakuda, A. Hayashi and M. Tatsumisago, Interfacial Observation between LiCoO2 Electrode and Li2S–P2S5 Solid Electrolytes of All-Solid-State Lithium Secondary Batteries Using Transmission Electron Microscopy, Chem. Mater., 2010, 22, 949–956 CrossRef CAS
- Y.-W. Byeon and H. Kim, Review on Interface and Interphase Issues in Sulfide Solid-State Electrolytes for All-Solid-State Li-Metal Batteries, Electrochem, 2021, 2, 452–471 CrossRef CAS
- L. M. Riegger, S. Mittelsdorf, T. Fuchs, R. Rueß, F. H. Richter and J. Janek, Evolution of the Interphase between Argyrodite-Based Solid Electrolytes and the Lithium Metal Anode–The Kinetics of Solid Electrolyte Interphase Growth, Chem. Mater., 2023, 35, 5091–5099 CrossRef CAS
- B. Pang, Y. Gan, Y. Xia, H. Huang, X. He and W. Zhang, Regulation of the Interfaces between Argyrodite
Solid Electrolytes and Lithium Metal Anode, Front. Chem., 2022, 10, 837978 CrossRef CAS PubMed
- N. Lee, J. Lee, T. Lee, J. Oh, I. Hwang, G. Seo, H. Kim and J. W. Choi, Rationally Designed Solution-Processible Conductive Carbon Additive Coating for Sulfide-Based All-Solid-State Batteries, ACS Appl. Mater. Interfaces, 2023, 15, 34931–34940 CrossRef CAS PubMed
- D. H. S. Tan, E. A. Wu, H. Nguyen, Z. Chen, M. A. T. Marple, J.-M. Doux, X. Wang, H. Yang, A. Banerjee and Y. S. Meng, Elucidating Reversible Electrochemical Redox of Li6PS5Cl Solid Electrolyte, ACS Energy Lett., 2019, 4, 2418–2427 CrossRef CAS
- L. Peng, H. Ren, J. Zhang, S. Chen, C. Yu, X. Miao, Z. Zhang, Z. He, M. Yu, L. Zhang, S. Cheng and J. Xie, LiNbO3-coated LiNi0.7Co0.1Mn0.2O2 and chlorine-rich argyrodite enabling high-performance solid-state batteries under different temperatures, Energy Storage Mater., 2021, 43, 53–61 CrossRef
- Y.-G. Lee, S. Fujiki, C. Jung, N. Suzuki, N. Yashiro, R. Omoda, D.-S. Ko, T. Shiratsuchi, T. Sugimoto, S. Ryu, J. H. Ku, T. Watanabe, Y. Park, Y. Aihara, D. Im and I. T. Han, High-energy long-cycling all-solid-state lithium metal batteries enabled by silver–carbon composite anodes, Nat. Energy, 2020, 5, 299–308 CrossRef CAS
- W. He, N. Ahmad, S. Sun, X. Zhang, L. Ran, R. Shao, X. Wang and W. Yang, Microscopic Segregation Dominated Nano-Interlayer Boosts 4.5 V Cyclability and Rate Performance for Sulfide-Based All-Solid-State Lithium Batteries, Adv. Energy Mater., 2023, 13, 2203703 CrossRef CAS
- A. Banik, Y. Liu, S. Ohno, Y. Rudel, A. Jiménez-Solano, A. Gloskovskii, N. M. Vargas-Barbosa, Y. Mo and W. G. Zeier, Can Substitutions Affect the Oxidative Stability of Lithium Argyrodite Solid Electrolytes?, ACS Appl. Energy Mater., 2022, 5, 2045–2053 CrossRef CAS
- T. Hwang, Y.-J. Lee, S. R. Lee, Y.-C. Ha, M. Cho, S.-M. Lee and K. Cho, The crucial role of oxygen substitution in argyrodite solid electrolytes from the bulk to the surface under atmospheric conditions, J. Mater. Chem. A, 2022, 10, 16908–16919 RSC
- Z. Sun, Y. Lai, N. lv, Y. Hu, B. Li, L. Jiang, J. Wang, S. Yin, K. Li and F. Liu, Insights on the Properties of the O-Doped Argyrodite Sulfide Solid Electrolytes (Li6PS5−xClOx, x = 0–1), ACS Appl. Mater. Interfaces, 2021, 13, 54924–54935 CrossRef CAS PubMed
- J. S. Kim, S. Jung, H. Kwak, Y. Han, S. Kim, J. Lim, Y. M. Lee and Y. S. Jung, Synergistic halide-sulfide hybrid solid electrolytes for Ni-rich cathodes design guided by digital twin for all-solid-state Li batteries, Energy Storage Mater., 2023, 55, 193–204 CrossRef
- Z. Zeng, J. Cheng, Y. Li, H. Zhang, D. Li, H. Liu, F. Ji, Q. Sun and L. Ci, Composite cathode for all-solid-state lithium batteries: progress and perspective, Mater. Today Phys., 2023, 32, 101009 CrossRef CAS
- K. Yoon, J.-J. Kim, W. M. Seong, M. H. Lee and K. Kang, Investigation on the interface between Li10GeP2S12 electrolyte and carbon conductive agents in all-solid-state lithium battery, Sci. Rep., 2018, 8, 8066 CrossRef PubMed
- F. Strauss, D. Stepien, J. Maibach, L. Pfaffmann, S. Indris, P. Hartmann and T. Brezesinski, Influence of electronically conductive additives on the cycling performance of argyrodite-based all-solid-state batteries, RSC Adv., 2020, 10, 1114–1119 RSC
- S. W. Park, G. Oh, J.-W. Park, Y.-C. Ha, S.-M. Lee, S. Y. Yoon and B. G. Kim, Graphitic Hollow Nanocarbon as a Promising Conducting Agent for Solid-State Lithium Batteries, Small, 2019, 15, 1900235 CrossRef
- A. R. Kathribail, A. Rezqita, D. Lager, R. Hamid, Y. Surace, M. Berecibar, J. Van Mierlo, A. Hubin, M. Jahn and J. Kahr, High-Performance Amorphous Carbon Coated LiNi0.6Mn0.2Co0.2O2 Cathode Material with Improved Capacity Retention for Lithium-Ion Batteries, Batteries, 2021, 7, 69 CrossRef CAS
- X. Zhang, M. Hou, A. G. Tamirate, H. Zhu, C. Wang and Y. Xia, Carbon coated nano-sized LiMn0.8Fe0.2PO4 porous microsphere cathode material for Li-ion batteries, J. Power Sources, 2020, 448, 227438 CrossRef CAS
- S. Liu, L. Zhou, J. Han, K. Wen, S. Guan, C. Xue, Z. Zhang, B. Xu, Y. Lin, Y. Shen, L. Li and C.-W. Nan, Super Long-Cycling All-Solid-State Battery with Thin Li6PS5Cl-Based Electrolyte, Adv. Energy Mater., 2022, 12, 2200660 CrossRef CAS
- F. Pedregosa, G. Varoquaux, A. Gramfort, V. Michel, B. Thirion, O. Grisel, M. Blondel, P. Prettenhofer, R. Weiss, V. Dubourg, J. Vanderplas, A. Passos, D. Cournapeau, M. Brucher, M. Perrot and É. Duchesnay, Scikit-learn: Machine Learning in Python, Journal of Machine Learning Research, 2011, 12, 2825–2830 Search PubMed
- G. Yang, R. Tao, C. J. Jafta, C. Shen, S. Zhao, L. He, I. Belharouak and J. Nanda, Investigating Multiscale Spatial Distribution of Sulfur in a CNT Scaffold and Its Impact on Li–S Cell Performance, J. Phys. Chem. C, 2021, 125, 13146–13157 CrossRef CAS
- F. Wu, Y. Yan, R. Wang, H. Cai, W. Tong and H. Tang, Synthesis of LiNi1/3Mn1/3Co1/3O2@graphene for lithium-ion batteries via self-assembled polyelectrolyte layers, Ceram. Int., 2017, 43, 7668–7673 CrossRef CAS
- G. Yang, X. Li, Y. Cheng, M. Wang, D. Ma, A. P. Sokolov, S. V. Kalinin, G. M. Veith and J. Nanda, Distilling nanoscale heterogeneity of amorphous silicon using tip-enhanced Raman spectroscopy (TERS) via multiresolution manifold learning, Nat. Commun., 2021, 12, 578 CrossRef CAS
- A. Mills, W.-Y. Tsai, T. Brahmbhatt, E. C. Self, B. L. Armstrong, D. T. Hallinan, J. Nanda and G. Yang, Navigating the complexities of solvent and binder selection for solution processing of sulfide solid-state electrolytes, MRS Commun., 2023, 13, 1063–1070 CrossRef CAS
- R. Rajagopal, Y. Subramanian, Y. J. Jung, S. Kang and K.-S. Ryu, Rapid Synthesis of Highly Conductive Li6PS5Cl Argyrodite-Type Solid Electrolytes Using Pyridine Solvent, ACS Appl. Energy Mater., 2022, 5, 9266–9272 CrossRef CAS
- Y. H. Jung, C. H. Lim and D. K. Kim, Graphene-supported Na3V2(PO4)3 as a high rate cathode material for sodium-ion batteries, J. Mater. Chem. A, 2013, 1, 11350–11354 RSC
- K. G. Araño, G. Yang, B. L. Armstrong, T. Aytug, M. S. Chambers, E. C. Self, H. M. Meyer III, J. Quinn, J. F. Browning, C. Wang and G. M. Veith, Carbon Coating Influence on the Formation of Percolating Electrode Networks for Silicon Anodes, ACS Appl. Energy Mater., 2023, 6, 11308–11321 CrossRef
- J. S. Nam, W. To A. Ran, S. H. Lee, T. H. L. Vuong, H. Jo, J.-H. Lee, S. M. Hwang and Y.-J. Kim, Densification and charge transport characterization of composite cathodes with single-crystalline LiNi0.8Co0.15Al0.05O2 for solid-state batteries, Energy Storage Mater., 2022, 46, 155–164 CrossRef
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta00674g |
| ‡ These authors contribute to this work equally. |
| This journal is © The Royal Society of Chemistry 2024 |