
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA DNA biosensor integrating surface hybridization, thermo-responsive coating, laminar-flow technology and localized photothermal effect for efficient electrochemical detection of nucleic acids†
Ludovica
Maugeri
a,
Giorgia
Fangano
a,
Angelo
Ferlazzo
 *b,
Giuseppe
Forte
a,
Antonino
Gulino
b and
Salvatore
Petralia
*ac
*b,
Giuseppe
Forte
a,
Antonino
Gulino
b and
Salvatore
Petralia
*ac
aDepartment of Drug and Health Science, University of Catania, Via Santa Sofia 64, 95125 Catania, Italy. E-mail: salvatore.petralia@unict.it
bDepartment of Chemical Sciences, University of Catania, Via Santa Sofia 64, 95125 Catania, Italy. E-mail: angelo.ferlazzo@unict.it
cInstitute of Biomolecular Chemistry, CNR, Via Paolo Gaifami 18, 95126 Catania, Italy
First published on 14th October 2024
Abstract
A hybrid electrochemical DNA biosensor that integrates various technologies, such as laminar flow, surface hybridization, DNA-microarray, thermo-responsive nanocoating and localized photothermal heating, is presented here. A photothermal module based on gold nanostructures photoactivated by a green-light source (532 nm) was developed for easy temperature management. The hybridization product is electrochemically detected by a three-planar-microelectrode system upon dsDNA denaturation. Performances of the hybrid biosensor were investigated by detection of the cDNA target, resulting in a sensitivity of about 2.62 μA nM−1 cm−2 and a limit of detection of 1.5 nM, as a function of the capture probe sequence. The findings facilitate the integration of multiple technologies, enabling the development of low-cost and point-of-care detection systems for molecular analysis.
Introduction
Miniaturization and integration of nucleic acid analysis are critical steps toward the development of a platform for sample-in-answer-out diagnostic analysis.1 Commonly, molecular testing involves multifaceted procedures, complex laboratory infrastructures and skilled personnel. Despite the increasing number of new technologies and innovative diagnostic platforms, surface hybridization-based molecular testing is still the most selective approach for nucleic acid detection.2 The strength of the surface hybridization test is its capability to integrate the test into a microarray format for multiplexing analysis. Easy integration of the surface-microarray hybridization analysis into miniaturized devices allows the development of low-cost and easy-to-use portable systems. The high versatility of surface hybridization testing is also demonstrated by the variety of transduction methods (optical,3 impedance4 capacitive5 and electrochemical6) that can be used to read out the hybridization products. Although revolutionary molecular analysis approaches have recently been proposed, including the CRISPR-Cas technology,7 cooperative DNA triplex formation,8 and multiplexing analysis, the rapid detection and integration of pre-processing steps are still the main limitations for the development of miniaturized nucleic acid sensors. The integration of various analytical technologies and nanotechnology in a unique miniaturized system is the most effective approach to obtain a cost-effective, easy-to-use multiplexing analysis procedure, with high sensitivity and good specificity.9 In this work, an innovative DNA biosensor based on the integration of various technologies, such as laminar-flow analysis (LFA), DNA-surface hybridization, ssDNA-microarray, thermo-responsive coating, photothermal heating and electrochemical read-out, is presented. The used thermo-responsive coating is based on poly(N-isopropylacrylamide) (PNM), which has high chemical stability, low toxicity, and temperature-/pH-responsivity. It is largely used for biomedical applications, such as drug delivery, sensing, and catalyst agent. The thermo-responsive behaviour of PNM is based on its low critical solution temperature (LCST). Basically, at temperatures below the LCST value (<33 °C), the PNM chains adopt a coil-extended hydrophilic conformation, and above the LCST value (>33 °C), a cooperative transition occurs and PNM adopts a hydrophobic globule conformation. In this hydrophobic collapsed structure, the number of hydrogen bonds decreases and the cargo molecules as well as solvent molecules are expelled from the chains.10,11 In detail, the proposed DNA biosensor consists of a cellulose strip for sample separation, an ssDNA-microarray deposited on a flexible, conductive and transparent ITO-PET substrate, a thermo-responsive coating made of poly(N-isopropylacrylamide) (PNM) for thermo-triggered electrochemical-probe release and a miniaturized photothermal heater activated by a source light (532 nm, power 2–5 W) (Scheme 1).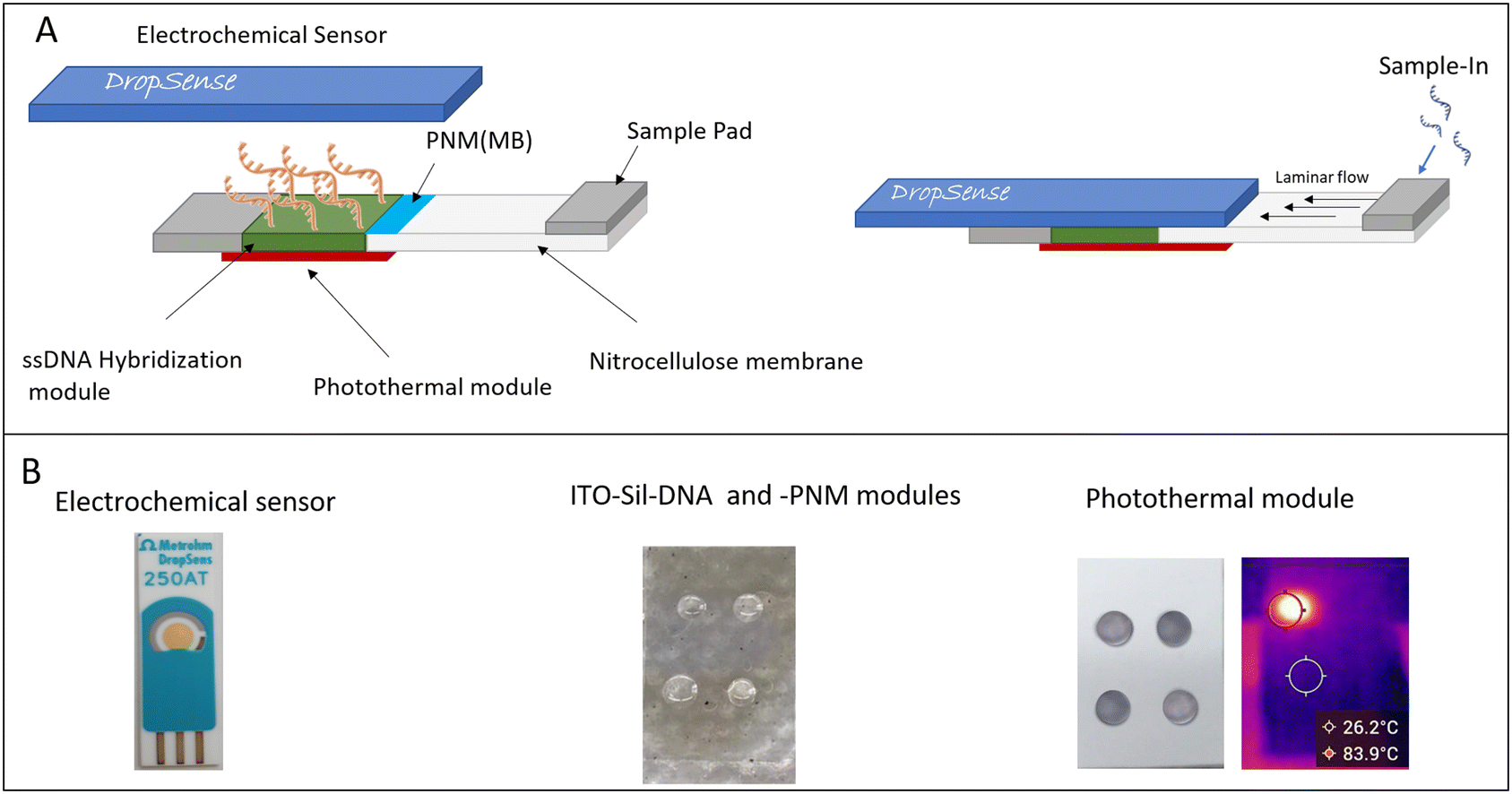 | ||
| Scheme 1 Schematic of DNA biosensor (A) and biosensor modules (B). | ||
Experimental section
Chemicals and materials
All reagents were purchased from Merck and used as received. The PES-backed nitrocellulose membrane and the MF1 glass fiber membrane were purchased from Cytiva and used as received; the PET-ITO substrate was purchased by Merck. Amino-modified ssDNA probes and targets were purchased from IDT in desalting purified forms. The DNA sequences are reported in Table S1.† The photothermal experiments were performed by irradiating each spot of the photothermal module with CW laser 532 nm (1.8 and 5 W) for different minutes. A standard FLIR infrared thermal imaging camera was used to record the temperature of spots during the light exposure. The DNA surface hybridization experiments were carried out using a specific target perfect match (25, 250 and 2500 nM) in 0.1 M KCl buffer. X-ray photoelectron spectra (XPS), measured at a take-off angle of 45° with respect to the surface sample holder, with a PHI 5000 Versa Probe II system (ULVAC-PHI, INC., main chamber base pressure 1 × 10−8 Pa), were excited with monochromatic X-ray radiation Al Kα (pass energy 5.85 eV).12,13 Calibration of the spectra was achieved by fixing the Ag 3d5/2 peak of a clean sample at 368.3 eV.13 Cyclovoltammetry (CV) measurements were performed using a Parstat potentiostat and 220AT gold screen-printed electrodes (high-temperature ink). A 0.1 M KCl solution with pH 7.0 was used for electrochemical measurements. CV tests were performed at a scanning speed of 10 mV s−1 in the potential range between −1 and 0.2 V. The sensitivity of the sensor was calculated as the ratio of the slope of the calibration line obtained from the maximum peak reduction current intensity to the geometric area of the used electrode (0.125 cm2).14,15 The limit of detection (LOD) was calculated as 3σ background/slope.16ITO-Sil-ssDNA hybridization module
The hybridization module is composed of a PET-ITO substrate properly functionalized with a reactive epoxy-silane (Sil) coating. In particular, the ITO substrates were processed as follows: (i) cleaning and activation with UV-O3 treatment for 600 s to remove organic residues from the surface and to increase the hydroxyl group density at the surface; (ii) vapor phase silanization process to produce reactive epoxy-silane monolayer at the ITO surface. In detail, the PET-ITO substrates were incubated in a volume of 10 mL of GPS (3-glycidyloxypropyltrimethoxysilane) in a vacuum oven for 4 h at 110 ± 1 °C. Further, the curing process was performed at 110 °C for 1 h. To confirm the effectiveness of the UV-O3 and epoxy silanization treatments, water contact angle (CA) measurements, FTIR-ATR and XPS analyses were performed. The ssDNA microarray fabrication was performed using the following procedure: an aliquot of 20 μL of a phosphate buffer (0.15 M at pH 9.32) solution of the amino-modified ssDNA probe (20 μM) was deposed on the ITO substrates to create a 4 × 4 microarray, and the substrates were incubated in the chamber at 45 °C, humidity 90% for 4 hours. Then, the substrates were rinsed with deionized water and dried using a nitrogen flow. To confirm the effectiveness of ssDNA anchoring the water contact angle (CA) measurements, FTIR-ATR and XPS analyses were performed.ITO-Sil-PNM module
ITO-Sil substrates were incubated with a phosphate buffer (0.15 M) solution of amino-modified PNM (50 μM) at pH 9.32 in a chamber at 45 °C, humidity 90% for 4 hours. Then, the substrates were rinsed with deionized water and dried using nitrogen flow. In order to confirm the effectiveness of PNM anchoring the water contact angle (CA) measurements, FTIR-ATR and XPS analyses were performed. The MB entrapping (ITO-Sil-PNM/MB) was performed by incubating the ITO-Sil-PNM substrate with an aqueous solution of MB 1 mM overnight at room temperature. Then, the substrates were rinsed with deionized water to remove the excess MB and dried under nitrogen flow.Photothermal module
An aliquot of 20 μL of citrate stabilized gold-nanoparticles (Au NPs) was spotted on the cellulose PS-backing membrane to form the photothermal spot-heater. Four different spots were created, and then the photothermal module was glued to the bottom of the hybridization module (Scheme 1B). The localized photothermal effect was generated by irradiating the spots with a laser source at 532 nm under various laser powers. The temperature was monitored by a thermocamera (FLIR). The AuNPs were prepared by dissolving an aliquot of 75 μL of HAuCl4 (5 × 10−2 M) in 37.5 mL of deionized Milli-Q water; after 20 min of stirring at a temperature of 70 ± 5 °C, an aliquot of 2.5 mL of sodium citrate (28.5 mg) was added. After 1 h of the reaction, the red-purple dispersion of AuNPs was centrifuged at 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 5 min, and the precipitated AuNPs were purified by washing with deionized Milli-Q-water and centrifugation.
000 rpm for 5 min, and the precipitated AuNPs were purified by washing with deionized Milli-Q-water and centrifugation.
Laminar flow module
The laminar flow module is composed of PES-backing nitrocellulose and MF1 membrane (sample-pad). The membranes were cut using a standard cutter.Results and discussion
Integrated DNA biosensor
The hybrid DNA biosensor was properly designed to integrate the laminar-flow technology, surface-hybridization reaction, ssDNA-microarray system, localized photothermal effect and electrochemical measurements for specific and efficient nucleic acid recognition. The starting sample was collected and purified by a laminar-flow module, connected to the ITO-ssDNA-microarray (ITO-Sil-ssDNA), and ITO-Sil-PNM modules designed for specific DNA capturing by surface-hybridization and for thermo-released methylene blue (MB), respectively, to act as the electrochemical probe. An innovative photothermal module, composed of Au-nanoparticle spots, was mounted on the bottom of the hybridization module to manage the temperature of the biosensor. The hybridization is revealed and quantified by a redox-label probe MB thermo-released by the ITO-Sil-PNM module upon the light stimuli. The MB probe is a specific dsDNA intercalator. The amount of MB released by denaturation of ITO-Sil-dsDNA/MB is related to the amount of the hybridization product (dsDNA) and quantitatively measured by a three-printed-electrodes sensor mounted on the top of the microarray module (Scheme 1). The formation of the dsDNA/MB complex has been the subject of extensive investigation in the literature.17 The findings indicate that the MB interacts with double-stranded DNA predominantly through a semi-intercalation binding mode, confirming that the MB, as well as other ligands such as ethidium bromide, engage in a multimodal binding with DNA.17ITO-Sil-DNA microarray
The ITO-Sil-ssDNA microarray substrate was prepared as described in the Experimental section. In particular, it is composed of a 2 × 2 DNA microarray containing four ssDNA probes printed onto the PET-ITO-Sil surface (Fig. S1†). More in detail, the PET-ITO substrate was functionalized with the epoxy-silane layer (ITO-Sil) using a two steps process based on the UV-O3 activation and the vapor-phase silanization process developed in our laboratory.2 The effectiveness of the epoxy-silane layer formation was confirmed by XPS analysis and by water contact angle measurements.Fig. S2† panel A shows the XP spectrum of the ITO-Silan sample in the C 1s binding energy region. Three experimental peaks are evident. A careful deconvolution of the experimental spectrum required three Gaussians at: 285.0 eV (magenta line, relative intensity 41%) due to the C–C, C–H states and some adventitious carbon present in all air-exposed materials; 286.6 eV due to the C–O levels of the ether and epoxide groups of the silane (dark yellow line, relative intensity 52%); and 289.1 eV due to some minor surface carbonate (dark cyan line, relative intensity 7%). The analysis thus confirms the presence of the silane bound to the ITO substrate.18
Fig. S2† panel A also shows the XP spectrum of the ITO-Silan sample in the O 1s binding energy region. Two peaks are evident at 530.2 and 532.3 eV but the deconvolution of the experimental spectrum required three Gaussians at: 530.1 eV (magenta line), characteristic of the In2O3 and SnO2 metal–oxygen states; 531.8 eV (cyan line), attributed to the O–Si group, and at 532.8 eV (wine line) due to the O–C levels.19 Fig. S2A† shows the XP spectrum of ITO-Silan in the Si 2p binding energy region that consists of a broad peak centred at 102.7 eV, due to the Si–O group of the silane.20 Fig. S2† panel A shows the XP spectrum of the ITO-Silan sample in the In 3d binding energy region. This spectrum is composed of the typical In 3d5/2,3/2 spin–orbit components located at 444.8 and 452.3 eV which closely matches the binding energy of In3+ in In2O3. The deconvolution required two spin–orbit doublets at 444.2–445.2 eV (3d5/2) magenta line and at 451.7–452.7 eV (3d3/2) dark yellow, in agreement with the typical XPS of ITO.21,22Fig. 2A finally shows the XP spectrum of the ITO-Silan sample in the Sn 3d binding energy region. The Sn 3d5/2,3/2 spin–orbit components, located at 487.0 and 495.4 eV, are typical of the ITO.23
Static water contact angle measurements (CA) performed at different process steps confirm the effective silanization process of the ITO surface. In detail, after the O3-plasma process, the surface exhibits a high wettability (CA < 10°) to indicate the effective cleaning and oxidation processes. Contrastingly, after the vapor phase epoxy-silanization, an increase in surface hydrophobicity was observed (CA values 56.1° ± 1.3°) to indicate the formation of the epoxy-layer coating (ITO-Sil). The DNA-microarray was printed onto the ITO-Sil substrate by depositing a volume of 2 μL of amino-terminated ssDNA (20 μM, in a buffer solution of sodium phosphate at pH 9.2) on the substrate surface. The ITO-Sil-DNA substrates were incubated at 45 °C/90% RH for 4 h; after three water rinses to remove the excess DNA, the substrate was dried under nitrogen flow and stored. The effectiveness of the DNA anchoring was confirmed by XPS analysis.
Fig. S2B† shows the XP spectrum of ITO-Silan-DNA in the C 1s binding energy region. Three experimental peaks at 285.0, 286.5 and 288.9 eV are evident. A careful deconvolution of the experimental spectrum required five Gaussians at: 285.0 eV (magenta line) due to the C–C and C–H states of the aliphatic chains (relative intensity 56%); 285.8 eV (dark yellow line) due to the amine group (C–N) of the DNA (relative intensity 4%); 286.6 eV (dark cyan line) due to the C–O states (relative intensity 31%), (C–OH, C–O–C and C–O–P groups); 287.8 eV (wine line) due to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O levels (relative intensity 4%), and 289.0 eV (navy line) given by the N–(CO)–N group (relative intensity 5%).24 Fig. S2B† also shows the XP spectrum of the ITO-Silan-DNA in the O 1s binding energy region, which consists of two peaks at 529.8 and 531.9 eV. The spectral fitting required four Gaussians at: 530.2 eV (magenta line) due to the ITO substrate (vide supra); 531.3 eV (dark yellow line) due to the DNA PO group;25 531.7 eV (dark cyan line) attributable to O–Si (vide supra) and OC levels, and at 532.8 eV (wine line) attributable to the DNA C–O–C, C–O–P, C–OH groups. Fig. S2B† also shows the XP spectrum of ITO-Silan-DNA in the N 1s binding energy region, which consists of a peak at 400.2 eV. The deconvolution required two Gaussians at: 399.3 eV (magenta line) due to the imine group (NC–) and 400.4 eV (dark yellow line) due to the contribution of amines, carbamides, and urethanes (N–CO–N, –NH2, –N–COO) from the DNA bases. Fig. S2B† shows the XP spectrum of the ITO-Silan-DNA in the P 2p binding energy region, which consists of a broad peak at 133.6 eV, attributable to the presence of P in the DNA backbone, where the P 2p3/2,1/2 spin–orbit splitting, as expected, is not appreciable. The XPS of the ITO-DNA sample exhibits the signals for In 3d, Sn 3d and Si 2p states, which agree well with the analogues of the ITO-silane sample.
O levels (relative intensity 4%), and 289.0 eV (navy line) given by the N–(CO)–N group (relative intensity 5%).24 Fig. S2B† also shows the XP spectrum of the ITO-Silan-DNA in the O 1s binding energy region, which consists of two peaks at 529.8 and 531.9 eV. The spectral fitting required four Gaussians at: 530.2 eV (magenta line) due to the ITO substrate (vide supra); 531.3 eV (dark yellow line) due to the DNA PO group;25 531.7 eV (dark cyan line) attributable to O–Si (vide supra) and OC levels, and at 532.8 eV (wine line) attributable to the DNA C–O–C, C–O–P, C–OH groups. Fig. S2B† also shows the XP spectrum of ITO-Silan-DNA in the N 1s binding energy region, which consists of a peak at 400.2 eV. The deconvolution required two Gaussians at: 399.3 eV (magenta line) due to the imine group (NC–) and 400.4 eV (dark yellow line) due to the contribution of amines, carbamides, and urethanes (N–CO–N, –NH2, –N–COO) from the DNA bases. Fig. S2B† shows the XP spectrum of the ITO-Silan-DNA in the P 2p binding energy region, which consists of a broad peak at 133.6 eV, attributable to the presence of P in the DNA backbone, where the P 2p3/2,1/2 spin–orbit splitting, as expected, is not appreciable. The XPS of the ITO-DNA sample exhibits the signals for In 3d, Sn 3d and Si 2p states, which agree well with the analogues of the ITO-silane sample.
Photothermal module
The photothermal module was designed to induce a localized temperature increase through the photothermal effect of Au-nanoparticle spots, upon green laser light (532 nm) photoexcitation. The photothermal module, composed of 2 × 2 photothermal Au-spots, was glued on the bottom of the ITO-Sil-DNA-microarray module, and the temperature was tuned by the laser power. A starting temperature of about 20 °C was recorded with the laser OFF (Fig. 1A). To promote the surface hybridization and MB photothermal release, the temperature of 46 °C was reached using a laser power value of 1.8 W, while the dsDNA denaturation phase at a temperature of about 85 °C was reached increasing the laser power value at 5.0 W. As expected, the temperature of the photothermal module was found to be directly proportional to the laser power (Fig. S6†). | ||
| Fig. 1 DNA biosensor: A) representative thermographs of the photothermal module during different phases: laser-OFF, hybridization and MB release phase (laser ON, power 1.8 W) and dsDNA denaturation phase (laser ON, power 5 W). B) ITO-Sil-DNA module: surface-hybridization/MB interaction, dsDNA/MB denaturation and MB electrochemical detection. | ||
ITO-Sil-DNA: hybridization reaction and electrochemical detection
Fig. 1B illustrates the main steps of the ITO-Sil-DNA module. In detail, it shows the surface-hybridization process with MB–DNA interaction, the dsDNA/MB denaturation with MB releasing, and the final MB electrochemical detection.To ascertain the capability of the ITO-Sil-DNA-microarray module to recognize nucleic acids by a specific surface hybridization reaction, the ITO-Sil-DNA-microarray samples were incubated with cDNA (2 μM) in the presence of the methylene blue redox-probe (MB). The hybridization temperature of about 45 °C was induced by a localized photothermal effect, irradiating the photothermal module as described above. After 10 min of the hybridization reaction, the substrates were rinsed with water to eliminate the excess of cDNA and MB. Then, an electrochemical printed sensor was mounted on the top of the ITO-Sil-dsDNA surface with a volume of 200 μL of KCl buffer. Then, the localized temperature was photothermally increased to about 85 °C to promote the dsDNA denaturation and the MB release. The electrochemical CV measurements confirmed the effective hybridization reaction by observing the electrochemical MB signal (CV curve in Fig. 1B). In fact, the anodic oxidation peak, and cathodic reduction peak related to MBox and MBrid species, respectively, were observed.
ITO-Sil-PNM module
The ITO-Sil-PNM module was manufactured as described in the Experimental section. The characterization was performed by XPS investigation, spectroscopic measurements and AFM analysis. Fig. 2A shows the XP spectrum of the ITO-Sil PNM in the C 1s binding energy region. Two experimental peaks at 285.0 and 286.4 eV are evident. A careful deconvolution of the experimental spectrum showed four Gaussians at: 285.0 eV due to the C–C and C–H states (magenta line); 285.8 eV due to the amine group (C–N) of the polymer (dark yellow line); 286.4 eV due to the C–O of the silane (dark cyan line) (vide supra); and 287.7 eV due to the –N–CO amide levels of the PNM polymer (wine line). The relative intensities of the above-mentioned Gaussians are in the 6:1:4:1 ratio and perfectly match the stoichiometry of the PNM polymer.26
 | ||
| Fig. 2 ITO-Sil-PNM characterization: A) Al Kα excited XPS of the ITO-Sil-PNM sample in the C 1s binding energy region: the magenta, dark yellow, dark cyan, and wine lines refer to the 285, 285.8, 286.4, and 287.7 eV Gaussian components, respectively; in the O 1s binding energy region: the magenta, dark yellow, and dark cyan lines refer to 531.6, 532.9, and 533.8 eV Gaussian components, respectively; and in the N 1s binding energy regions. The blue line represents the background and the red line superimposed on the experimental black profile refers to the sum of the Gaussian components. B) ATR-FTIR spectra for PNM and ITO-Sil-PNM. | ||
Fig. 2A shows the XP spectrum of ITO-Sil-PNM in the O 1s binding energy region, which consists of two peaks at 531.6 and 533.5 eV. The deconvolution produced three Gaussians at: 531.6 eV due to O–Si and OC groups of the silane and PNM polymer (magenta line); 532.9 eV due to the O–C levels (dark yellow line), and 533.8 eV due to the presence of some surface H2O (dark cyan line). Fig. 2A shows in addition the XP spectrum of ITO-PNM in the N 1s binding energy region, which consists of a peak at 399.7 eV due to the N–CO amide group.27 The XP spectrum of the ITO-Sil-PNM sample for Si 2p states is shown in the ESI† (Fig. S3).
Fig. 2B illustrates the ATR-FTIR spectra for the ITO-Sil-PNM and for PNM for comparison. The presence of diagnostic peaks for PNM at: 3283.3 cm−1 (N–H, stretching), 2968.2 cm−1 (C–H, stretching), 1638.8 cm−1 (CO stretching, amide I), 1541.8 cm−1 (CO stretching, amide II), 1375.5 cm−1 (C–H, bending) and 1132.2 cm−1 (C–C, stretching), suggests the effective PNM layer formation.
ITO-Sil-PNM: photothermal MB release
Our experiments confirm that the ITO-Sil-PNM substrate exhibits an excellent photothermal-responsive effect through the well-known coil-to-globule transition of the PNM layer.28 In detail, at temperature above the LCST value (∼33 °C) the PNM chain changes from the hydrophilic-coil-structure to the hydrophobic-globule-structure, resulting in changes of the surface hydrophobicity inducing the release of the cargo loading on it (Fig. 3A). The surface coil-to-globule transition was photothermally induced by light irradiation (laser 532 nm) of the photothermal module as above described. The surface coil-to-globule transition of the ITO-Sil-PNM surface was investigated by the static water contact angle method at different temperatures (Fig. 3B). The increase of the C.A. values (44.7°, 56.9°, 48.9°, 50.36°, 52.62° to 57.15 °) with the increase of temperature (21, 30, 33, 37, 40 to 46 °C) confirms the effective surface coil-to-globule transition of the PNM structure. | ||
| Fig. 3 ITO-Sil-PNM substrate: A) surface coil-to-globule transition mechanism induced by the photothermal effect; B) C.A. at different temperature values; C) optical absorption spectra of MB loaded onto ITO-Sil-PNM substrate and MB solution; D) absorbance values at 665 nm of MB released versus the incubation time at 45 °C and 25 °C (inset: the absorption spectra of water solution at 45 °C); and E) CV cycles of the MB release process in buffer KCl 0.1 M, 25 °C (scan rate 0.01 V s−1) from MB loaded ITO-Sil-PNM substrate (black line) and from the MB-loaded ITO/Sil substrate (red line). | ||
To assess the capability of the ITO-Sil-PNM substrate to efficiently perform a cargo load and release process, the ITO-Sil-PNM substrate (0.5 × 2 cm) samples were incubated with an aqueous solution of MB for 6 h at room temperature. The effective MB entrapment at the PNM coating was confirmed by the optical absorption spectra of the MB-loaded ITO-Sil-PNM substrate (Fig. 3C). The absorption spectra report the diagnostic MB absorption band centred at 665 nm. While no absorption peak was observed for the substrate ITO-Sil-PNM without MB (Fig. S7†). Then, the MB release was investigated by incubating the MB-loaded ITO-Sil-PNM in a volume of 2 mL of deionized water, at the temperature of 45 °C (above the LCST value) for 30 min. The effective MB release process was confirmed by measuring the optical absorption at 665 nm after 5, 10, 20, 30 and 40 min of heating (black line Fig. 3D). For comparison, the MB release from MB-loaded ITO-Sil-PNM substrate was investigated at 25 °C (below the LCST value). In this case, no absorption at 665 nm was recorded, to corroborate with the thermo-responsive loading-releasing mechanism of the ITO-Sil-PNM surface. In addition, the photothermally induced MB release from the ITO-Sil-PNM substrate was investigated by electrochemical measurements. In detail, the photothermal module was mounted on the bottom of the MB-loaded ITO-Sil-PNM substrate, then a three-electrode printed electrochemical sensor was mounted on the top, and a volume of 200 μL of KCl buffer 0.1 M was added as an electrochemical buffer. The temperature of the substrate was increased to 45 °C by a localized photothermal effect through laser irradiation (0.18 W) and the CV measurements were performed after 5 min of irradiation. The diagnostic reduction and oxidation peaks of the released MB at about −0.4 V and 0.3 V were recorded (Fig. 3E, black line), to confirm the effective photothermal release of MB from the ITO-Sil-PNM substrate. For comparison, MB loading/photothermal release processes were conducted on the ITO-Sil substrates (without the PNM coating) under the same experimental conditions. In this case, no CV signals were recorded upon localized photothermal heating (Fig. 3E red line). These results clearly show that the role of PNM is crucial for the MB release process and that the used electrode is capable of detecting the presence of MB. In fact, the bare electrode shows an excellent response to the presence of 2.5 μM of MB in a 0.1 M KCl solution, as shown in Fig. S4,† confirming the capability of the printed-electrode to detect oxidation and reduction of MB at potentials of about −0.35 and −0.41 V, respectively. The same result was obtained by performing a test on the device in the presence of MB but in the absence of cDNA and this showed that the PNM was able to release the MB and that the electrode was capable of detecting it as in the tests performed in the solution (Fig. 3E).
Molecular modelling studies were performed to investigate the MB–PNM interaction during the coil-to-globule transition. In detail, a 32-mer PNM system was assembled by CHARMM-GUI and the MB molecule was physiosorbed onto the polymer chains. The resulting system was placed at the center of a 7 × 5 × 5 nm box, with water molecules, and subjected to equilibration through a procedure consisting of 20000 steps of minimization using the steepest descent algorithm. Then, MD simulation investigations were performed at 315 K and three structures were selected as system configurations for subsequent 200 ns MD simulations. Atomic coordinates were saved with intervals of 10, 100 and 150 ns using a step of 1.0 ns, thus obtaining three structures called PNM–MB1, PNM–MB2 and PNM–MB3. These structures were optimized, and used for energy calculations. The energy difference among the optimized geometries selected from MD simulations indicated that the initial configuration, PNM–MB1 at 10 ns has lower energy (0.00 kcal mol−1) after 100 ns with PNM still in the coil-form, an increase of energy was obtained (15.22 kcal mol−1) to indicate a strong interaction between the PNM and the MB, while at 150 nm with PNM in globule-form structure, the interaction between the PNM polymer and MB weakens (7.34 kcal mol−1) and the MB molecule start to desorb from the polymer after 150 ns (Fig. 4). These data suggest that the release of MB from the PNM coating is primarily driven by the transition of the PNM chain from the coil-to-globular structure.
 | ||
| Fig. 4 PNM–MB molecular modelling calculations; PNM in ball and stick representation, MB in CPK representation: optimized geometries of PNM–MB adduct for three different geometries: A) PNM–MB1 at 10 ns; B) PNM–MB2 at 100 ns; C) PNM–MB3 at 150 ns, all simulations were performed at 315 K, water molecules are omitted for clarity. | ||
DNA biosensor testing
To demonstrate the efficiency of the DNA biosensor, experiments were performed under different conditions using the experimental setup illustrated in Fig. S5.† Firstly, to ascertain the effectiveness of the photothermal module, aliquots of cDNA (2.5 μM) were loaded on the DNA biosensor, and then electrochemical measurements were performed after photothermal denaturation phases (laser 5 W 80 °C) for 2 and 4 minutes. For comparison, experimental controls were performed without the photothermal heating phase (no laser). In detail, an aliquot of 200 μL of a buffer solution containing cDNA (2.5 μM) was loaded onto the sample PAD and the PNM-module was photothermally activated for 5 min to induce the thermo-release of the MB probe (laser at 1.8 W). After 10 min of hybridization, a volume of 500 μL of the KCl solution was loaded on the sample pad to eliminate the excess of cDNA and MB from the microarray area. Then, the spot-1 containing the specific probe was photothermally activated for 2 and 4 minutes with laser 5 W; the localized temperature increased to about 80 °C to promote dsDNA denaturation and MB release. The electrochemical CV responses are reported in Fig. 5A (red line 2 min laser and blue line 4 min laser). The results clearly indicate that after 2 and 4 min of photothermal heating (laser 5 W), the electrochemical signals arise according to the denaturation of dsDNA and release of the MB probe (Fig. 5A, red and blue lines). For comparison, control experiments were performed without the photothermal heating phase (Fig. 5A, black line NO laser). In this case, no electrochemical signal was observed to confirm that no DNA denaturation and no MB release occurred without the photothermal heating process with laser at 5 W. Similarly, no CV signal was observed in the experiments conducted with laser 1.8 W (5 min) irradiation, indicating that no DNA release occurs at a temperature of 45 °C. It is noteworthy that the obtained data revealed a higher peak intensity (Ip) during the reduction phase of MB than during the oxidation phase. This led to considering the reduction Ip for the construction of the calibration curve obtained from measurements with different amounts of DNA. To assess the specificity, all spots including P2 (negative control), P3 (replicated specific probe) and P4 (unspecific probe) were tested using 4 min of photothermal-denaturation time for each spot. The results reported in Fig. 5B show CV signals during the interrogation of P1 (2.21 ± 0.2 μA) and P3 (1.96 ± 0.25 μA) spots. While slight signals were recorded during the interrogation of the P2 (0.16 ± 0.01 μA) and P4 (0.35 ± 0.05 μA) spots, confirming the excellent specificity (>90%) of DNA biosensor for the detection of specific CDNA target. | ||
| Fig. 5 DNA biosensor CV experiments: A) CV cycles for the detection of cDNA 250 nM in KCl 0.1 M (scan rate 0.01 V s−1; 25 °C) at different photothermal conditions: no laser, laser 5 W for 2 min and laser 5 W for 4 min; B) specificity tests, peaks current at −0.4 V for each investigated spots: P1 (specific probe), P2 (negative control), P3 (specific probe) and P4 (aspecific probe). | ||
To ascertain the sensitivity of the DNA biosensor, different amounts of cDNA (0, 25, 250 and 2500 nM) were tested. The results depicted in Fig. 6A, confirm the good electrochemical response at various cDNA amounts. Measurements show the presence of two peaks for oxidation and reduction of the MB (Fig. 6A). This phenomenon was not noticed when the sensor was tested in the absence of DNA. The presence of the oxidation peak at −0.35 V and the reduction peak at −0.40 V indicates the presence of free MB released from the DNA double helix. The oxidation peak at −0.2 V and reduction peak at −0.53 V can also be attributed to the MB/cDNA interaction.29 Indeed, MB has a strong interaction between the proton transfer, during the reduction, and the biomolecule by influencing the apparent rate of the first electron transfer step. Specifically, the hydrogen bonding interactions between DNA and MB influence the protonation reaction of the radical intermediate. When high concentrations are present, the overall electron transfer kinetics of the system increase. The acceleration of the first phase of electron transfer (cathodic phase) causes a reduction in the cleavage of the peak in the oxidation wave and an increase in the cleavage in the reduction wave (anodic peak). This electrochemical phenomenon, due to the transfer of the second electron, is still limited by the protonation rate. In fact, the shift towards oxidation and reduction potentials is due to the interaction of MB with cDNA. Based on what has been reported in the literature, it is possible that the denaturation of the DNA double helix releases the MB that continues to maintain an interaction with the cDNA single strand. This causes an alteration in the ability to transfer electrons from the MB to the electrode, thus observing a wider electrochemical window with higher oxidation and reduction potentials.29,30 Indeed, this phenomenon is more evident in the curve related to a higher cDNA concentration. All of this confirms that the MB intercalates between the double helix and is subsequently released, which can be an excellent nucleic acid tracking system. The calibration curve in Fig. 6B shows an excellent linear range between the peak current intensity and the logarithm of the concentration, with a linear range of Y = 0.328X − 0.34; R2 = 0.9969. A limit of detection value (LoD) of about 1.5 nM and sensitivity of about 2.62 μA nM−1 cm−2 for the detection of cDNA were calculated.
 | ||
| Fig. 6 DNA biosensor sensitivity test: A) CV cycles of DNA biosensor for the detection of different amounts of cDNA (0, 25, 250 and 2500 nM) in KCl 0.1 M (scan rate 0.01 V s−1; 25 °C); B) log–log linear response of the peak current (μA) at −0.4 V versus cDNA (nM), (Y = 0.328X − 0.34; R2 = 0.9969). | ||
To assess the recovery of the assay test, samples of DNA (P1 probe, 2500 nM) were tested. A signal of about 0.75 μA corresponding to a concentration of about 2350 nM was recorded, indicating a final recovery of 94%. Dry storage stability at +5 °C was tested within 2 months. The response of the biosensor decreased by about 5–7% from its initial response after 2 months of storage. A long-term stability study is in progress and the results will be published.
A comparison study between the proposed DNA biosensor and the various biosensors reported in the literature is shown in Table 1. In particular, the sensing strategy, limit of detection value, and the capability to be integrated into a point-of-care (PoC) system are compared.
Drozd and colleagues have published an SPRi-based biosensing platform for the detection of specific DNA sequences.31 They have investigated different DNA immobilization and labelling strategies reaching a limit of detection of 1.96 nM. The sensing approach was successfully integrated into a sensor array device suitable for point-of-care systems.29 In 2020 Xu and coworkers reported potentiometric DNA sensors using surface plasmon resonance analyses for the detection of DNA by hybridization reactions. Despite a good limit of detection of 0.6 nM, the proposed approach is still not ready for integration in the point-of-care system.32 Yesil and coworkers have developed an electrochemical DNA biosensor based on avidin–biotin interaction integrated on a graphite electrode (PGA/PGE) for the detection of specific DNA sequences. The DNA biosensor shows a linear response in the 1.5–12.5 nM range and a detection limit of 1.3 nM. The integration of this DNA biosensor into a point-of-care system requires more optimisation.33 Recently, El Aamri and coworkers reported an innovative colorimetric DNA biosensor based on the reaction between phenol and sugar. The sensing approach was tested in a 96-well plate reaching a limit of detection of 200 nM within a linear range of 0.2–1 μM.34 Also in this case, the integration of this DNA biosensor into a point-of-care system requires further optimisation.
In summary, the DNA biosensor proposed in this work represents a pioneering integration of multiple technologies including microarray surface hybridization, thermo-responsive coating and localized photothermal effect, in a miniaturized, portable and cost-effective system for the detection of DNA sequences with a limit of detection of 0.15 nM.
Conclusions
This work presents a hybrid DNA biosensor for the electrochemical detection of nucleic acids, through an effective integration of various technologies such as paper-based laminar flow, surface-hybridization, thermo-responsive nanocoating, electrochemical readout and photothermal heating.The capability to detect specific oligonucleotides with a LoD of about 1.5 nM, sensitivity of value of about 2.62 μA nM−1 cm−2 and good specificity (>90%) are demonstrated. The aptitude to recognize DNA at a low cost and the robustness of this assay point to a diagnostic solution in a portable and low-cost point-of-care format for the detection of nucleic acids. Further validation of human DNA will be performed to ascertain the assay performance with real genomic samples.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
L. Maugeri: investigation, G. Fangano: investigation, A. Ferlazzo: investigation and edit the original manuscript, A. Gulino investigation and edit the original manuscript, G. Forte investigation, S. Petralia: supervisor, investigation, edit the original manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has been funded by the European Union (NextGeneration EU), through the MUR-PNRR project SAMOTHRACE (CUP: E63C22000900006). The BRIT laboratory of UNICT is acknowledged for the XPS facility.References
- I. Handipta Mahardika, S. Naorungroj, W. Khamcharoen, S. Kin, N. Rodthongkum, O. Chailapakul and K. Shin, Adv. NanoBiomed Res., 2023, 3, 2300058 CrossRef.
- G. Ventimiglia and S. Petralia, BioNanoScience, 2013, 3, 428–450 CrossRef.
- M. F. Santangelo, E. L. Sciuto, S. A. Lombardo, A. C. Busacca, S. Petralia, S. Conoci and S. Libertino, IEEE J. Sel. Top. Quantum Electron., 2016, 22, 6900307 Search PubMed; M. Guarnaccia, G. Gentile, E. Alessi, S. Petralia and S. Cavallaro, Genomics, 2014, 103, 177–182 CrossRef CAS PubMed.
- J. A. Ribeiro and P. A. S. Jorge, Sens. Actuators Rep., 2024, 8, 100205S CrossRef.
- S. Petralia, T. Cosentino, F. Sinatra, M. Favetta, P. Fiorenza, C. Bongiorno, E. L. Sciuto, S. Conoci and S. Libertino, Sens. Actuators, B, 2017, 252, 492–502 CrossRef CAS.
- J. Wu, H. Liu and W. Chen, et al. , Nat. Rev. Bioeng., 2023, 1, 346–360 CrossRef.
- M. M. Kaminski, O. O. Abudayyeh, J. S. Gootenberg, F. Zhang and J. J. Collins, CRISPR-based diagnostics, Nat. Biomed. Eng., 2021, 5, 643–656 CrossRef CAS PubMed.
- S. Petralia, E. L. Sciuto, M. L. Di Pietro, M. G. Grimaldi and S. Conoci, Analyst, 2017, 142, 2090–2093 RSC.
- S. Petralia, T. Barbuzzi and G. Ventimiglia, Mater. Sci. Eng., C, 2012, 32, 848–850 CrossRef CAS; M. A. Messina, C. Meli, S. Conoci and S. Petralia, Analyst, 2017, 142, 4629–4632 RSC.
- G. M. L. Consoli, M. L. Giuffrida, S. Zimbone, L. Ferreri, L. Maugeri, M. Palmeri, C. Satriano, G. Forte and S. Petralia, ACS Appl. Mater. Interfaces, 2023, 15, 5732–5743 CrossRef CAS.
- G. M. L. Consoli, M. L. Giuffrida, C. Satriano, T. Musumeci, G. Forte and S. Petralia, Chem. Commun., 2022, 58, 3126 RSC.
- D. Briggs and J. T. Grant, Surface Analysis by Auger and X-Ray Photoelectron Spectroscopy, IMP, Chichester, UK, 2003 Search PubMed.
- A. Gulino, Anal. Bioanal. Chem., 2013, 405, 1479–1495 CrossRef CAS.
- G. Greczynski and L. Hultman, Angew. Chem., Int. Ed., 2020, 59, 5002–5006 CrossRef CAS.
- A. Ferlazzo, A. Gulino and G. Neri, Environ. Sci.: Adv., 2024, 3, 1392–1399 CAS.
- V. Bressi, C. Celesti, A. Ferlazzo, T. Len, K. Moulaee, G. Neri, R. Luque and C. Espro, Environ. Sci.: Nano, 2024, 11, 1245–1258 RSC.
- P. O. Vardevanyan, A. P. Antonyan, M. A. Parsadanyan, M. A. Shahinyan and L. A. Hambardzumyan, J. Appl. Spectrosc., 2013, 80, 595–599 CrossRef CAS.
- M. G. Kim, M. G. Kanatzidis, A. Facchetti and T. J. Marks, Nat. Mater., 2011, 10, 382–388 CrossRef CAS.
- J. Balaji, S. H. Roh, T. N. J. I. Edison, H. Y. Jung and M. G. Sethuraman, J. Mol. Liq., 2020, 302, 112551 CrossRef CAS.
- A. Gulino, G. G. Condorelli, P. Mineo and I. Fragalà, Nanotechnology, 2005, 16, 2170 CrossRef CAS PubMed.
- C. Han, L. Mazzarella, Y. Zhao, G. Yang, P. Procel, M. Tijssen and M. Zeman, ACS Appl. Mater. Interfaces, 2019, 11, 45586–45595 CrossRef CAS PubMed.
- M. Mirzaee and A. Dolati, J. Sol-Gel Sci. Technol., 2015, 75, 582–592 CrossRef CAS.
- D. A. Cristaldi and A. Gulino, ChemSusChem, 2013, 6, 1031–1036 CrossRef CAS PubMed.
- M. B. Hahn, P. M. Dietrich and J. Radnik, Commun. Chem., 2021, 4, 50 CrossRef CAS.
- Q. Xie, Y. Li, Z. Lv, H. Zhou, X. Yang, J. Chen and H. Guo, Sci. Rep., 2017, 7, 3316 CrossRef.
- M. A. Cole, N. H. Voelcker, H. Thissen and H. J. Griesser, Biomaterials, 2009, 30, 1827–1850 CrossRef CAS PubMed.
- J. S. Won, J. M. Lee, P. G. Lee, H. Y. Choi, T. J. Kwak and S. G. Lee, J. Mater. Sci., 2022, 1–17 Search PubMed.
- G. Forte, G. Consiglio, C. Satriano, L. Maugeri and S. Petralia, Colloids Surf., B, 2022, 217, 112628 CrossRef CAS.
- C. G. Pheeney and J. K. Barton, Langmuir, 2012, 28, 7063–7070 CrossRef CAS.
- J. D. Mahlum, M. A. Pellitero and N. Arroyo-Curras, J. Phys. Chem. C, 2021, 125, 9038–9049 CrossRef CAS.
- M. Drozd, M. D. Pietrzak and E. Malinowska, Front. Chem., 2018, 6, 173 CrossRef PubMed.
- X. Xu, A. Makaraviciute, E. Abdurakhmanov, F. Wermeling, S. Li, U. H. Danielson, L. Nyholm and Z. Zhang, ACS Sens., 2020, 5(1), 217–224 CrossRef CAS PubMed.
- M. Yesil, S. Donmex and F. Arslan, J. Chem. Sci., 2016, 128, 1823–1829 CrossRef CAS.
- M. El Aamri, Y. Khalki, H. Mohammadi and A. Amine, Biosensors, 2023, 13, 853 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sd00288a |
| This journal is © The Royal Society of Chemistry 2024 |