
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHeptacyclic aromatic hydrocarbon isomers with two azulene units fused†
Jianwen
Guo‡
,
Fangxin
Du‡
,
Bo
Yu‡
,
Pengcheng
Du
,
Haoyuan
Li
 ,
Jianhua
Zhang
and
Hanshen
Xin
*
,
Jianhua
Zhang
and
Hanshen
Xin
*
School of Microelectronics, Shanghai University, 201800, Shanghai, China. E-mail: xinhanshen@shu.edu.cn
First published on 28th June 2024
Abstract
Azulene, known for its unique electronic properties and structural asymmetry, serves as a promising building block for the design of novel non-benzenoid polycyclic aromatic hydrocarbons (PAHs). Herein, we present the synthesis, characterization, and physical properties of three diazulene-fused heptacyclic aromatic hydrocarbons, 8,17-dioctyldiazuleno[2,1-a:2′,1′-h]anthracene (trans configuration), 16,18-dioctyldiazuleno[2,1-a:1′,2′-j]anthracene (cis configuration) and 3,18-dioctyldiazuleno[2,1-a:1′,2′-i]phenanthrene (zigzag configuration). Three compounds are configurational isomers with different fusing patterns of aromatic rings. All three isomers exhibit pronounced aromaticity, as revealed by nuclear magnetic resonance spectroscopy and theoretical calculations. They exhibit characteristics of both azulene and benzenoid PAHs and are much more stable than their all-benzene analogues. The optical and electrochemical properties of these three isomers were investigated through UV-vis absorption spectra and cyclic voltammetry, revealing distinct behaviors influenced by their molecular configurations. Furthermore, the isomer in trans configuration exhibits promising semiconducting properties with a hole mobility of up to 0.22 cm2 V−1 s−1, indicating its potential in organic electronics applications.
Introduction
Polycyclic aromatic hydrocarbons (PAHs) have been widely used as semiconducting materials due to their distinctive electronic and self-assembling properties.1–3 Graphene, one of the most famous two-dimensional materials, which is composed of a series of alternating benzene rings, has attracted widespread attention due to its unique photoelectric and magnetic properties.4–6 Seven-membered and five-membered rings constructed entirely of sp2 carbons are considered defects in graphene and its nanofragments.7,8 Numerous reports suggest that the presence of seven-membered or five-membered rings can significantly affect the physical and chemical properties, including optoelectronic properties, magnetism, chemical stability, and mechanical strength of PAHs, like those found in graphene nanofragments.8–10 Therefore, the introduction of non-traditional conjugated structural units such as seven-membered or five-membered rings is deemed an effective method to modulate the physicochemical properties and functions of PAHs.11–13 The design and synthesis of PAHs containing seven-membered14–19 or five-membered11,20–22 rings, such as azulene-fused compounds,23–29 and the investigation of their properties and functions are becoming an emerging interdisciplinary frontier attracting widespread attention.Azulene,30 an aromatic bicyclic isomer of naphthalene, exhibits a substantial dipole moment of 1.08 D.23,31 Furthermore, the non-mirror-related frontier molecular orbitals (FMOs) of azulene result in a narrow HOMO–LUMO energy gap and effectively increase the gap from the first singlet excited state (S1) to the second singlet excited state (S2).32 On account of these distinctive features, azulene and its derivatives have been recognized as potential building blocks for the construction of organic electronic materials,33–37 with a hypothesized crucial role in modulating the chemical and electronic properties of PAHs.38–44 For example, Chi's group designed and synthesized a linear heptacyclic aromatic compound with two fused azulene units,45 as shown in Fig. 1a. This heptacyclic aromatic compound is an isomer of heptacene (or dibenzo[a,l]pentacene), but it exhibits better air stability compared to its all benzene structural isomers.46 However, limited methods39 and annulation strategies exacerbate the challenges associated with synthesis and design of azulene in PAHs; thus research on azulene-fused PAHs remains relatively scarce.
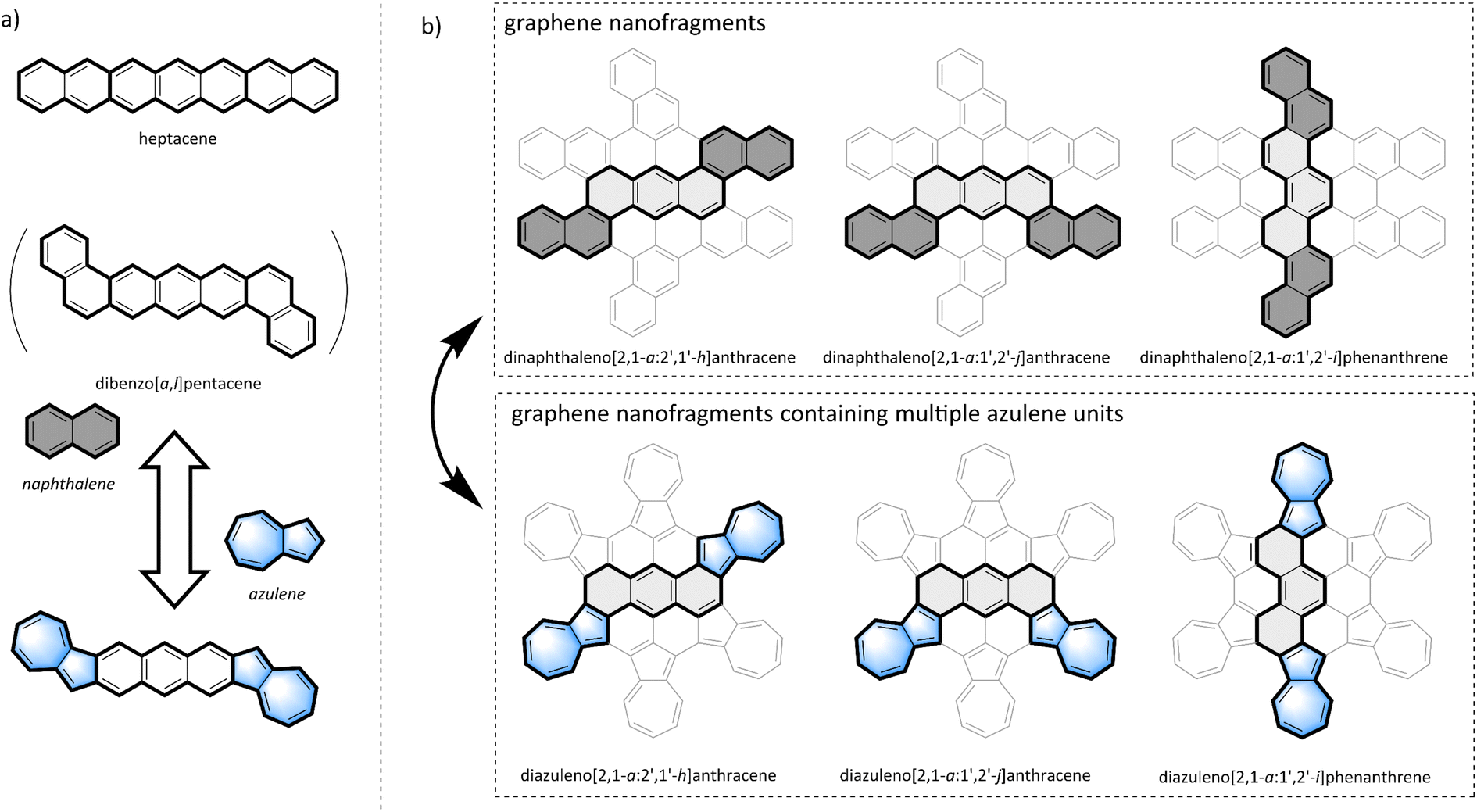 | ||
| Fig. 1 (a) Chemical structures of heptacene, dibenzo[a,l]pentacene, and their azulene-fused isomers. (b) Chemical structure of graphene nanofragments containing three isomers of heptacyclic aromatic hydrocarbons (top layer) and their azulene-fused isomers (bottom). | ||
In the graphene nanofragments shown in the top layer of Fig. 1b, there are three isomers with seven fused benzene rings: dinaphthaleno[2,1-a:2′,1′-h]anthracene (trans configuration), dinaphthaleno[2,1-a:1′,2′-j]anthracene (cis configuration), and dinaphthaleno[2,1-a:1′,2′-i]phenanthrene (zigzag configuration). Variations in condensation patterns result in distinct spatial configurations, impacting the distribution of electrons and the structure of energy levels. These alterations are directly linked to the optical, electrical, and chemical reactivity characteristics of the molecules.5 However, due to limitations in synthesis methods and stability issues, only dinaphthaleno[2,1-a:1′,2′-i]phenanthrene has been synthesized,47 and studies on the other two isomers have not yet been reported. As isomers of the graphene nanofragments shown in the bottom layer of Fig. 1b, which contains multiple azulene units fused together, there also exist three isomers with seven condensed aromatic rings: diazuleno[2,1-a:2′,1′-h]anthracene (trans configuration), diazuleno[2,1-a:1′,2′-j]anthracene (cis configuration), and diazuleno[2,1-a:1′,2′-i]phenanthrene (zigzag configuration). Researching the synthesis, properties, and functions of this series of azulene-fused heptacyclic aromatic hydrocarbons is not only important for exploring non-benzene PAHs but also provides insights into the synthesis and application of their all-benzene PAH isomers.
In this study, we have designed and synthesized three isomers featuring the aforementioned azulene-fused heptacyclic aromatic structures (Scheme 1). Octyl groups were introduced as solubilizing chains to enhance the solubility of these PAHs, resulting in the creation of 8,17-dioctyldiazuleno[2,1-a:2′,1′-h]anthracene (1), 16,18-dioctyldiazuleno[2,1-a:1′,2′-j]anthracene (2) and 3,18-dioctyldiazuleno[2,1-a:1′,2′-i]phenanthrene (3). Compounds 1 and 2 are cis–trans configurational isomers, with two azulene units fused at both ends of an anthracene core, while compound 3 has two azulene units condensed onto a phenanthrene core, forming a zigzag configuration. Chemical shift changes in the 1H NMR spectra among the three isomers 1–3 revealed structural properties arising from different configurations. Spectroscopic analyses, electrochemical investigations, and theoretical calculations have demonstrated that all three isomers manifest pronounced aromaticity and good redox properties. However, the differences in the fusion patterns of the aromatic rings, leading to distinct configurations, markedly affect their energy states and electron distribution profiles. Moreover, 1 shows p-type semiconducting behaviour with a high average hole mobility of 0.16 cm2 V−1 s−1 and holds great potential as a semiconductor.
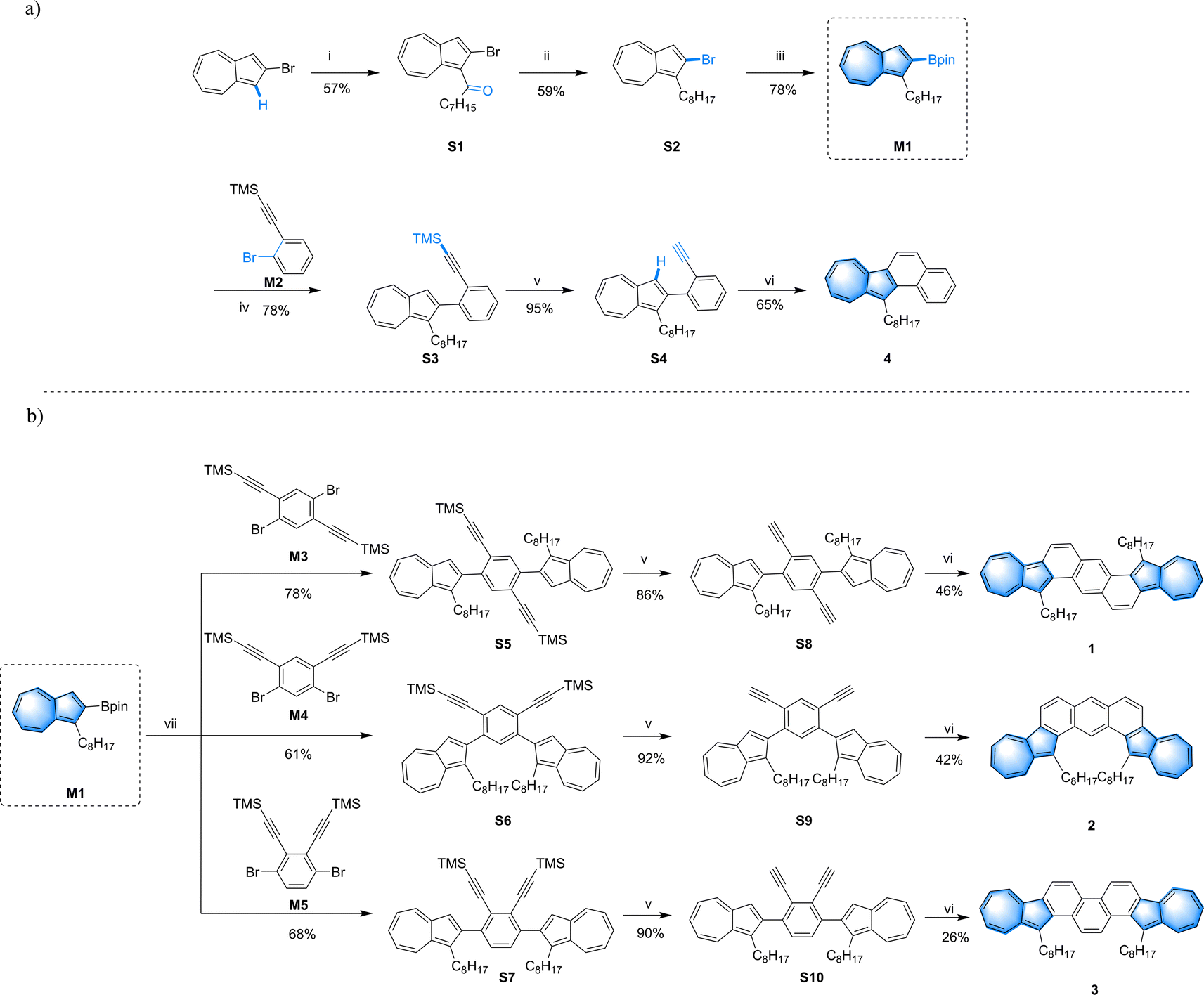 | ||
| Scheme 1 Chemical structures and synthetic routes of compound 4 (a), and compounds 1–3 (b): (i) AlCl3, octanoyl chloride, DCM, 0 °C; (ii) BH3·THF, BF3·Et2O, THF, 0 °C to r.t.; (iii) B2Pin2, Pd(dppf)Cl2, KOAc, DMSO, 80 °C; (iv) Pd(PPh3)4, K2CO3, toluene/EtOH/H2O, 85 °C; (v) KF, DMF/H2O, rt.; (vi) PtCl2, toluene, 85 °C; (vii) Pd2(dba)3, XPhos, K2CO3, THF/H2O, 85 °C. | ||
Results and discussion
The synthetic routes for compounds 1–3 are depicted in Scheme 1. At the outset of the project, we strategically designed and synthesized the key intermediate M1. And monobromide M2 was employed as a substrate to investigate the model reaction (Scheme 1a). Compound S1 was synthesized via Friedel–Crafts acylation between 2-bromoazulene and octanoyl chloride, resulting in a yield of 57%. This was followed by a reduction using borane and boron trifluoride, providing compound S2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 48 with a moderate yield of 59%. Subsequently, the key intermediate M1 was prepared through a conventional Pd-catalyzed reaction between S2 and bis(pinacolato)diboron, achieving a yield of 78%. After that, the model compound 4 was smoothly synthesized from intermediate M1 by applying a Suzuki–Miyaura coupling reaction, followed by TMS cleavage and an annulation reaction. Since the Suzuki coupling reaction of M1 with dibromobenzene derivatives (M3,49M450 and M5) was not as efficient as the reaction between M1 and M2, a series of conditions were assessed to optimize the yield. As shown in Scheme 1b, by adding XPhos as a ligand, S5 was obtained through coupling M3 with two equivalents of M1 in a mixed solvent of THF and H2O, affording a yield of 78%. Under the same conditions, M1 could react with M4 and M5 respectively, producing S6 and S7 with acceptable yields. Subsequently, by treating TMS-protected precursors S5–S7 with KF, the desilylation products S8–S10 were obtained with yields of 86%, 92%, and 90% accordingly. Finally, the target molecules were successfully synthesized through a Pt-catalyzed double annulation reaction.51 Compounds 1 and 2 were synthesized with medium yields of 46% and 42% respectively, while compound 3 was obtained with a lower yield of 26% via the same procedure. This lower yield may be attributed to the presence of two adjacent alkynyl groups in o-diphenylacetylene, which might hinder the coordination step in the Pt-catalyzed reaction, leading to unexpected byproducts and a reduced yield.
48 with a moderate yield of 59%. Subsequently, the key intermediate M1 was prepared through a conventional Pd-catalyzed reaction between S2 and bis(pinacolato)diboron, achieving a yield of 78%. After that, the model compound 4 was smoothly synthesized from intermediate M1 by applying a Suzuki–Miyaura coupling reaction, followed by TMS cleavage and an annulation reaction. Since the Suzuki coupling reaction of M1 with dibromobenzene derivatives (M3,49M450 and M5) was not as efficient as the reaction between M1 and M2, a series of conditions were assessed to optimize the yield. As shown in Scheme 1b, by adding XPhos as a ligand, S5 was obtained through coupling M3 with two equivalents of M1 in a mixed solvent of THF and H2O, affording a yield of 78%. Under the same conditions, M1 could react with M4 and M5 respectively, producing S6 and S7 with acceptable yields. Subsequently, by treating TMS-protected precursors S5–S7 with KF, the desilylation products S8–S10 were obtained with yields of 86%, 92%, and 90% accordingly. Finally, the target molecules were successfully synthesized through a Pt-catalyzed double annulation reaction.51 Compounds 1 and 2 were synthesized with medium yields of 46% and 42% respectively, while compound 3 was obtained with a lower yield of 26% via the same procedure. This lower yield may be attributed to the presence of two adjacent alkynyl groups in o-diphenylacetylene, which might hinder the coordination step in the Pt-catalyzed reaction, leading to unexpected byproducts and a reduced yield.
At room temperature, diazulene-fused isomers 1–3 with two octyl chains show good solubility in dichloromethane and aromatic solvents (>7 mg mL−1), exhibiting pronounced insolubility in methanol, a characteristic that could be utilized in purification and characterization. The chemical structures of isomers 1–3 were fully characterized by nuclear magnetic resonance (NMR) spectroscopy and high-resolution mass spectrometry (HRMS). Thermogravimetric analysis (TGA) was performed under a nitrogen atmosphere to investigate the thermal properties of isomers 1–3. As shown in Fig. S1,† TGA curves reveal the high 5 wt% decomposition temperatures of these compounds, indicating good thermodynamic stability. It can be observed that isomer 3 exhibits a higher stability (383 °C) compared to isomers 1 (308 °C) and 2 (243 °C). This may be attributed to the higher rigidity of the zigzag conformation of isomer 3, while the cis conformation of isomer 2 exhibits lower rigidity, with higher steric hindrance between adjacent alkyl chains. As shown in Table S1 and Fig. S2,† both the single point energy calculations and independent gradient model (IGM)52,53 results also indicate that 3 has greater thermal stability than 2 and 3 (see the ESI† for details).
As depicted in Fig. 2, the 1H chemical shifts within the aromatic region for isomers 1–3, as well as model compound 4, were presented for comparison. Notably, isomer 1 features a singlet at 9.46 ppm for the 1Hh, a characteristic attributed to its centrosymmetric structure. In contrast, isomer 2, which possesses a C2-symmetric structure, shows a splitting of the signal in the corresponding position into two sets (1Hh′ and 1Hi′). Significantly, the 1Hi′ experiences a remarkable downfield shift to 10.43 ppm, an effect likely due to deshielding by the ring current in the bay region (Fig. 3d). When compared to isomers 1 and 2, the chemical shifts for the seven-membered rings in isomer 3 (1Ha′′, 1Hb′′, 1Hc′′, 1Hd′′, and 1He′′) shift to a higher field, indicating a decrease in aromaticity for the seven-membered rings. Specifically, in compound 3, the chemical shifts for the 1Hf′′ and 1Hg′′ protons show a significant downfield shift compared to the analogous protons in compounds 1, 2, and 4. This indicates that the benzene rings fused to the azulene units in compound 3 exhibit increased aromaticity compared to those in the other compounds, a conclusion that is supported by the nucleus-independent chemical shifts (NICS) calculations shown in Fig. 3c. Moreover, the central benzene ring in compound 3 has the smallest chemical shift for 1Hh′ among the diazulene-fused isomers and possesses the largest NICS value, indicating lower aromaticity.
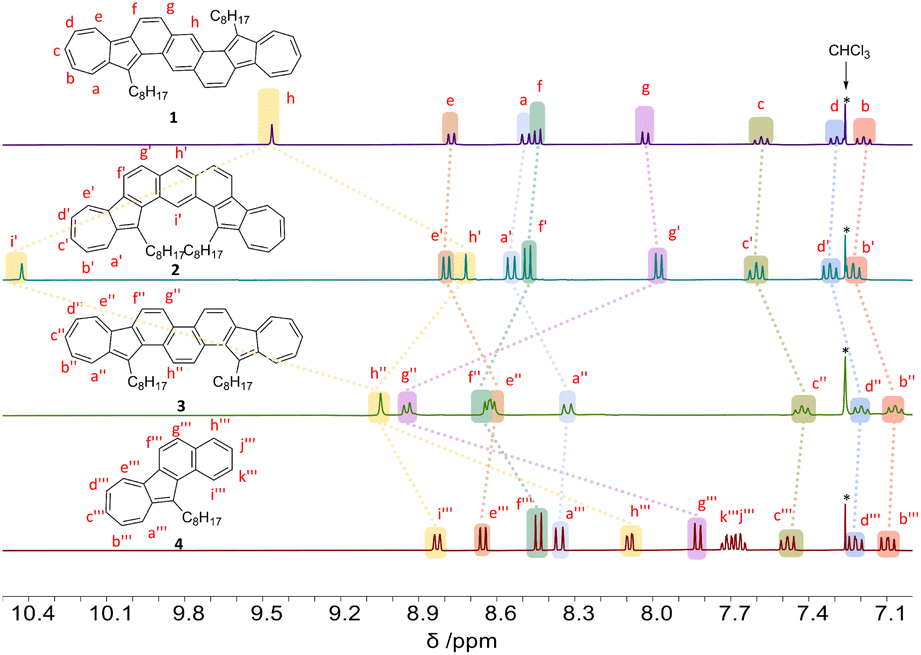 | ||
| Fig. 2 1H NMR (400 MHz, CDCl3, 298 K) spectra of the aromatic regions for compounds 1, 2, 3, and 4. All protons are labelled. | ||
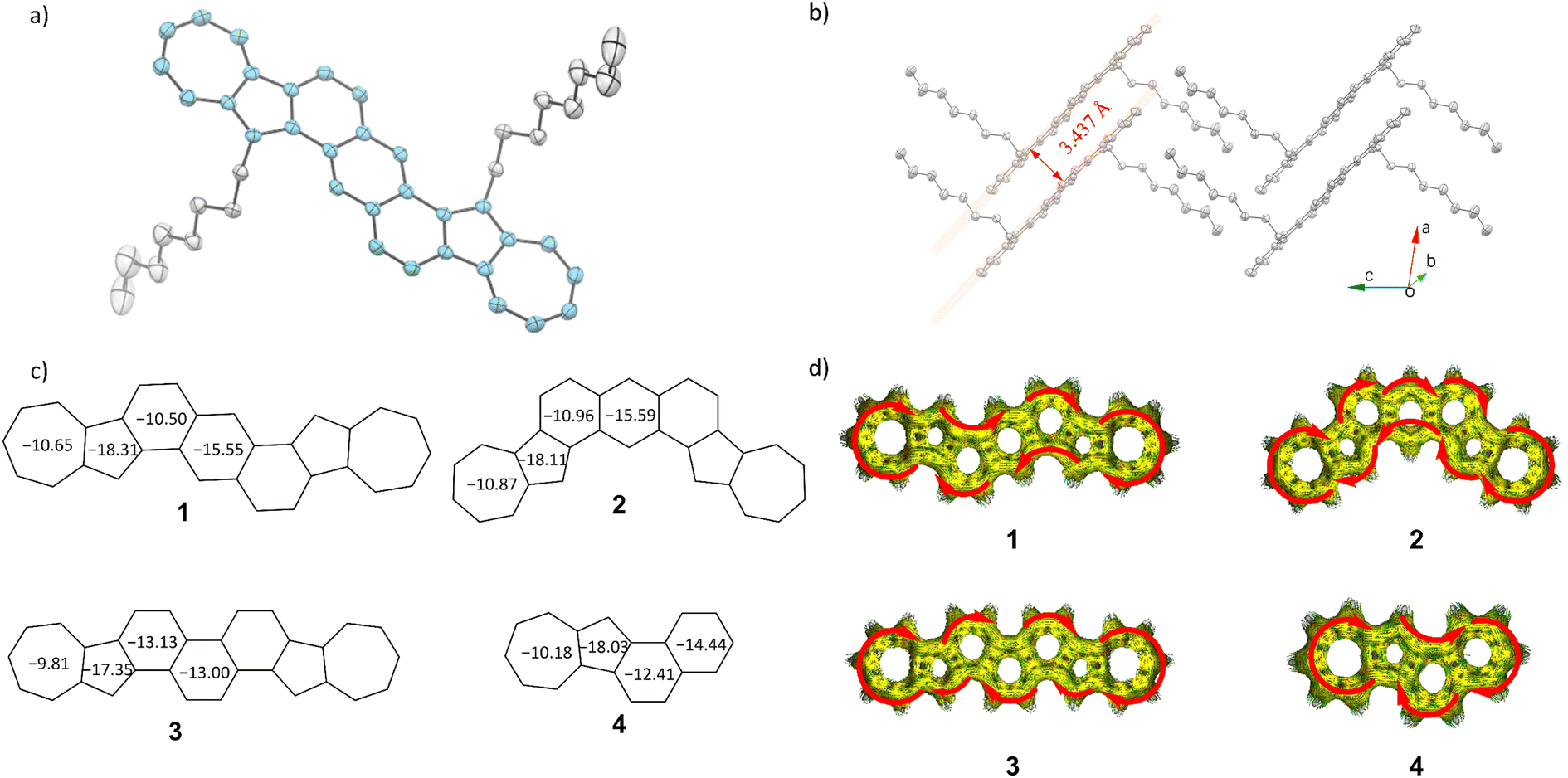 | ||
| Fig. 3 (a) Thermal ellipsoids of compound 1 (probability level = 50%). (b) Packing structure of compound 1. (c) Calculated NICS(1)zz values for compounds 1–4. (d) Calculated ACID plots for compounds 1–4. | ||
The diazulene-fused structure of compound 1 was further confirmed by single crystal X-ray analysis, which was crystalized in the mixed solvents of ethylbenzene and o-dichlorobenzene by slowly cooling down to −20 °C. Unfortunately, compounds 2 and 3 with two ipsilateral exsolution chains are difficult to crystalize perfectly for analysis, which may be due to steric hindrance. The single crystal of compound 1 exhibits a triclinic crystal system, P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group, with each unit cell containing only one molecule (Table S2 and Fig. S3†).
space group, with each unit cell containing only one molecule (Table S2 and Fig. S3†).
As shown in Fig. S3b,† crystallographic analysis revealed that the conjugated skeleton of compound 1 exhibits good planarity, with the maximum dihedral angle between any fused rings being 2.05°. Within each stacked column, all adjacent conjugated planes maintain a consistent π–π distance of 3.437 Å between levels (Fig. 3b). The good planarity and short packing distance may endow compound 1 with satisfactory charge mobility. As shown in Fig. S3b,† the average bond length within the skeleton of compound 1 is shorter than the expected length for a conjugated single bond between two sp2 carbon atoms (1.45 Å) falling instead within the range of aromatic double bonds for sp2 carbons (1.38–1.40 Å). This observation strongly supports and confirms the excellent aromaticity exhibited by compound 1.54 To understand the electronic structure and aromaticity of azulene-fused PAHs, NICS(1)zz (isotropic chemical shielding surface at 1 Å of the Z axis)55 and anisotropy of the induced current density (ACID) calculations56 were carried out as shown in Fig. 3. All fused rings have negative NICS(1)zz values (−9.81 to −18.31 ppm), indicating their aromatic characters. The average NICS(1)zz values of all azulene-fused PAHs are more negative than those of their all benzene-fused isomers (Fig. S4†), indicating that the incorporation of azulene units in the conjugated skeleton gently enhances its aromaticity. The strong aromaticity of these azulene-fused PAHs is also supported by the ACID plots (Fig. 3d), which display a clockwise ring current of the π electrons along the periphery. According to the NICS(1)zz values, compared with isomer 1 and isomer 2, isomer 3 has lower aromaticity in the azulene units and the central benzene ring, while the benzene rings adjacent to the azulene units have higher aromaticity. This result is well consistent with the NMR results. Since the aromaticity of the central rings of acenes could be overestimated using NICS values,57,58 which may bother the study of molecular aromatics, we conducted additional analyses including multi-center index (MCI),59 fluctuation index (FLU),60 harmonic oscillator model of aromaticity (HOMA),61 and normalized multicenter bond order (NCBO)62 to provide a more comprehensive assessment of the aromatic nature of compounds 1–4. As shown in Table S3,† the results from MCI, FLU, HOMA, and NCBO all indicate that the aromaticity of the central ring in compounds 1–3 is consistent with the NICS(1)zz values.
The optical and electrochemical properties of three diazulene-fused heptacyclic dyes 1–3 and the tetracene analogue 4 were investigated using UV-vis absorption spectra and cyclic voltammetry (CV) measurements. The corresponding data are summarized in Table 1. As shown in Fig. 4a, the absorption spectra obtained in dichloromethane show that compounds 1–3 exhibit strong absorbance from 300 to 600 nm, accompanied by weaker absorption bands from 600 to 900 nm, as anticipated. The strong absorbance arises from the π–π* transition of the conjugated system, while the weaker absorption bands from 600 to 900 nm represent a characteristic “azulene type” structured absorption profile, originating from the S0 to S1 transitions of azulene moieties. Compared to compound 4 with lower degrees of conjugation, the absorption of compounds 1, 2, and 3 was enhanced and a remarkable bathochromic shift has been observed. Notably, within the strong absorbance range, isomer 2 exhibits two distinct absorption bands (300–400 nm with a peak at 352 nm; ε = 5.29 × 104 M−1 cm−1, and 400–550 nm with peaks at 417, 439, and 466 nm), in contrast to 1 (peak at 402 nm; ε = 4.49 × 104 M−1 cm−1) and 3 (peak at 378 nm; ε = 5.51 × 104 M−1 cm−1). This difference may stem from the unique cis configuration of isomer 2, which facilitates specific electronic transition pathways. This observation aligns with time-dependent density functional theory (TD-DFT) calculations (Fig. S5†). Furthermore, compared to isomers 1 and 2, isomer 3 exhibits fewer absorption peaks in the 300–500 nm range, with only a shoulder peak at 427 nm. This may be due to the zigzag configuration having greater structural rigidity compared to the trans and cis configurations, which reduces the coupling between electronic transitions and molecular vibrational states. Besides, in the zigzag configuration, π electrons may be more evenly distributed across the molecular framework. This result is consistent with NICS(1)zz calculations, which show a more uniform distribution of values across the rings in isomer 3 (Fig. 3c). As shown in Fig. S6,† the absorption of compounds 1, 2, and 4 in the film is like their absorption in dichloromethane, with their maximum absorption wavelength red-shifted by about 6 nm; whereas the absorption of compound 3 in the film, compared to its solution absorption, has significantly blue-shifted by about 50 nm. These observations indicate that compound 3 tends to form H-aggregates in a solid state, while the other molecules tend to form J-aggregates.63 Although azulene exhibits atypical fluorescence that follows the anti-Kasha rule,22 we studied the emission properties of compounds 1–4 and observed no detectable fluorescence. As shown in Fig. S7,† compounds 1, 2, and 3 exhibit good photostability (half-life time t1/2: 25 days, 34 days, and 26 days, respectively) under ambient air and light conditions as compared to their all benzene-fused counterparts and other azulene-fused PAHs.45 The electrochemical properties of three diazulene-fused heptacyclic aromatics 1–3 and the model compounds 4 were studied with cyclic voltammetry (CV) and differential pulse voltammetry (DPV) in DCM. As shown in Fig. 4, isomers 1–3, with two azulenes fused, unambiguously exhibit two oxidation waves and two reduction waves, whereas compound 4, with one azulene fused, only shows one oxidation wave and one reduction wave. Based on the peaks of the DPV, the HOMO/LUMO energy levels of compounds 1–3 were calculated respectively and are listed in Table 1. The HOMO/LUMO energies for isomers 1, 2, and 3 were estimated to be −4.92/−2.91, −4.99/−2.87, and −4.98/−2.89 eV, respectively. Consequently, the electrochemical gap for isomers 1, 2, and 3 was calculated to be 2.01, 2.13, and 2.11 eV, respectively. The results indicate that, although all three isomers consist of two azulene units and three benzene rings as 30π electron systems, different configurations have a significant impact on their redox properties.
| Cp. ID | λ absmax | λ onset | E ox | E red | E cvHOMO | E cvLUMO | E cvg | E optg | E calHOMO | E calLUMO | E calg | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a Measured in 1 × 10−5 M DCM solutions. b Measured in nBu4NPF6 solution vs. Ag/AgCl (calibration using ferrocene), Eox/red acquired from DPV curves. c Calculated from EHOMO/LUMO = −(Eox/red + 4.8), Ecvg = EcvLUMO − EcvHOMO, Eoptg = 1240/λonset. d Estimated from DFT calculations. | |||||||||||||
| 1 | 402 and 751 | 935 | 0.12 | 0.39 | −2.14 | −1.89 | −4.92 | −2.91 | 2.01 | 1.32 | −4.52 | −2.23 | 2.29 |
| 2 | 352, 417, and 682 | 912 | 0.19 | 0.45 | −2.16 | −1.93 | −4.99 | −2.87 | 2.13 | 1.36 | −4.64 | −2.08 | 2.56 |
| 3 | 378 and 748 | 918 | 0.19 | 0.39 | −2.14 | −1.91 | −4.98 | −2.89 | 2.11 | 1.35 | −4.59 | −2.20 | 2.39 |
| 4 | 321 and 660 | 435 | 0.27 | −2.00 | −5.07 | −2.80 | 2.27 | 2.85 | −4.77 | −2.03 | 2.74 | ||
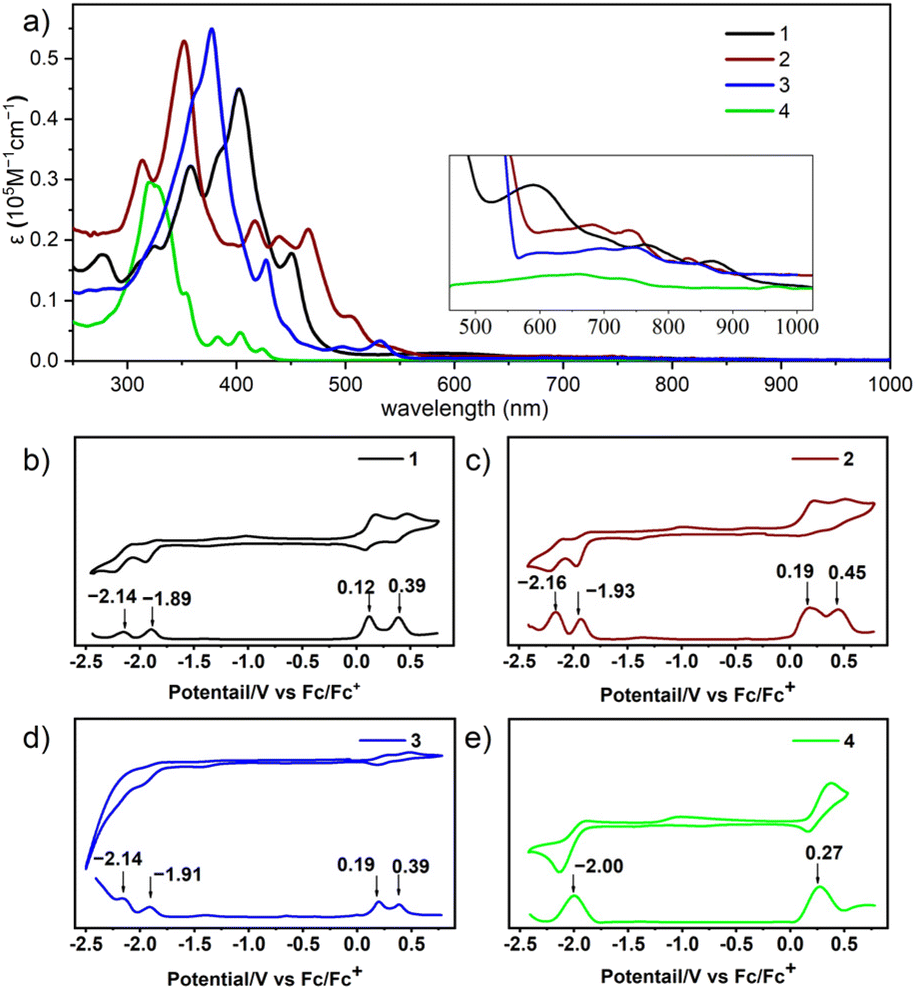 | ||
| Fig. 4 (a) UV-vis absorption (10−6 M in DCM). Cyclic voltammetry (CV) curves and differential pulse voltammetry (DPV) curves of (b) 1, (c) 2, (d) 3, and (e) 4, measured in 0.1 M solution of nBu4NPF6 in DCM. | ||
DFT calculations were carried out to investigate the structural and electronic properties of azulene-fused PAHs using the RB3LYP/6-31G(d,p) level of theory. The calculated data are shown in the ESI,† and the conclusions are listed in Table 1. The energy gaps estimated from calculations are larger than those obtained from DPV measurements and UV-vis spectra. Although the results are slightly varied, the data are consistent with the experiment results in trend. Consistent with the single crystal diffraction analysis results of compound 1, all azulene-fused PAHs exhibit planar geometry with two octyl chains distributed on both sides of the scaffold plane. The electron density distribution of both HOMOs and LUMOs is delocalized over the whole backbone. As shown in Fig. 5, azulene moieties possess more contribution to LUMOs, while central backbones contribute significantly to HOMOs. It is worth noting that the HOMO electron density distribution of isomer 3 is more uniformly delocalized across the entire skeleton compared to isomers 1 and 2, which results in a more singular path and energy change for electron transitions. Therefore, the UV-vis absorption spectrum of isomer 3 exhibits fewer fine peaks (Fig. 4a). And the NICS(1)zz value of six-membered rings in isomer 3 is more equal.
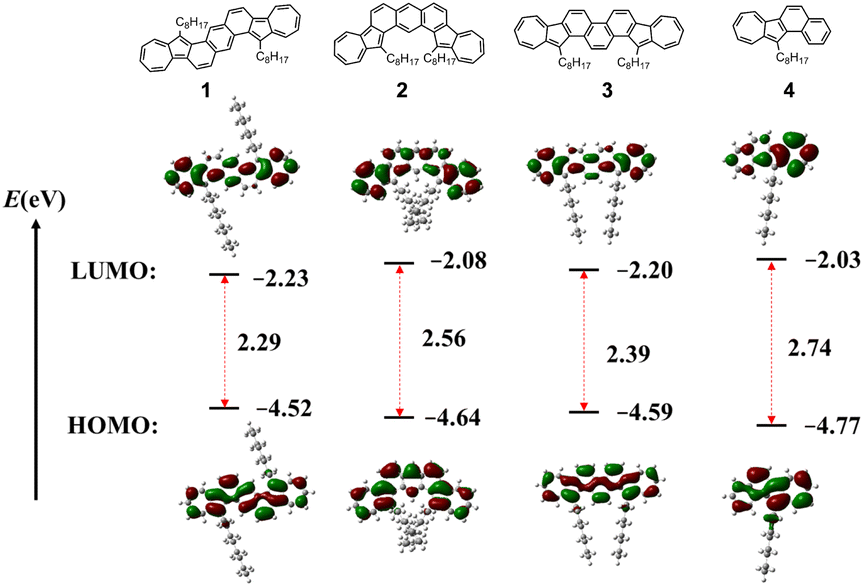 | ||
| Fig. 5 Frontier molecular orbitals and their energies of compounds 1–4, obtained using DFT calculations. | ||
Azulene is an acid-sensitive compound, which can be protonated by strong acids such as trifluoroacetic acid (TFA) and trifluoromethanesulfonic acid (TfOH).24,64–70 This protonation disrupts the conjugated system by attaching a proton, resulting in the formation of a stable azulenium cation. The original structure can be restored by adding a base. To clarify the protonation behaviour of diazulene-fused PAHs 1–3, UV-vis absorption spectroscopy and 1H NMR analyses were conducted. As illustrated in Fig. S8,† with an increase in TFA equivalents, the intensity of characteristic peaks of the three isomers in the 300–500 nm range gradually decreases, while a new absorption peak that gradually strengthens appears in the long-wavelength area. Notably, the absorption spectra of protonated 1–3 also show significant differences. The new absorption peak for 12+ appears in the 500–800 nm range, for 22+ in the 600–900 nm range, and for 32+ in the 450–600 nm range; therefore, the colours of 12+, 22+, and 32+ in dichloromethane solution are purple, amaranth, and orange respectively. These results indicate that upon acid addition, all three isomers form azulenium cations (Fig. 6c), and the azulenium cations remain conjugated with the fused benzene rings. However, due to different connection positions (12+vs.22+) and different fused configurations (anthracene vs. phenanthrene), the π electron structures of 12+, 22+, and 32+ are distinctly different. This result can also be further confirmed using fluorescence spectra, as shown in Fig. 6b, where the emission peak of 12+ is at 650 nm, that of 22+ is at 640 nm, and that of 32+ shifts to 580 nm. As shown in Fig. S9,† the changed chemical shift can be restored by adding TEA, indicating that the protonated molecules have good stability.
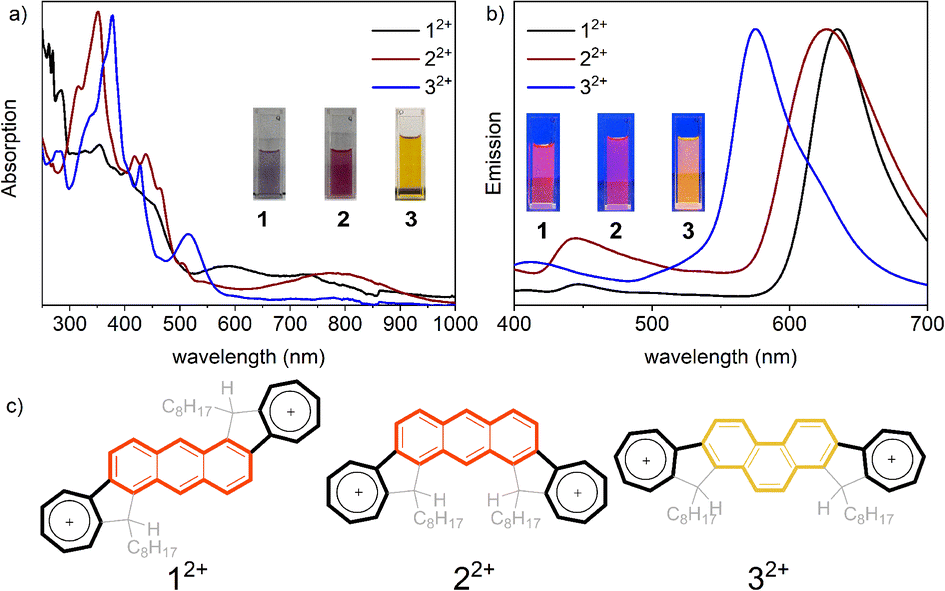 | ||
| Fig. 6 Protonation behaviour: (a) UV-vis absorption and (b) emission of protonated compounds 12+, 22+, and 32+. Insets show photos of the solutions. (c) The structure of protonated compounds. | ||
Based on its conjugated π structure, short π–π stacking distance (3.437 Å), and the suitable configuration with two alkyl chains distributed on both sides, isomer 1 was selected for investigation of its charge transport properties in organic field effect transistors (OFETs). Thin film transistors of 1 were fabricated by drop-casting toluene solutions (0.5 mg mL−1) onto octadecyl trichlorosilane (OTS)-treated Si/SiO2 substrates. Au source/drain electrodes were deposited on the thin film by vacuum evaporation, resulting in a bottom gate contact (BGBC) device structure. As shown in Fig. 7, devices based on isomer 1 function as typical p-type semiconductors, which correlates well with the energy of its frontier orbitals. The average hole mobility (μh) extracted from the transfer curves is 0.16 cm2 V−1 s−1 with a current on/off ratio (Ion/Ioff) of 104–105 and a low threshold voltage (Vth) of less than 5 V. Furthermore, the highest μh is 0.22 cm2 V−1 s−1, ranking among the highest hole mobilities for PAH-based thin film OFETs.20,22,71–76 These results demonstrate that compound 1 is a promising material for organic semiconductors.
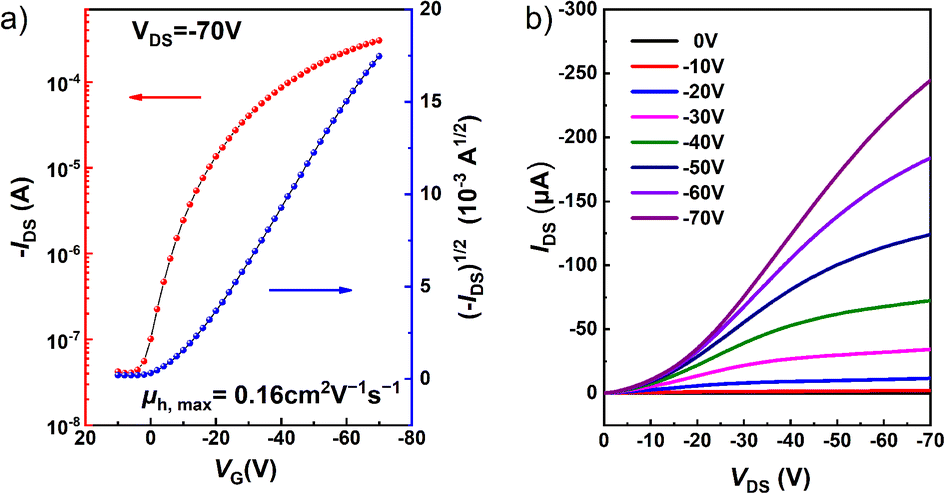 | ||
| Fig. 7 (a) Transfer and (b) output curves of the OEFT device of 1. | ||
Conclusions
In summary, three diazulene-fused heptacyclic aromatic hydrocarbons were designed, synthesized, and characterized. The structures of the three isomers 1–3 were unambiguously confirmed by nuclear magnetic resonance (NMR) spectroscopy and high-resolution mass spectrometry (HRMS). Compound 1 was further confirmed by single crystal X-ray diffraction analysis. The isomers 1–3 all exhibited excellent thermal and ambient stability, as well as good redox properties. Nuclear independent chemical shift (NICS) and anisotropy of induced current density (ACID) calculations have shown that all three isomers exhibit strong aromaticity. UV-visible absorption, electrochemical voltammetry, and theoretical calculations demonstrated that the different configurations of these isomers result in distinct optical and electrochemical properties. Additionally, isomer 1 demonstrates promising performance in OFETs with a hole mobility of up to 0.22 cm2 V−1 s−1, making it among the best PAHs for thin film OFETs. This showcases the potential of diazulene-fused PAHs in future electronic devices. Our studies not only expand the library of PAHs but also provide valuable insights into designing materials with distinct properties for specific applications.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
H. Xin designed the study and supervised the project. J. Guo, F. Du, and B. Yu conducted the experiments. P. Du, H. Li, and J. Zhang collected and analysed the data. All authors discussed the results and co-wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was financially supported by the National Natural Science Foundation of China (No. 22101170) and Shanghai University. We thank Prof. Xike Gao and Dr Junjun Xiang at SIOC for helpful discussions. We thank Wenwen Zhang and Jinheng Pan (Mass Spectrometry & Metabolomics Core Facility of Westlake University) for MS analysis.Notes and references
- J. E. Anthony, Chem. Rev., 2006, 106, 5028–5048 CrossRef CAS PubMed.
- A. Artigas, D. Hagebaum-Reignier, Y. Carissan and Y. Coquerel, Chem. Sci., 2021, 12, 13092–13100 RSC.
- T. Dumslaff, Y. Gu, G. M. Paternò, Z. Qiu, A. Maghsoumi, M. Tommasini, X. Feng, F. Scotognella, A. Narita and K. Müllen, Chem. Sci., 2020, 11, 12816–12821 RSC.
- J. Wu, W. Pisula and K. Müllen, Chem. Rev., 2007, 107, 718–747 CrossRef CAS PubMed.
- X.-Y. Wang, X. Yao and K. Müllen, Sci. China: Chem., 2019, 62, 1099–1144 CrossRef CAS.
- L. Ruan, W. Luo, H. Zhang, P. Liu, Y. Shi and P. An, Chem. Sci., 2024, 15, 1511–1519 RSC.
- H. J. Park, J. Meyer, S. Roth and V. Skákalová, Carbon, 2010, 48, 1088–1094 CrossRef CAS.
- T. Björkman, S. Kurasch, O. Lehtinen, J. Kotakoski, O. V. Yazyev, A. Srivastava, V. Skakalova, J. H. Smet, U. Kaiser and A. V. Krasheninnikov, Sci. Rep., 2013, 3, 3482 CrossRef PubMed.
- B. P. Klein, S. E. Harman, L. Ruppenthal, G. M. Ruehl, S. J. Hall, S. J. Carey, J. Herritsc, M. Schmid, R. J. Maurer, R. Tonner, C. T. Campbell and J. M. Gottfried, Chem. Mater., 2020, 32, 1041–1053 CrossRef CAS.
- C. M. Cruz, S. Castro-Fernández, E. Maçôas, A. Millán and A. G. Campaña, Synlett, 2019, 30, 997–1002 CrossRef CAS.
- K. Kawasumi, Q. Zhang, Y. Segawa, L. T. Scott and K. Itami, Nat. Chem., 2013, 5, 739–744 CrossRef CAS PubMed.
- Y. Fei and J. Liu, Adv. Sci., 2022, 9, 2201000 CrossRef CAS PubMed.
- Q. Fan, D. Martin-Jimenez, D. Ebeling, C. K. Krug, L. Brechmann, C. Kohlmeyer, G. Hilt, W. Hieringer, A. Schirmeisen and J. M. Gottfried, J. Am. Chem. Soc., 2019, 141, 17713–17720 CrossRef CAS PubMed.
- T. Maekawa, H. Ueno, Y. Segawa, M. M. Haley and K. Itami, Chem. Sci., 2016, 7, 650–654 RSC.
- J. Wang, F. G. Gámez, J. Marín-Beloqui, A. Diaz-Andres, X. Miao, D. Casanova, J. Casado and J. Liu, Angew. Chem., Int. Ed., 2023, 62, e202217124 CrossRef CAS PubMed.
- R. Liu, Y. Fu, F. Wu, F. Liu, J. J. Zhang, L. Yang, A. A. Popov, J. Ma and X. Feng, Angew. Chem., Int. Ed., 2023, 62, e202219091 CrossRef CAS PubMed.
- S. Zank, J. M. Fernandez-Garcia, A. J. Stasyuk, A. A. Voityuk, M. Krug, M. Sola, D. M. Guldi and N. Martin, Angew. Chem., Int. Ed., 2022, 61, e202112834 CrossRef CAS PubMed.
- J. P. Mora-Fuentes, M. D. Codesal, M. Reale, C. M. Cruz, V. G. Jiménez, A. Sciortino, M. Cannas, F. Messina, V. Blanco and A. G. Campaña, Angew. Chem., Int. Ed., 2023, 62, e202301356 CrossRef CAS PubMed.
- I. R. Márquez, S. Castro-Fernández, A. Millán and A. G. Campaña, Chem. Commun., 2018, 54, 6705–6718 RSC.
- L. Qin, Y. Y. Huang, B. Wu, J. Pan, J. Yang, J. Zhang, G. Han, S. Yang, L. Chen, Z. Yin, Y. Shu, L. Jiang, Y. Yi, Q. Peng, X. Zhou, C. Li, G. Zhang, X. S. Zhang, K. Wu and D. Zhang, Angew. Chem., Int. Ed., 2023, 62, e202304632 CrossRef CAS PubMed.
- Q. Jiang, T. Tao, H. Phan, Y. Han, T. Y. Gopalakrishna, T. S. Herng, G. Li, L. Yuan, J. Ding and C. Chi, Angew. Chem., Int. Ed., 2018, 57, 16737–16741 CrossRef CAS PubMed.
- X. Yang, X. Shi, N. Aratani, T. P. Gonçalves, K.-W. Huang, H. Yamada, C. Chi and Q. Miao, Chem. Sci., 2016, 7, 6176–6181 RSC.
- H. Xin and X. Gao, ChemPlusChem, 2017, 82, 945–956 CrossRef CAS PubMed.
- E. Amir, M. Murai, R. J. Amir, J. S. C. Jr., M. L. Chabinyc and C. J. Hawker, Chem. Sci., 2014, 5, 4483–4489 RSC.
- K. Horii, R. Kishi, M. Nakano, D. Shiomi, K. Sato, T. Takui, A. Konishi and M. Yasuda, J. Am. Chem. Soc., 2022, 144, 3370–3375 CrossRef CAS PubMed.
- J. Ma, Y. Fu, E. Dmitrieva, F. Liu, H. Komber, F. Hennersdorf, A. A. Popov, J. J. Weigand, J. Liu and X. Feng, Angew. Chem., Int. Ed., 2020, 59, 5637–5642 CrossRef CAS PubMed.
- K. Biswas, Q. Chen, S. Obermann, J. Ma, D. Soler-Polo, J. Melidonie, A. Barragán, A. Sánchez-Grande, K. Lauwaet, J. M. Gallego, R. Miranda, D. Écija, P. Jelínek, X. Feng and J. I. Urgel, Angew. Chem., Int. Ed., 2024, 63, e202318185 CrossRef CAS PubMed.
- N. Ogawa, Y. Yamaoka, H. Takikawa, K. I. Yamada and K. Takasu, J. Am. Chem. Soc., 2020, 142, 13322–13327 CrossRef CAS PubMed.
- Y. Liang, S. Wang, M. Tang, L. Wu, L. Bian, J. Liang, Z. B. Tang, J. Liu, A. Guan and Z. Liu, Angew. Chem., Int. Ed., 2023, 62, e202218839 CrossRef CAS PubMed.
- A. G. Anderson and B. M. Steckler, J. Am. Chem. Soc., 1959, 81, 4941–4946 CrossRef CAS.
- D. M. Lemal and G. D. Goldman, J. Chem. Educ., 1988, 65, 923–925 CrossRef CAS.
- D. Dunlop, L. Ludvíková, A. Banerjee, H. Ottosson and T. Slanina, J. Am. Chem. Soc., 2023, 145, 21569–21575 CrossRef CAS PubMed.
- B. Hou, Z. Zhou, C. Yu, X.-S. Xue, J. Zhang, X. Yang, J. Li, C. Ge, J. Wang and X. Gao, ACS Macro Lett., 2022, 11, 680–686 CrossRef CAS PubMed.
- F. Schwarz, M. Koch, G. Kastlunger, H. Berke, R. Stadler, K. Venkatesan and E. Lörtscher, Angew. Chem., Int. Ed., 2016, 55, 11781–11786 CrossRef CAS PubMed.
- A. Diaz-Andres, J. Marín-Beloqui, J. Wang, J. Liu, J. Casado and D. Casanova, Chem. Sci., 2023, 14, 6420–6429 RSC.
- M. H. Garner, J. T. Blaskovits and C. Corminboeuf, Chem. Sci., 2023, 14, 10458–10466 RSC.
- J. Spengler, C. Zhu, K. Shoyama and F. Würthner, Chem. Sci., 2023, 14, 10861–10866 RSC.
- T. Shoji, T. Okujima and S. Ito, Int. J. Mol. Sci., 2020, 21, 7087 CrossRef CAS PubMed.
- H. Xin, B. Hou and X. Gao, Acc. Chem. Res., 2021, 54, 1737–1753 CrossRef CAS.
- C. Duan, J. Zhang, J. Xiang, X. Yang and X. Gao, Angew. Chem., Int. Ed., 2022, 61, e202201494 CrossRef CAS PubMed.
- S. Wang, M. Tang, L. Wu, L. Bian, L. Jiang, J. Liu, Z. B. Tang, Y. Liang and Z. Liu, Angew. Chem., Int. Ed., 2022, 61, e202205658 CrossRef CAS PubMed.
- H. Xin, J. Li, X. Yang and X. Gao, J. Org. Chem., 2019, 85, 70–78 CrossRef PubMed.
- H. Xin, J. Li, R.-Q. Lu, X. Gao and T. M. Swager, J. Am. Chem. Soc., 2020, 142, 13598–13605 CrossRef CAS PubMed.
- C. Duan, J. Zhang, S. Cai, J. Xiang, X. Yang and X. Gao, Eur. J. Org Chem., 2023, 26, e202201347 CrossRef CAS.
- A. Ong, T. Tao, Q. Jiang, Y. Han, Y. Ou, K. W. Huang and C. Chi, Angew. Chem., Int. Ed., 2022, 61, e202209286 CrossRef CAS PubMed.
- M. M. Payne, S. R. Parkin and J. E. Anthony, J. Am. Chem. Soc., 2005, 127, 8028–8029 CrossRef CAS PubMed.
- F. B. Mallory, K. E. Butler, A. C. Evans, E. J. Brondyke, C. W. Mallory, C. Yang and A. Ellenstein, J. Am. Chem. Soc., 1997, 119, 2119–2124 CrossRef CAS.
- B. Yu, P. Du, J. Guo, H. Xin and J. Zhang, Chin. Chem. Lett., 2024, 35, 109321 CrossRef CAS.
- K. Takimiya, Y. Konda, H. Ebata, N. Niihara and T. Otsubo, J. Org. Chem., 2005, 70, 10569–10571 CrossRef CAS PubMed.
- D. T. Y. Bong, E. W. L. Chan, R. Diercks, P. I. Dosa, M. M. Haley, A. J. Matzger, O. S. Miljanic and K. P. C. Vollhardt, Org. Lett., 2004, 6, 2249–2252 CrossRef CAS PubMed.
- M. B. Goldfinger and T. M. Swager, J. Am. Chem. Soc., 1994, 116, 7895–7896 CrossRef CAS.
- T. Lu and Q. Chen, J. Comput. Chem., 2022, 43, 539–555 CrossRef CAS.
- C. Lefebvre, G. Rubez, H. Khartabil, J. C. Boisson, J. Contreras-Garcia and E. Henon, Phys. Chem. Chem. Phys., 2017, 19, 17928–17936 RSC.
- R. B. Campbell, J. M. Robertson and J. Trotter, Acta Crystallogr., 1962, 15, 289–290 CrossRef CAS.
- P. v. R. Schleyer, C. Maerker, A. Dransfeld, H. Jiao and N. J. R. v. E. Hommes, J. Am. Chem. Soc., 1996, 118, 6317–6318 CrossRef CAS PubMed.
- D. Geuenich, K. Hess, F. Köhler and R. Herges, Chem. Rev., 2005, 105, 3758–3772 CrossRef CAS PubMed.
- G. Portella, J. Poater, J. M. Bofill, P. Alemany and M. Solà, J. Org. Chem., 2005, 70, 2509–2521 CrossRef CAS PubMed.
- T. M. Krygowski, M. K. Cyrañski, Z. Czarnocki, G. Häfelingerb and A. R. Katritzky, Tetrahedron Lett., 2000, 56, 1783–1796 CrossRef CAS.
- M. Giambiagi, M. S. d. Giarnbiagi and K. C. Mundirn, Struct. Chem., 1990, 1, 423–427 CrossRef CAS.
- E. Matito, M. Duran and M. Solà, J. Chem. Phys., 2005, 122, 14109 CrossRef PubMed.
- J. Kruszewski and T. M. Krygowski, Tetrahedron Lett., 1972, 13, 3839–3842 CrossRef.
- E. Matito, Phys. Chem. Chem. Phys., 2016, 18, 11839–11846 RSC.
- F. Würthner, T. E. Kaiser and C. R. Saha-Möller, Angew. Chem., Int. Ed., 2011, 50, 3376–3410 CrossRef PubMed.
- K. H. Grellmann, E. Heilbronner, P. Seiler and A. Wellerla, J. Am. Chem. Soc., 1968, 16, 4238–4242 CrossRef.
- N. Kihara, H. Nakayama and T. Fukutomi, Macromolecules, 1997, 30, 6385–6387 CrossRef CAS.
- E. Amir, R. J. Amir, L. M. Campos and C. J. Hawker, J. Am. Chem. Soc., 2011, 133, 10046–10049 CrossRef CAS PubMed.
- M. Murai, E. Amir, R. J. Amira and C. J. Hawker, Chem. Sci., 2012, 3, 2721–2725 RSC.
- K. Tsurui, M. Murai, S. Y. Ku, C. J. Hawker and M. J. Robb, Adv. Funct. Mater., 2014, 24, 7338–7347 CrossRef CAS.
- M. Murai, S.-Y. Ku, N. D. Treat, M. J. Robb, M. L. Chabinyca and C. J. Hawker, Chem. Sci., 2014, 5, 3753–3760 RSC.
- A. G. Lvov and A. Bredihhin, Org. Biomol. Chem., 2021, 19, 4460–4468 RSC.
- K. Yamamoto, Y. le, N. Tohnai, F. Kakiuchi and Y. Aso, Sci. Rep., 2018, 8, 17663 CrossRef PubMed.
- H. Lee, Y. Zhang, L. Zhang, T. Mirabito, E. K. Burnett, S. Trahan, A. R. Mohebbi, S. C. B. Mannsfeld, F. Wudlb and A. L. Briseno, J. Mater. Chem. C, 2014, 2, 3361–3366 RSC.
- K. Y. Cheung, X. Xu and Q. Miao, J. Am. Chem. Soc., 2015, 137, 3910–3914 CrossRef CAS PubMed.
- S. H. Pun, Y. Wang, M. Chu, C. K. Chan, Y. Li, Z. Liu and Q. Miao, J. Am. Chem. Soc., 2019, 141, 9680–9686 CrossRef CAS PubMed.
- D. Geng, K. Wang, L. Li, K. Myny, A. Nathan, J. Jang, Y. Kuo and M. Liu, Nat. Electron., 2023, 6, 963–972 CrossRef CAS.
- K. Sekine, J. Schulmeister, F. Paulus, M. Rudolph, J. Zaumseil, K. P. Goetz, F. Rominger and A. S. K. Hashmi, Chem.–Eur. J., 2019, 25, 216–220 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2336236. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc02566k |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |