
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceWith or without a co-solvent? highly efficient ultrafast phenanthrenequinone-electron rich alkene (PQ-ERA) photoclick reactions†
Anna M.
Doze‡
 ab,
Youxin
Fu‡§
a,
Mariangela
Di Donato
cd,
Michiel F.
Hilbers
e,
Gert
Luurtsema
b,
Philip H.
Elsinga
b,
Wybren Jan
Buma
ef,
Wiktor
Szymanski
*agh and
Ben L.
Feringa
*a
ab,
Youxin
Fu‡§
a,
Mariangela
Di Donato
cd,
Michiel F.
Hilbers
e,
Gert
Luurtsema
b,
Philip H.
Elsinga
b,
Wybren Jan
Buma
ef,
Wiktor
Szymanski
*agh and
Ben L.
Feringa
*a
aCentre for Systems Chemistry, Stratingh Institute for Chemistry, Faculty of Science and Engineering, University of Groningen, Nijenborgh 4, 9747 AG Groningen, The Netherlands. E-mail: w.szymanski@umcg.nl; b.l.feringa@rug.nl
bDepartment of Nuclear Medicine and Molecular Imaging, University of Groningen, University Medical Centre Groningen, Hanzeplein 1, 9713 GZ Groningen, The Netherlands
cLENS (European Laboratory for Non-Linear Spectroscopy), via N. Carrara 1, 50019 Sesto Fiorentino (FI), Italy
dICCOM-CNR, via Madonna del Piano 10, 50019 Sesto Fiorentino (FI), Italy
eVan ’t Hoff Institute for Molecular Sciences, University of Amsterdam Science Park 904, 1098 XH Amsterdam, The Netherlands
fInstitute for Molecules and Materials, FELIX Laboratory, Radboud University, Toernooiveld 7c, 6525 ED Nijmegen, The Netherlands
gDepartment of Radiology, Medical Imaging Center, University of Groningen, University Medical Centre Groningen, Hanzeplein 1, 9713 GZ Groningen, The Netherlands
hDepartment of Medicinal Chemistry, Photopharmacology and Imaging, Groningen Research Institute of Pharmacy, University of Groningen, Antonius Deusinglaan 1, 9713 AV, Groningen, The Netherlands
First published on 26th June 2024
Abstract
The light-induced photocycloaddition of 9,10-phenanthrenequinone (PQ) with electron-rich alkenes (ERA), known as the PQ-ERA reaction, is a highly attractive photoclick reaction characterized by its operational simplicity and high biocompatibility. One essential aspect of photoclick reactions is their high rate, however the limited solubility of PQs often requires the use of a co-solvent. Evaluating the effect of different co-solvents on the PQ-ERA reaction and their influence on the reaction rate, we discovered that sulfur-containing compounds, in particular the frequently used solubilizing co-solvent DMSO, quench the triplet state of the PQ. These experimental results, supported by nanosecond-microsecond and ultrafast transient absorption data, show that even minimal amounts of DMSO result in a decreased lifetime of the reactive triplet state, essential for the photoclick reaction. Without DMSO as co-solvent, exceptionally high photoreaction quantum yields (ΦP up to 93% with only 1 equivalent ERA) and complete conversion in seconds can be achieved. With these outstanding efficiencies, the PQ-ERA reaction can be used without excess ERA and at low light intensities, facilitating photoclick transformations in various future applications.
1 Introduction
The use of photochemical activation procedures in click chemistry has led to the development of a new category of light-induced reactions, known as “photoclick chemistry”.1–4 Besides the advantageous properties of conventional click chemistry (operationally simple, fast, high yielding and selective), these reactions offer a high degree of spatial resolution and temporal control.5–10 Therefore, photoclick reactions are excellent transformations for diverse applications ranging from surface functionalization, polymer conjugation and photo-crosslinking to protein labelling and bioimaging.11–14The uniquely high efficiency of photoclick reactions is exemplified by the [4 + 2] cycloaddition between 9,10-phenanthrenequinones (PQs) and electron rich alkenes (ERAs).14–17 This reaction was first reported in 1944 by Schönberg,15 but was recently re-introduced by Zhang and coworkers.17 Taking advantage of ERAs (vinyl ethers, enamines) in PQ-ERA photocycloadditions, our group discovered that in this photoclick system, which simultaneously introduces a fluorescent moiety, the reaction rates, photoreaction quantum yields (ΦP up to 65%) and fluorescence quantum yields (ΦF up to 97%) can be drastically increased.14,18–20
The PQ-ERA proceeds selectively in a range of organic solvents such as acetonitrile (MeCN), toluene, dichloromethane, and ethyl acetate, while the use of ethereal solvents and alcohols leads to formation of side products.14,16 The PQ-ERA can also be performed in PBS-buffer, making it relevant for biological applications.17,18 However, as with many other photoclick reactions, the limited solubility of its substrates (especially PQs) in polar media often requires the use of small amounts of solubilizing additives.11,17,18,21–24 Considering the previously reported effects of organic solvents on the PQ-ERA, we were curious to explore the effects of such co-solvents on this photoclick reaction.
Herein, we report for the first time a study of the effect of various solubilizing co-solvents on the reaction kinetics of the PQ-ERA photoclick reaction. Strikingly, it was found that the commonly used co-solvent dimethyl sulfoxide (DMSO) has a significant effect on these kinetics (Scheme 1). The lifetime of the PQ-triplet state decreases in the presence of DMSO, leading to a 5-fold decrease in reaction rate. Without DMSO as co-solvent, extraordinary photoreaction quantum yields (up to 93%) can be achieved with typical reaction times of seconds.
 | ||
| Scheme 1 Comparison of two conditions for photocycloaddition of 9,10-phenanthrenequinone (PQ) with electron-rich alkenes (ERAs), cyan: with DMSO, brown: without DMSO. One of the possible isomers formed is shown as an example for clarity. kobs: observed first-order reaction rate; and ΦP: photoreaction quantum yield. | ||
2 Results and discussion
We initiated this research by a systematic evaluation of the influence of various additives on the PQ-ERA reaction rate (in acetonitrile (MeCN) as the main solvent), including common alcohols (methanol (MeOH), ethanol (EtOH), isopropyl alcohol (IPA)), ethereal solvents (tetrahydrofuran (THF), dioxane), amide solvents (dimethyl formamide (DMF), dimethyl acetamide (DMA)), sulfur-bearing additives (DMSO, dimethyl sulfide (DMS), sulfolane), and water. Unsubstituted PQ and N-boc-2,3-dihydro-1H-pyrrole PY (10 eq., Fig. 1A) were chosen as model compounds for this PQ-ERA reaction study, because of their accessibility and relatively fast reaction rate. It has to be noted that the reaction also proceeds efficiently with lower equivalents of PY.19 The progress of the reaction was monitored by UV-Vis absorption spectroscopy, which provided the reaction rate constants kobs(390) of the photoclick processes (Fig. 1B and C).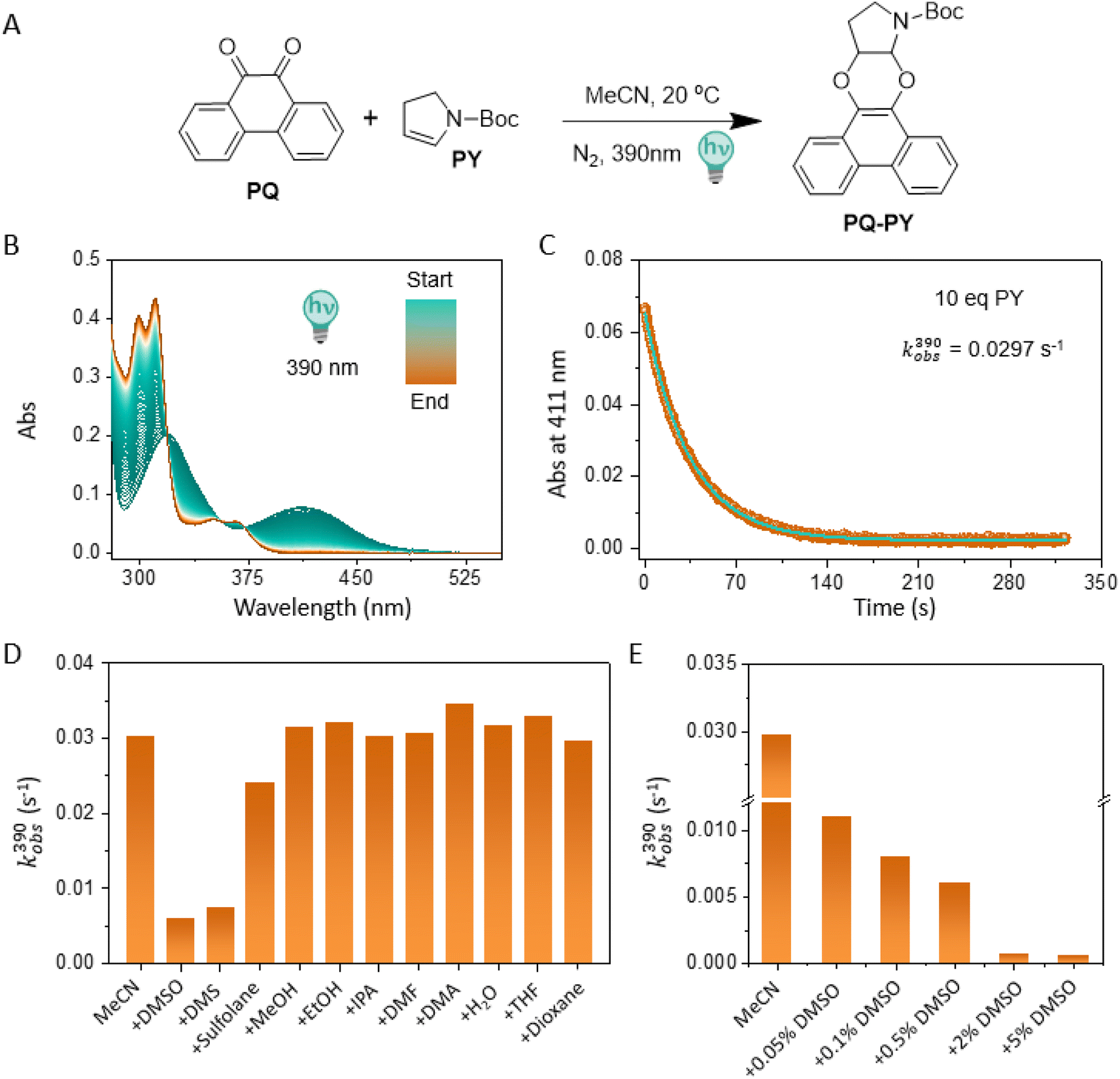 | ||
| Fig. 1 Photocycloaddition reaction between 9,10-phenanthrenequinone (PQ) and 2,3-dihydropyrrole (PY) in the presence of co-solvents and additives. (A) Reaction scheme; (B) time-resolved UV-Vis absorption spectra (0 s → 320 s); (C) UV-Vis kinetic traces (at λ = 411 nm) of the photocycloaddition between PQ and PY in pure MeCN; (D) kinetic analysis of photocycloaddition between PQ and PY with various additives. These include DMSO, DMS, sulfolane, MeOH, EtOH, IPA, DMF, DMA, H2O, THF and dioxane, respectively and, (E) concentration-dependent kinetic analysis of PQ-PY photoclick reaction in MeCN with addition of different amounts of DMSO. For all of the measurements, the sample was irradiated with a 390 nm LED and the absorbance at 411 nm was followed over time by UV-Vis absorption spectroscopy (1 cm cuvette, 2.5 mL MeCN sample volume, sample interval 1 s). PQ-PY formation was fitted to an exponential rise to the maximum equation, y = (y0 − a)ekobs× t + b, to give kobs. | ||
Remarkably, the PQ-PY reaction in pure MeCN showed a rate constant of 0.0297 s−1, a substantial enhancement (∼5 times) compared to previous reports (0.0062 s−1 with 0.5% (v/v) DMSO addition).19 This indicates that the commonly used co-solvent DMSO has a significant effect on the reaction kinetics. Intriguingly, the phenomenon observed upon DMS addition was similar to that of DMSO. Upon the addition of other solvents, including the sulfur-bearing sulfolane, minimal alterations in the kobs(390) were found (Fig. 1D, for detailed information see ESI, Section 2.1, Fig. S2†). The results were confirmed by HPLC analysis (see ESI, Section 3, Fig. S13–S22†).
The large change in reaction kinetics, caused by the addition of a small amount of DMSO, is striking, as DMSO is often used as co-solvent in biomedical studies because of its low toxicity and effective solubilizing properties.25,26 Therefore, we continued by conducting a detailed study on the effect of DMSO in the PQ-ERA photoclick reaction. Increasing amounts (0% to 5%, v/v) of DMSO were introduced, followed by the determination of kobs(390) within this concentration range. The results unveiled a notable trend in decreasing kobs(390) upon increasing the DMSO content (Fig. 1E, for detailed information see ESI, Section 2.1, Fig. S3†). For instance, with a DMSO addition of 5%, the kobs(390) plummeted to 0.0006 s−1, a stark reduction of approximately 50-fold in comparison to the pure MeCN solvent system (0.0297 s−1). Although the effects of increasing amounts of DMS were similar to that of DMSO (for detailed information, see ESI, Section 2.1, Fig. S4†), we focused on DMSO due to its common use as co-solvent.
Reported indications of DMSO influencing the triplet states of p-quinones made us realize that potentially such an effect could also occur with the o-quinone PQ studied here.27,28 To investigate the generality of this DMSO-effect on the PQ-ERA photoclick reaction, we examined a range of PQ derivatives with varying substituents (depicted in Fig. 2A, ranging from electron-withdrawing groups to electron-donating groups). The kobs(390) for these photocycloaddition reactions was quantified (for detailed information, see ESI, Section 2.1, Fig. S5†) under both DMSO and DMSO-free conditions. Notably, as illustrated in Fig. 2B, the kobs(390) values for all PQ derivatives were significantly higher without DMSO co-solvent, aligning with the trend observed for the unsubstituted PQ.
 | ||
| Fig. 2 Overview of the first order reaction rate (kobs) and photoreaction quantum yield (ΦP) of the PQ-ERA photoclick reaction system. (A) Structures of the investigated PQs; (B) The kobs(390) and (C) ΦP of PQ-ERA photoclick reaction. Cyan: with DMSO, orange: DMSO-free, respectively. NB: kobs and ΦP are measured under different conditions (for more detailed information see ESI,† Section 2.1 and 2.2). | ||
These results are also reflected by the extraordinary high photoreaction quantum yields (ΦP) of these PQ-ERA reactions. The outcomes were evident across diverse PQ derivatives, engaged in reactions with PY, as illustrated in Fig. 2A (with and without DMSO co-solvent, respectively). Particularly noteworthy is the ΦP value demonstrated by PQ in the DMSO-free context, achieving an impressive 86% with the addition of 10 equivalents of PY (Fig. 2C; for detailed information, see ESI, Section 2.2, Fig. S7–S12†).
Interestingly, the ΦP of the PQ-ERA photoclick reactions in the absence of DMSO co-solvent become largely independent on electron-donating or electron-withdrawing nature of the substituents on the PQ molecule, showcasing uniformly high values of approximately 80%. It has to be noted that the effect of DMSO seems smaller for PQ-Ph-OMe, which also has a high kobs(390) and ΦP in the presence of DMSO (for detailed information see ESI, Section 2.1, Fig. S6†).
The high ΦP values reported here, surpass the efficacy of our previously introduced fast PQs-VEs/PY systems, with ΦP (PQs-VEs/PY) ranging from 0.6% to 65%,14,18 with an exceptional case of 98%,19 as well as other documented photoclick reactions.4 In comparison to other photoclick processes such as o-naphthoquinone methides (o-NQMs)-ene hetero-Diels–Alder cycloaddition (ΦP = 20%),29 photoinduced tetrazole-alkene cycloaddition (ΦP = 0.29% to 24%),30,31 and photoinduced sydnone-alkene/alkyne cycloadditions (ΦP = 10% to 25%),21,32 the present system with a ΦP of 83% establishes a groundbreaking benchmark.
2.1 Transient absorption spectroscopy
Intrigued by these results, we set out to investigate the cause of these large changes in kinetics upon introduction of DMSO as a co-solvent. It is known that the formation of the triplet state of PQ is essential for the photocycloaddition to occur.19 Our previous studies demonstrated that unsubstituted PQ undergoes a rapid intersystem crossing (ISC), producing triplet states with a high quantum yield.19 We hypothesized that the reduced reaction rate upon addition of DMSO could be related to either the formation or the lifetime of the triplet state of PQ.To investigate if DMSO influences the ISC process, we measured the transient absorption spectra of PQ in the sub-picosecond to nanosecond timescale (ultrafast setup) with and without addition of DMSO. The triplet absorption spectrum of PQ presents two positive signatures (a peak at about 460 nm and a double peak at 650–680 nm) that have been attributed to two different triplet states with 3nπ* and 3ππ* nature reaching fast equilibration.19 After excitation with 400 nm light, a transient spectrum attributed to the triplet state rises in about 9 ps (τ2, see Table 1). The rise of this triplet state was not influenced by addition of different amounts of DMSO, up to 5% (Table 1 and ESI, Section 4.1, Fig. S23†). From these data, it was concluded that the rate of ISC is not affected by the addition of DMSO.
| DMSO content (v/v) in MeCN | τ 1 | τ 2 | τ 3 | τ k |
|---|---|---|---|---|
| a The fastest constant (τ1) refers to a singlet excited state relaxation process; τ2 represents the ISC rate, while τ3 is the triplet lifetime. τk is associated with the formation of ketyl radical. b From ultrafast measurements (sub-ps to 1.5 ns). c From ns to μs measurements. d From ultrafast measurements. | ||||
| 0% | 1.0 ps | 8.8 ps | 3.8 μs | 85 μs |
| 0.05% | 2.1 ps | 8.7 ps | 126 ns | — |
| 0.1% | 1.0 ps | 9.1 ps | 79.5 ns | — |
| 0.5% | 2.0 ps | 8.6 ps | 20.9 ns | — |
| 1% | 1.5 ps | 8.6 ps | 10.9 ns | — |
| 2% | 2.3 ps | 8.5 ps | 5.9 ns | — |
| 5% | 1.9 ps | 9.0 ps | 2.2d ns | — |
Next, we measured the transient absorption spectra of PQ in the nanosecond to millisecond timescale (ns setup), to examine the potential effect of DMSO on the lifetime of the triplet state. Our previous research confirmed that excitation of PQ leads to the presence of two long-lived species, the lowest-lying triplet state, which decays on a time scale of a few microseconds (τ3), and a ketyl radical, which disappears on a time scale of about 80 μs (τk).19 Upon the addition of 0.05% (v/v) DMSO we observed a 20-fold decrease in the lifetime of the triplet state as compared to DMSO-free conditions, lowering τ3 from 3.8 μs to 126 ns. Interestingly, formation of the ketyl radical was not observed anymore. Increasing the DMSO concentration resulted in a further reduction of τ3, to only several nanoseconds upon the addition of 2% (v/v) DMSO. The decreased triplet lifetime upon the addition of >0.5% (v/v) DMSO is already noticeable from measurements performed with the ultrafast setup (<1 ps to 1.5 ns), as shown in Fig. 3. Further details about the transient absorption measurements are reported in ESI (Section 4.1, Fig. S24†).
 | ||
| Fig. 3 Transient absorption measurements performed in the sub-ps to 1.5 ns time interval upon excitation at 400 nm on PQ dissolved in (A) MeCN and, (B) MeCN with addition of 2% (v/v) DMSO. (C) Kinetic traces recorded at 456 nm and 680 nm for PQ dissolved in MeCN, measured with the ns setup (10 ns to 40 μs time interval). (D) Kinetic traces recorded at 456 nm and 680 nm for PQ dissolved in MeCN with addition of 2% (v/v) DMSO. Time points up to 1.5 ns have been acquired using the ultrafast setup, time points in the 10–50 ns range have been acquired using the ns setup (see ESI† Section 4 for further information). | ||
Additionally, we have found that τ3 lowered with increasing excitation power, indicating occurrence of quenching at high irradiation energies. Interestingly, upon addition of 0.05% (v/v) DMSO, the τ3 did not depend on the excitation power anymore (ESI, Section 4.1, Fig. S25†). This indicates that the triplet does not exist long enough to recombine via triplet–triplet annihilation.
Plotting of the triplet state lifetimes according to the Stern–Volmer quenching equation showed a good correlation and the corresponding quenching rate constant was determined to be 5.97 ×108 M−1 s−1 (ESI, Section 4.2, Fig. S27†). This is in the same order of magnitude as other quenching mechanisms and indicates an intermolecular deactivation process upon the addition of DMSO. This quenching rate constant explains the differences in the experimentally observed quantum yields and rate constants.
The quenching effect of DMSO on PQ-Ph-OCH3 seems to be less pronounced than for that of other substituted PQs (for detailed information see ESI, Section 2.1, Fig. S6†). To explain this observation, we measured the transient absorption spectra of the various PQs (for detailed information see ESI, Section 4.1, Fig. S26†). In all cases, increased occupation of the 3ππ* triplet state was observed, likely due to the asymmetric nature of these substituted PQs. However, compared to native PQ, the absorption band of this 3ππ* was either blue-shifted (PQ-Ph-CN, PQ-Ph-CF3), comparable (PQ-Ph-CH3) or red-shifted (PQ-Ph-OCH3). As an electron transfer mechanism is suggested for DMSO quenching of para-quinones,27 which is consistent with the minimal effect of sulfolane on the photoclick, we hypothesize that the redox potential of the 3ππ* of PQ-Ph-OCH3 is decreased compared to that of other PQs, making it less sensitive towards triplet quenching by DMSO.
2.2 Optimized PQ-ERA photoclick conditions
Having established that DMSO quenches the triplet state of PQ, we were able to rationalize the significant accelerations observed in our PQ-ERA photoclick process without the use of DMSO as co-solvent. This inspired us to investigate the kinetics and ΦP of the PQ-ERA reaction using PQ-3TP (see Scheme 1, R = 2-thienyl), which we found to be the most efficient PQ-derivative so far. Our prior research indicated its enhanced reaction kinetics compared to standard PQs due to the advantageous generation of the reactive triplet state. We initially reported a ΦP of 65% using 1 equivalent of PY. Remarkably, without DMSO as co-solvent, this ΦP could be increased to 93% (For detail information see ESI, Section 2.2, Fig. S12†). This is near-quantitative conversion with equivalent amounts of PQ and PY, which further underscores the essential role of a long-lived triplet state. To the best of our knowledge, this achievement stands as the highest ΦP among all known photoclick reactions.18,19Turning our attention to the fundamental work conducted by Schönberg and colleagues almost 80 years ago,15,33 we aimed for a comparison to benchmark the advancements offered by our newly devised system (For detail information, see ESI, Movie 1†). We compared our optimized system featuring PQ-3TP, PY (1 eq), and pure MeCN as solvent, against the original conditions involving PQ, stilbene, and benzene as solvent. The initial reaction requires 9 days of exposure to sunlight, in the intense Egyptian sun.33 In contrast, we performed our reaction behind a glass window, utilizing the (limited) sunlight as the driving force on a February day in Groningen (The Netherlands). We were excited to observe the completion of the reaction in a mere 10 seconds (>77000 times faster than the original report, see ESI, Movie 1†), unequivocally highlighting that contemporary limitations in reaction time or light intensity are no longer barriers to advance PQ-ERA photoclick chemistry.
3 Conclusion
In summary, we have explored the impact of various solubilizing additives on the reaction kinetics of the PQ-ERA photoclick reaction. Notably, we found a substantial influence of the commonly employed co-solvent DMSO on these kinetics. The use of minimal quantities of DMSO leads to a remarkable decrease of the reaction speed (up to 5 times slower), accompanied by a drop in photoreaction quantum yield (ΦP), from 80% to 65%. Employing transient absorption spectroscopy, we established that the DMSO quenches the triplet state of PQ. Using this knowledge, we evaluated the use of PQ-3TP in the PQ-ERA system without co-solvents. Addition of only 1 equivalent of PY leads to an outstanding ΦP of 93% and reaction times of merely seconds to completion. These findings are testimony of the extraordinary characteristics of the PQ-ERA photoclick reaction system. We anticipate that the ultrafast PQ-ERA photoclick reaction, showcased in this work, holds promise beyond the depicted chemical scenarios. Its potential advantages are expected to extend to diverse applications, including surface photo-patterning, biomacromolecule labeling, and photochemical crosslinking. Studies along these lines are now ongoing in our laboratories.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
A. M. D., Y. F., M. D. D., G. L., P. H. E., W. J. B., W. S., and B. L. F. conceived the project and designed phenanthrenequinone derivatives. B. L. F. and W. S. guided the research. Y. F. and A. M. D. synthesized all phenanthrenequinone derivatives. A. M. D. and Y. F. performed UV-Vis experiments. M. D. D. performed the Femtosecond Transient Absorption Spectroscopy measurements. A. M. D., Y. F., M. F. H., and W. J. B. performed the Nanosecond Transient Absorption Spectroscopy measurements. A. M. D., Y. F., M. D. D., G. L., P. H. E., W. J. B., W. S., and B. L. F. wrote the manuscript with support and contributions from all authors.Conflicts of interest
There are no competing conflicts of interests to declare.Acknowledgements
We gratefully acknowledge the generous financial support from the Horizon 2020 Framework Program (ERC Advanced Investigator Grant No. 694345 to BLF), the Ministry of Education, Culture and Science of the Netherlands (Gravitation Program No. 024.001.035 to BLF). MDD gratefully acknowledges support from the European Union's Horizon 2020 research and innovation program under grant agreement no. 871124 Laserlab-Europe.References
- B. D. Fairbanks, L. J. Macdougall, S. Mavila, J. Sinha, B. E. Kirkpatrick, K. S. Anseth and C. N. Bowman, Chem. Rev., 2021, 121, 6915–6990 CrossRef CAS PubMed.
- M. Montalti, A. Credi, L. Prodi and M. T. Gandolfi, Handbook of Photochemistry, CRC Press, Boca Raton, 2006 Search PubMed.
- M. A. Tasdelen and Y. Yagci, Angew. Chem., Int. Ed., 2013, 52, 5930–5938 CrossRef CAS PubMed.
- G. S. Kumar and Q. Lin, Chem. Rev., 2021, 121, 6991–7031 CrossRef CAS PubMed.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS PubMed.
- N. K. Devaraj and M. G. Finn, Chem. Rev., 2021, 121, 6697–6698 CrossRef CAS PubMed.
- A. Battigelli, B. Almeida and A. Shukla, Bioconjugate Chem., 2022, 33, 263–271 CrossRef CAS PubMed.
- C. W. Tornøe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057–3064 CrossRef PubMed.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS PubMed.
- W. Song, Y. Wang, J. Qu and Q. Lin, J. Am. Chem. Soc., 2008, 130, 9654–9655 CrossRef CAS PubMed.
- C. E. Hoyle and C. N. Bowman, Angew. Chem., Int. Ed., 2010, 49, 1540–1573 CrossRef CAS PubMed.
- L. Zhang, X. Zhang, Z. Yao, S. Jiang, J. Deng, B. Li and Z. Yu, J. Am. Chem. Soc., 2018, 140, 7390–7394 CrossRef CAS PubMed.
- A. A. Poloukhtine, N. E. Mbua, M. A. Wolfert, G. J. Boons and V. V. Popik, J. Am. Chem. Soc., 2009, 131, 15769–15776 CrossRef CAS PubMed.
- Y. Fu, H. Helbert, N. A. Simeth, S. Crespi, G. B. Spoelstra, J. M. van Dijl, M. van Oosten, L. R. Nazario, D. van der Born, G. Luurtsema, W. Szymanski, P. H. Elsinga and B. L. Feringa, J. Am. Chem. Soc., 2021, 143, 10041–10047 CrossRef CAS PubMed.
- A. Schönberg and A. Mustafa, Nature, 1944, 153, 195 CrossRef.
- D. Fong, A. Lang, K. Li and A. Adronov, Macromolecules, 2020, 53, 1760–1766 CrossRef CAS.
- J. Li, H. Kong, L. Huang, B. Cheng, K. Qin, M. Zheng, Z. Yan and Y. Zhang, J. Am. Chem. Soc., 2018, 140, 14542–14546 CrossRef CAS PubMed.
- Y. Fu, N. A. Simeth, R. Toyoda, R. Brilmayer, W. Szymanski and B. L. Feringa, Angew. Chem., Int. Ed., 2023, 62, e202218203 CrossRef CAS PubMed.
- Y. Fu, G. Alachouzos, N. A. Simeth, M. Di Donato, M. F. Hilbers, W. J. Buma, W. Szymanski and B. L. Feringa, Chem. Sci., 2023, 14, 7465–7474 RSC.
- Y. Fu, K. Wu, G. Alachouzos, N. A. Simeth, T. Freese, M. Falkowski, W. Szymanski, H. Zhang and B. L. Feringa, Adv. Funct. Mater., 2023, 33, 2306531 CrossRef CAS.
- J. Gao, Q. Xiong, X. Wu, J. Deng, X. Zhang, X. Zhao, P. Deng and Z. Yu, Commun. Chem., 2020, 3, 29 CrossRef CAS PubMed.
- P. An, T. M. Lewandowski, T. G. Erbay, P. Liu and Q. Lin, J. Am. Chem. Soc., 2018, 140, 4860–4868 CrossRef CAS PubMed.
- Z. Li, L. Qian, L. Li, J. C. Bernhammer, H. V. Huynh, J. S. Lee and S. Q. Yao, Angew. Chem., Int. Ed., 2016, 55, 2002–2006 CrossRef CAS PubMed.
- A. Di Guo, D. Wei, H. J. Nie, H. Hu, C. Peng, S. T. Li, K. N. Yan, B. S. Zhou, L. Feng, C. Fang, M. Tan, R. Huang and X. H. Chen, Nat. Commun., 2020, 11, 1–13 CrossRef PubMed.
- J. Catalán, C. Díaz and F. García-Blanco, J. Org. Chem., 2001, 66, 5846–5852 CrossRef PubMed.
- K. V. Balakin, Y. A. Ivanenkov, A. V. Skorenko, Y. V. Nikolsky, N. P. Savchuk and A. A. Ivashchenko, J. Biomol. Screening, 2004, 9, 22–31 CrossRef CAS PubMed.
- H. Görner, Photochem. Photobiol., 2006, 82, 71 CrossRef PubMed.
- M. Durmuş and T. Nyokong, Spectrochim. Acta, Part A, 2008, 69, 1170–1177 CrossRef PubMed.
- S. Arumugam and V. V. Popik, J. Am. Chem. Soc., 2011, 133, 5573–5579 CrossRef CAS PubMed.
- G. S. Kumar, S. Racioppi, E. Zurek and Q. Lin, J. Am. Chem. Soc., 2022, 144, 57–62 CrossRef CAS PubMed.
- S. Jiang, Z. Wu, H. Liu, J. Deng, X. Zhang, Z. Yao, Y. Zheng, B. Li and Z. Yu, ChemPhotoChem, 2020, 4, 327–331 CrossRef CAS.
- X. Zhang, X. Wu, S. Jiang, J. Gao, Z. Yao, J. Deng, L. Zhang and Z. Yu, Chem. Commun., 2019, 55, 7187–7190 RSC.
- A. Schönberg and A. Mustafa, J. Chem. Soc., 1944, 387 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc01810a |
| ‡ A. M. Doze and Y. Fu contributed equally to this project. |
| § Current address: College of Science, Nanjing Forestry University, Nanjing, 210037, P. R. China. |
| This journal is © The Royal Society of Chemistry 2024 |