
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA tricycloquinazoline based 2D conjugated metal–organic framework for robust sodium-ion batteries with co-storage of both cations and anions†
Dan
Chen‡
ac,
Linqi
Cheng‡
b,
Weiben
Chen‡
ac,
Heng-Guo
Wang
 *b,
Fengchao
Cui
b and
Long
Chen
*a
*b,
Fengchao
Cui
b and
Long
Chen
*a
aState Key Laboratory of Supramolecular Structure and Materials, College of Chemistry, Jilin University, Changchun, 130012, China. E-mail: longchen@jlu.edu.cn
bKey Laboratory of Polyoxometalate and Reticular Material Chemistry of Ministry of Education, Faculty of Chemistry, Northeast Normal University, Changchun, 130024, China. E-mail: wanghg061@nenu.edu.cn
cDepartment of Chemistry, Tianjin Key Laboratory of Molecular Optoelectronic Science, Tianjin University, Tianjin, 300072, China
First published on 18th June 2024
Abstract
There is growing interest in 2D conjugated metal–organic frameworks (2D c-MOFs) for batteries due to their reversible redox chemistry. Nevertheless, currently reported 2D c-MOFs based on n-type ligands are mostly focused on the storage of cations for batteries. Herein, we successfully synthesize nitrogen-rich and electron-deficient p-type ligand-based Ni3(HATQ)2 assembled from 2,3,7,8,12,13-hexaaminotricycloquinazoline (HATQ), and the ion co-storage feature of cations and anions in sodium ion batteries (SIBs) is demonstrated for 2D c-MOFs for the first time. The redox chemistry from the p-type ligand and π–d hybridization center endows the Ni3(HATQ)2 cathode with high capacity and good rate performance, especially excellent capacity retention of 95% after 1000 cycles. These findings provide a promising avenue for the exploration of other p-type multidentate chelating ligands toward new 2D c-MOFs and expand the application of 2D c-MOFs in energy storage systems.
Introduction
The development of clean and sustainable energy sources is driving the progress of emerging energy storage devices and technologies, which would have the advantage of overcoming the ever-increasing cost of lithium-ion batteries (LIBs).1–4 Among these technologies, sodium ion batteries (SIBs) are expected to become promising alternatives to lithium ion batteries (LIBs) due to their cost-effectiveness, comparable electrochemical performance and abundant sodium resources.5,6 However, the common electrode materials that are suitable for LIBs may exhibit sluggish reaction kinetics and rapid capacity decay in SIBs due to the larger radius of Na+ compared to Li+.7,8 In this context, organic electrode materials seem to be a promising candidate due to their flexible structure designability as well as their cost effectiveness and resource abundance. Unfortunately, their low electrical conductivity and high dissolution in electrolytes seriously reduce the utilization of redox active sites and cycle stability.9,10 Therefore, the exploration of structurally adaptive and promising conductive electrode materials with multi-redox active sites for SIBs is highly desired.As a distinct category of conductive coordination polymers (CCPs), two-dimensional conjugated metal–organic frameworks (2D c-MOFs) are constructed by coordination of transition-metal ions with planar conjugated ligands containing ortho-substituted functional groups (–OH, –NH2, and SH, Fig. 1a and b).11–16 The unique square-planar linkages (MX4, M = metal) facilitate the delocalization of charge carriers in 2D conjugated networks, which immensely enhances electronic transport.17–20 While a plethora of MO4 linkage-based 2D c-MOFs have been explored, varying functional groups from –OH to –NH2 and –SH in the same ligands usually result in tunable electrical conductivity and coordination ability due to the discrepancy in the atom radius and π–d hybridization between the ligands and transition-metal ions.21–25 Specifically, several recent studies have demonstrated that MX4 units based on benzene or triphenylene ligands can serve as crucial redox active centers.26–28 Furthermore, redox-active ligands based on tetraminobenzoquinone and hexaazatrinaphthylene in 2D c-MOFs have been reported, which integrate dual-redox centers.26,29–31 However, these reported MX4 linked organic ligands are mainly based on n-type organic compounds that serve as electron acceptors with intercalation of cations, limiting high throughput screening of electrode materials. By comparison, tricycloquinazoline (TQ),32–35 as a nitrogen-rich and electron-deficient heteroaromatic-conjugated ligand core, can not only increase the acidity of the coordination functional groups, resulting in higher crystalline 2D c-MOFs, but also act as a p-type organic compound that serves as the electron donor with intercalation of anions,36 expanding the active charge carriers of 2D c-MOFs. Therefore, the integration of p-type TQ ligands can construct artificial bipolar 2D c-MOFs that can achieve the co-storage of cations and anions. However, as far as we know, 2D c-MOFs based on p-type ligands as cathode materials for advanced SIBs with novel cation/anion co-storage modes have not been reported.
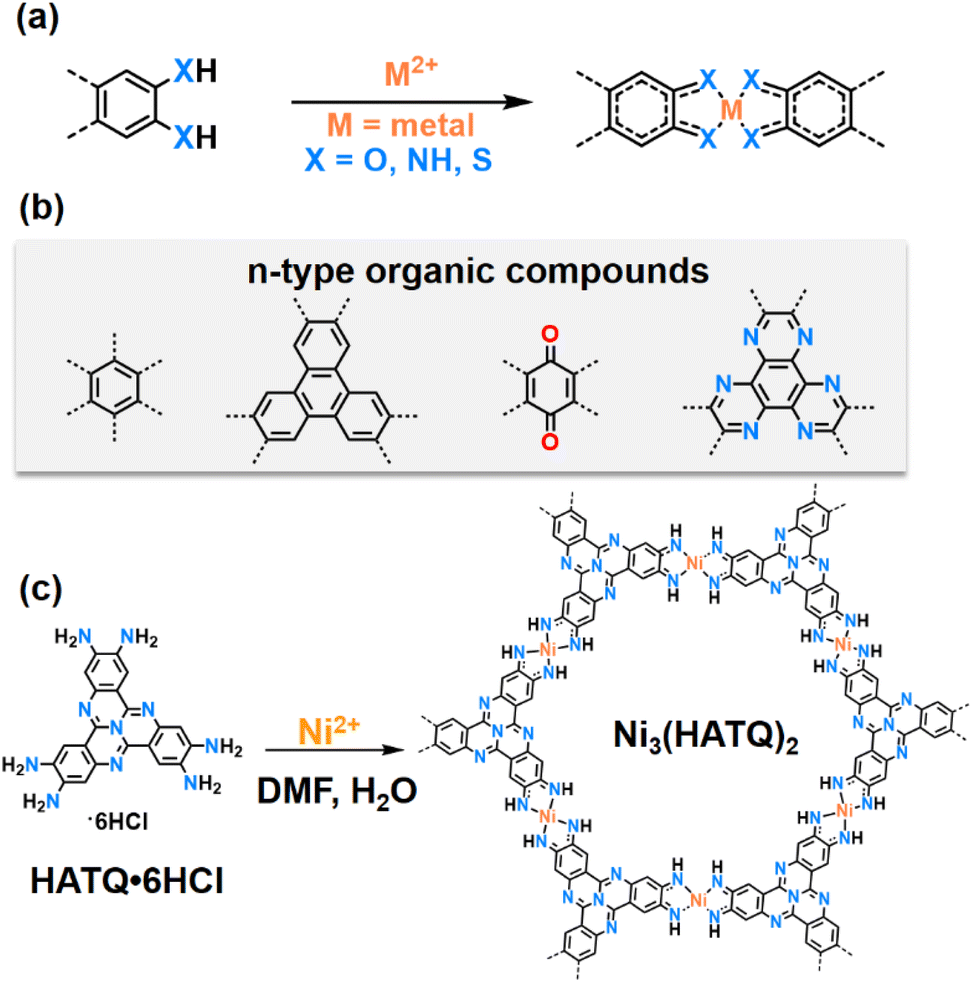 | ||
| Fig. 1 The coordination of transition-metal ions with different planar conjugated ligands and design strategy of Ni3(HATQ)2. (a) The representative coordination process of square-planar transition-metal ions with ortho-substituted organic conjugated ligands. (b) The typical n-type organic structures in 2D c-MOFs. (c) The synthetic route of Ni3(HATQ)2. | ||
In this work, we reported the synthesis of hexaaminotricycloquinazoline (HATQ)-based 2D c-MOF Ni3(HATQ)2 (Fig. 1c) that acted as a bipolar active material for advanced SIBs for the first time. Interestingly, the bipolar framework material consists of n-type MN4 active centers, aiming to store cations, and p-type TQ blocks, aiming to store anions. As expected, Ni3(HATQ)2 was then investigated as a cathode of SIBs, exhibiting a high reversible capacity of 115.1 mA h g−1, good rate performance with 77.1 mA h g−1 at 2 A g−1, and long-term cycling stability with 95% capacity retention after 1000 cycles, which is superior to its Ni3(HITP)2 (HITP = hexaiminotriphenylene) counterpart. In addition, all-organic SIBs were assembled by pairing the Ni3(HATQ)2 cathode with a sodium terephthalate anode, which shows the feasibility of practical application. Remarkably, the co-storage features of cations and anions were revealed by both experimental and theoretical investigations. These investigations may pave the way for the design and construction of novel 2D c-MOFs with the co-storage mode.
Results and discussion
HATQ was ingeniously designed and newly prepared in high yields (Schemes S1–S5†). More specifically, the pivotal hexabromotricycloquinazoline (HBTQ) precursor was first prepared through bromination and trimerization reactions. Subsequently, the diphenylmethanimine-substituted tricycloquinazoline was obtained by the Buchwald–Hartwig coupling reaction using HBTQ and diphenylmethanimine, followed by hydrolysis to yield brown precipitates (Fig. S1–S4†).37 The chemical compositions of the target product were verified to be hexaaminotricycloquinazoline hexahydrochloride (HATQ·6HCl) by using 1H and 13C NMR, high-resolution mass spectra, elemental analysis, thermogravimetric analysis (TGA), and X-ray photoelectron spectroscopies (XPS) (Fig. S5–S8†).Ni3(HATQ)2 was constructed by coordination of HATQ·6HCl with Ni2+ in square-planar geometry upon solvothermal synthesis using N,N-dimethylformamide (DMF) and water as the mixed solvent under basic conditions at 85 °C for three days (Fig. S9 and Scheme S6†). The resulting precipitate was thoroughly washed with N,N-dimethylacetamide, water, methanol and acetone, suggesting its solvent stability. Detailed characterization studies were performed to evaluate the fine structure of Ni3(HATQ)2. Fourier transform infrared (FT-IR) spectra showed that the featured peaks of amino (ca. 3190–3350 cm−1) apparently disappeared (Fig. S10†). The peaks at ca. 505 and ca. 3630 cm−1 were assigned to Ni–N and –NH bands, respectively, indicating the formation of coordination bonds between Ni2+ and NH of the HATQ ligand.38,39 Besides, the other characteristic peaks in the TQ core also existed in Ni3(HATQ)2, further suggesting successful formation of Ni3(HATQ)2. Meanwhile, the elemental compositions of Ni3(HATQ)2 were disclosed by XPS. As shown in Fig. S11,† these featured peaks in the full spectrum were assigned to be C, N, O and Ni elements. Among them, the existence of the O 1s peak in the spectrum may be attributed to trapped H2O in the porous skeletons.23,40–42 Compared to high-resolution C 1s and N 1s spectra of HATQ·6HCl in Fig. S8,† the Ni ion coordinated with N results in the evident change of binding energies in C (C–N) and N (N–C) of Ni3(HATQ)2. The absence of Cl− signal peaks and extraneous Na+ counterions suggests that Ni3(HATQ)2 is neutral.23 Two characteristic peaks at 855.6 and 873.4 eV in the high-resolution spectra of the Ni element are assigned to Ni 2p3/2 and Ni 2p1/2 with evident satellite peaks at 860 and 880 eV, suggesting that the Ni element could be in the divalent state. Furthermore, the local structure of Ni3(HATQ)2 was investigated by X-ray absorption fine-structure (XAFS) using Ni foil and NiO as the reference.43,44 As shown in Fig. 2a, the collected Ni K-edge X-ray absorption near edge structure (XANES) profile of Ni3(HATQ)2 resembled that of NiO and mismatched with Ni foil, suggesting the domination of the +2-oxidation state of Ni. Furthermore, the coordination environments of Ni in Ni3(HATQ)2, NiO and Ni foil were evaluated using wavelet transformed extended X-ray absorption fine structure (EXAFS) (Fig. S12c and S13†). The maximum peaks at ca. 3.3 Å−1 of the k-space value and ca. 1.7 Å of the R-space value were close to that of Ni–O contribution in the NiO spectrum (Fig. S12b†), which was attributed to the Ni–N bonds, without apparent peaks of the Ni–Ni bond at ca. 2.2 Å of the R-space value, indicating the absence of residual metallic Ni clusters in Ni3(HATQ)2. Additionally, the coordinated bond parameters of Ni3(HATQ)2 were further investigated based on fitting of experimental Ni K-edge EXAFS oscillation k2χ(k) as shown in Fig. S12a and b.† The first coordination peak of Ni3(HATQ)2 with ca. 2.12 Å of the interatomic distances was ascribed to the Ni–N bond with an average coordination number of ca. 4.5 (Table S1†). Thus, the XAFS spectra strongly support the formation of square-planar NiN4 coordination units in Ni3(HATQ)2. The crystalline structure of Ni3(HATQ)2 has been determined through a combination of powder X-ray diffraction (PXRD) measurements and theoretical simulation. The experimental PXRD profile of Ni3(HATQ)2 displays four distinct peaks at 4.00°, 8.05°, 10.60° and 27.55° which are assigned to be (100), (200), (210) and (001) facets, respectively (Fig. 2b). Meanwhile, the simulated XRD profile and the Pawley refined data of eclipsed AA stacking mode can match well with the experimental data, with a small difference (Rwp = 3.08% and Rp = 2.48%, Fig. 2c). The unit cell parameters for Ni3(HATQ)2 are determined to be a = b = 25.91 Å, c = 3.42 Å, α = β = 90°, and γ = 120°. In contrast, the XRD profiles of staggered AB stacking mode cannot reproduce the experimental one (Fig. S14†). Furthermore, the permanent porosities of Ni3(HATQ)2 were evaluated using the nitrogen adsorption–desorption isotherm at 77 K (Fig. 2d). The Brunauer–Emmett–Teller surface area of Ni3(HATQ)2 was determined to be 513 m2 g−1 and the pore size was calculated to be 2.24 nm, which matches perfectly with the theoretical value (2.24 nm).20 As shown in Fig. S15,† the field emission scanning electron microscopy (FE-SEM) images exhibited nanorod-like morphology of Ni3(HATQ)2. Furthermore, the high-resolution transmission electron microscopy images exhibited distinct lattice fringes, with 2.08 nm of the corresponding channels being close to the theoretical and experimental pore size values (Fig. S16†). As shown in Fig. S17,† the solid-state ultraviolet diffuse reflection spectra of Ni3(HATQ)2 displayed broader absorption compared to the precursor, and the optical bandgap was calculated to be 1.52 eV using Tauc plots, which indicated that an extended π-conjugated skeleton is favorable for modulating the optical properties. Notably, the electrical conductivity is a unique identity of 2D c-MOFs. The conductivity value of bulk Ni3(HATQ)2 was 2.12 mS m−1 at room temperature, which is beneficial for electrochemical devices (Fig. S18†). Furthermore, thermogravimetric analysis revealed that the framework decomposes at ca. 400 °C (Fig. S19†), indicating good thermal stability.
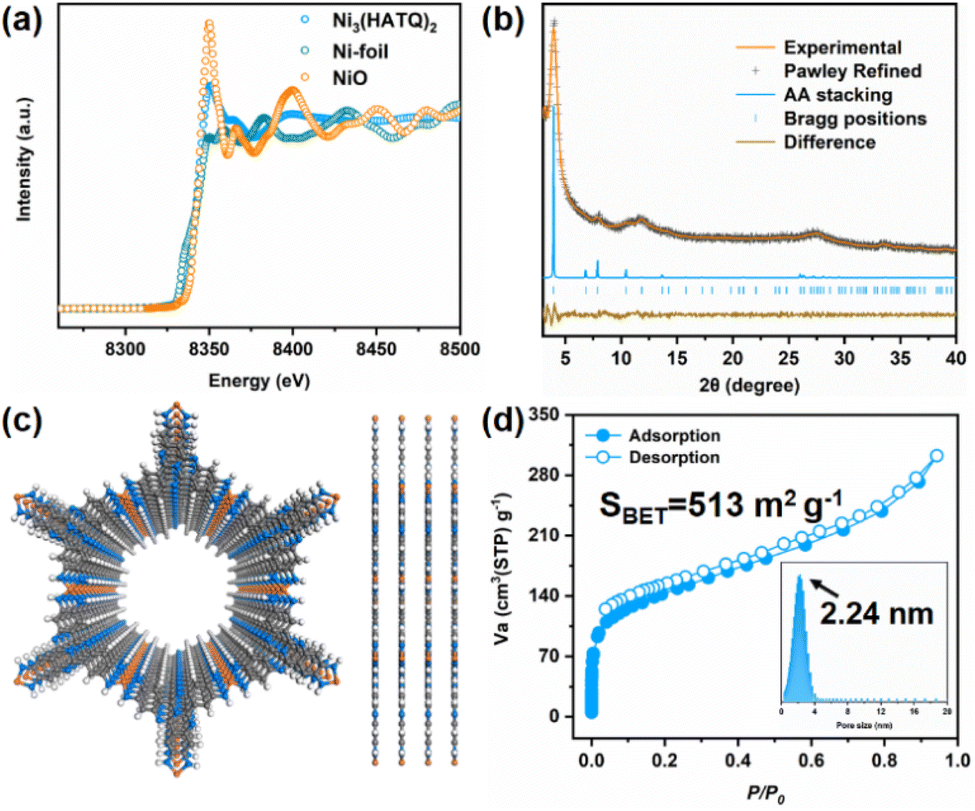 | ||
| Fig. 2 Characterization of the valence state, crystallinity, simulated structure and porosity of Ni3(HATQ)2. (a) XANES of NiO, Ni foil and Ni3(HATQ)2. (b) Experimental and Pawley refined XRD profiles of Ni3(HATQ)2. (c) The structural model. (d) Nitrogen-sorption isotherms of Ni3(HATQ)2. | ||
For comparison, Ni3(HITP)2 was also synthesized and characterized (Scheme S7, Fig. S20 and S21†). As shown in Fig. S20,† Ni3(HITP)2 exhibited a feature of crystallinity. Additionally, elemental compositions of Ni3(HITP)2 were disclosed by XPS as shown in Fig. S21.† First, in the full spectrum, these featured peaks were assigned to be C, N, O and Ni elements. Similar to Ni3(HATQ)2, the existence of the O 1s peak may be attributed to trapped H2O.23,40–42 Two characteristic peaks at 855.5 and 872.7 eV in the high-resolution spectra of the Ni element are assigned to Ni 2p3/2 and Ni 2p1/2, suggesting that the Ni element could be in the divalent state.
Benefiting from its unique crystalline feature, Ni3(HATQ)2 is supposed to exhibit potential application in SIBs. First, coin-type half batteries were assembled using pure metallic sodium (counter electrode) and Ni3(HATQ)2 (cathode material) to evaluate the electrochemical performance. Cyclic voltammetry (CV) curves were measured at a scan rate of 0.1 mV s−1 with a voltage ranging from 1.0 to 3.6 V. As shown in Fig. 3a, the first CV curve showed a reductive peak at 1.57 V, which may be attributed to the reduction of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond in the coordination center, and two oxidation peaks at 1.63 and 2.45 V, which may be ascribed to the oxidation of C–N–Na. Distinct redox peaks at 2.73 and 3.21 V were manifested as anion desertion and insertion.28 These results suggest multiple electron participation in charge and discharge processes.
N bond in the coordination center, and two oxidation peaks at 1.63 and 2.45 V, which may be ascribed to the oxidation of C–N–Na. Distinct redox peaks at 2.73 and 3.21 V were manifested as anion desertion and insertion.28 These results suggest multiple electron participation in charge and discharge processes.
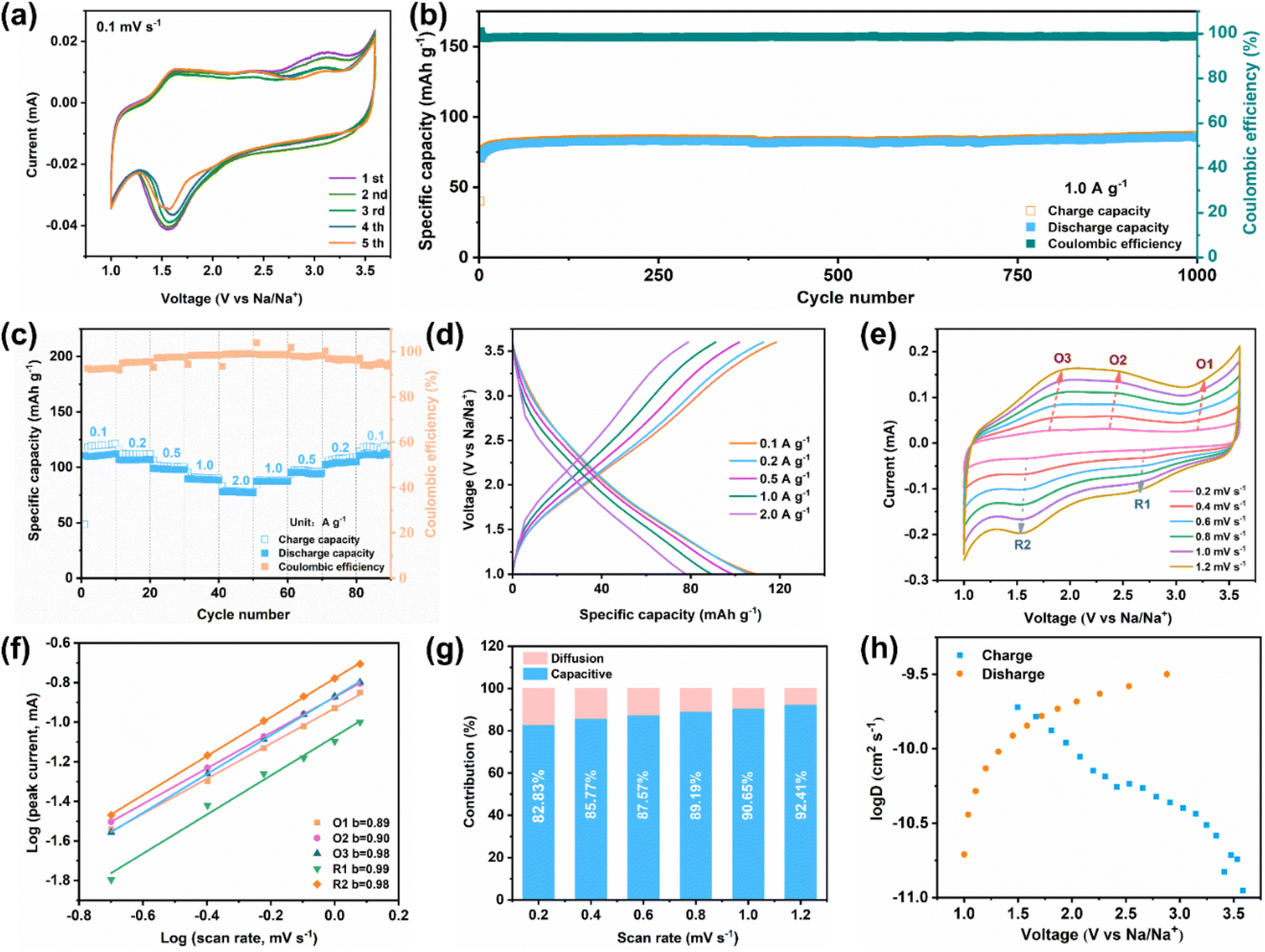 | ||
Fig. 3 Different electrochemical performances of the Ni3(HATQ)2 cathode in SIBs. (a) CV curves of Ni3(HATQ)2 at 0.1 mV s−1. (b) Long-term cycling stability of Ni3(HATQ)2 measured at 1.0 A g−1. (c) Rate capability of Ni3(HATQ)2. (d) GCD profiles of Ni3(HATQ)2 at different current densities. (e) CV curves of Ni3(HATQ)2 measured at different scan rates. (f) The relationship between logarithm peak current and the logarithm peak scan rate. (g) The percentage of diffusion and capacitive contributions at different scan rates. (h) Corresponding log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) D (D = Na+ diffusion coefficient) at the discharge/charge state of Ni3(HATQ)2. D (D = Na+ diffusion coefficient) at the discharge/charge state of Ni3(HATQ)2. | ||
Meanwhile, the redox peaks became more stable with increasing scan cycles, indicating the excellent reversibility of the Ni3(HATQ)2-based cathode. Significantly, reasonable selection of test procedures has a non-negligible effect on the electrochemical performance. As shown in Fig. S22,† the Ni3(HATQ)2-based cathode when charged to 3.6 V first shows higher initial capacity and better cycle performance than that discharged to 1.0 V, which might be attributed to the efficient intercalation of anions. In addition, a reasonable voltage range is crucial for stimulating capacity and maintaining stability. As shown in Fig. S23,† by comparing the different voltage ranges (1.0–3.5 V, 1.0–3.6 V and 1.0–3.8 V), it is clear that changing the upper limit of the voltage range can improve the capacity, but it may lead to the decomposition of the electrolyte, resulting in poor cycle stability. Furthermore, lowering the lower voltage limit can also increase the capacity, but the low voltage is not suitable for the cathode (Fig. S24†). In addition, decreasing mass loading also benefits the enhanced capacity (Fig. S25†).
Additionally, at 0.2 A g−1, Ni3(HATQ)2 exhibits a reversible specific capacity of 115.1 mA h g−1 after 150 cycles (Fig. S26a†).Furthermore, compared with HATQ·6HCl, the Ni3(HATQ)2-based cathode exhibited superior redox ability and electrochemical performance (Fig. S26†). More importantly, the Ni3(HATQ)2-based cathode demonstrated distinctly superior performance to Ni3(HITP)2 (ref. 45) at 1.0 A g−1 (Fig. S27†). Furthermore, the capacity of Ni3(HATQ)2 is superior to many other MOF-based cathodes and other inorganic cathodes (Table S2†) indicating the importance of electrode material selection and design. Fig. 3b shows that the excellent capacity retention (around 97%) and coulombic efficiency (CE) (nearly 100%) were retained at a higher current density of 1.0 A g−1 after 1000 cycles, revealing good long-term cycle stability. Moreover, the rate performance on the Ni3(HATQ)2 cathode was measured (Fig. 3c and d), with current densities ranging from 0.1 to 2.0 A g−1. The discharge specific capacities were about 108.1, 105.7, 98.0, 88.6 and 77.1 mA h g−1, respectively. Upon returning to a current density of 0.1 A g−1, the capacity was recovered to 107.8 mA h g−1 with a restoration ratio of 99%. Furthermore, the reaction kinetics were analyzed through CV curves at different scan rates from 0.2 to 1.2 mV s−1. As shown in Fig. 3e, the profiles of different CV curves are highly similar, while the corresponding peak area gradually increases when the scan rate aggrandizes.46 According to the equation i = avb, b values were determined by fitting the plot of log(i) and log(v) (see the ESI†). As shown in Fig. 3f, the calculated b values of O1, O2, O3, R1 and R2 were inclined to 1, revealing that the battery process is mainly controlled by capacitive behavior.
Furthermore, the ratio of diffusion to capacitive control is quantitatively analyzed using the Dunn equation. Fig. 3g and S28† show that the capacitive contribution is obviously increased with enhanced scan rates, and the ratio of capacitive control is up to 92.41% at a scan rate of 1.2 mV s−1, indicating that the capacitive effect is the dominant contribution to battery capacity. Additionally, the Na+ diffusion coefficient was determined by the galvanostatic intermittent titration technique (GITT), and the results showed that Ni3(HATQ)2 exhibits a high Na+ diffusion coefficient (D) with logD values ranging from −9.0 to −11.0 cm2 s−1 (Fig. 3h). These results indicate that Ni3(HATQ)2 features a fast kinetic process, which is beneficial for its electrochemical performance. Besides, Fig. S29† depicts the schematic diagram and electrochemical performance of the coin-type full cell when the anode is sodium terephthalate (Na2TP), in which the Ni-TABQ‖Na2TP cell also shows desirable cycling performance with a reversible specific capacity of 56.3 mA h g−1 after 160 cycles at 0.2 A g−1, indicating the feasibility of practical application.
To further understand the ion storage mechanism of Ni3(HATQ)2, ex situ FT-IR, XPS and EPR spectra were obtained to investigate the structural variation at selected voltage stages during charge/discharge processes (Fig. 4a). As shown in the ex situ FT-IR spectra in Fig. 4b, the corresponding peaks of the C–N bond gradually increased and the CN bond stepwise weakened during discharge processes, indicating the generation of a C–N–Na group. Upon charging, the intensity of C–N bond gradually diminished and CN bond gradually enhanced. Specifically, evident peaks of PF6− (ca. 868 cm−1) were detected in the ex situ FT-IR spectra at the charging state, revealing certain interaction between the anion and Ni3(HATQ)2 during the redox process.30 To further confirm the transformation of the functional groups, XPS spectra were measured. The XPS spectra of N(1s) showed that CN, C–N, and N-PF6 exhibit similar variation tendencies with FT-IR spectra (Fig. 4c). Besides, the XPS P(2p) spectra exhibited an overwhelming signal upon charging to 3.6 V, which was distinguished from that of the discharge state (Fig. 4d). Compared with the pristine state, Ni(2p) spectra demonstrated negligible transformation during cycling (Fig. 4e), indicating that Ni(II) is immutable as a cathode material. These experimental data reveal co-storage of Na+ and PF6− in Ni3(HATQ)2 during discharge/charge processes, which is beneficial for capacity promotion. The EPR signal increased during charging processes, which may be caused by the augmented the CN bond (in the coordination center). In contrast, the signal peak decreased upon discharging to 1.0 V, indicating the generation of C–N–Na and formation of a radical intermediate during Na ion extraction/insertion processes (Fig. 4f). Moreover, these results indicate that the Na ion binding site is probably the coordinated nitrogen atoms instead of the Ni metal centers without valence state change. Meanwhile, Fig. S30† shows that the crystallinity of Ni3(HATQ)2 was well maintained at both charge and discharge states and the SEM image of charging to 2.8 V exhibited the intrinsic nanorod morphology, suggesting the structural stability of Ni3(HATQ)2 during the redox process.
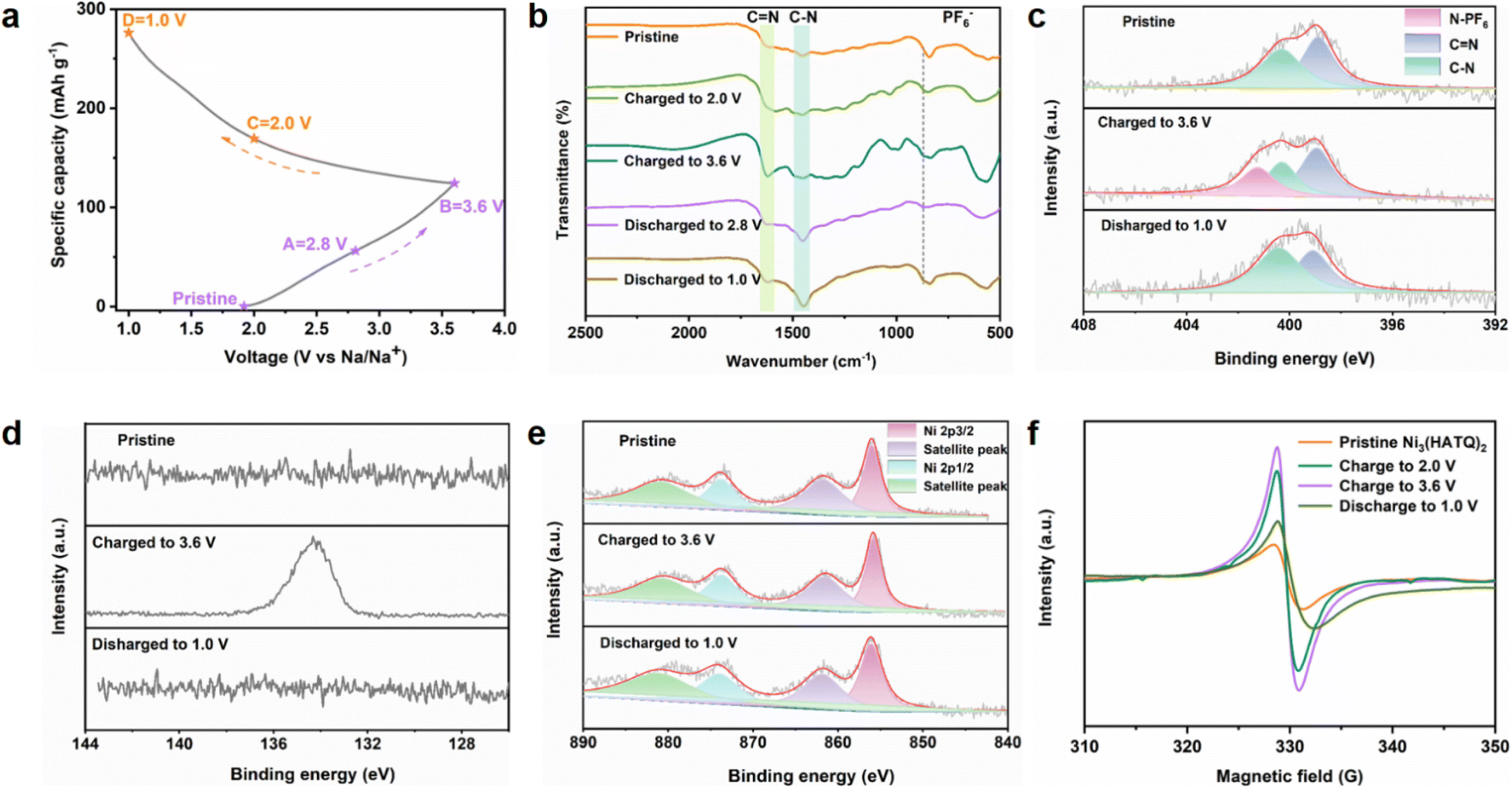 | ||
| Fig. 4 XPS measurements and EPR spectra for investigating the ion storage mechanism of Ni3(HATQ)2. (a) Charge–discharge profiles and corresponding collections of voltage stages for ex situ investigation. (b) The ex situ FT-IR spectra. (c) The high resolution XPS N(1s) spectra. (d) The high resolution XPS P(2p) spectra. (e) The high resolution XPS Ni(2p) spectra and (f) EPR spectra of the Ni3(HATQ)2-based cathode at different charge and discharge voltages. | ||
To further investigate the co-storage mechanism of cations and anions, DFT calculations are conducted. The electronic band structure and density of states (DOS) reveal that Ni3(HATQ)2 possesses metallic electronic band structures, as evidenced by the non-zero density of states at Fermi energy (Fig. 5a).47,48 This metallic property facilitates the transmission of electrons to the active sites. Additionally, molecular electrostatic potential (MESP) is determined to predict the possible active site for binding Na+ and PF6−. The minimum value on the van der Waals surface (blue region) is centered at nitrogen elements, indicating that the CN bond (coordination center) might attract Na ions more easily (Fig. 5b). In addition, Ni3(HATQ)2 exhibits heterogeneous charge distribution, with the N atoms of NiN4 units being more negative than that of the TQ cores, which is beneficial for the preferential binding of Na ions. In contrast, the nitrogen elements of TQ ligand tend to act as the desirable active sites for binding PF6− (Fig. 5c). The subsequent differential charge explains that the charge density between Ni3(HATQ)2 and Na ions is increased, indicating a strong electronic interaction in the NiN4 unit (Fig. 5d and S31†).48 Meanwhile, the calculated binding energy verifies that Na+ and PF6− are inclined to interact with the NiN4 core and TQ motif, respectively. As well, the calculated reaction potentials of Na+/PF6− intercalation are 1.00 V and 2.75 V, respectively, which correspond to the experimental potentials (Fig. 5e). Finally, the structural evolution progress is calculated to further confirm the redox site and the binding number of Na ions (Fig. 5f). The data indicate that six Na ions and two PF6− can be respectively extracted/inserted at the coordination bond and nitrogen elements of the TQ ligand during the charge/discharge process based on the repetitive optimized cell. Overall, the calculated data are consistent with the experimental data and provide further insights into the cation and anion storage mechanism of Ni3(HATQ)2.
 | ||
| Fig. 5 DFT calculations for exploring the co-storage mechanism of cations and anions of Ni3(HATQ)2. (a) Electronic band structure and DOS of Ni3(HATQ)2. (b) MESP of the optimized cell of Ni3(HATQ)2. (c) The calculated 2D charge density. (d) Differential charge distribution of the Ni3(HATQ)2 repetitive unit. (e) The calculated reaction potential and binding energy of Na+/PF6− interacting with different reaction sites based on Ni3(HATQ)2. (f) Structural evolution during the insertion/desertion of PF6− and sodium/de-sodium processes. | ||
Conclusions
In summary, we successfully developed the facile synthesis of hexaaminotricycloquinazoline (HATQ) ligand as well as corresponding 2D c-MOF: Ni3(HATQ)2. Benefited by co-existence of p-type ligands (TQ) and an n-type coordination core (NiN4), bipolar Ni3(HATQ)2 exhibits high discharge/charge capacities, excellent rate performance, and fast ion diffusion properties, when employed as a cathode material in SIBs. Moreover, 99% of the initial capacity retains after 150 cycles at a current density of 0.2 A g−1 and 95% of the restoration ratio at a higher current density of 1 A g−1 after 1000 cycles, revealing good long-term cycle stability. The experimental and theoretical results reveal that Na+ binding sites are located on the N of NiN4 linkages and the p-type TQ ligand could simultaneously accommodate PF6−. Overall, the current work highlights that reasonably developing novel p-type ligands to form functional 2D c-MOFs will provide a new pathway to simultaneously store cations and anions to further optimize battery performance.Data availability
All experimental supporting data and procedures are available in the ESI.†Author contributions
L. Chen conceived, designed, and guided the whole project; L. Q. Cheng and H. G. Wang performed the electrochemical experiments; D. Chen and W. B. Chen conducted the experiments and wrote the paper; F. C. Cui carried out the theoretical electrochemical calculation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (51973153 and 52172186). The authors are grateful to have access to the beamline 4B9A of the Beijing Synchrotron Radiation Facility (BSRF).References
- L. Kong, M. Liu, H. Huang, Y. Xu and X.-H. Bu, Adv. Energy Mater., 2022, 12, 2100172 CrossRef CAS.
- J. Li, X. Jing, Q. Li, S. Li, X. Gao, X. Feng and B. Wang, Chem. Soc. Rev., 2020, 49, 3565–3604 RSC.
- G. Zhou, L. Xu, G. Hu, L. Mai and Y. Cui, Chem. Rev., 2019, 119, 11042–11109 CrossRef CAS PubMed.
- P. Zhang, F. Wang, M. Yu, X. Zhuang and X. Feng, Chem. Soc. Rev., 2018, 47, 7426–7451 RSC.
- J.-Y. Hwang, S.-T. Myung and Y.-K. Sun, Chem. Soc. Rev., 2017, 46, 3529–3614 RSC.
- Y. Huang, L. Zhao, L. Li, M. Xie, F. Wu and R. Chen, Adv. Mater., 2019, 31, 1808393 CrossRef PubMed.
- P. K. Nayak, L. Yang, W. Brehm and P. Adelhelm, Angew. Chem., Int. Ed., 2018, 57, 102–120 CrossRef CAS PubMed.
- S.-W. Kim, D.-H. Seo, X. Ma, G. Ceder and K. Kang, Adv. Energy Mater., 2012, 2, 710–721 CrossRef CAS.
- Q. Zhao, A. K. Whittaker and X. S. Zhao, Mater, 2018, 11, 2567 CrossRef PubMed.
- X. Yin, S. Sarkar, S. Shi, Q.-A. Huang, H. Zhao, L. Yan, Y. Zhao and J. Zhang, Adv. Funct. Mater., 2020, 30, 1908445 CrossRef CAS.
- M. D. Allendorf, R. Dong, X. Feng, S. Kaskel, D. Matoga and V. Stavila, Chem. Rev., 2020, 120, 8581–8640 CrossRef CAS PubMed.
- M. Wang, R. Dong and X. Feng, Chem. Soc. Rev., 2021, 50, 2764–2793 RSC.
- M. Hmadeh, Z. Lu, Z. Liu, F. Gándara, H. Furukawa, S. Wan, V. Augustyn, R. Chang, L. Liao, F. Zhou, E. Perre, V. Ozolins, K. Suenaga, X. Duan, B. Dunn, Y. Yamamto, O. Terasaki and O. M. Yaghi, Chem. Mater., 2012, 24, 3511–3513 CrossRef CAS.
- X. Huang, S. Zhang, L. Liu, L. Yu, G. Chen, W. Xu and D. Zhu, Angew. Chem., Int. Ed., 2018, 57, 146–150 CrossRef CAS PubMed.
- Z. Wu, J. Xie, Z. J. Xu, S. Zhang and Q. Zhang, J. Mater. Chem. A, 2019, 7, 4259–4290 RSC.
- C. Cong and H. Ma, Small, 2023, 19, 2207547 CrossRef CAS PubMed.
- M. Wang, H. Shi, P. Zhang, Z. Liao, M. Wang, H. Zhong, F. Schwotzer, A. S. Nia, E. Zschech, S. Zhou, S. Kaskel, R. Dong and X. Feng, Adv. Funct. Mater., 2020, 30, 2002664 CrossRef CAS.
- Y. Lu, Y. Zhang, C.-Y. Yang, S. Revuelta, H. Qi, C. Huang, W. Jin, Z. Li, V. Vega-Mayoral, Y. Liu, X. Huang, D. Pohl, M. Položij, S. Zhou, E. Cánovas, T. Heine, S. Fabiano, X. Feng and R. Dong, Nat. Commun., 2022, 13, 7240 CrossRef CAS PubMed.
- D. Feng, T. Lei, M. R. Lukatskaya, J. Park, Z. Huang, M. Lee, L. Shaw, S. Chen, A. A. Yakovenko, A. Kulkarni, J. Xiao, K. Fredrickson, J. B. Tok, X. Zou, Y. Cui and Z. Bao, Nat. Energy, 2018, 3, 30–36 CrossRef CAS.
- C. Qian, Q.-Y. Qi, G.-F. Jiang, F.-Z. Cui, Y. Tian and X. Zhao, J. Am. Chem. Soc., 2017, 139, 6736–6743 CrossRef CAS PubMed.
- L. S. Xie, G. Skorupskii and M. Dincă, Chem. Rev., 2020, 120, 8536–8580 CrossRef CAS PubMed.
- D. Herebian, E. Bothe, F. Neese, T. Weyhermüller and K. Wieghardt, J. Am. Chem. Soc., 2003, 125, 9116–9128 CrossRef CAS PubMed.
- D. Sheberla, L. Sun, M. A. Blood-Forsythe, S. Er, C. R. Wade, C. K. Brozek, A. Aspuru-Guzik and M. Dincă, J. Am. Chem. Soc., 2014, 136, 8859–8862 CrossRef CAS PubMed.
- G. Wu, J. Huang, Y. Zang, J. He and G. Xu, J. Am. Chem. Soc., 2017, 139, 1360–1363 CrossRef CAS PubMed.
- M. G. Campbell, S. F. Liu, T. M. Swager and M. Dincă, J. Am. Chem. Soc., 2015, 137, 13780–13783 CrossRef CAS PubMed.
- B. Wang, J. Li, M. Ye, Y. Zhang, Y. Tang, X. Hu, J. He and C. C. Li, Adv. Funct. Mater., 2022, 32, 2112072 CrossRef CAS.
- F. Wang, Z. Liu, C. Yang, H. Zhong, G. Nam, P. Zhang, R. Dong, Y. Wu, J. Cho, J. Zhang and X. Feng, Adv. Mater., 2020, 32, 1905361 CrossRef CAS PubMed.
- Y. Chen, Q. Zhu, K. Fan, Y. Gu, M. Sun, Z. Li, C. Zhang, Y. Wu, Q. Wang, S. Xu, J. Ma, C. Wang and W. Hu, Angew. Chem., Int. Ed., 2021, 60, 18769–18776 CrossRef CAS PubMed.
- L. Wang, Y. Ni, X. Hou, L. Chen, F. Li and J. Chen, Angew. Chem., Int. Ed., 2020, 59, 22126–22131 CrossRef CAS PubMed.
- Q. Jiang, P. Xiong, J. Liu, Z. Xie, Q. Wang, X.-Q. Yang, E. Hu, Y. Cao, J. Sun, Y. Xu and L. Chen, Angew. Chem., Int. Ed., 2020, 59, 5273–5277 CrossRef CAS PubMed.
- J. Yin, N. Li, M. Liu, Z. Li, X. Wang, M. Cheng, M. Zhong, W. Li, Y. Xu and X.-H. Bu, Adv. Funct. Mater., 2023, 33, 2211950 CrossRef CAS.
- O. Buyukcakir, R. Yuksel, Y. Jiang, S. H. Lee, W. K. Seong, X. Chen and R. S. Ruoff, Angew. Chem., Int. Ed., 2019, 58, 872–876 CrossRef CAS PubMed.
- J.-H. Dou, M. Q. Arguilla, Y. Luo, J. Li, W. Zhang, L. Sun, J. L. Mancuso, L. Yang, T. Chen, L. R. Parent, G. Skorupskii, N. J. Libretto, C. Sun, M. C. Yang, P. V. Dip, E. J. Brignole, J. T. Miller, J. Kong, C. H. Hendon, J. Sun and M. Dincă, Nat. Mater., 2021, 20, 222–228 CrossRef CAS PubMed.
- J. Liu, D. Yang, Y. Zhou, G. Zhang, G. Xing, Y. Liu, Y. Ma, O. Terasaki, S. Yang and L. Chen, Angew. Chem., Int. Ed., 2021, 60, 14473–14479 CrossRef CAS PubMed.
- J. Yan, Y. Cui, M. Xie, G.-Z. Yang, D.-S. Bin and D. Li, Angew. Chem., Int. Ed., 2021, 60, 24467–24472 CrossRef CAS PubMed.
- Z. Yang, T. Wang, H. Chen, X. Suo, P. Halstenberg, H. Lyu, W. Jiang, S. M. Mahurin, I. Popovs and S. Dai, ACS Energy Lett., 2021, 6, 41–51 CrossRef CAS.
- L. Chen, J. Kim, T. Ishizuka, Y. Honsho, A. Saeki, S. Seki, H. Ihee and D. Jiang, J. Am. Chem. Soc., 2009, 131, 7287–7292 CrossRef CAS PubMed.
- H. Bahron, S. S. Khaidir, A. M. Tajuddin, K. Ramasamy and B. M. Yamin, Polyhedron, 2019, 161, 84–92 CrossRef CAS.
- V. Torabi, H. Kargar, A. Akbari, R. Behjatmanesh-Ardakani, H. Amiri Rudbari and M. Nawaz Tahir, J. Coord. Chem., 2018, 71, 3748–3762 CrossRef CAS.
- Y. Jiang, I. Oh, S. H. Joo, O. Buyukcakir, X. Chen, S. H. Lee, M. Huang, W. K. Seong, S. K. Kwak, J.-W. Yoo and R. S. Ruoff, J. Am. Chem. Soc., 2019, 141, 16884–16893 CrossRef CAS PubMed.
- Y. Yue, P. Cai, X. Xu, H. Li, H. Chen, H.-C. Zhou and N. Huang, Angew. Chem., Int. Ed., 2021, 60, 10806–10813 CrossRef CAS PubMed.
- J. Xu, W. Zhang, D. Geng, Y. Liu, H. Wang, N. Tang and G. Yu, Adv. Electron. Mater., 2015, 1, 1500084 CrossRef.
- C. Li, W. Ju, S. Vijay, J. Timoshenko, K. Mou, D. A. Cullen, J. Yang, X. Wang, P. Pachfule, S. Brückner, H. S. Jeon, F. T. Haase, S.-C. Tsang, C. Rettenmaier, K. Chan, B. R. Cuenya, A. Thomas and P. Strasser, Angew. Chem., Int. Ed., 2022, 61, e202114707 CrossRef CAS PubMed.
- J.-D. Yi, D.-H. Si, R. Xie, Q. Yin, M.-D. Zhang, Q. Wu, G.-L. Chai, Y.-B. Huang and R. Cao, Angew. Chem., Int. Ed., 2021, 60, 17108–17114 CrossRef CAS PubMed.
- T. Chen, J.-H. Dou, L. Yang, C. Sun, N. J. Libretto, G. Skorupskii, J. T. Miller and M. Dincă, J. Am. Chem. Soc., 2020, 142, 12367–12373 CrossRef CAS PubMed.
- H. Li, M. Tang, Y. Wu, Y. Chen, S. Zhu, B. Wang, C. Jiang, E. Wang and C. Wang, J. Phys. Chem. Lett., 2018, 9, 3205–3211 CrossRef CAS PubMed.
- Y.-X. Liu, H. Zhang and X.-L. Cheng, Chem. Phys., 2023, 568, 111837 CrossRef CAS.
- H.-g. Wang, Q. Li, Q. Wu, Z. Si, X. Lv, X. Liang, H. Wang, L. Sun, W. Shi and S. Song, Adv. Energy Mater., 2021, 11, 2100381 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc00932k |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |