
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConstruction of an electron-transfer channel via Cu–O–Ni to inhibit the overoxidation of Ni for durable methanol oxidation at industrial current density†
Han
Tian‡
a,
Xiaohan
Wang‡
abc,
Wenshu
Luo
ac,
Rundong
Ma
a,
Xu
Yu
ac,
Shujing
Li
b,
Fantao
Kong
a,
Xiangzhi
Cui
 *abc and
Jianlin
Shi
ac
*abc and
Jianlin
Shi
ac
aShanghai Institute of Ceramics, Chinese Academy of Sciences, Shanghai 200050, P. R. China. E-mail: cuixz@mail.sic.ac.cn
bSchool of Chemistry and Materials Science, Hangzhou Institute for Advanced Study, University of Chinese Academy of Sciences, Hangzhou 310024, P. R. China
cCenter of Materials Science and Optoelectronics Engineering, University of Chinese Academy of Sciences, Beijing 100049, P.R. China
First published on 7th June 2024
Abstract
The electrocatalytic methanol oxidation reaction (MOR) is a viable approach for realizing high value-added formate transformation from biomass byproducts. However, usually it is restricted by the excess adsorption of intermediates (COad) and overoxidation of catalysts, which results in low product selectivity and inactivation of the active sites. Herein, a novel Cu–O–Ni electron-transfer channel was constructed by loading NiCuOx on nickel foam (NF) to inhibit the overoxidation of Ni and enhance the formate selectivity of the MOR. The optimized NiCuOx-2/NF demonstrated excellent MOR catalytic performance at industrial current density (E500 = 1.42 V) and high faradaic efficiency of ∼100%, as well as durable formate generation up to 600 h at ∼500 mA cm−2. The directional electron transfer from Cu to Ni and enhanced lattice stability could alleviate the overoxidation of Ni(III) active sites to guarantee reversible Ni(II)/Ni(III) cycles and endow NiCuOx-2/NF with high stability under increased current density, respectively. An established electrolytic cell created by coupling the MOR with the hydrogen evolution reaction could produce H2 with low electric consumption (230 mV lower voltage at 400 mA cm−2) and concurrently generated the high value-added product of formate at the anode.
Introduction
Methanol (MeOH) is the simplest alcohol, and it can be easily produced by chemical or biomass industrial synthesis.1 Compared with other organic molecules, the price of methanol (∼350 $ per ton) is relatively cheap and can be selectively oxidized to high value-added products such as formic acid (FA) and formate salts (∼1300 $ per ton),2 which are important chemicals in rubber and pharmaceutical industries. Compared with the traditional industrial method with large amounts of energy consumed, the generation of formic acid (formate) through an electrocatalytic methanol oxidation reaction (MOR) driven by renewable energy is a green and zero-carbon emission strategy.3,4 Moreover, the electrochemical thermodynamic potential of the MOR (0.103 V) to FA is much lower than that of the oxygen evolution reaction (OER; 1.23 V),5 which can also be used to replace the OER and couple with production of H2 in the electrocatalytic water-splitting system, achieving low electric consumption for hydrogen production and generating high value-added formate at the anode.6,7 Unfortunately, the MOR usually suffers from being strongly absorbed by intermediates (COad), leading to further oxidization to low-value CO2 at high current densities (>100 mA cm−2), and losing its economic value.8 Therefore, it is of great importance to develop highly selective MOR catalysts.Precious metal catalysts have been considered to be efficient catalysts toward the MOR, but scarce reserves, high prices and especially the susceptibility to poisoning by the intermediate COad greatly restrict their wide application.9 In contrast, earth-abundant transition metal-based catalysts show great potential towards the MOR. Among them, Ni-based electrocatalysts have attracted great attention thanks to their cost-effectiveness, low overpotential and formate production with high faradaic efficiency (>90%).10–12 In an electrochemical process, nickel-based catalysts often undergo electrochemical oxidation, hydroxylation and surface reconstruction to generate electrocatalytic activity. It has been widely believed that Ni or Ni oxides first form Ni(OH)2 in an alkaline environment, which is transformed further into active NiOOH with the increase in potential.13 The strong electron-gaining ability of Ni3+ in the formed NiOOH can efficiently oxidize methanol molecules and then be reduced back into Ni2+, thus realizing a reversible cycle of Ni2+/Ni3+ in the MOR.14 Unfortunately, the further development of Ni-based electrocatalysts in methanol economy is restricted due to the following three reasons. (1) The excess adsorption of intermediates during the MOR poisons Ni active sites,15 thus limiting the continuous conversion from methanol to formate. (2) The overoxidation of Ni species, such as the transformation from Ni3+ to Ni4+ at high voltages, destroys the reversible Ni2+/Ni3+ cycle, resulting in a dramatic decrease in activity.16 (3) The current density of the MOR is still low, which is not suitable for industrial production (>300 mA cm−2). Thus, developing durable Ni-based catalysts with high formate selectivity at high current densities is a big challenge.
It has been reported that Ma–O–Mb units with different metals can establish interface-bonding channels to enhance charge transfer and modulate the charge state of the surface adsorbate to activate reaction intermediates.17 Considering the special characteristics of Cu, such as half-fully occupied 4s orbitals, a flexible hybrid spin state and abundant chemical valences,18,19 we proposed the construction of Cu–O–Ni units. Among the latter, on the one hand, Cu serves as an electron donor to generate directional electron transfer from Cu to Ni through O atoms to lower the excess accumulation of intermediate COad at Ni active sites. On the other hand, Cu inhibits the overoxidation of Ni3+ species during the MOR to stabilize the metastable high-valence Ni3+ active sites.20 Besides, through the constructed Cu–O–Ni electron-transfer channel, the dehydrogenation ability of Ni is enhanced, leading to excellent MOR activity at high current density.
Specifically, we constructed a novel Cu–O–Ni unit to inhibit the overoxidation of Ni (Ni3+ → Ni4+) by introducing Cu, in which the bimetallic oxide (NiCuOx) was loaded on nickel foam (NF) as an efficient MOR electrocatalyst via a two-step method involving electrochemical deposition and thermal treatment. The optimal NiCuOx-2/NF electrocatalyst with an electrochemical deposition time of 2000 s exhibited excellent catalytic activity towards the MOR at an industrial-scale current density of 500 mA cm−2 with only 1.42 V (vs. RHE) and high formate faradaic efficiencies of ∼100%. Impressively, the NiCuOx-2/NF catalyst could catalyze methanol oxidation stably up to 600 h at a high current density of ∼500 mA cm−2, which is superior to reported Ni-based MOR catalysts. Based on XPS, electrochemical results and operando impedance measurements, we revealed that the introduction of Cu could generate an electron-transfer channel from Cu to Ni, which consequently promoted the CH3OH* dehydrogenation ability of Ni and stabilized Ni3+ active sites. Besides, the strong Cu–O–Ni interaction at oxide interfaces also avoids the overoxidation of Ni species (Ni3+ → Ni4+) to a certain extent, making it possible to catalyze methanol effectively and stably at industrial-grade current densities (>300 mA cm−2). By coupling the MOR with a cathodic HER, the integrated two-electrode system using NiCuOx-2/NF as both anode and cathode catalysts could reach a current density of 400 mA cm−2 at 2.02 V, which is lower than that of the electrolytic water splitting by ∼230 mV, thereby consolidating the application potential in commercial H2 production and biomass increment conversion.
Results and discussion
Synthesis and characterizations
NiCuOx nanostructures were grown on NF by a two-step method based on electrochemical deposition, followed by further heat treatment (Fig. 1a). First, the pretreated NF was immersed and successively deposited electrochemically in H2SO4 solutions with NiCl2˙6H2O and CuCl2˙6H2O as Ni and Cu sources, respectively. Then, the resultant NiCu/NF precursor was heated slightly in a muffle furnace to obtain NiCuOx/NF. To study the effect of different amounts of Cu doping, NiCuOx samples were prepared by changing the electrodeposition time from 1000 s to 4000 s in CuCl2 solutions.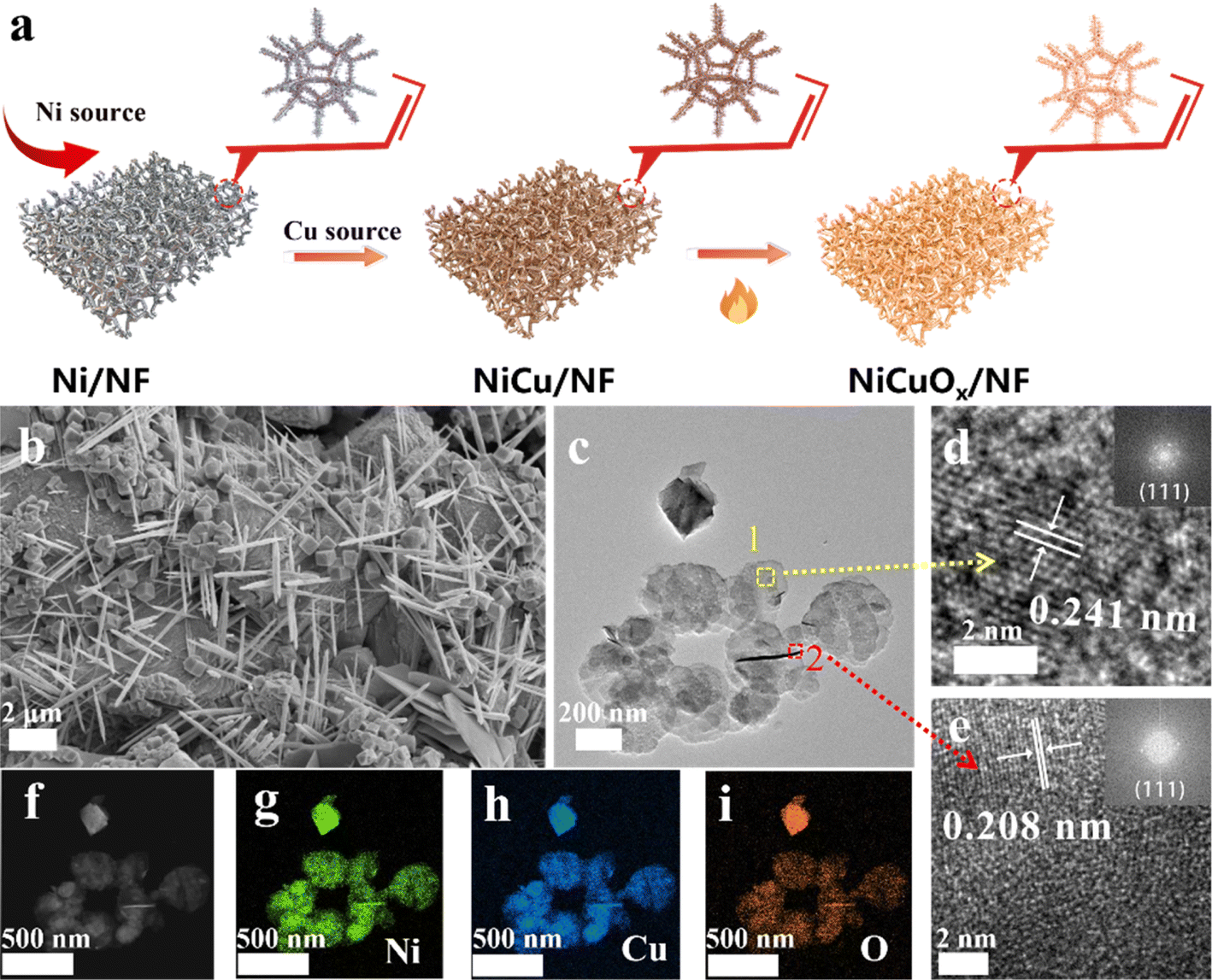 | ||
| Fig. 1 (a) Synthesis of NiCuOx/NF (schematic). (b) SEM, (c) TEM, (d and e) HRTEM images and (f–i) EDS elemental mappings of NiCuOx-2/NF. | ||
Scanning electron microscopy (SEM) images (Fig. 1b) showed that NiCuOx-2/NF with an electrodeposition time of 2000 s consisted of both nanoneedles and cubic nanoparticles, which covered the surface of NF densely and uniformly. NiCuOx-1/NF (Fig. S1†) showed a similar structure to NiCuOx-2/NF, but was not fully grown due to the insufficient electrodeposition time. As the electrodeposition time increased further, the nanoneedles gradually disappeared and only cubic and agglomerated nanoparticles were observed for NiCuOx-3/NF (Fig. S2†) compared with the flat or irregular morphology of control samples of CuOx/NF (Fig. S3†) and Ni/NF (Fig. S4†). Furthermore, the needle-like and cubic morphologies were also confirmed by transmission electron microscopy (TEM) images (Fig. 1c). The TEM images (Fig. S5 and S6†) of control samples showed similar evolution trends to those in the SEM images. The lattice spacings of 2.41 Å and 2.08 Å in high-resolution TEM (HRTEM) images (Fig. 1d and e) corresponded with the (111) plane of NiCuOx and (111) plane of NiCu alloy, respectively. It has been reported that the (111) crystal planes of Cu and Ni can enhance the MOR activity of catalysts. Thus, the exposure of (111) crystal faces in NiCuOx/NF facilitated the adsorption and dissociation of methanol molecules and enhanced the high MOR electrocatalytic activity. Besides, the high-angle annular dark field (HAADF) image and corresponding energy dispersive spectroscopy (EDS) mappings demonstrated the uniform distribution of Ni, Cu and O elements in both nanoneedles and nanoparticles of NiCuOx-2/NF (Fig. 1f–i). The content ratio of Ni to Cu in NiCuOx gradually decreased with the longer electrodeposition time of Cu source, and the specific data are displayed in Tables S1 and S2.†
X-ray diffraction (XRD) patterns were recorded to identify the crystalline structure of as-prepared catalysts. Ni/NF showed three obvious characteristic peaks with 2θ of 44.5°, 51.8° and 76.4° corresponding to cubic-phase Ni (PDF#87-0712) (Fig. S7†).21 After introduction of the Cu source (Fig. 2a), the diffraction peaks at 43.5° and 44.6° could be assigned to the cubic phases of NiCu alloy and NiCuOx oxide, respectively. The slight shifts to a low angle of the corresponding characteristic peaks (PDF#85-1326, PDF#73-1519) for NiCuOx, as well as the elemental mapping analysis stated above confirmed the generation of Cu–Ni lattice mixing. The atom substitution of Cu to Ni for pristine NiO lattice led to the generation of Cu–O–Ni units. Besides, the intensity ratio of NiCuOx to NiCu increased with a longer electrodeposition time, revealing that the introduction of Cu could facilitate the generation of oxidized Ni species. The diffraction peaks at 28.7° and 47.6° attributed to cubic CuCl (PDF#77-2383) disappeared after the electrochemical test. The XRD pattern of CuOx/NF (Fig. S8†) showed similar diffraction peaks to those of NiCuOx.
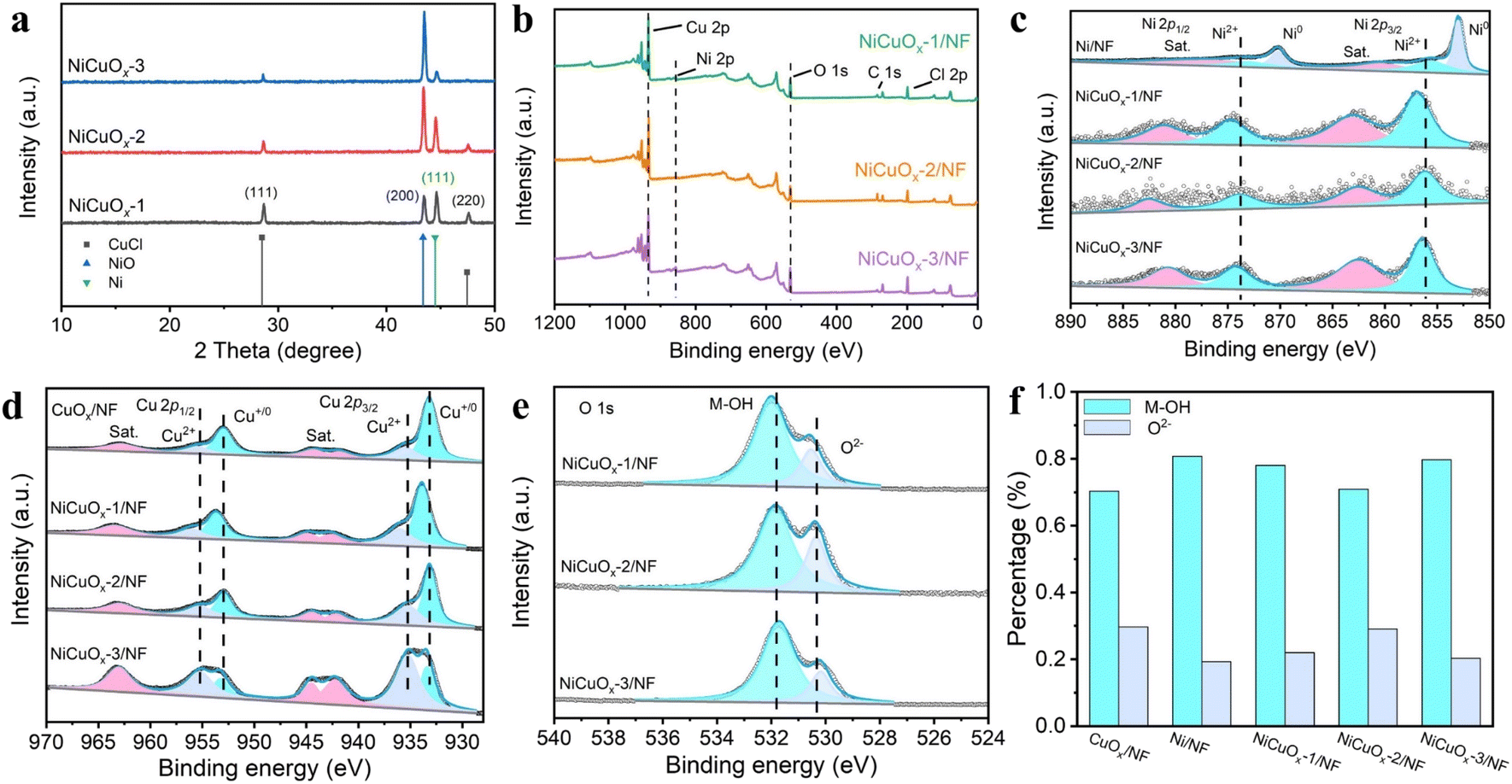 | ||
| Fig. 2 (a) XRD patterns for NiCuOx-1, NiCuOx-2 and NiCuOx-3. Survey XPS spectrum (b), high-resolution XPS Ni 2p (c), Cu 2p (d), O 1s (e) and corresponding histograms (f) of M-OH/O2− sub-bands for as-prepared catalysts. | ||
The chemical compositions and oxidation states of as-synthesized catalysts were probed using the X-ray photoelectron spectroscopy (XPS). As shown in Fig. 2b, Cu, Ni, O, C and Cl elements could be observed for NiCuOx-1/NF, NiCuOx-2/NF and NiCuOx-3/NF, data which were consistent with XRD results.
Fig. 2c shows the XPS Ni 2p spectra for Ni/NF, NiCuOx-1/NF, NiCuOx-2/NF and NiCuOx-3/NF. Ni/NF mainly demonstrated the metal Ni0 with corresponding binding energies at about 853 and 870.2 eV assigned to Ni0 2p3/2, and Ni0 2p1/2, respectively. After Cu introduction, the chemical valence of Ni in NiCuOx/NF composites increased to Ni(II), and the binding energies at 856.1 and 873.3 eV were attributed to Ni2+ 2p3/2 and Ni2+ 2p1/2, respectively.22 Among NiCuOx/NF composites, NiCuOx-2/NF exhibited a slightly negative shift in Ni 2p spectra, indicating the local electronic transfer from Cu to Ni. The Cu 2p spectra in Fig. 2d display the characteristic sub-bands at the binding energies of 933.2, 935.2, 953 and 955.2 eV, which were assigned to Cu+/0 2p3/2, Cu2+ 2p3/2, Cu+/0 2p1/2 and Cu2+ 2p1/2, respectively.23 The specific ratios of Cu2+ to Cu+/0 in the material gradually increased as the amount of Cu loading increased, which is consistent with the result of Ni 2p XPS spectra. Further, the O 1s spectra (Fig. 2e, S10 and S11†) of as-prepared catalysts were deconvoluted into two sub-bands at ∼530.4 and ∼531.8 eV, which corresponded to lattice oxygen (metal-bonded O2−) and surface-chemisorbed oxygen (oxygen in M–OH), respectively.24 NiCuOx-2 showed the highest proportion of O2− (Fig. 2f), which is beneficial for lattice stability under high current densities.
During the MOR, Ni2+ species in Ni-based materials tend to be in situ electrochemically oxidized to form active Ni3+ with high MOR catalytic activity. Unfortunately, as the current density increases to industrial grade, active Ni3+ can undergo further oxidation, leading to a reduction in catalytic activity. According to the XPS results, the introduction of Cu in NiCuOx-2/NF to construct a Cu–O–Ni unit could stabilize the lattice and prevent the further oxidation of Ni3+ during the MOR, leading to the ultra-long MOR stability of NiCuOx-2/NF.
Electrocatalytic performance evaluation
The MOR performances of as-prepared catalysts were investigated in 1 M KOH solution with different concentrations of MeOH using linear sweep voltammetry (LSV). First, the onset oxidation potentials of the MOR (Fig. 3a) for NiCuOx-2/NF decreased significantly after the addition of MeOH to the electrolyte. The MOR current density increased significantly with increasing of the concentration of MeOH up to 1 M. As the concentration increased to 2 M, the current density for the MOR began to decrease owing to excess adsorption of methanol molecules. Therefore, 1 M of MeOH was chosen as the additive concentration for subsequent MOR measurements. The cyclic voltammetry (CV) curves in Fig. 3b suggested that all of the catalysts had one pair of reversible redox peaks corresponding to the Ni2+/Ni3+ transformation.25 The sharp increase in current densities above ∼1.2 V matched well with the Ni(II) → Ni(III)OOH oxidation, and the backward scanning peak of NiCuOx/NF corresponding to Ni(III) reduction shifted to the lower potential compared with Ni/NF. More impressively, the lower reduction potential of Ni(III) → Ni(II) in NiCuOx-2/NF was responsible to the higher current density and better MOR performance. | ||
| Fig. 3 (a) Polarization curves of NiCuOx-2/NF at different concentrations of methanol in KOH (1 M). (b) CV curves, (c) LSV curves and (d) corresponding Tafel plots of samples (with 100% iR compensation). (e) Nyquist plots measured in KOH (1 M) + MeOH (1 M). (f) FE and averaged generation rates of formate at the anode. (g) Chronoamperometry (I–t) curves of NiCuOx-2/NF at a constant potential of 1.63 V (vs. RHE). (h) Comparisons of test duration and corresponding current density between this work and catalysts reported in the literature.26–32 | ||
The LSV curves in Fig. 3c suggested that NiCuOx-2/NF exhibited the best MOR performance in as-prepared catalysts. NiCuOx-2/NF needed only 1.42 V to reach an industrial-scale current density of 500 mA cm−2, which was lower than that of Ni/NF, CuOx/NF, NiCuOx-1/NF and NiCuOx-3/NF by 153, 104, 50 and 16 mV, respectively. This result indicated that the amount of Cu species introduced in NiCuOx-2/NF with electrochemical deposition of 2000 s was optimal for the MOR performance, and that excessive Cu species would cover the catalyst surface and inhibit the oxidation of Ni(II) to active Ni(III). The onset potentials (∼1.3 V) of three NiCuOx/NF species were close to that of the Ni(II)/Ni(III) redox couple, confirming that Ni(III) was the active species for the MOR. Moreover, the Tafel slope of NiCuOx-2/NF was 87.2 mV dec−1 (Fig. 3d), which was much lower than that of CuOx/NF (100 mV dec−1), Ni/NF (116.1 mV dec−1), NiCuOx-1/NF (91.1 mV dec−1) and NiCuOx-1/NF (107.2 mV dec−1), implying the fast MOR kinetics of NiCuOx-2/NF. Fig. 3e reveals the electrochemical impedance spectroscopy (EIS) data. The resistance to electron transfer increased in the order Ni/NF > NiCuOx-1/NF > NiCuOx-3/NF > NiCuOx-2/NF, which was in good agreement with the LSV results stated above. The lower resistance and faster charge transport/diffusion ability of NiCuOx-2/NF contributed to the more significant enhancement of MOR catalytic activity.
Furthermore, 1H and 13C nuclear magnetic resonance (NMR) spectroscopy were carried out to identify the products of MOR. Formate was the main product after chronoamperometric (i–t) tests for 1 h (Fig. S12†), and the faradaic efficiency (FE) for methanol-to-formate oxidation at different current densities was also investigated. The FE of MOR for NiCuOx-2/NF was ∼100% at different current densities and >96.8% even at a high current density of 500 mA cm−2, suggesting the ultrahigh selectivity of NiCuOx-2/NF (Fig. 3f). Furthermore, the formate concentrations generated by the NiCuOx-2/NF anode at different current densities were investigated. The average generation rate of formate was 83.09, 107.14, 148.75 and 184.03 mmol cm−2 h−1 at a current density of 200, 300, 400 and 500 mA cm−2, respectively.
Specifically, the durability of the operation and prospect of industrial application of NiCuOx-2/NF was estimated by chronoamperometry at an especially high current density (∼500 mA cm−2) under 1.63 V (vs. RHE, without iR-compensation). Encouragingly, the current density did not experience severe degradation after the stability test of up to 600 h, indicating the superior stability of NiCuOx-2/NF for the MOR. After the stability test, there was no obvious change in the main crystalline phases of NiCu alloy and NiCuOx in NiCuOx-2/NF except for the disappearance of the diffluent NiCl phase (Fig. S13†). The slight shift to a low angle was attributed to the introduction of more Cu in the NiCuOx lattice. The emergence of high-valence Cu2+ and Cu+ peaks in Raman spectrum (Fig. S14†) suggested a deeper alloying effect of Cu during the MOR.33 SEM and TEM images of post-testing NiCuOx-2/NF showed a morphology of nanoplates, and no impurity phase was observed (Fig. S15 and S16†). Besides, the XPS results after the stability test (Fig. S17†) showed that high-valence Ni4+ species were not formed in NiCuOx-2/NF. Hence, the formation of Ni4+ was suppressed by the introduction of Cu species. The addition of Cu as the charge-compensation agent could inhibit the overoxidation of Ni and promote lattice stability for the NiCuOx-2/NF catalyst. Compared with reported self-supporting nickel-based MOR catalysts, the designed NiCuOx-2/NF obtained in our study was extremely competitive and showed superior stability for the longest period of time at industrial-grade current densities (Fig. 3h and Table S3†).
Insight into the MOR mechanism
Operando electrochemical impedance spectroscopy (EIS) was employed to reveal the electrochemical mechanism of NiCuOx-2/NF during MOR under different potentials. In the low-frequency region (0.01–10 Hz), the peak is usually regarded as nonhomogeneous charge distribution associated with the surface oxidizing species on the electrode surface (OER/MOR).34,35 First, from the Bodes plots of NiCuOx-2/NF (Fig. 4a and b), an apparent peak could be found at a potential of 1.25 V during the MOR, lower than that during the OER (1.5 V), indicating faster MOR kinetics than for the OER. The Nyquist plots of NICuOx-2/NF (Fig. S18†) also showed a smaller semicircle, denoting the lower impedance and more rapid electronic transfer towards the MOR. Impressively, the peak at 1.25 V shifted slowly to a higher frequency and lower phase angle with increasing the potential to 1.50 V (Fig. 4c), which was because the reaction kinetics of methanol oxidation are superior to the OER under low potentials .5 As the polarization potential changed to >1.50 V, the frequency peak decreased with increasing voltage, leading to a decrease in Faraday resistance and an increase in the surface reaction rate. | ||
| Fig. 4 Reaction mechanism of catalysts. (a)–(c) Bode plots of NiCuOx-2/NF for the OER (a) and MOR (b) at different potentials. (d) Multi-potential step curves of NiCuOx-2/NF in KOH solution (1 M) without and with MeOH (1 M). In all panels, the orange-, yellow-, and light blue-shaded areas indicate application of a constant voltage of 1.62 V (vs. RHE), an open-circuit process and a constant voltage of 0.57 V (vs. RHE), respectively. (e) The MOR reaction scheme at different potentials on NiCuOx-2/NF. | ||
Furthermore, the MOR and OER measurements of multipotential steps were performed on NiCuOx-2/NF to investigate the MOR active sites and catalytic mechanism (Fig. 4d). First, the initial applied potential for the NiCuOx-2/NF anode was set to 1.62 V (vs. RHE) to produce abundant NiIII–OOH species. Then, the applied potential was switched to 0.57 V (vs. RHE) after entering an open-circuit state for 90 s. Under OER conditions, NiCuOx-2/NF showed significant reduction currents at 0.57 V (vs. RHE), suggesting non-spontaneous reduction of Ni3+ in the absence of methanol. In contrast, when methanol was added in the open-circuit state, only weak reduction currents appeared at 0.57 V (vs. RHE), suggesting that the addition of methanol promoted the transition of Ni3+ to Ni2+ under MOR conditions. In addition, when methanol was added prior to the measurement, higher current densities were observed at 1.62 V (vs. RHE) compared with the data obtained under OER conditions, confirming the occurrence of the MOR. After the open circuit, only a weak current density was detected at 0.57 V (vs. RHE), indicating that the generated Ni(III) had been consumed simultaneously under MOR conditions. Therefore, we concluded that the MOR process of NiCuOx-2/NF involved the reversible Ni(II)/Ni(III) cycle (Fig. 4e): (i) the Ni(II) species in NiCuOx-2/NF are oxidized to high-valence Ni(III) under the oxidation potential; (ii) methanol is oxidized to formic acid by the real active site Ni(III), and the accumulated Ni(III) is reduced back to initial Ni(II) concurrently. At the same time, the FE of methanol to formic acid increases steadily with increasing voltage and remains stable after reaching a maximum value until 1.50 V (vs. RHE), which agreed well with the result shown in Fig. 4c. Besides, through combining the electrochemical test results, the introduction of Cu could avoid the overoxidation of Ni species (Ni3+ → Ni4+), thus maintaining the reversible Ni(II)/Ni(III) cycle and realizing superior MOR activity, especially under high current densities.
Co-electrolysis in MeOH–water solution
The NiCuOx-2/NF sample also showed HER catalytic ability in KOH (1 M) with/without containing methanol (1 M) (Fig. S19†). Hence, a MeOH–water solution electrolyser in the two-electrode system was constructed with NiCuOx-2/NF simultaneously used as the catalyst at both the cathode and anode. As exemplified in Fig. 5a, the MOR occurred at the anode to produce high value-added products, and the HER occurred at the cathode to generate H2. To reach a current density of 400 mA cm−2, the MeOH–water electrolysis cell required a cell voltage of only 2.02 V. This was lower (by ∼230 mV) than that in the electrolyte without MeOH (2.25 V), suggesting a significantly reduced electric consumption by coupling the MOR with the HER and valued chemical productions at the anode simultaneously. | ||
| Fig. 5 (a) Scheme for a two-electrode electrolyzer for concurrent electrolytic production of hydrogen and value-added chemicals, and LSV curves of NiCuOx-2/NF in KOH (1.0 M) with and without addition of methanol (1 M). (b) FEs of generated formate at the anode after operating at 200, 300, 400 and 500 mA cm−2 for 1 h. (c) Chronoamperometry (I–t) curves by NiCuOx-2/NF-equipped MOR‖HER at a constant potential of 2.03 V. | ||
In order to determine the oxidation products and calculate the corresponding FE, the MeOH–water electrolysis cell catalyzed by NiCuOx-2/NF catalysts was operated at a current density of 200, 300, 400, and 500 mA cm−2, respectively, and the liquid samples were collected after running for 1 h. It is noteworthy that FE could be maintained >96% at high current densities, even at 400 and 500 mA cm−2 (Fig. 5b). According to the long-term chronoamperometry test at 2.03 V (Fig. 5c), the NiCuOx-2/NF equipped two-electrode system could stand for 360 h without an obvious decrease in current density. Hence, it could find application as a bifunctional electrocatalyst in H2 production and biomass increment conversion.
Conclusions
A novel Cu–O–Ni charge-transfer channel was developed in NiCuOx bimetallic oxide to boost MOR electrocatalytic activity. The as-prepared NiCuOx-2/NF catalyst exhibited an excellent electrocatalytic MOR performance. As low as 1.42 V (vs. RHE) was needed to achieve a high current density of 500 mA cm−2. FE of ∼100% for formate generation and superior stability up to 600 h at industrial current densities were observed. The high MOR performance of NiCuOx-2/NF was because the overoxidation of active site Ni(III) could be effectively inhibited by the introduction of Cu through the formed Cu–O–Ni electron-transfer channel to maintain the reversible Ni(II)/Ni(III) transformation. On the other hand, the electronic transfer from Cu to Ni alleviated the accumulation of intermediate COad at Ni sites, which led to the remarkable catalytic activity and stability at an increased current density. Besides, an established electrolytic cell created by coupling the MOR with the HER with NiCuOx-2/NF as both anode and cathode catalysts outputs elicited a current density of 400 mA cm−2 at 2.02 V, which is ∼230 mV lower than that obtained by the splitting of pure water. This work provides a new perspective for the design of efficient and stable MOR electrocatalysts at high current density, and also promotes the development for energy-saving H2 production by coupling the oxidation of small molecules in biomass.Data availability
The data underlying this study are available in the published article and its ESI.†Author contributions
H. T.: data curation, investigation, methodology, data analysis, funding acquisition, and writing (original draft). X. W.: data curation, investigation, and data analysis. W. L., data curation and investigation. R. M. and X. Y.: data curation. S. L. and F. K.: investigation. X. C.: conceptualization, funding acquisition, supervision, and writing (review and editing). S. J.: conceptualization and supervision.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors gratefully acknowledge the support of this research by National Natural Science Foundation of China (52172110), the “Scientific and Technical Innovation Action Plan” Hong Kong, Macao and Taiwan Science & Technology Cooperation Project of Shanghai Science and Technology Committee (21520760500), “Super postdoctoral Incentive Program” of Shanghai Municipal Human Resources and Social Security Bureau (2021411) and Special Research Assistant Grant Project from Chinese Academy of Sciences.References
- G. Fu, X. Kang, Y. Zhang, X. Yang, L. Wang, X. Z. Fu, J. Zhang, J. L. Luo and J. Liu, Nano-Micro Lett., 2022, 14, 200 CrossRef CAS.
- C. Tang, Y. Zheng, M. Jaroniec and S. Z. Qiao, Angew. Chem., Int. Ed., 2021, 60, 19572–19590 CrossRef CAS.
- W. Luo, H. Tian, Q. Li, G. Meng, Z. Chang, C. Chen, R. Shen, X. Yu, L. Zhu, F. Kong, X. Cui and J. Shi, Adv. Funct. Mater., 2023, 34, 2306995 CrossRef.
- N. Han, Y. Wang, H. Yang, J. Deng, J. Wu, Y. Li and Y. Li, Nat. Commun., 2018, 9, 1320 CrossRef.
- X. Wei, Y. Li, L. Chen and J. Shi, Angew. Chem., Int. Ed., 2021, 60, 3148–3155 CrossRef CAS.
- Y. Hao, D. Yu, S. Zhu, C.-H. Kuo, Y.-M. Chang, L. Wang, H.-Y. Chen, M. Shao and S. Peng, Energy Environ. Sci., 2023, 16, 1100–1110 RSC.
- J. Hao, J. Liu, D. Wu, M. Chen, Y. Liang, Q. Wang, L. Wang, X.-Z. Fu and J.-L. Luo, Appl. Catal., B, 2021, 281, 119510 CrossRef CAS.
- S. Li, R. Ma, J. Hu, Z. Li, L. Liu, X. Wang, Y. Lu, G. E. Sterbinsky, S. Liu, L. Zheng, J. Liu, D. Liu and J. Wang, Nat. Commun., 2022, 13, 2916 CrossRef CAS.
- X. Wang, S. Xi, W. S. V. Lee, P. Huang, P. Cui, L. Zhao, W. Hao, X. Zhao, Z. Wang, H. Wu, H. Wang, C. Diao, A. Borgna, Y. Du, Z. G. Yu, S. Pennycook and J. Xue, Nat. Commun., 2020, 11, 4647 CrossRef CAS.
- Y. Qi, Y. Zhang, L. Yang, Y. Zhao, Y. Zhu, H. Jiang and C. Li, Nat. Commun., 2022, 13, 4602 CrossRef CAS.
- Y. Xu, M. Liu, M. Wang, T. Ren, K. Ren, Z. Wang, X. Li, L. Wang and H. Wang, Appl. Catal., B, 2022, 300, 120753 CrossRef CAS.
- C. Cao, D. D. Ma, J. Jia, Q. Xu, X. T. Wu and Q. L. Zhu, Adv. Mater., 2021, 33, e2008631 CrossRef.
- B. Zhu, B. Dong, F. Wang, Q. Yang, Y. He, C. Zhang, P. Jin and L. Feng, Nat. Commun., 2023, 14, 1686 CrossRef CAS.
- B. Mondal, N. Karjule, C. Singh, R. Shimoni, M. Volokh, I. Hod and M. Shalom, Adv. Energy Mater., 2021, 11, 2101858 CrossRef CAS.
- J. Li, R. Wei, X. Wang, Y. Zuo, X. Han, J. Arbiol, J. Llorca, Y. Yang, A. Cabot and C. Cui, Angew. Chem., Int. Ed., 2020, 59, 20826–20830 CrossRef CAS.
- B. Zhao, J. Liu, C. Xu, R. Feng, P. Sui, L. Wang, J. Zhang, J. L. Luo and X. Z. Fu, Adv. Funct. Mater., 2020, 31, 2008812 CrossRef.
- L. Wang, L. Song, Z. Yang, Y. M. Chang, F. Hu, L. Li, L. Li, H. Y. Chen and S. Peng, Adv. Funct. Mater., 2022, 33, 2210322 CrossRef.
- Y. Zhang, B. Zhou, Z. Wei, W. Zhou, D. Wang, J. Tian, T. Wang, S. Zhao, J. Liu, L. Tao and S. Wang, Adv. Mater., 2021, 33, e2104791 CrossRef.
- T. Wang, L. Tao, X. Zhu, C. Chen, W. Chen, S. Du, Y. Zhou, B. Zhou, D. Wang, C. Xie, P. Long, W. Li, Y. Wang, R. Chen, Y. Zou, X.-Z. Fu, Y. Li, X. Duan and S. Wang, Nat. Catal., 2021, 5, 66–73 CrossRef.
- M. Li, X. Deng, K. Xiang, Y. Liang, B. Zhao, J. Hao, J. L. Luo and X. Z. Fu, ChemSusChem, 2020, 13, 914–921 CrossRef CAS.
- Z. Chang, G. Meng, Y. Chen, C. Chen, S. Han, P. Wu, L. Zhu, H. Tian, F. Kong, M. Wang, X. Cui and J. Shi, Adv. Mater., 2023, 35, e2304508 CrossRef.
- Y. Liu, Z. Jiang and Z. J. Jiang, Adv. Funct. Mater., 2023, 33, 2302883 CrossRef CAS.
- H. Huo, J. Wang, Q. Fan, Y. Hu and J. Yang, Adv. Energy Mater., 2021, 11, 2102447 CrossRef CAS.
- H. Tian, X. Yu, W. Huang, Z. Chang, F. Pei, J. Zhou, N. Dai, G. Meng, C. Chen, X. Cui and J. Shi, Small, 2023, 19, e2303061 CrossRef.
- X. Deng, M. Li, Y. Fan, L. Wang, X.-Z. Fu and J.-L. Luo, Appl. Catal., B, 2020, 278, 119339 CrossRef CAS.
- B. Zhao, J.-W. Liu, Y.-R. Yin, D. Wu, J.-L. Luo and X.-Z. Fu, J. Mater. Chem. A, 2019, 7, 25878–25886 RSC.
- C. Xiao, L. Cheng, Y. Wang, J. Liu, R. Chen, H. Jiang, Y. Li and C. Li, J. Mater. Chem. A, 2022, 10, 1329–1335 RSC.
- Q. Yang, C. Zhang, B. Dong, Y. Cui, F. Wang, J. Cai, P. Jin and L. Feng, Appl. Catal., B, 2021, 296, 120359 CrossRef CAS.
- M. Li, X. Deng, Y. Liang, K. Xiang, D. Wu, B. Zhao, H. Yang, J.-L. Luo and X.-Z. Fu, J. Energy Chem., 2020, 50, 314–323 CrossRef.
- B. Zhao, J. Liu, X. Wang, C. Xu, P. Sui, R. Feng, L. Wang, J. Zhang, J.-L. Luo and X.-Z. Fu, Nano Energy, 2021, 80, 105530 CrossRef CAS.
- T. Wang, X. Cao, H. Qin, X. Chen, J. Li and L. Jiao, J. Mater. Chem. A, 2021, 9, 21094–21100 CAS.
- K. Xiang, D. Wu, X. Deng, M. Li, S. Chen, P. Hao, X. Guo, J. L. Luo and X. Z. Fu, Adv. Funct. Mater., 2020, 30, 1909610 CrossRef CAS.
- H. Li, P. Wei, D. Gao and G. Wang, Curr. Opin. Green Sustainable Chem., 2022, 34, 100589 CrossRef CAS.
- W. Chen, L. Xu, X. Zhu, Y. C. Huang, W. Zhou, D. Wang, Y. Zhou, S. Du, Q. Li, C. Xie, L. Tao, C. L. Dong, J. Liu, Y. Wang, R. Chen, H. Su, C. Chen, Y. Zou, Y. Li, Q. Liu and S. Wang, Angew. Chem., Int. Ed., 2021, 60, 7297–7307 CrossRef CAS.
- B. Zhou, C.-L. Dong, Y.-C. Huang, N. Zhang, Y. Wu, Y. Lu, X. Yue, Z. Xiao, Y. Zou and S. Wang, J. Energy Chem., 2021, 61, 179–185 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc00842a |
| ‡ H. T. and X. W. contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |