
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence2D hexagonal assembly of a dipolar rotor with a close interval of 0.8 nm using a triptycene-based supramolecular scaffold†
Takejiro
Ogawa
ab,
Fumitaka
Ishiwari‡
 *a,
Fatin
Hajjaj
a,
Yoshiaki
Shoji
abc,
Takashi
Kajitani
ade,
Koji
Yazawa
f,
Takahiro
Ohkubo
g and
Takanori
Fukushima
*abc
*a,
Fatin
Hajjaj
a,
Yoshiaki
Shoji
abc,
Takashi
Kajitani
ade,
Koji
Yazawa
f,
Takahiro
Ohkubo
g and
Takanori
Fukushima
*abc
aLaboratory for Chemistry and Life Science, Institute of Innovative Research, Tokyo Institute of Technology, 4259 Nagatsuta, Midori-ku, Yokohama 226-8501, Japan. E-mail: ishiwari@chem.eng.osaka-u.ac.jp; fukushima@res.titech.ac.jp
bDepartment of Chemical Science and Engineering, School of Materials and Chemical Technology, Tokyo Institute of Technology, 4259 Nagatsuta, Midori-ku, Yokohama 226-8501, Japan
cResearch Center for Autonomous Systems Materialogy (ASMat), Institute of Innovative Research, Tokyo Institute of Technology, 4259 Nagatsuta, Midori-ku, Yokohama, Kanagawa 226-8501, Japan
dOpen Facility Development Office, Open Facility Center, Tokyo Institute of Technology, 4259 Nagatsuta, Midori-ku, Yokohama 226-8501, Japan
eRIKEN SPring-8 Center, 1-1-1 Kouto, Sayo, Hyogo 679-5148, Japan
fJEOL Ltd, 3-1-2 Musashino, Akishima, Tokyo 196-8558, Japan
gDepartment of Applied Chemistry and Biotechnology, Graduate School of Engineering, Chiba University, 1–33 Yayoi-cho, Inage-ku, Chiba 263-8522, Japan
First published on 13th June 2024
Abstract
Controlling the rotation of carbon–carbon bonds, which is ubiquitous in organic molecules, to create functionality has been a subject of interest for a long time. In this context, it would be interesting to explore whether cooperative and collective rotation could occur if dipolar molecular rotors were aligned close together while leaving adequate space for rotation. However, it is difficult to realize such structures as bulk molecular assemblies, since molecules generally tend to assemble into the closest packing structure to maximize intermolecular forces. To tackle this question, we examined an approach using a supramolecular scaffold composed of a tripodal triptycene, which has been demonstrated to strongly promote the assembly of various molecular and polymer units into regular “2D hexagonal packing + 1D layer” structures. We found that a molecule (1) consisting of a dipolar 1,2-difluorobenzene rotor sandwiched by two 10-ethynyl-1,8,13-tridodecyloxy triptycenes, successfully self-assembles into the desired structure, where the dipolar rotor units align two-dimensionally at a close interval of approximately 0.8 nm while having a degree of freedom for rotational motion. Here we describe the self-assembly behavior of 1 in comparison with the general trend in molecular self-assembly, as well as the motility of the two-dimensionally aligned molecular rotors investigated using solid-state 19F-MAS NMR spectroscopy.
Introduction
The rotation of carbon–carbon bonds is one of the most fundamental motions within organic molecules, and for a long time many attempts have been made to control it to create functionality. To this end, it may be promising to construct solid-state materials with an ordered alignment of dipolar molecular rotors capable of responding to the application of an external electric field. In particular, special attention has focused on two-dimensional (2D) assemblies of dipolar molecular rotors arranged in close proximity, so that dipole–dipole interactions can operate effectively. Indeed, many experimental and theoretical studies have been carried out on the construction of such 2D systems using molecular crystals, mesoporous silicates, metal–organic frameworks, and films and their transfer-monolayer on a solid surface, seeking the emergence of characteristic phase transition behaviors.1–7 Examples of these materials include molecular crystals, mesoporous silicates, metal–organic frameworks, and monolayers on solid substrates, led by Garcia-Garibay,1–3,8 Sozzani,4,9 Akutagawa,10 Setaka,11 Hla,12 and Michl and Kaleta.13–17 Nonetheless, material designs that can achieve a close 2D assembly of dipolar molecular rotors while ensuring adequate space to allow them to rotate remain a difficult challenge.1–3 For instance, Garcia-Garibay and coworkers reported the formation of a single-crystalline 2D hexagonal assembly of apolar benzene rotors, where a 1,4-diethynyl phenyl unit is sandwiched between two bulky triptycene units having multiple alkyl substituents as a stator (A, Fig. 1a). However, solvent molecules are incorporated to fill the space around the rotor units.18 In molecular systems B and C, which are prototypes of A, space-filling structures have been observed (Fig. 1b), where the rotors and stator units are interdigitated to hamper rotational motions. Michl, Kaleta, and coworkers reported the successful formation of a 2D assembly of a dipolar rotor without interdigitation as a monolayer film on a solid substrate by applying the Langmuir–Blodgett (LB) technique to amphiphilic molecular system D (Fig. 1c).14–17 Obviously, in the bulk-state molecular self-assembly that proceeds under thermodynamic control, densely packed structures are generally formed to maximize the enthalpy gain from intermolecular forces. Structures that provide space to allow the rotation of molecular rotors may be formed kinetically rather than thermodynamically. However, since such structures are metastable, a particular molecular design would be required to sustain them with sufficient lifetime and thermal stability.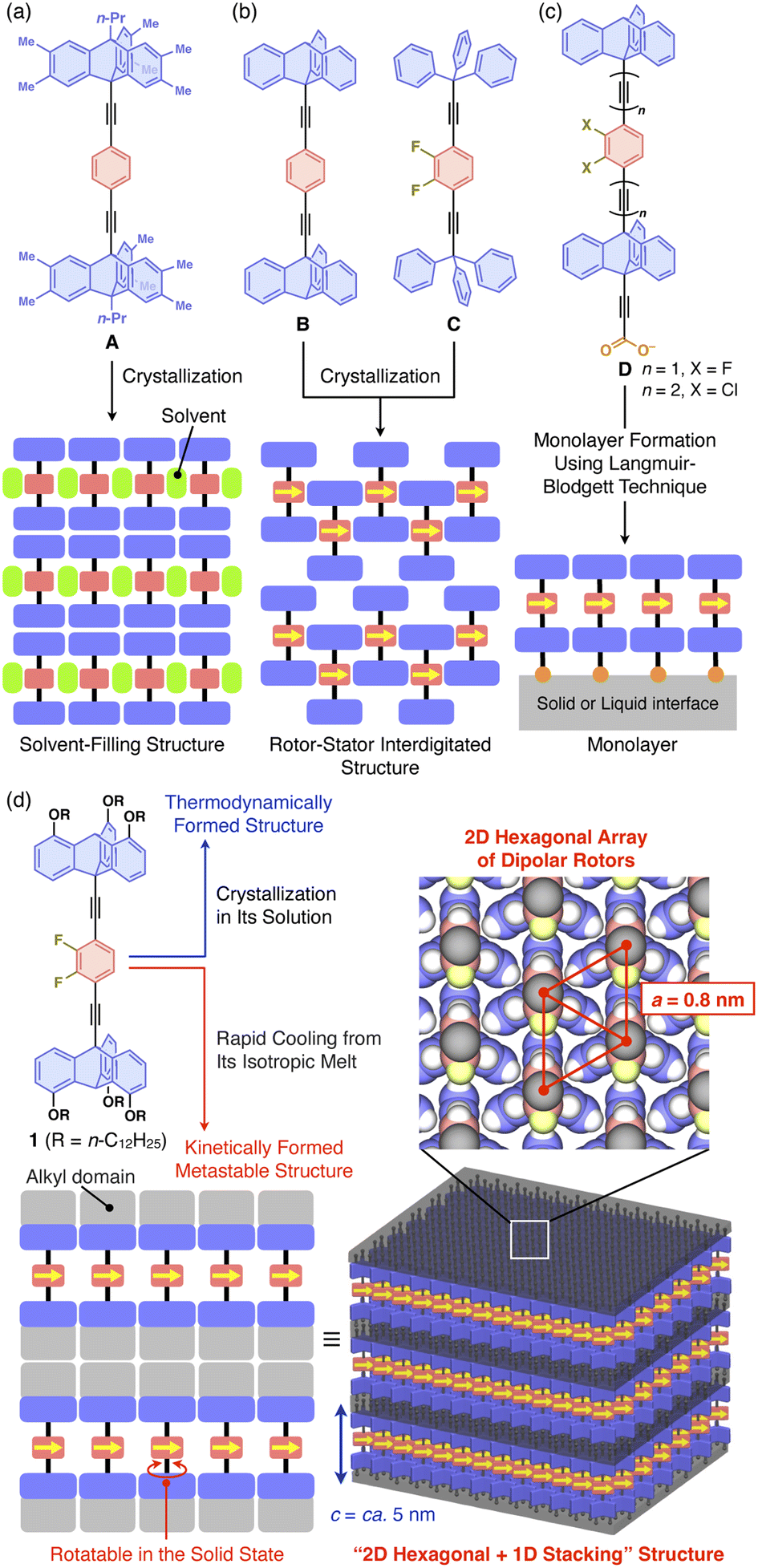 | ||
| Fig. 1 (a–c) Chemical structures of previously reported molecular rotors by Garcia-Garibay et al. (A–C)1–3,8 and Michl, Kaleta et al. (D)14–17 and schematic illustrations of their assembly structures. (d) Chemical structure of the molecular rotor designed in this study, consisting of a 2,3-difluorobenzene and two tripodal triptycene units as dipolar rotor and stator parts, respectively, and its assembly structure formed via a heating/cooling process in the bulk state. | ||
In this study, we developed 1 (Fig. 1d), which combines the molecular rotor designs of Garcia-Garibay1,2,18 and Michl and Kaleta14–17 with our original finding that a particular type of triptycene can strongly induce 2D self-assembly. We have demonstrated that 1,8,13-substituted triptycene derivatives, which can be referred to as tripodal triptycenes, exhibit excellent self-assembling ability, forming robust “2D hexagonal + 1D lamellar” structures through nested packing of their phenylene blades.19–22 These tripodal triptycenes have also been shown to serve as a supramolecular scaffold23 to align functional molecular units such as C60 (ref. 20) and even high-molecular-weight polymers24–26 into 2D + 1D structures with hexagonal lattice parameters of approximately 0.8 nm. A detailed investigation of the self-assembling behavior of 1 revealed that the rotation of its rotor unit is inhibited in the thermodynamically stable structure. On the other hand, 1 can also form a kinetically metastable structure, which allows the rotor unit to rotate with a low activation barrier. This metastable structure is robust and long-lasting at room temperature.
Results and discussion
Synthesis and self-assembly behavior
Compound 1 (Fig. 1d) was synthesized by the Pd-catalyzed Sonogashira–Hagihara coupling reaction of 2,3-difluoro-1,4-diiodobenzene and 10-ethynyl-1,8,13-tridodecyloxy triptycene.20 The chemical composition of 1 was unambiguously characterized by NMR and FT-IR spectroscopy, as well as high-resolution APCI-TOF mass spectrometry (ESI†). Since the signal patterns of 1, observed by 1H, 13C, and 19F NMR spectroscopy in solution (Fig. S1–S3, ESI†), are characteristic of a C3 symmetric structure, the dipolar 1,2-difluorobenzene unit freely rotates faster than the NMR timescale.When compound 1 was suspended in chloroform, heated under reflux to completely dissolve, and then allowed to cool to room temperature, a microcrystalline powdery material formed. 1H NMR spectroscopy in o-dichlorobenzene-d4, thermogravimetric analysis (TGA), and elemental analysis confirmed that the recrystallized powdery material did not contain the solvent molecules used in recrystallization. Unfortunately, the powder X-ray diffraction (PXRD) pattern of as-recrystallized 1 was different from that expected if a “2D hexagonal + 1D lamellar” structure had been formed (Fig. 2a). Presumably, similar to previously reported molecular rotor systems,1,2,8 self-assembly of 1 proceeding via thermodynamic equilibrium states would not result in the desired 2D + 1D structure that provides adequate space for the dipolar rotor unit to rotate.
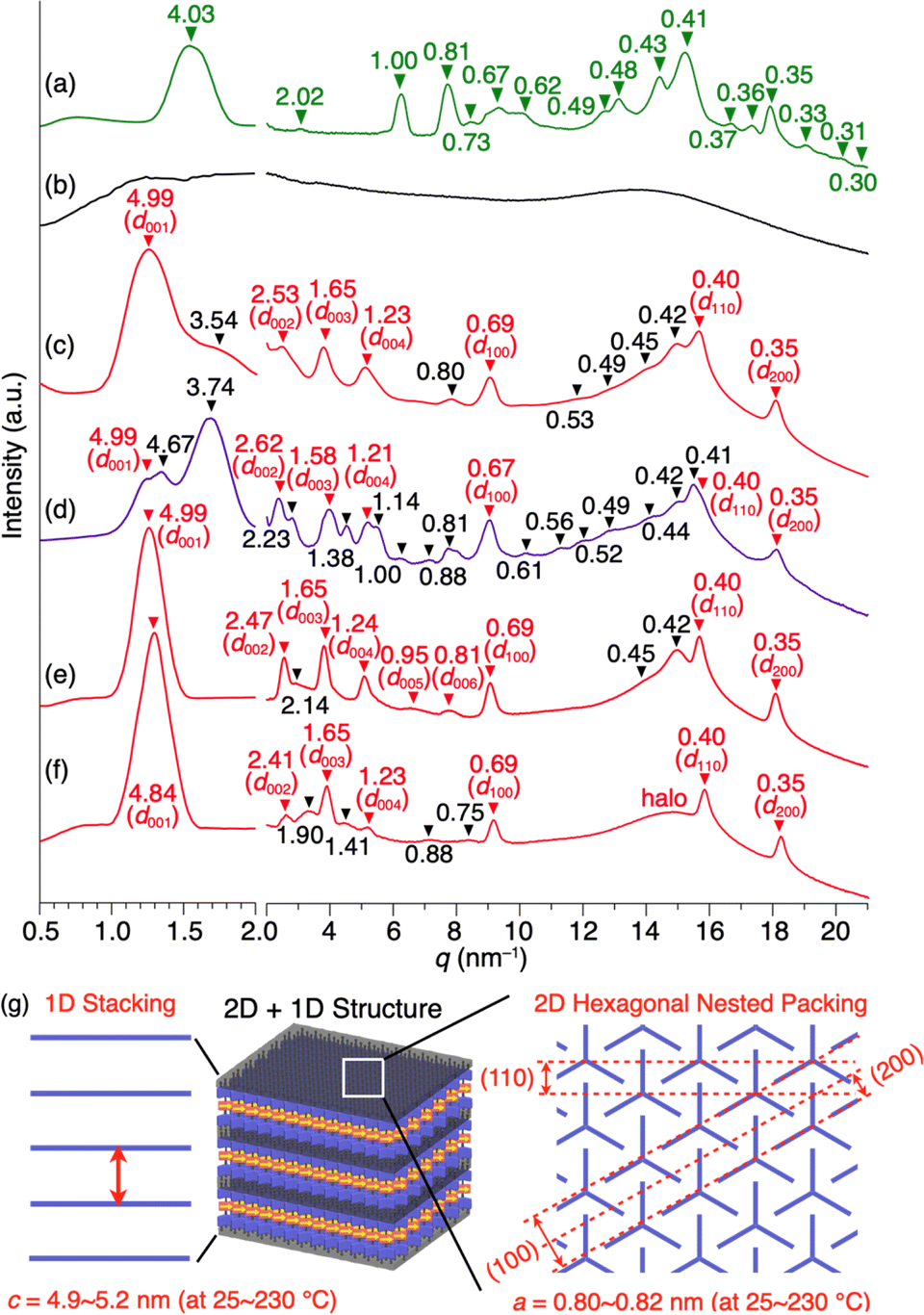 | ||
| Fig. 2 PXRD patterns of 1 measured in a glass capillary with a diameter of 1.5 mm. The d-spacing values are given in nm. (a) As-recrystallized sample at 25 °C, and (b) isotropic melt at 270 °C, and bulk samples at 25 °C after being heated once to their melting point and then cooled at a rate of (c) 10 °C min−1, (d) 2 °C min−1, or (e) 30 °C min−1. In (e), the peaks observed at q = 9.13, 15.8, and 18.2 nm−1 correspond to d-spacings = 0.69, 0.40, and 0.35 nm, respectively, that arise from diffraction from the (100), (110), and (200) planes of a 2D hexagonal array formed by nested packing of the triptycene units. The hexagonal lattice parameter is calculated to be 0.80 nm. The peaks observed at q = 1.26, 2.55, 3.80, 5.07, 6.61, and 7.76 nm−1 corresponding to d-spacings = 4.99, 2.46, 1.65, 1.24, 0.95, and 0.81 nm, respectively, are due to diffraction from the (00n) planes (n = 1–6) of a lamellar structure of one-dimensionally aligned 2D triptycene arrays. The interlayer spacing (4.9 nm) agrees well with the length of the longer axis of 1. (f) A bulk sample at 25 °C after being heated once to its melting point and then cooled rapidly (ca. 200 °C min−1) by removal from the heater. (g) Schematic illustration of the 2D + 1D structure of 1. | ||
Compound 1 exhibits phase transition behavior in the bulk state. In differential scanning calorimetry (DSC), upon heating at a rate of 10 °C min−1, as-recrystallized 1 melts at 232 °C after undergoing two endothermic processes at 167 and 219 °C (Fig. 3a).27 Upon subsequent cooling, two exothermic processes occur at 229 and 208 °C. When as-recrystallized 1 is heated once to melt, its original structural information is erased. Thus, in the second heating/cooling cycle, the two endothermic peaks in the first heating process disappear, and after melting, the DSC profile becomes identical to that observed in the first cooling process.
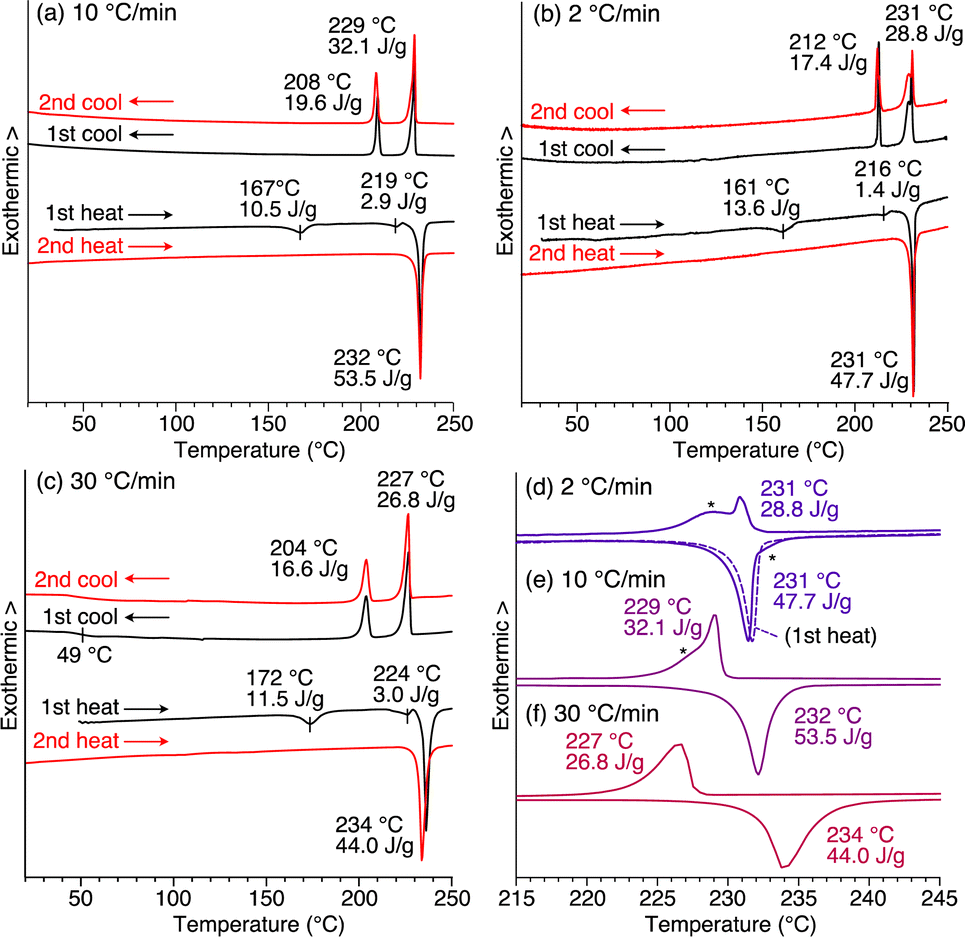 | ||
| Fig. 3 DSC profiles of 1 measured using as-recrystallized samples at scan rates of (a) 10 °C min−1, (b) 2 °C min−1, and (c) 30 °C min−1 under a constant nitrogen flow (50 mL min−1). Black and red curves represent the data obtained in the first and second heating/cooling cycles, respectively. Enlarged DSC profiles ranging from 215 to 245 °C in the second heating/cooling cycle at scan rates of (d) 2.0 °C min−1, (e) 10 °C min−1, and (f) 30 °C min−1. The fact that no side peaks are detected for the first heating process [broken curve in (d)], indicates that when 1 undergoes a heating/cooling process, structures different from that in the as-recrystallized sample result. | ||
Controlled assembly into the 2D + 1D structure
Fig. 2b and c show the PXRD patterns of 1, measured at a higher temperature (270 °C) than its melting point (232 °C) and at 25 °C after cooling from its hot melt at a rate of 10 °C min−1. As expected, the hot melt of 1 is isotropic and displays a featureless XRD pattern (Fig. 2b). Notably, the assembly of 1 upon cooling from its isotropic melt has a structure much different from that formed in as-recrystallized 1. The resulting PXRD pattern (Fig. 2c) shows a peak set (red triangle) as the major diffraction, which is typically observed for tripodal triptycene derivatives assembled into 2D + 1D structures (Fig. 2g). However, the presence of diffraction peaks (black triangle), which cannot be attributed to this structure, indicates that the assembly of 1 is not uniform and contains unidentified structures.To ensure that 1 assembles into the desired 2D + 1D structure, we reduced the cooling rate from the isotropic melt to 2 °C min−1, as a slower cooling rate is usually associated with greater structural regularity. However, a more complex PXRD pattern resulted, where diffraction peaks arising from these unidentified structures became more pronounced (Fig. 2d). To our surprise, when the cooling rate was increased to 30 °C min−1, the diffraction peaks converged almost completely to those arising from the 2D + 1D structure, allowing us to fully index the observed diffraction (Fig. 2e). Furthermore, even when a sample of the isotropic melt was removed from the heater and cooled rapidly to room temperature, a well-defined PXRD pattern (Fig. 2f) was obtained, which was nearly identical to that shown in Fig. 2e. A closer look at Fig. 2e and f indicates that the PXRD pattern, measured after rapid cooling, features higher integrity in diffraction in the region overlapping the halo (q = 13–17 nm−1), and two new weak diffraction peaks are detected between q = 14–15 nm−1. The absence of diffraction peaks due to the unidentified structure (black reversed triangle in Fig. 2c) indicates that the structural order is likely to be better in the rapidly cooled sample. The 2D + 1D structure of 1 thus obtained was found to be maintained even upon cooling to −180 °C, as well as on standing at 25 °C for at least 6 months.
The cooling rate-dependent structuring behavior of 1 was confirmed by DSC analysis. As shown in Fig. 3, while the DSC profiles measured at three different scan rates (2, 10, and 30 °C min−1) are similar overall, the temperature of each corresponding peak is slightly shifted. In organic solids with low molecular mobility, phase-transition temperatures often change depending on the DSC scan rate. However, a closer look at the endothermic and exothermic peaks in the higher temperature range revealed that side peaks (indicated with asterisks) coexist in the profiles obtained at scan rates of 2 and 10 °C min−1, while such side peaks are absent at a scan rate of 30 °C min−1 (Fig. 3d–f). Based on these results and those from the PXRD experiments, the side peaks can be attributed to the unidentified structures that generate upon slow cooling from the isotropic melt of 1.
By tracing the phase transition of 1 using synchrotron PXRD, we found that the 2D + 1D structure of 1 is formed even after the second endothermic process in the first heating. Fig. 4 shows variable-temperature (VT) synchrotron PXRD patterns of 1, measured during rapid (>30 °C min−1) heating/cooling cycles. In a temperature range of 25–150 °C, which is lower than the first endothermic process (172 °C in DSC, Fig. 3c), multiple diffraction peaks characteristic of crystalline materials can be seen. After the first endothermic process, the number of peaks decreases, and the XRD pattern between q = 3–20 nm−1 changes, while the intense peak in the small-angle region (q = 1.63 nm−1) remains almost unchanged. When temperatures exceed the temperature of the second endothermic process (224 °C in DSC, Fig. 3c), a drastic change occurs, generating a typical structure, which is attributed to a 2D sheet-like triptycene assembly with nested hexagonal packing and a 1D layer of the 2D sheets. Notably, the PXRD pattern at this stage exhibits higher-order diffraction peaks even from the (008) planes. Upon further heating, 1 undergoes a phase transition into its isotropic melt, giving a featureless XRD pattern.
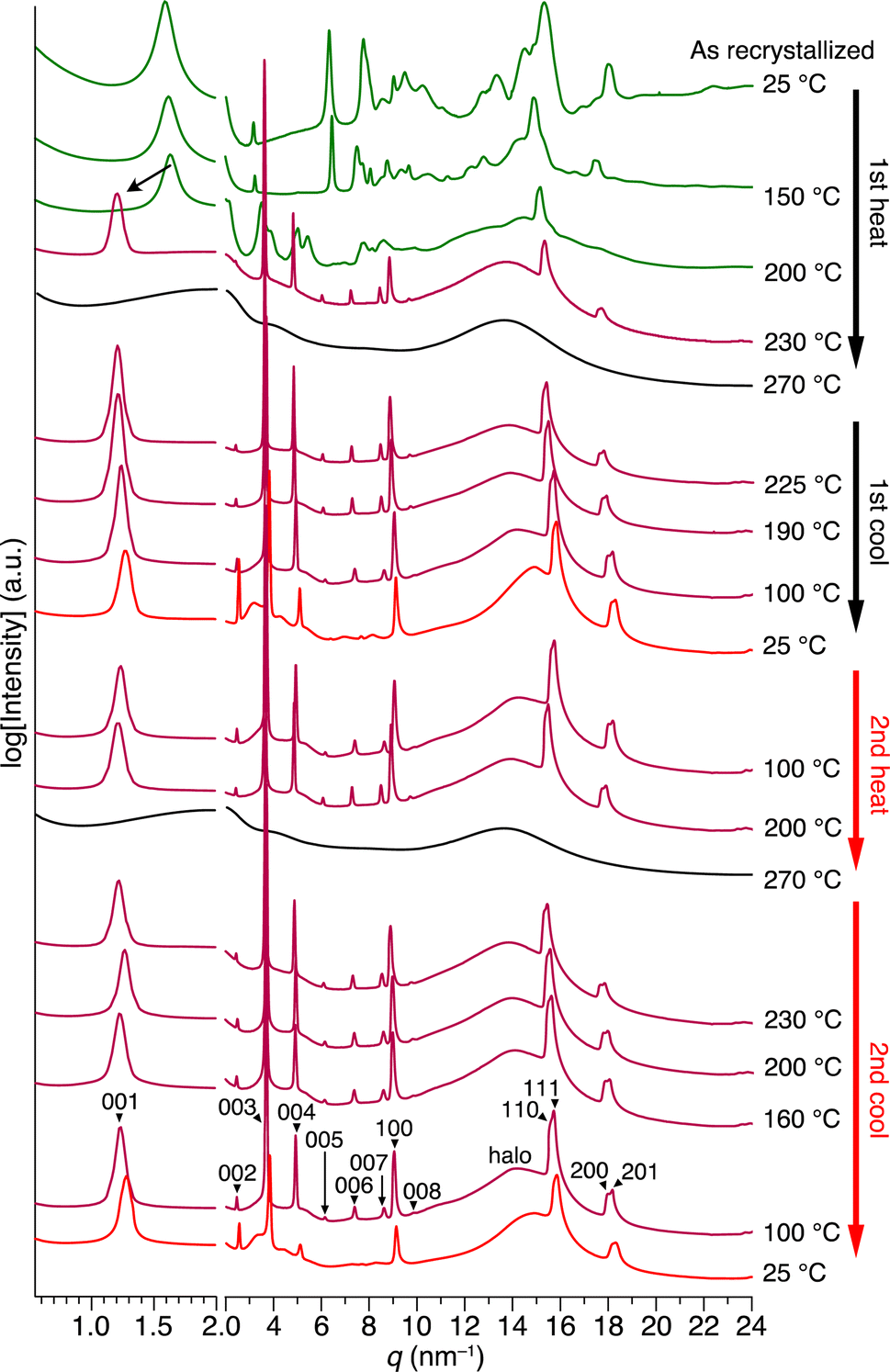 | ||
| Fig. 4 VT synchrotron PXRD profiles obtained for as-recrystallized 1 in a glass capillary with a diameter of 1.5 mm, when heated and cooled repeatedly at a rate of approximately 30 °C min−1. The detailed peak assignments are shown in Fig. S7 and Table S1 (ESI†). | ||
When the isotropic melt is cooled to a temperature (e.g., 225 °C) lower than the temperature of the first exothermic process (227 °C in DSC, Fig. 3c), the diffraction pattern characteristic of the 2D + 1D assembly of 1 again appears. Although a clear exothermic feature is seen at 204 °C in DSC, the pattern remains unchanged during the cooling process reaching e.g., 100 °C. We consider that the exotherm is due to a phase transition into a slightly different structure from the high-temperature phase, while essentially preserving the 2D + 1D order. For example, a structure in which the molecules are slightly tilted toward the 2D plane, as seen in the single-crystal structure of 1,8,13-tris(dodecyloxy)triptycene,19 can be assumed. Meanwhile, all of the peaks are slightly shifted to the wide-angle region, most likely due to thermal shrinkage of the material. Thus, the hexagonal parameter (a) of 0.82 nm and layer spacing (c) of 5.2 nm at 230 °C are changed to 0.80 nm and 4.9 nm at 25 °C, respectively (Fig. S7 and Table S1, ESI†). At 25 °C, while higher-order diffraction peaks arising from the (005)–(008) planes become unclear (see also Fig. 2f), the main diffraction peaks, including those from the 2D hexagonal triptycene assembly, are maintained. According to the DSC profile (Fig. 3c), 1 exhibits a glass-transition feature at around 49 °C under rapid cooling conditions. Presumably, the long alkyl chains vitrify to lower the structural regularity of the 1D layer. Upon heating again at 100 °C, the resulting PXRD pattern is identical to that observed at the same temperature in the first cooling process and is maintained until the melting point. The change in PXRD patterns in the second cooling process completely reproduces that of the first cooling process. The above observations clearly indicate that heating/cooling 1 in the solid state results in the formation of a 2D + 1D structure, featuring two-dimensionally aligned dipolar rotors with approximately 0.8 nm intervals, which is so robust that it is maintained unless the material melts.
Structural transitions during self-assembly
With all the experimental results in mind, we propose the structuring behavior of 1 as shown in Fig. 5. As-recrystallized 1 features a crystalline assembly formed via thermodynamic equilibrium in solution. Given the fact that solvent molecules are not incorporated in the crystal, the difluorobenzene and triptycene units should be interdigitated with each other to maximize enthalpy gain from intermolecular forces. When this crystalline material is heated, it undergoes a phase transition through the second endothermic process (224 °C in DSC, Fig. 3c), resulting in the well-defined 2D + 1D structure, where nested packing of the triptycene blades occurs, simultaneously releasing the rotor units to form 2D arrays on their own. In this state, due to the difference in excluded volume between the difluorobenzene rotor and triptycene stator units, a space could be created around the rotor to allow for its rotation. The key to achieve uniform formation of the 2D + 1D structure is to cool the isotropic melt of 1 rapidly at a rate greater than 30 °C min−1, otherwise structurally unidentified assemblies concomitantly form. Thus, it is reasonable to consider that the 2D + 1D structure is a kinetically generated metastable form of 1. Once formed, the 2D + 1D structure of 1 is maintained at room temperature for a long time even at temperatures lower than the melting point.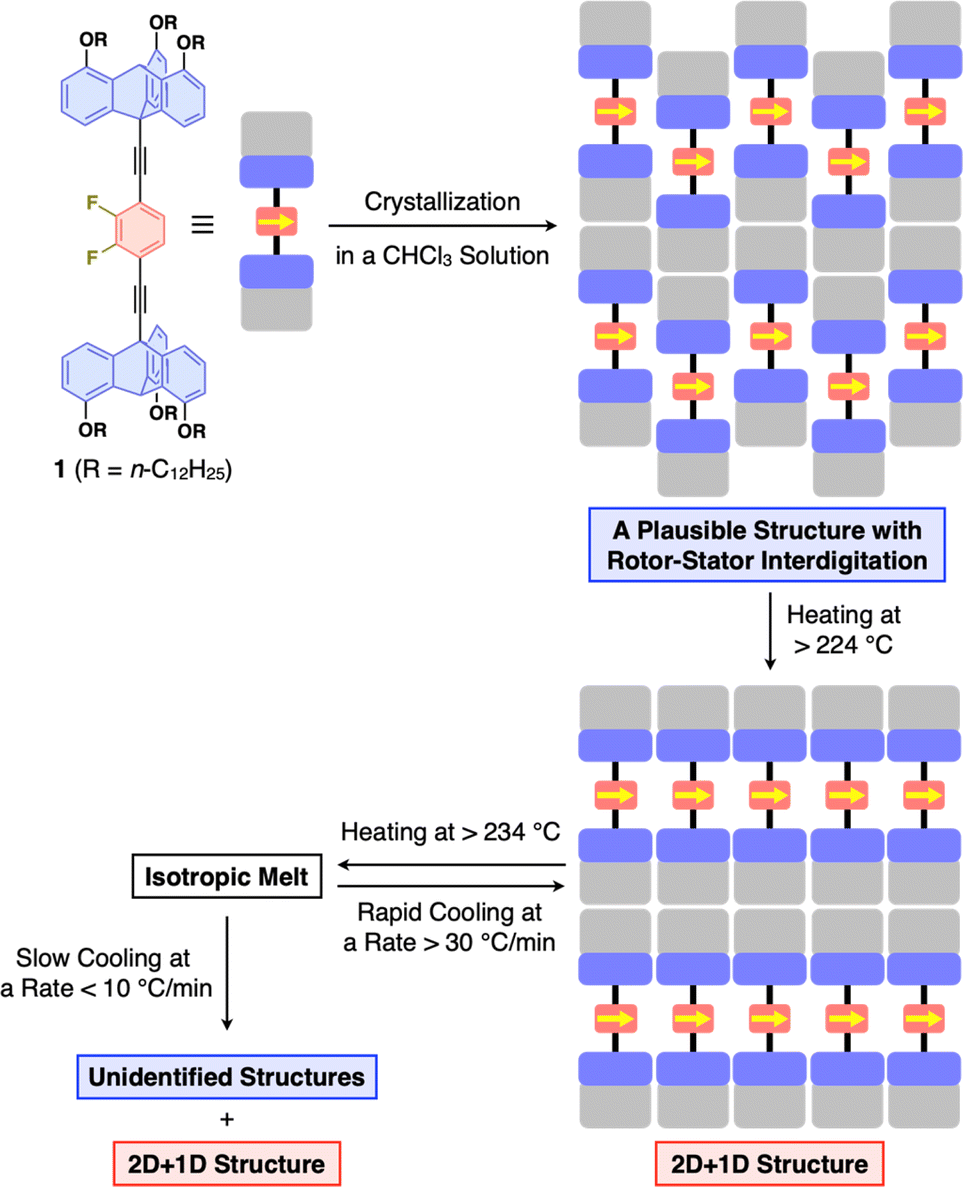 | ||
| Fig. 5 Schematic illustration of the overall structuring behavior of 1. | ||
Dynamic behavior of the rotor units
To verify if the 2D + 1D assembly of 1 indeed possesses space around the rotor units, we measured solid-state 19F-MAS NMR spectra to gain insight into the dynamic behavior of the difluorobenzene rotor moieties. The as-recrystallized powder sample of 1 at 25.0 °C showed multimodal 19F NMR signals at around −133 ppm with a spin-lattice relaxation time (T1) of 35.8 s (Fig. 6a and S8, ESI†), which are due to the presence of a heterogeneous chemical environment around the rotor moieties. It is reasonable to consider that the rotor units in as-recrystallized 1 are closely packed together, hampering their dynamic motions (Fig. 5). Upon heating the sample to 115 °C, the signal became sharp, and at 145 °C, the maximum operating temperature of the instrument, it split into three signals. When the sample was subsequently cooled to 25 °C, these signals did not coalesce. These complicated and irreversible changes in 19F NMR spectroscopy may be related to the endothermic phase transition at around 160–170 °C observed in DSC (Fig. 3), although the change in the assembly structure of 1 associated with this phase transition is unknown.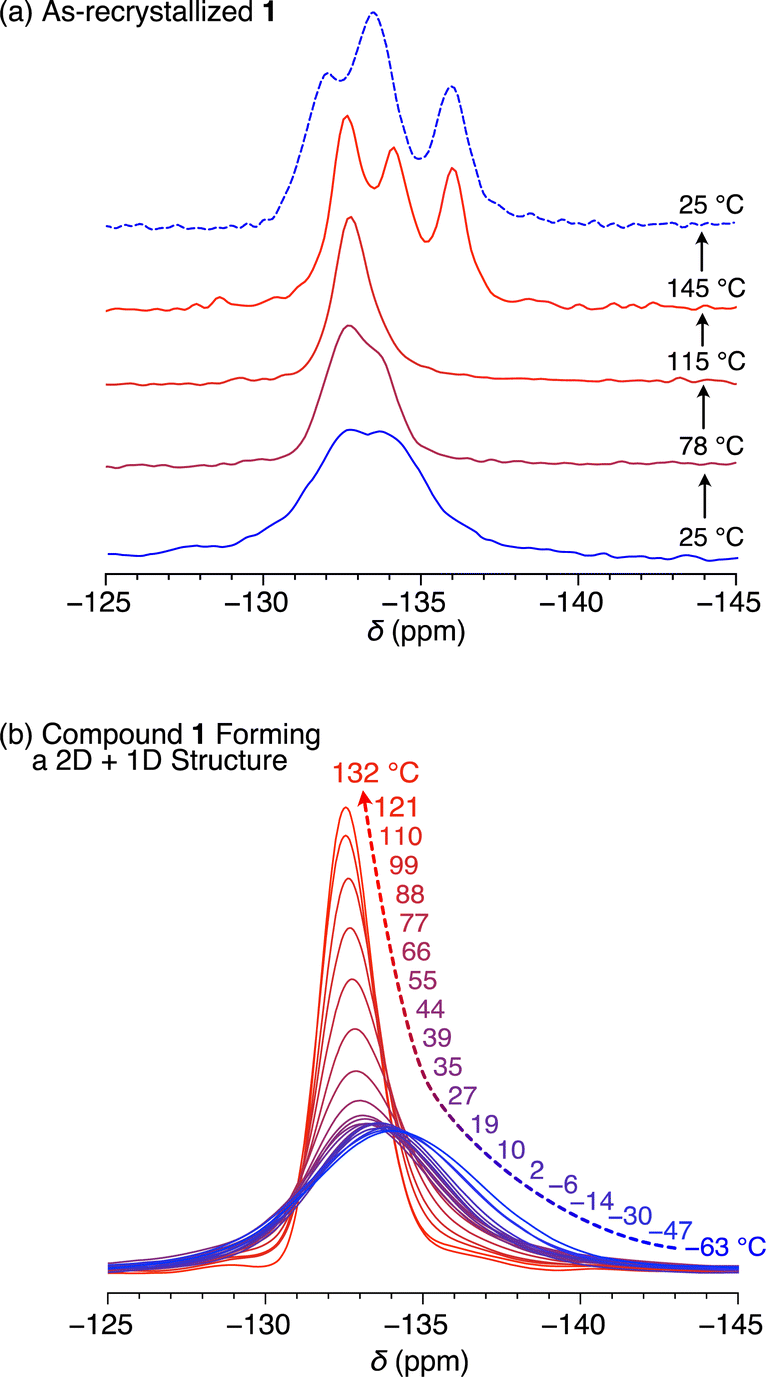 | ||
| Fig. 6 Solid-state VT 19F-MAS NMR spectra of (a) as-recrystallized and (b) thermally treated 1 having a 2D + 1D structure. | ||
Fig. 6b shows the temperature dependence from −63 to 132 °C of the 19F NMR signal of a solid sample of 1 featuring the 2D + 1D structure (Fig. S9, ESI†). Even at −63 °C, the 19F NMR signal at −133 ppm was unimodal with a T1 value of 0.67 s (Fig. S10, ESI†). This value is considerably smaller than that observed for the as-recrystallized powder sample even at 25 °C (35.8 s), clearly indicating much greater motility based on rotational and/or flipping motion of the rotor moieties in the 2D + 1D assembly, compared with those in as-recrystallized 1. With increasing temperature, the 19F NMR signal became sharper (Fig. S9, ESI†), particularly above the glass-transition temperature (49 °C, Fig. 3c). Notably, unlike in the case of as-recrystallized 1, this spectral change was reversible during the heating and cooling processes, with T1 displaying complex behavior and values ranging from 0.67 s (at −63 °C) to 1.05 s (at 2 °C).
Fig. 7a illustrates Arrhenius-type plots of 1/T1. There are at least two peaks, suggesting the presence of multiple motional modes corresponding to the Larmor frequency (470 MHz) of the 19F nucleus of the rotor units. Fitting the profile with two Kubo–Tomita functions17,28–30 results in relatively small activation energies (Ea) of 2.0 kcal mol−1 and 3.3 kcal mol−1 for the lower (blue curve) and higher (red curve) temperature regions, respectively.31 However, compared with examples of phenylene rotor dynamics in a metal–organic framework (MOF-5, Ea = 11.3 ± 2 kcal mol−1)29 and a porous organic crystal (Ea = 6.7 kcal mol−1),30 the values of activation energy are relatively low, and considering also that the change in T1 is small, the Larmor frequency of the 19F nucleus may not precisely capture the motion of the rotor units. The linewidth of the 19F NMR spectrum is mainly due to 1H–19F or 19F–19F dipole interactions, which affect motion in the order of several tens of kHz. Thus, T1ρ was measured using a spin-lock frequency of 50 kHz at various temperatures (Fig. S9 and S11, ESI†), and Arrhenius-type plotting was carried out (Fig. 7b). The obtained T1ρ values change between 6.8 ms (at 132 °C) and 0.84 ms (at 44 °C), and a unimodal peak is observed at around 45 °C with a shoulder at around 10 °C and a tailing feature in the lower temperature region (<−30 °C). Fitting the profile with two Kubo–Tomita functions gives Ea values of 5.5 kcal mol−1 and 5.7 kcal mol−1 for the lower (blue curve) and higher (red curve) temperature regions, respectively.31 Note that these Ea values are low enough to allow for motion of the rotor units at ambient to moderate temperatures.
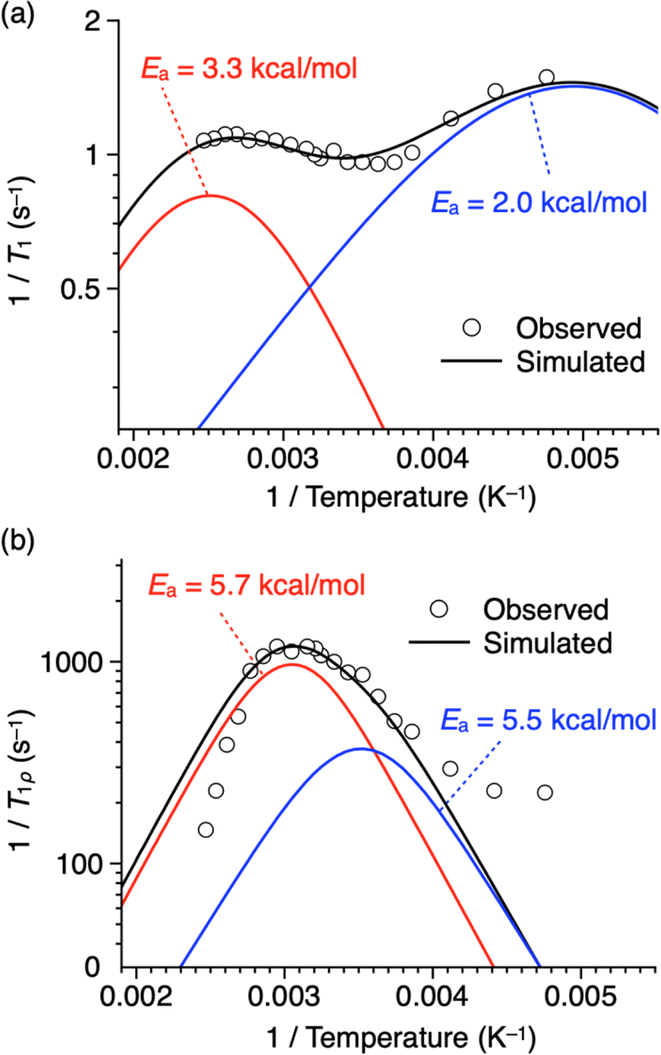 | ||
| Fig. 7 Plots of (a) 1/T1 and (b) 1/T1ρversus reciprocal of temperature of thermally treated 1 having a 2D + 1D structure (white circles with a black border) fitted with a linear combination (black curves) of two Kubo–Tomita functions28 (red and blue curves). Fitting parameters are shown in Tables S2 and S3 (ESI).† | ||
Although it is difficult to completely assign the motional modes of the dipolar rotors from NMR data alone, we presume that the Ea values obtained from the analysis of T1 and T1ρ reflect rapid motions including vibration and slow rotor dynamics such as rotation, respectively. Of particular note is the fact that a clear temperature dependence is observed for the linewidth and T1ρ of the 19F NMR signal, which suggests a kHz-order rotational motion of the two-dimensionally close-aligned difluorobenzene rotors, in which dipole–dipole interactions possibly operate. Importantly, the experimental results, including the structural analysis based on XRD and solid-state 19F-MAS NMR spectroscopy, are all consistent and indicate that the desired 2D dipolar rotor assembly illustrated in Fig. 1d has been achieved using the tripodal triptycene scaffold.
Calculation of the rotational barrier in a model system
To estimate the degree of motility of the rotor unit in the 2D assembly of 1, we performed density functional theory (DFT) calculations based on a simplified model (1′, Fig. 8a), where the dodecyloxy groups of 1 were replaced by hydrogen atoms, and a 2 × 2 × 1 super-cell (Z = 4) was constructed using the hexagonal lattice parameter (a = 0.7961 nm) obtained from the PXRD experiments (at 25 °C, Fig. 4 and Table S1, ESI†). We placed a 1.5 nm-thick slab layer (vacuum layer) between the 2D hexagonal arrays of triptycene under periodic boundary conditions, resulting in c = 3.5 nm (for details, see the ESI†). The rotor unit of a molecule located at the center of the 2D hexagonal lattice (molecule d in Fig. 8a) was manually rotated in 5-degree increments (rotational angle θ = 0–360°, Fig. 8a), while the angles of the other rotor units were fixed.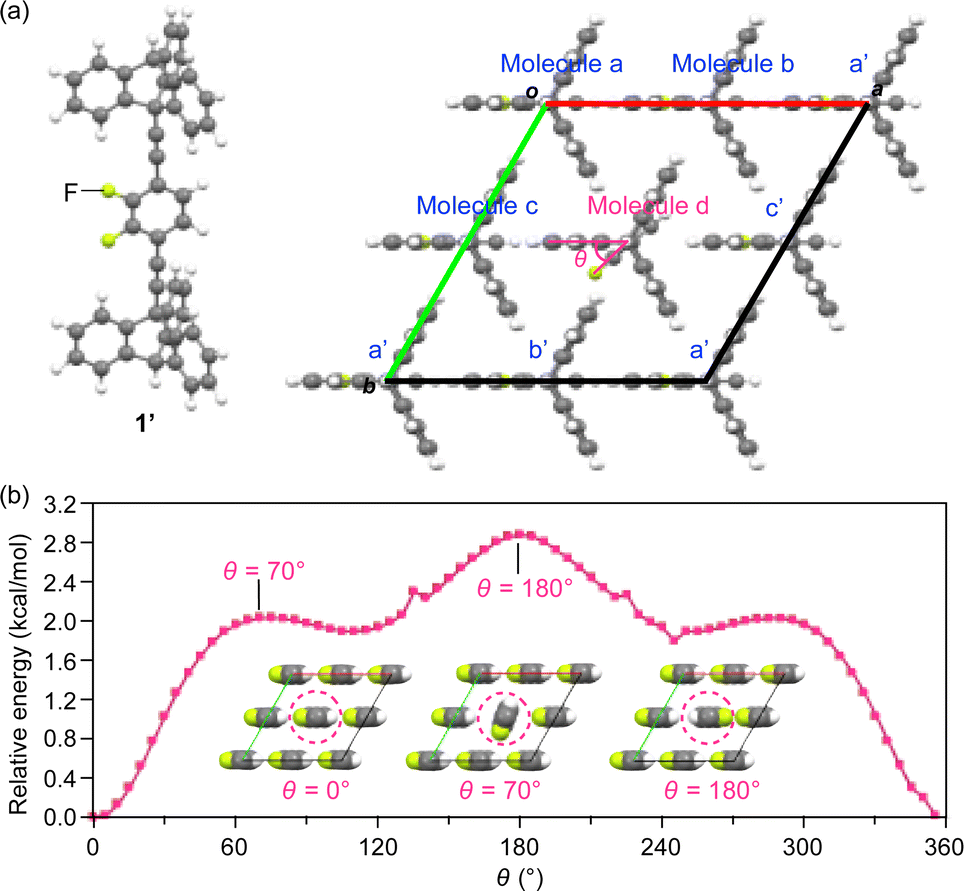 | ||
| Fig. 8 (a) Structure of a model (c-axis projection) with a 2 × 2 × 1 super-cell of 1′ (Z = 4) for DFT calculations. (b) Plots of relative energy versus the rotational angle (θ in molecule d, Fig. 8a) of the rotor units of 1′ located at the center of the 2D hexagonal lattice (molecule d), calculated under periodic boundary conditions. Insets: the arrangement of the rotors at selected rotational angles (see also Movie S1, ESI†). | ||
As shown in Fig. 8b, all the rotor units in the most stable structure (θ = 0°) are aligned in the same direction (inset, left). The energy profile contains three maxima at θ = 70, 180 and 290° and three saddle points at θ = 0, 110 and 245°. The largest barrier was determined to be 2.85 kcal mol−1 at θ = 180°, where the fluorine atoms of adjacent rotor units are in close proximity to each other (inset, right). This value roughly agrees with the Ea values obtained from the solid-state 19F NMR measurements (Fig. 7). The rather small rotation barrier estimated from these calculations indicates that the two-dimensionally arranged rotor unit has a large degree of motility that is enough to allow even a 360° rotation. It is interesting to note that there is a clear angular dependence in the rotational barriers (Fig. 8b); the most stable and unstable structures are found in the geometries where the orientation of the dipole moment of the rotating unit is parallel and antiparallel, respectively, to those of the other rotor units. This suggests the possibility that cooperative rotational motion can occur, and we have begun to address this issue.
Conclusions
If dipolar molecular rotor units are aligned close together while leaving adequate space for rotation, do they exhibit cooperative and collective rotation through dipole–dipole interaction? To examine this question, it is necessary to construct such structures in a bulk solid material that would allow measurement of its physical properties. However, in reality, this is difficult to achieve since molecules, when assembling, tend to form the closest packing so as to maximize intermolecular forces. In the present study, through careful investigation of the self-assembly behavior of newly designed 1, we have demonstrated that the use of a tripodal triptycene-based supramolecular scaffold can achieve a desired structure, in which 2D hexagonal arrays of the triptycene units and 2D arrays of the dipolar rotor units are alternatingly aligned to form 1D layers. Here, the triptycene units also serve as a stator. The key to this achievement was finding that this structure can be obtained when a bulk sample of 1 is heated once to its isotropic melt and then cooled very rapidly, rather than relying on self-assembly from the thermodynamic equilibrium state in solution, i.e., recrystallization. Although the resulting assembly of 1 featuring the 2D + 1D structure may lie in a kinetically formed metastable state, it can be maintained at room temperature for at least 6 months, and even upon heating for a short time at temperatures lower than its melting point. Furthermore, as confirmed by solid-state 19F-MAS NMR spectroscopy, the dipolar rotor has motility with an activation energy (Ea) of ca. 5.7 kcal mol−1 in the solid state. At the same time, this indicates that the desired assembly structure in which the dipole rotors are closely aligned in a 2D structure while maintaining their dynamic properties has been achieved. The DFT calculations for a simplified model further suggest that the rotor unit in the 2D assembly has sufficient degrees of freedom to rotate even 360°. We believe that this work is an important step toward exploring the dynamic behavior of dipole rotors aligned with 2D correlations. We are currently investigating the detailed physical properties of this material to demonstrate unique functions and phenomena arising from the coupling of rotational molecular motion and dipole–dipole interactions.Data availability
All experimental data associated with this work are available in the ESI.†Author contributions
T. F. conceived the project; F. I., Y. S. and T. F. designed the experiments; Take. O., F. H., Y. S. and F. I. carried out the synthesis and characterization of the materials; Take. O., F. I., Y. S. and T. K. performed the X-ray diffraction experiments and analysed the data; Take. O., F. I., Y. S. and K. Y. performed the solid-state NMR measurement and analysed the data; Taka. O. performed DFT calculations; Take. O., F. I., Y. S., Taka. O and T. F. co-wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by JSPS KAKENHI (JP21H05024 and JP21H04690 for T. F.), the Japan Science and Technology Agency (JST) CREST (JPMJCR18I4 for T. F.), and Grant-in-Aid for Transformative Research Areas (A) “Condensed Conjugation” (JP20H05868 for T. F.) from MEXT. This work was also supported in part by the Research Program of “Five-Star Alliance” in “NJRC Mater. & Dev.”. We thank the Materials Analysis Division, Open Facility Center, Tokyo Institute of Technology, for their support with the NMR measurements and elemental analysis. Computational resources were provided by the Supercomputer Fugaku provided by the RIKEN Center for Computational Science (project ID: hp240036).Notes and references
- C. S. Vogelsberg and M. A. Garcia-Garibay, Chem. Soc. Rev., 2012, 41, 1892–1910 RSC.
- M. E. Howe and M. A. Garcia-Garibay, J. Org. Chem., 2019, 84, 9835–9849 CrossRef CAS.
- I. Liepuoniute, M. J. Jellen and M. A. Garcia-Garibay, Chem. Sci., 2020, 11, 12994–13007 RSC.
- A. Comotti, S. Bracco and P. Sozzani, Acc. Chem. Res., 2016, 49, 1701–1710 CrossRef CAS.
- V. M. Rozenbaum, V. M. Ogenko and A. A. Chuľko, Sov. Phys. Usp., 1991, 34, 883–902 CrossRef.
- V. M. Rozenbaum, J. Exp. Theor. Phys. Lett., 1996, 63, 662–667 CrossRef.
- K. Kim and N. S. Sullivan, Phys. Rev. B: Condens. Matter Mater. Phys., 1997, 55, R664 CrossRef CAS.
- R. D. Horansky, L. I. Clarke, E. B. Winston, J. C. Price, S. D. Karlen, P. D. Jarowski, R. Santillan and M. A. Garcia-Garibay, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 74, 054306 CrossRef.
- S. Bracco, M. Beretta, A. Cattaneo, A. Comotti, A. Falqui, K. Zhao, C. Rogers and P. Sozzani, Angew. Chem., Int. Ed., 2015, 54, 4773–4777 CrossRef CAS.
- T. Akutagawa, H. Koshinaka, D. Sato, S. Takeda, S.-I. Noro, H. Takahashi, R. Kumai, Y. Tokura and T. Nakamura, Nat. Mater., 2009, 8, 342–347 CrossRef CAS.
- T. Tsuchiya, Y. Inagaki, K. Yamaguchi and W. Setaka, J. Org. Chem., 2021, 86, 2423–2430 CrossRef CAS.
- Y. Zhang, H. Kersell, R. Stefak, J. Echeverria, V. Iancu, U. G. E. Perera, Y. Li, A. Deshpande, K.-F. Braun, C. Joachim, G. Rapenne and S.-W. Hla, Nat. Nanotechnol., 2016, 11, 706–712 CrossRef CAS.
- J. Kaleta, P. I. Dron, K. Zhao, Y. Shen, I. Císařová, C. T. Rogers and J. Michl, J. Org. Chem., 2015, 80, 6173–6192 CrossRef CAS.
- J. Kaleta, E. Kaletová, I. Císařová, S. J. Teat and J. Michl, J. Org. Chem., 2015, 80, 10134–10150 CrossRef CAS.
- J. Kaleta, J. Wen, T. F. Magnera, P. I. Dron, C. Zhu and J. Michl, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 9373–9378 CrossRef CAS.
- E. Kaletová, C. S. Hurtado, I. Císařová, S. J. Teat and J. Kaleta, ChemPlusChem, 2022, 87, e202200023 CrossRef.
- J. Perego, C. X. Bezuidenhout, S. Bracco, S. Piva, G. Prando, C. Aloisi, P. Carretta, J. Kaleta, T. P. Le, P. Sozzani, A. Daolio and A. Comotti, Angew. Chem., Int. Ed., 2023, 62, e202215893 CrossRef CAS.
- M. A. Garcia-Garibay and C. E. Godinez, Cryst. Growth Des., 2009, 9, 3124–3128 CrossRef CAS.
- N. Seiki, Y. Shoji, T. Kajitani, F. Ishiwari, A. Kosaka, T. Hikima, M. Takata, T. Someya and T. Fukushima, Science, 2015, 348, 1122–1126 CrossRef CAS.
- F. K. C. Leung, F. Ishiwari, T. Kajitani, Y. Shoji, T. Hikima, M. Takata, A. Saeki, S. Seki, Y. M. A. Yamada and T. Fukushima, J. Am. Chem. Soc., 2016, 138, 11727–11733 CrossRef CAS.
- T. Yokota, T. Kajitani, R. Shidachi, T. Tokuhara, M. Kaltenbrunner, Y. Shoji, F. Ishiwari, T. Sekitani, T. Fukushima and T. Someya, Nat. Nanotechnol., 2018, 13, 139–144 CrossRef CAS.
- M. Kondo, T. Kajitani, T. Uemura, Y. Noda, F. Ishiwari, Y. Shoji, T. Araki, S. Yoshimoto, T. Fukushima and T. Sekitani, Sci. Rep., 2019, 9, 9200 CrossRef.
- F. Ishiwari, Y. Shoji and T. Fukushima, Chem. Sci., 2018, 9, 2028–2041 RSC.
- F. Ishiwari, G. Okabe, H. Ogiwara, T. Kajitani, M. Tokita, M. Takata and T. Fukushima, J. Am. Chem. Soc., 2018, 140, 13497–13502 CrossRef CAS.
- Y. Chen, F. Ishiwari, T. Fukui, T. Kajitani, H. Liu, X. Liang, K. Nakajima, M. Tokita and T. Fukushima, Chem. Sci., 2023, 14, 2431–2440 RSC.
- J. Yu, A. Itagaki, Y. Chen, T. Fukui, F. Ishiwari, T. Kajitani and T. Fukushima, Macromolecules, 2023, 56, 4556–4565 CrossRef CAS.
- We have confirmed that the two weak endothermic peaks are not due to the evaporation of residual solvent by thermogravimetric analysis (Fig. S6, ESI†).
- R. Kubo and K. Tomita, J. Phys. Soc. Jpn., 1954, 9, 888–919 CrossRef CAS.
- C. S. Vogelsberg, F. J. Uribe-Romo, A. S. Lipton, S. Yang, K. N. Houk, S. Brown and M. A. Garcia-Garibay, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 13613–13618 CrossRef CAS.
- A. Comotti, S. Bracco, A. Yamamoto, M. Beretta, T. Hirukawa, N. Tohnai, M. Miyata and P. Sozzani, J. Am. Chem. Soc., 2014, 136, 618–621 CrossRef CAS.
- Since peak maxima of the 1/T1 and 1/T1ρ profiles in the lower temperature regions are not clear, there is a possibility that the fitting for this temperature range is not strictly accurate. In the T1ρ profile, the tailing observed in the lower temperature region may be due to motions with lower activation energies, other than rotation and/or flipping. Similar tailing behaviors have been reported previously in ref. 29.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc02750g |
| ‡ Present address: Department of Applied Chemistry, Graduate School of Engineering, Osaka University, 2-1 Yamadaoka, Suita, Osaka 565-0871, Japan. |
| This journal is © The Royal Society of Chemistry 2024 |