
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAsymmetric catalytic [1,3]- or [3,3]-sigmatropic rearrangement of 3-allyloxy-4H-chromenones and their analogues†
Yi
Li‡
,
Lichao
Ning‡
,
Qi
Tang
,
Kexin
Lan
,
Bingqian
Yang
,
Qianchi
Lin
,
Xiaoming
Feng
 * and
Xiaohua
Liu
*
* and
Xiaohua
Liu
*
Key Laboratory of Green Chemistry & Technology, Ministry of Education, College of Chemistry, Sichuan University, Chengdu 610064, China. E-mail: liuxh@scu.edu.cn; xmfeng@scu.edu.cn
First published on 19th June 2024
Abstract
A highly efficient asymmetric [1,3]- and [3,3]-O-to-C sigmatropic rearrangement of 3-allyloxy-4H-chromenones and their analogues was developed. Chiral N,N′-dioxide complexes of 3d late transition metal complexes enabled two mechanistically different processes, giving a series of optically active 2,2-disubstituted chromane-3,4-diones and 2-allyl-3-hydroxy-4H-chromen-4-ones as well as their related compounds in excellent yield and enantioselectivity. Systemic mechanistic studies and DFT calculation revealed the nature of the vinyl ether unit of the substrate, which biased regioselectivity via a stepwise tight ion pair pathway and a concerted pericyclic pathway, respectively. The enantioselectivity of the two processes is also disclosed.
Introduction
O-to-C-sigmatropic rearrangements represent an important method to construct sterically congested carbon stereocenters,1 providing convenient synthetic routes to complicated natural products and related compounds. In the past two decades, Lewis-acid-catalyzed sigmatropic rearrangement developed rapidly.2 Lewis acids accelerate the concerted Claisen rearrangement through a charge-delocalized transition state. Alternatively, a suitable Lewis acid could induce the heterolytic cleavage of the C–O bond to concomitantly form stabilized electrophilic and nucleophilic fragments, performing non-concerted [1,3] O-to-C rearrangement, which was earlier reported by Yamamoto,3 Grieco,4 Gansäuer,5 and Rovis6 (Scheme 1a). The regioselectivity of O-to-C-sigmatropic rearrangement shows bias to both acidity and steric hindrance of the Lewis acid catalysts, and the intrinsic property of the allyl vinyl ethers (Scheme 1a). Catalytic asymmetric versions of the [3,3]- and [1,3]-rearrangements have made achievements in recent years,7,8 but the chiral catalytic system which is tolerant to both [1,3]- and [3,3]-rearrangements is elusive due to the diverse reaction pathways. | ||
| Scheme 1 Lewis-acid-catalyzed regioselective [1,3]- and [3,3]-O-to-C rearrangement. | ||
The natural-product-inspired synthesis plays an important role in enriching chemical space and exploring bioactive compounds.9 Substituted 3-hydroxyl pyranones and benzopyranones are popular structural motifs in natural products, such as flavonol, lawsone, kojic acid, and allomaltol (Scheme 1b). Rearrangements of 3-allyloxyflavone or O-allyl kojic acid have been designed for the synthesis of sanggenon-type natural products by Porco10 and Hou,11 and the tricyclic core of 1α-alkyldaphnanes by Wender and co-workers.12 There are rare examples related to the asymmetric catalytic C2-functionalization of 3-hydroxychromenones: one is a chiral Pybox-Sc(III)-complex-catalyzed formal [3,3]-rearrangement to construct 3,4-chromanediones by Porco and co-workers,13 and another is the chiral NHC-initiated formation of an α,β-unsaturated acyl azolium intermediate to perform Coates–Claisen rearrangement by Bode's group14 and Rafiński's group.15 Based on the above-mentioned studies, as well as Rovis's and Ishihara's16 vigorous work on asymmetric catalytic [1,3]-rearrangement, we envision that suitable chiral Lewis acids and substitution on allyl vinyl ethers would enable [1,3]- and [3,3]-rearrangements of allyloxyflavones to construct C2-functionalized 3-hydroxychromenones and their analogues. As a continuation of the project of asymmetric rearrangements by chiral N,N′-dioxide-metal complexes,17 we herein disclose both rearrangement processes via substrate and catalyst modification under mild reaction conditions. A series of optically active 2,2-disubstituted chromane-3,4-diones were obtained efficiently via cobalt-complex-catalyzed asymmetric [1,3]-rearrangement, while a 2-allyl-3-hydroxy-4H-chromen-4-one series could be afforded via nickel-complex-catalyzed asymmetric [3,3]-rearrangement. The key structure could be extended to kojic acid, allomaltol and lawsone varieties. DFT calculation revealed the regioselectivity tendency in the two cases, and demonstrated the enantioselectivity profile in the rearrangements.
Results and discussion
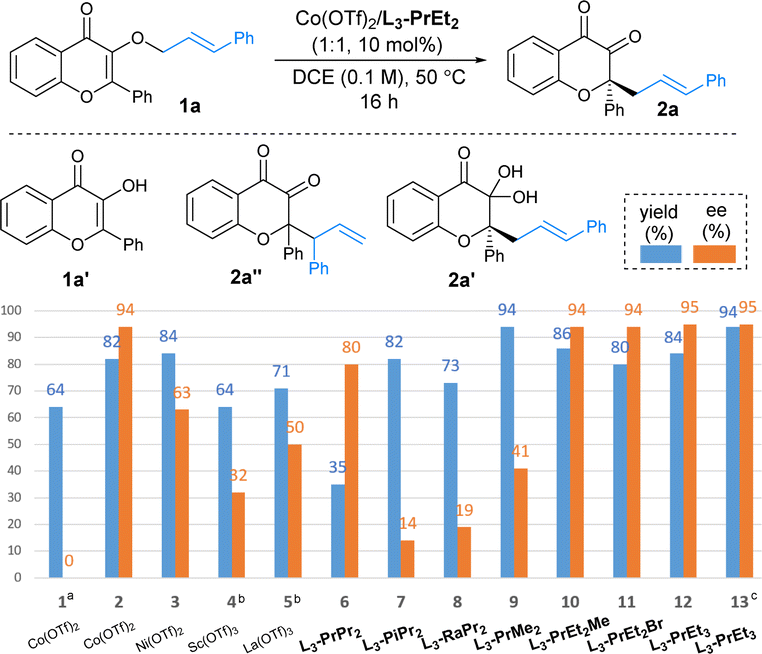
To develop an enantioselective [n,3]-O-to-C sigmatropic rearrangement of a 3-allyloxy-flavonol derivative, we began by searching for the reaction conditions of the rearrangement of E-cinnamyl flavonol ether 1a (Table 2). We found that the reaction in the presence of 10 mol% of Co(OTf)2 in DCE at 50 °C led to the generation of a [1,3]-rearrangement product diketone 2a in 64% yield after 16 hours. Additionally, high regioselectivity was observed and none of the [3,3]-rearrangement product 2a′′ was formed. At the same time, flavonol 1a′ was detected via deallylation, which indicates that the [1,3]-rearrangement probably takes place via a non-concerted route. It was also found that there is an equilibrium between diketone 2a and ketal 2a′ in solvent;18 thus the total yield of 2a and 2a′ was determined by 1H NMR experiments using CHBr2CHBr2 as an internal standard. Employing chiral N,N′-dioxide-metal complexes to induce enantioselectivity manifested that both metal salts and the substructures of the ligands had a dramatic influence on the reactivity and the presence of L3-PrEt2 (entry 2). L-Proline-based ligands resulted in better ee values than L-pipecolic acid or L-ramipril (entries 6–8); while 2,6-diethyl substitutions at the anilines of the ligand are beneficial to enantioselectivity in comparison with methyl or isopropyl groups (entries 2, 6, and 9). An array of L3-PrEt2 analogues produced by installing a para-substitution could give good yield with high enantioselectivity (entries 10–12), and 1,3-rearrangement product 2a could be obtained in 94% yield and 95% ee after 24 hours in the presence of a L3-PrEt3/Co(OTf)2 complex catalyst (entry 13).

|
a Unless otherwise noted, all reactions were carried out with Co(OTf)2/ligand (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 10 mol%), 1a (0.1 mmol) in DCE (0.1 M) at 50 °C; the yield of the mixture 2a/2a′ was determined by 1H NMR spectra using CHBr2CHBr2 as internal standard; ee determined by UPC2 analysis.
b Without ligand.
c At 35 °C.
d 24 h. :1, 10 mol%), 1a (0.1 mmol) in DCE (0.1 M) at 50 °C; the yield of the mixture 2a/2a′ was determined by 1H NMR spectra using CHBr2CHBr2 as internal standard; ee determined by UPC2 analysis.
b Without ligand.
c At 35 °C.
d 24 h.
|
|---|
|
The scope of enantioselective [1,3]-rearrangement is reasonably broad with respect to flavonol ethers bearing different substitutions at the aryl backbone (Table 3). For the convenience of characterization, the products were treated with benzene-1,2-diamine to form quinoxaline 3 in one pot after the standard catalytic reaction. Highly efficient [1,3]-rearrangement products (3a–3q) were afforded in good yields (71–98%) and enantioselectivities (81 to >99% ee). Generally, electron-withdrawing and electron-donating groups at the 6-, 7- or 8-positions (3b–3i) did not significantly affect the outcomes. Benzo[h]chromen-4-one derivative 3j was formed in 86% yield with >99% ee. An array of flavones bearing a substituted aryl group at the C2-position served as suitable reactants in the [1,3]-rearrangement, and the desired quinoxalines (3k–3q) were delivered smoothly in good yield (71–98%) with 81–97% ee. The absolute configuration of product 3k was determined to be S according to an X-ray crystallographic analysis.19 Subsequently, we turned our attention to different allyl substitutions (3r–3y). Noteworthily, the reactions of these substrates are electron-sensitive, and para-halo-substituted cinnamyl groups (3r–3u) undermined the reactivity and enantioselectivity in comparison with para- and meta-methyl substituted ones (3v–3w) or a heteroaromatic ring substituted one (3x). The reaction of phenylbut-2-enyl substituted ether also worked to give the product 3y in medium yield (43%) with 94% ee, accompanied by byproduct flavonol 1a′. In addition, benzyl flavonol ethers with electron-donating substituents underwent [1,3]-rearrangement smoothly, giving the corresponding products 3z and 3aa in the presence of L3-PrPr2/Co(OTf)2 catalyst. Moreover, not only lawsone but also allomaltol-derived allylic ethers performed the rearrangement well, giving important derivatives (3ab and 3ac) with excellent results.
|
a Unless otherwise noted, all reactions were carried out with Co(OTf)2/L3-PrEt3 (1:1, 10 mol%), 1 (0.1 mmol) in DCE (0.1 M) at 50 °C for 24 h; then o-phenylenedianmine (1.5 equiv.) at 30 °C for 15 min. Isolated yield of 3. ee determined by UPC2 analysis.
b 72 h.
c At 40 °C.
d At 30 °C.
e
L3-PrPr2 as the ligand.
|
|---|

|
By switching the flavonol-derived ether 1a into 3-hydroxychromone derivative E-4a in the presence of an L3-PrEt3/Co(OTf)2 complex, the [3,3]-rearrangement product 5a was formed in 78% yield with 45% ee (Table 4, entry 1) instead of [1,3]-rearrangement product 5a′. Further optimizing the reaction conditions showed that the results with a complex of Ni(OTf)2 were superior to those of other classic Lewis acids, such as Sc(OTf)3 or La(OTf)3 (Table 4, entries 1–4). Reinvestigation of the N,N′-dioxide (Table 1) confirmed that L3-TQPr2 gave better enantioselectivity and yield than the ligands bearing other amino acid backbones (Table 4, entries 5–8). When the alkyl chain between the two amine-oxides changed from three carbons (L3-TQPr2) to four carbons (L4-TQPr2), the enantioselectivity increased to 88% ee (entry 9). Finally, the combination of L4-TQPr2 with Ni(NTf2)2 well promoted the [3,3]-rearrangement at 30 °C, affording 2-substituted 3-hydroxychromone 5a in 92% yield, with 92% ee (entry 10).
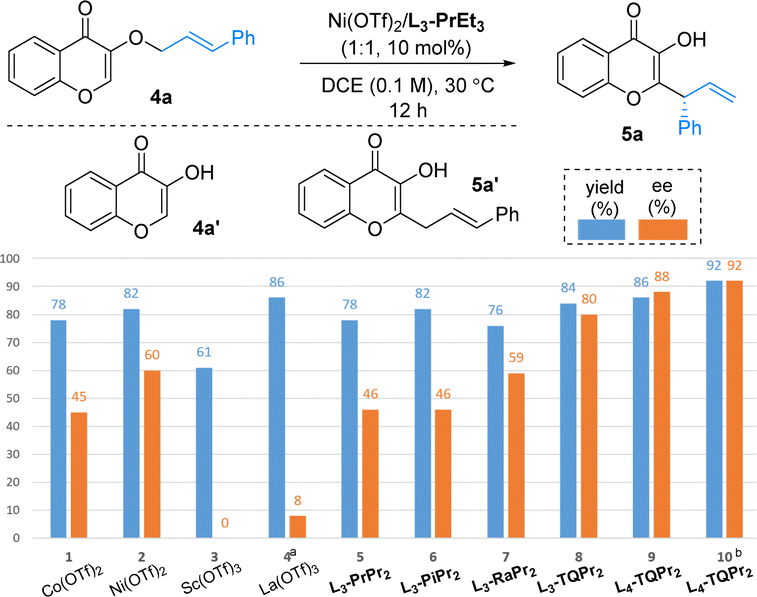
Subsequently, the scope of enantioselective [3,3]-rearrangement was systematically investigated (Table 5). It gave access to a wide range of 2-(1-phenylallyl)-substituted 3-hydroxyl-4H-chromen-4-ones (5b–5h) in good yields (70–92%) with good enantioselectivities (83–92% ee). Unlike [1,3]-rearrangement, the different substituents at the cinnamyl groups had little influence on yield or ee value in [3,3]-rearrangement. Good yields (83–90%) and enantioselectivities (84–87% ee) were obtained when cinnamyl groups were used bearing different substituents at the para- and meta-positions of the aryl group (5i–5m). The ortho-methyl substituted ether 4n reacted smoothly to afford 5n in 85% yield with medium enantioselectivity (68% ee). A 3,4-dichlorophenyl-containing substrate was also compatible to give 5o in 61% yield with 90% ee. We also tested 3-cyclohexylallyl ether 4o, which showed a lower reactivity (47% yield) but good enantioselectivity (95% ee) after a longer reaction time. Furthermore, other important compounds derived from kojic acid (5q), allomaltol (5r) and lawsone (5s) could be efficiently constructed in good yields (64–87%) and enantioselectivities (74–92% ee).

The allyl flavonol ether 1ad which has been studied by Porco's group10 was also tested, and the desired product 3ad was obtained with excellent yield (85%) and enantioselectivity (>99% ee) (Scheme 2a). The two kinds of rearrangement can be easily scaled up, as demonstrated for 1a (3.0 mmol) in Scheme 2b and 4a (4.5 mmol) in Scheme 2c. Other chromone derivatives could be obtained through synthetic elaboration (Scheme 2d). Besides transformation into quinoxaline, a mixture of diketone 2a and ketal 2a′ could also be smoothly converted to imidazole 6 in 98% yield, and to diol 7via selective reduction with NaBH4, as well as 2-phenyl-2-alkyl substituted diketone 8 and its ketal form upon reduction. 1-Phenylpropanyl substituted 3-hydroxychromone 9 could be obtained after reduction by H2 and Pd/C, which was converted to known methyl 2-phenylbutanoate 10 by successive oxidative fragmentation (RuCl3/NaIO4) and esterification reactions. The absolute configuration of 10 was assigned by comparison of the optical rotation with known data;20 thus the absolute configuration of product 5a was assigned as R. Additionally, 2,3-disubstituted 4H-chromen-4-one 12 could be readily obtained via Suzuki coupling, forming the triflate derivative 11.
 | ||
| Scheme 2 The gram-scale experiment and synthetic elaborations. | ||
In order to understand the mechanistic profile of the regioselective rearrangement, we set out control experiments (Scheme 3). First, the effect of E/Z cinnamyl substitution on the performance was investigated. It was found that without a catalyst, Z-dominated 1a (E/Z = 1/5) did not undergo the rearrangement (Scheme 3a condition A). While the same [1,3]-rearrangement E-products, which are the thermodynamically stable isomers, were found from Z-dominated 1a (E/Z = 1/5) with comparable yield and enantioselectivity by the use of a Co(OTf)2 or chiral L3-PrEt3/Co(OTf)2 catalyst (Scheme 3a, conditions B and C). It is notable that the E/Z configuration of the cinnamyl unit changed during the [1,3]-rearrangement, which is consistent with a non-concerted step. In contrast, [3,3]-rearrangement product 5a was observed from Z-dominated 4a (E/Z = 1/7), but with decreased conversion, especially in the presence of a chiral L4-TQPr2/Ni(NTf2)2 catalyst (Scheme 3b, conditions E and F). Z/E selectivity of the recovered reactant revealed the configuration preference for E-4a over Z-4a in the rearrangement, and in the presence of the chiral catalyst Z-4a was fully recovered. This indicates that the [3,3]-rearrangement likely undergoes a concerted pathway, and the cyclic transition states are under the influence of the interaction of the subunits of the reactants, as well as with the chiral catalysts. Next, Hammett plots were carried out to probe into the electronic effect of substitution on the cinnamyl group on the reactivity (Scheme 3c). For the [1,3]-rearrangement reaction, the Hammett plot of log(kX/kH) against substituent constant σp+ for the reaction of para-substituted cinnamyl flavonol ethers 1 gave a straight line with good correlation. The negative slope (ρ = −11.52) strongly suggests that the [1,3]-rearrangement is more likely to proceed through an ion-pair mechanism, and an electron-donating group accelerates the reaction, probably due to stabilizing the allyl cation intermediate. Instead, a relatively smaller slope (ρ = −0.15) was observed in Hammett studies of [3,3]-rearrangement,21 indicting concerted Claisen rearrangement. Furthermore, a crossover reaction was conducted with two different cinnamyl flavonol ethers for [1,3]-rearrangement (Scheme 3d), and no crossover products were detected, supporting a tight pair pathway.
 | ||
| Scheme 3 Control experiments for mechanism study. | ||
DFT calculations were carried out to elucidate the substrate-dependent regioselective [1,3]- and [3,3]-rearrangement. The discussion here is based on the data calculated on the B3LYP-D3/def2-TZVP(SMD)//B3LYP-D3/6-31G(d)∼SDD(Co) level of theory (see the ESI† for more details). Firstly, we performed computations on [1,3]- and [3,3]-rearrangements catalyzed by Ni(OTf)2. For 2-phenyl substituted 1a (Scheme 4a), in the proposed chair-like transition state (PhTS1′), the steric repulsion between 2-aryl substituent and cinnamyl group caused the energy barrier of [3,3]-rearrangement to be much higher. However, a 2-aryl substituent could stabilize the negative charge of enolate PhINT2; the energy barrier for the [1,3]-rearrangement is lower than that of [3,3]-rearrangement (14.8 kcal mol−1vs. 18.2 kcal mol−1). Furthermore, the free energy profile of the enantioselective version in the case of the Ni(II)/L3-PrEt3-catalyzed [1,3]-rearrangement reaction of 1a is shown in Scheme 4c. The energy barrier for the formation of the S-product is 1.6 kcal mol−1 lower than that of the R-product due to the stronger π–π interaction between the Lewis-acid-stabilized enolate and an allyl cation pair intermediate. The expected ee value is 87%, consistent with the experimental results.
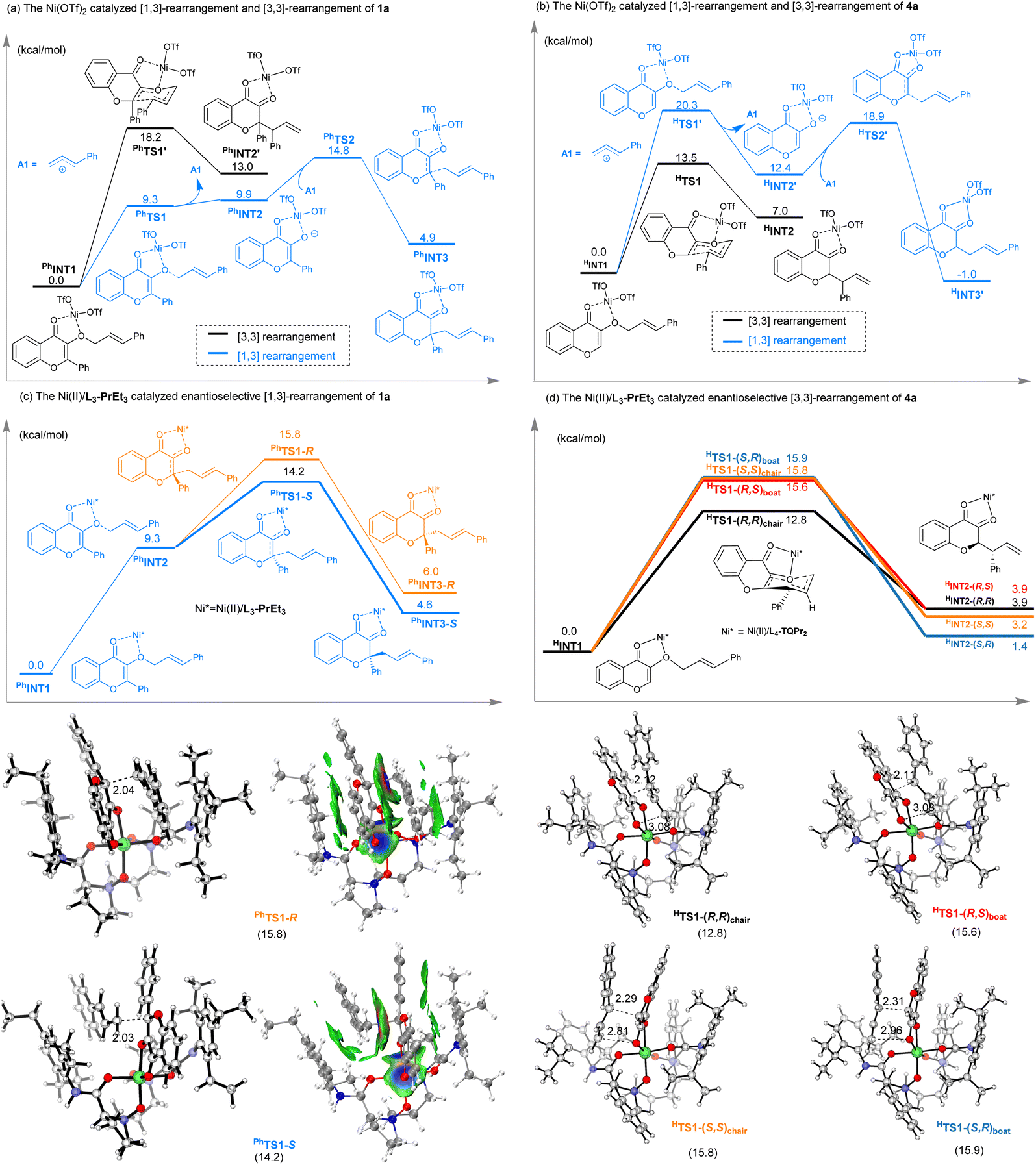 | ||
| Scheme 4 The possible catalytic cycle and asymmetric catalytic modes. | ||
Conversely, for 2-unsubstituted reactant 4a (Scheme 4b), the energy barrier for the [3,3]-rearrangement is lower than that of [1,3]-rearrangement (13.5 kcal mol−1vs. 20.3 kcal mol−1). In the case of the Ni(II)/L4-TQPr2-catalyzed enantioselective [3,3]-rearrangement reaction of 4a, four possible transition states to give the final product with one stereocenter are compared (Scheme 4d). The energy barrier for the formation of the R configuration via a chair-type transition state (PhTS1-(R,R)chair) is 2.8 kcal mol−1 lower than that of the S-configuration-based boat-type transition state (PhTS1-(R,S)boat).
Conclusions
In summary, we have achieved a regioselective asymmetric O-to-C-rearrangement of 3-allyloxy-4H-chromenones and their analogues. The [1,3]- and [3,3]-rearrangements are affected by substitution on the substrates, which resulted in biased steric hindrance and stability of the intermediates. Nonetheless, both processes could be realized in good yield and enantioselectivity after the chiral N,N′-dioxide/metal complex catalysts were adjusted slightly, meeting the requirements of the different mechanisms. The protocol enabled access to a number of 3-hydroxyl pyranone and benzopyranone derivatives. Systematic mechanistic studies and DFT calculation revealed the reasons for the regioselectivity and stereoselectivity, which provided guidance and reference for regioselective asymmetric O-to-C [1,3] and [3,3]-rearrangement of allyl vinyl ether. Further investigations on other types of asymmetric rearrangements are currently underway.Data availability
Further details of the experimental procedure, 1H, 13C{1H} and 19F{1H} NMR, HPLC spectra, X-ray crystallographic data and details of the computational studies are available in the ESI.†Author contributions
Y. L., Q. T., K. X. L. and Q. C. L. preformed the experiments. L. C. N. conducted the DFT calculation. B. Q. Y. reproduced the data. X. M. F. and X. H. L. supervised the project. Y. L. and X. H. L. co-wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We appreciate the National Natural Science Foundation of China (No. U19A2014 and 21921002), and Sichuan University (2020SCUNL204). We thank Dr Yuqiao Zhou (Sichuan University) for the assistance in X-ray analysis.Notes and references
- (a) C. M. Rojas, Molecular Rearrangement in Organic Synthesis, Wiley, Hoboken, 2015 CrossRef; (b) J. P. Zhu, Eur. J. Org Chem., 2019, 1964 Search PubMed.
- Y. B. Liu, X. H. Liu and X. M. Feng, Chem. Sci., 2022, 12290 RSC.
- K. Nonoshita, H. Banno, K. Maruoka and H. Yamamoto, J. Am. Chem. Soc., 1990, 112, 316 CrossRef CAS.
- P. A. Grieco, J. D. Clark and C. T. Jagoe, J. Am. Chem. Soc., 1991, 113, 5488 CrossRef CAS.
- (a) A. Gansäuer, D. Fielenbach and C. Stock, Adv. Synth. Catal., 2002, 344, 845 CrossRef; (b) A. Gansäuer, D. Fielenbach, C. Stock and D. Geich-Gimbel, Adv. Synth. Catal., 2003, 345, 1017 CrossRef.
- C. G. Nasveschuk and T. Rovis, Org. Lett., 2005, 7, 2173 CrossRef CAS.
- For asymmetric 3,3-rearrangement reaction examples: (a) L. Abraham, R. Czerwonka and M. Hiersemann, Angew. Chem., Int. Ed., 2001, 40, 4700 CrossRef CAS; (b) M. Rueping and A. P. Antonchick, Angew. Chem., Int. Ed., 2008, 47, 10090 CrossRef CAS; (c) M. Rueping and A. P. Antonchick, Angew. Chem., 2008, 120, 10244 CrossRef; (d) H. Ren and W. D. Wulff, J. Am. Chem. Soc., 2011, 133, 5656 CrossRef CAS . For asymmetric 1,3-rearrangement reaction examples:; (e) M. Terada and Y. Toda, J. Am. Chem. Soc., 2009, 131, 6354 CrossRef CAS; (f) M. Terada, T. Komuro, Y. Toda and T. Korenaga, J. Am. Chem. Soc., 2014, 136, 7044 CrossRef CAS; (g) A. B. Gade, P. N. Bagle, P. S. Shinde, V. Bhardwaj, S. Banerjee, A. Chande and N. T. Patil, Angew. Chem., Int. Ed., 2018, 57, 5735 CrossRef CAS.
- Y. L. Zhang and Y. S. Chen, Org. Chem. Front., 2022, 9, 4744 RSC.
- T. Rodrigues, D. Reker, P. Schneider and G. Schneider, Nat. Chem., 2016, 8, 531 CrossRef CAS.
- C. Qi, Y. Xiong, V. Eschenbrenner-Lux, H. Cong and J. A. Porco Jr, J. Am. Chem. Soc., 2016, 138, 798 CrossRef CAS.
- X. Sheng, X. Y. Jia, F. Tang, Y. Wang and A. J. Hou, Tetrahedron, 2017, 73, 3485 CrossRef CAS.
- P. A. Wender, N. D'Angelo, V. I. Elitzin, M. Ernst, E. E. Jackson-Ugueto, J. A. Kowalski, S. McKendry, M. Rehfeuter, R. Sun and D. Voigtlaender, Org. Lett., 2007, 9, 1829 CrossRef CAS.
- J. Marié, Y. Xiong, K. Min, A. R. Yeager, T. Taniguchi, N. Berova, S. E. Schaus and J. A. Porco Jr, J. Org. Chem., 2010, 75, 4584 CrossRef.
- (a) J. Kaeobamrung, J. Mahatthananchai, P. Zheng and J. W. Bode, J. Am. Chem. Soc., 2010, 132, 8810 CrossRef CAS; (b) J. Mahatthananchai, P. Zheng and J. W. Bode, Angew. Chem., Int. Ed., 2011, 50, 1673 CrossRef CAS; (c) J. Mahatthananchai, J. Kaeobamrung and J. W. Bode, ACS Catal., 2012, 2, 494 CrossRef CAS; (d) J. Mahatthananchai and J. W. Bode, Acc. Chem. Res., 2014, 47, 696 CrossRef CAS.
- K. Dzieszkowski, M. Słotwiński, K. Rafińska, T. M. Muzioł and Z. Rafiński, Chem. Commun., 2021, 57, 9999 RSC.
- L. Yao and K. Ishihara, Chem. Sci., 2019, 10, 2259 RSC.
- For related examples: (a) Y. B. Liu, H. P. Hu, H. F. Zheng, Y. Xia, X. H. Liu, L. L. Lin and X. M. Feng, Angew. Chem., Int. Ed., 2014, 53, 11579 CrossRef CAS; (b) Y. B. Liu, X. H. Liu, H. P. Hu, J. Guo, Y. Xia, L. L. Lin and X. M. Feng, Angew. Chem., Int. Ed., 2016, 55, 4054 CrossRef CAS; (c) J. Li, L. L. Lin, B. W. Hu, P. F. Zhou, T. Y. Huang, X. H. Liu and X. M. Feng, Angew. Chem., Int. Ed., 2017, 56, 885 CrossRef CAS; (d) H. F. Zheng, Y. Wang, C. R. Xu, X. Xu, L. L. Lin, X. H. Liu and X. M. Feng, Nat. Commun., 2018, 9, 1968 CrossRef; (e) Y. S. Chen, S. X. Dong, X. Xu, X. H. Liu and X. M. Feng, Angew. Chem., Int. Ed., 2018, 57, 16554 CrossRef CAS; (f) L. Wang, P. F. Zhou, Q. C. Lin, S. X. Dong, X. H. Liu and X. M. Feng, Chem. Sci., 2020, 11, 10101 RSC; (g) X. B. Lin, Z. Tan, W. K. Yang, W. Yang, X. H. Liu and X. M. Feng, CCS Chem., 2021, 3, 1423 CrossRef CAS; (h) L. Wang, Y. Q. Zhou, Z. S. Su, F. C. Zhang, W. D. Cao, X. H. Liu and X. M. Feng, Angew. Chem., Int. Ed., 2022, 61, e202211785 CrossRef CAS; (i) Y. Liu, Y. S. Chen, A. Y. Yihuo, Y. Zhou, X. H. Liu, L. L. Lin and X. M. Feng, ACS Catal., 2022, 12, 1784 CrossRef CAS; (j) L. F. Hu, J. Z. Li, Y. Y. Zhang, X. M. Feng and X. H. Liu, Chem. Sci., 2022, 13, 4103 RSC . For reviews on chiral N,N′-dioxides:; (k) X. H. Liu, L. L. Lin and X. M. Feng, Acc. Chem. Res., 2011, 44, 574 CrossRef CAS; (l) X. H. Liu, L. L. Lin and X. M. Feng, Org. Chem. Front., 2014, 1, 298 RSC; (m) X. H. Liu, H. F. Zheng, Y. Xia, L. L. Lin and X. M. Feng, Acc. Chem. Res., 2017, 50, 2621 CrossRef CAS; (n) X. H. Liu, S. X. Dong, L. L. Lin and X. M. Feng, Chin. J. Chem., 2018, 36, 791 CrossRef CAS; (o) Z. Wang, X. H. Liu and X. M. Feng, Aldrichimica Acta, 2020, 53, 3 CAS; (p) M. Y. Wang and W. Li, Chin. J. Chem., 2021, 39, 969 CrossRef CAS.
- The equilibrium between 2a and 2a′, for detail see ESI†
 .
. - CCDC 2204539 (for 3k) contain the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre.†.
- (a) G. Sarakinos and E. J. Corey, Org. Lett., 1999, 11, 1741 CrossRef; (b) Y. Zhang, Z. B. Han, F. Y. Li, K. L. Ding and A. Zhang, Chem. Commun., 2010, 46, 156 RSC; (c) J. Y. Mao, F. P. Liu, M. Wang, L. Wu, B. Zheng, S. Z. Liu, J. C. Zhong, Q. H. Bian and W. J. Patrick, J. Am. Chem. Soc., 2014, 136, 17662 CrossRef CAS; (d) F. Malmedy and T. Wirth, Chem.–Eur. J., 2016, 22, 16072 CrossRef CAS.
- N. F. O'Rourke and J. E. Wulff, Org. Biomol. Chem., 2014, 12, 1292 RSC.

Footnotes |
| † CCDC 2204539. For crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc02201g |
| ‡ Both authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |