
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA free-radical design featuring an intramolecular migration for a synthetically versatile alkyl–(hetero)arylation of simple olefins†
Dylan J.
Babcock
,
Andrew J.
Wolfram
,
Jaxon L.
Barney
,
Santino M.
Servagno
,
Ayush
Sharma
and
Eric D.
Nacsa
 *
*
The Pennsylvania State University, Department of Chemistry, University Park, PA 16802, USA. E-mail: nacsa@psu.edu
First published on 2nd February 2024
Abstract
A free-radical approach has enabled the development of a synthetically versatile alkyl–(hetero)arylation of olefins. Alkyl and (hetero)aryl groups were added concurrently to a full suite of mono- to tetrasubstituted simple alkenes (i.e., without requiring directing or electronically activating groups) for the first time. Key advances also included the introduction of synthetically diversifiable alkyl groups featuring different degrees of substitution, good diastereocontrol in both cyclic and acyclic settings, the addition of biologically valuable heteroarenes featuring Lewis basic nitrogen atoms as well as simple benzenes, and the generation of either tertiary or quaternary benzylic centers. The synthetic potential of this transformation was demonstrated by leveraging it as the key step in a concise synthesis of oliceridine, a new painkiller that received FDA approval in 2020.
Introduction
Transformations that form multiple C–C bonds can significantly expedite the synthesis of valuable organic compounds such as medicines, agrochemicals, materials, fragrances, and food products.1–17 Olefins are attractive substrates since they are ubiquitous in both chemical feedstocks and complex natural products, they can be installed in a variety of settings,18 and they are inherently well-suited to vicinal difunctionalization by spanning adjacent carbons. Intermolecular difunctionalizations forming two C–C bonds have thus received significant interest over the past 10–15 years.3–17,19–32Given the importance of aromatic moieties in bioactive compounds and the continued underrepresentation of sp3 content in pharmaceuticals,33,34 alkyl–arylations of olefins are critical tools in the synthetic arsenal. Transition-metal-catalyzed conjunctive couplings between an olefin, an aromatic partner, and an aliphatic partner have received the most attention to this end.35–47 Productive olefins, however, have overwhelmingly required either an activating group (conjugated π-bond or heteroatom) or a Lewis basic directing group (Fig. 1a, bottom) to promote olefin–catalyst binding, which restricts the alkenes that can be valorized and the products that can be obtained. To unlock the full potential of alkyl–arylations, they must equally engage simple olefins (Fig. 1a, top).
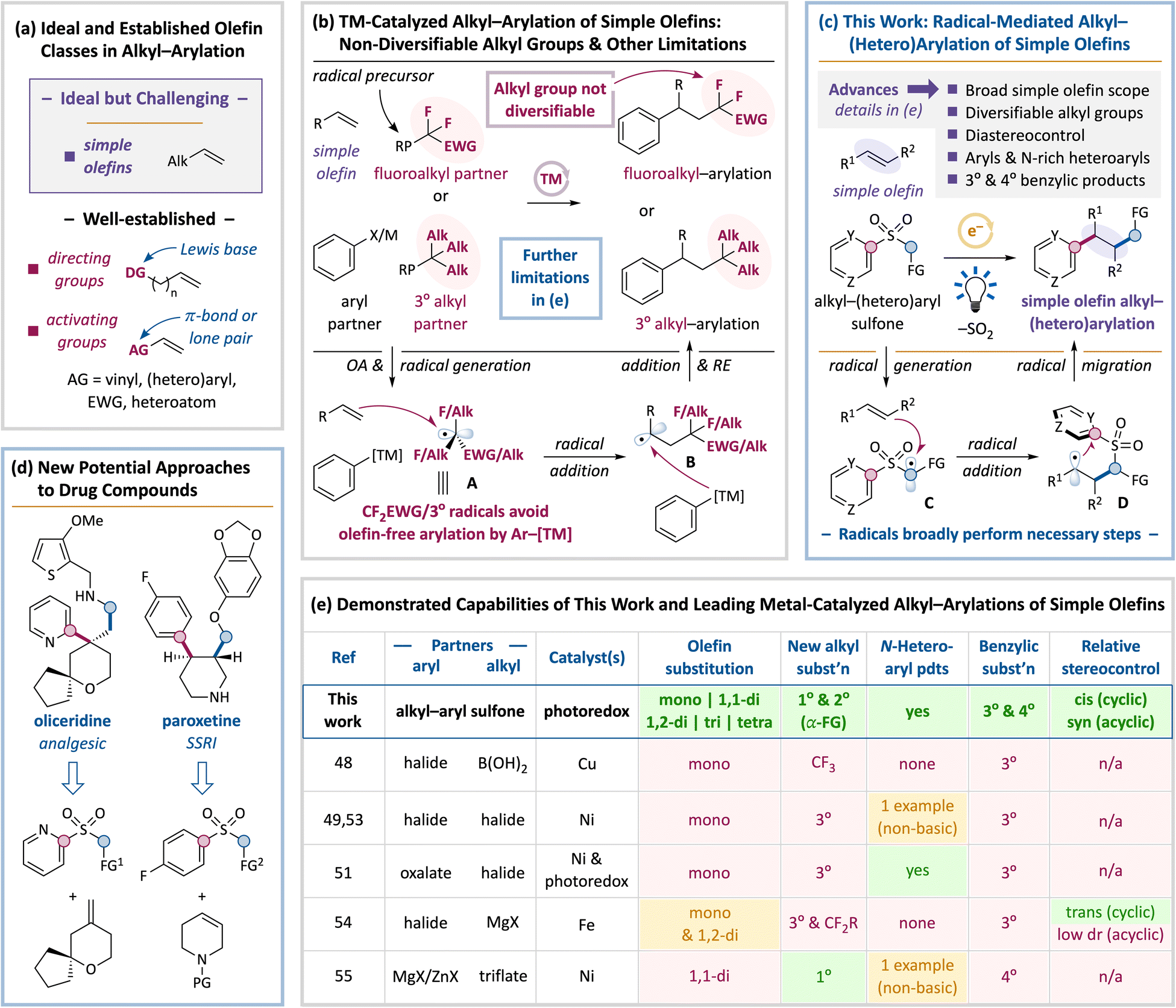 | ||
| Fig. 1 Comparison of leading transition-metal (TM)-catalyzed olefin alkyl–arylations to this work. (a) Simple olefins are ideal for alkyl–arylation but have proven challenging to engage. Most productive olefins feature directing or activating groups. (b) TM-catalyzed alkyl–arylations of simple olefins activate the alkyl partner as a radical and employ non-diversifiable tertiary or fluoroalkyl groups to avoid metal-mediated coupling of this radical to the aryl partner. (c) This work employs alkyl–aryl sulfones under free-radical conditions to deliver alkyl and aryl groups to olefins, which affords several synthetic advances. (d) Representative synthetic approaches to bioactive targets enabled by this work. (e) Comparison of key synthetic capabilities of this work to TM-catalyzed alkyl–arylations of simple olefins. | ||
Some metal-catalyzed alkyl–arylations of simple alkenes have thus been developed.48–55 With one exception,55 they all engage the alkyl partner as an alkyl radical (A, Fig. 1b), leveraging the better propensity of these radicals than of transition metals to add to simple alkenes. This step generates a new alkyl radical (B) that binds to the metal catalyst, forming the desired product after reductive elimination. To avoid the counterproductive olefin-free arylation of the alkyl partner, however, these strategies have only succeeded with tertiary or fluoroalkyl groups that are themselves reluctant to undergo metal-mediated arylation.56–58 These fully substituted alkyl groups are synthetically non-diversifiable, which limits the versatility of these alkyl–arylations (the non-radical method55 adds primary alkyl groups, but only restricted 1,1-disubstituted simple alkenes were productive, and its use of alkylmetal reagents compromises its functional-group tolerance).
Further key synthetic challenges have pervaded these alkyl–arylations of simple olefins48–55 (Fig. 1e). Beyond (1) the addition of non-diversifiable tertiary or fluoroalkyl groups that features only a single exception,55 (2) these systems are mostly restricted to monosubstituted simple alkenes. The only two exceptions are the aforementioned alkyl–arylation that only engages 1,1-disubstituted simple alkenes with alkylmetal reagents,55 as well as an Fe-catalyzed protocol that can employ mono- and 1,2-disubstituted congeners and that relies on aryl Grignard reagents.54 Moreover, only single, disparate reports describe (3) stereocontrol (affording trans-alkyl–aryl products for cyclic olefin substrates and proving unselective with an acyclic congener),54 (4) the introduction of biologically valuable N-heteroarenes,51,59 and (5) the generation of quaternary benzylic products55 (with this exception generating only quaternary products).
Design plan
We thus hypothesized that a completely free-radical approach60,61 could underpin a more versatile platform. As shown in Fig. 1c, we envisioned that simple alkyl–(hetero)aryl sulfones,62,63 straightforwardly prepared in 1–2 steps64,65 from alkyl halides and (hetero)aryl sulfinates or thiols, could add their alkyl and (hetero)aryl groups across an olefin under photoredox activation66–68 and extrude SO2. Mechanistically, electrophilic sulfone-derived alkyl radical C would add to the olefin, generating the desired C(sp3)–alkyl bond and new alkyl radical D. The latter intermediate would be well-poised for a radical migration (radical Smiles–Truce rearrangement) and desulfonylation to forge the C(sp3)–aryl bond.69–88 This design should enable several advances. (1) Desulfonylation from the alkyl fragment and the use of a second, simple functional group at this position should enable the introduction of synthetically diversifiable alkyl groups with multiple substitution patterns. (2) A wide scope of simple olefins should add to electrophilic radical C.89 (3) The cyclic intermediates generated by the intramolecular radical-mediated migration should provide opportunities to confer stereocontrol.77,90,91 (4) In addition to simple benzenes, the metal-free migratory C(sp3)–(hetero)aryl bond-forming step should also accommodate biologically privileged92 N-rich heteroaryl groups that often inhibit transition-metal catalysts. Finally, (5) elongated open-shell transition states should facilitate the generation of either tertiary or quaternary benzylic centers,93 which is also challenging for transition metals.New synthetic approaches to bioactive molecules potentially empowered by this approach are illustrated in Fig. 1d. The colored, bold bonds would be forged by the proposed alkyl–(hetero)arylation, and standard functional-group manipulations would complete the peripheries.
The detailed mechanistic design for the alkyl–(hetero)arylation of olefins (1) with alkyl–(hetero)aryl sulfones (2) to afford products 3 is illustrated in Fig. 2. Deprotonation of 2 (pKa[PhSO2(COPh)CH2] = 11.4 in DMSO)94 and single-electron oxidation of the resulting anion (Ered1/2 [PhSO2(COPh)CH˙/PhSO2(COPh)CH−] = +0.78 V vs. SCE in DMSO)94 by an excited-state photoredox catalyst (4, Ered1/2[*Ru(bpy)32+ (5)/Ru(bpy)3+ (6)] = +0.77 V vs. SCE in MeCN)95,96 would generate alkyl radical 7. Addition of simple olefin 1 to this electrophilic radical89 would form the first C–C bond and new alkyl radical 8. The latter intermediate would be well-poised for a [1,4]-(hetero)aryl migration, forging the second desired C–C bond via intermediate 9 and extruding SO2. Resulting electron-poor alkyl radical 10 (Ered1/2 [(EtCO)(Me)CH˙/(EtCO)(Me)CH−] = −0.55 V vs. SCE in DMSO)97 would react with reduced, ground-state photoredox catalyst (6, Ered1/2[Ru(bpy)32+ (4)/Ru(bpy)3+ (6)] = −1.33 V vs. SCE in DMSO)95 to generate an anion such as an enolate (pKa[(EtCO)(Me)CH] = 27.1 in DMSO),97 protonation of which would afford the desired alkyl–(hetero)aryl product.
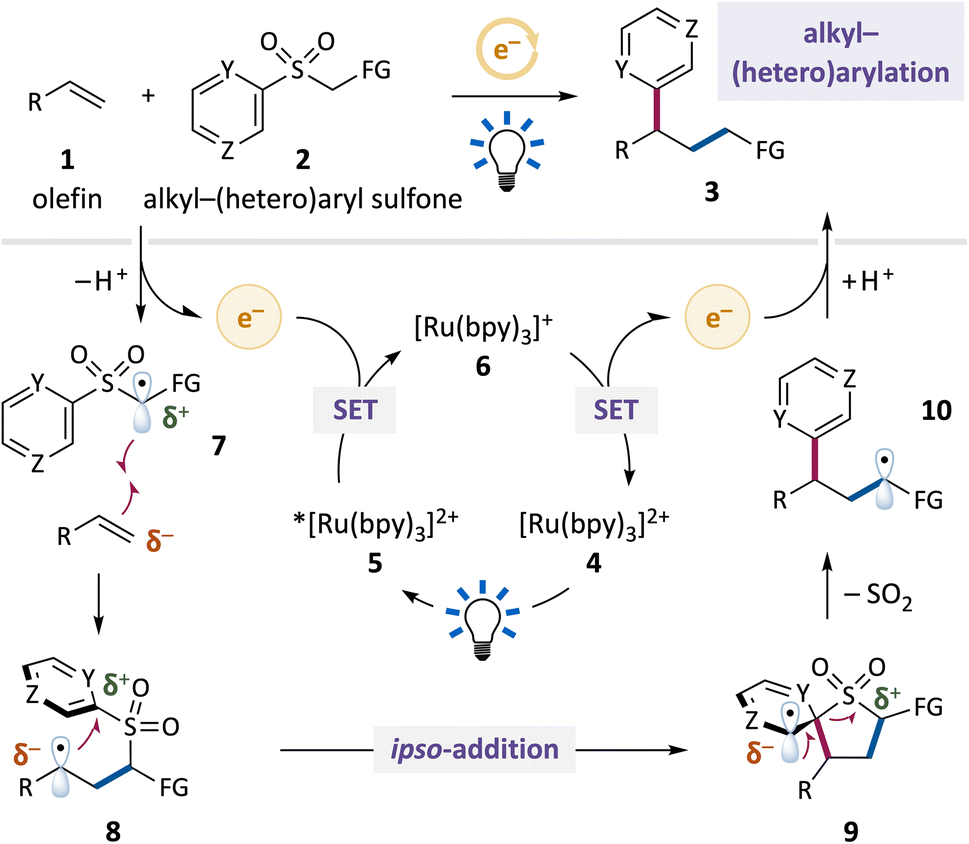 | ||
| Fig. 2 Mechanistic design of radical-mediated olefin alkyl–(hetero)arylation. See text for details. | ||
Results
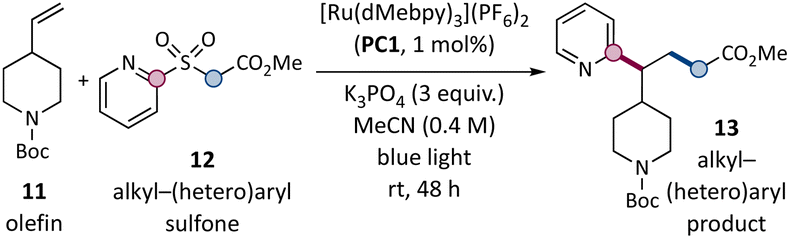
Model studies between simple olefin 11 and alkyl–(hetero)aryl sulfone 12 to afford 13 identified optimal conditions employing commercially available [Ru(dMebpy)3](PF6)2 (PC1) as the photocatalyst and K3PO4 as the base in MeCN (Table 1). Using a modest excess of the olefin (3 equiv.), the desired product was obtained in 81% yield after 48 h at ambient temperature (entry 1). [Ru(bpy)3](PF6)2 gave a slightly lower yield (entry 2, 71% yield). Common Ir-based photoredox catalysts (up to 74% yield, entries 3–6) were also competent, as long as they were not highly oxidizing (7% yield, entry 4) or reducing (23% yield, entry 6). 4CzIPN could also be used in this role (entry 7, 73% yield), enabling a fully transition-metal-free protocol. A selection of alternate inorganic or organic bases were unsuccessful (entries 8–10, 0–8% yields). A minimal decrease in efficiency occurred when using a smaller excess of olefin (entry 11, 2 equiv., 74% yield), and 51% yield was obtained at equimolar stoichiometry (entry 12). Other olefins, however, reacted efficiently in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometries (see synthesis of oliceridine below). Yields were unaffected when the mixture was not degassed or when the reaction was performed open to air (entries 13–14, 79–80% yields). The photoredox catalyst, light, and base were all essential (entries 15–17, 0% yield).
:1 stoichiometries (see synthesis of oliceridine below). Yields were unaffected when the mixture was not degassed or when the reaction was performed open to air (entries 13–14, 79–80% yields). The photoredox catalyst, light, and base were all essential (entries 15–17, 0% yield).
|
|
||
|---|---|---|
| Entry | Derivation from standard conditions | Yield (%) |
| a Olefin 11 (3 equiv.), sulfone 12 (0.4 mmol, 1 equiv.), K3PO4 (3 equiv.), and [Ru(dMebpy)3](PF6)2 (PC1, 1 mol%) were irradiated with blue light (440 nm) in MeCN (0.4 M in 12) at rt for 48 h with variations as noted. NMR yields. See ESI for detailed procedures. | ||
| 1 | None | 81 |
| 2 | [Ru(bpy)3](PF6)2 (4) photocatalyst | 71 |
| 3 | [Ir(dFCF3ppy)2(dtbbpy)]PF6 photocatalyst | 73 |
| 4 | [Ir(dFCF3ppy)2(dCF3bpy)]PF6 photocatalyst | 7 |
| 5 | [Ir(ppy)2(dtbbpy)]PF6 photocatalyst | 74 |
| 6 | fac-[Ir(ppy)]3 photocatalyst | 23 |
| 7 | 4CzIPN photocatalyst | 73 |
| 8 | K2CO3 base | 0 |
| 9 | K2HPO4 base | 0 |
| 10 | DBU base | 8 |
| 11 | 2 equiv. olefin | 74 |
| 12 | 1 equiv. olefin | 51 |
| 13 | No degassing | 78 |
| 14 | Open to air | 79 |
| 15 | No photocatalyst | 0 |
| 16 | No light | 0 |
| 17 | No base | 0 |
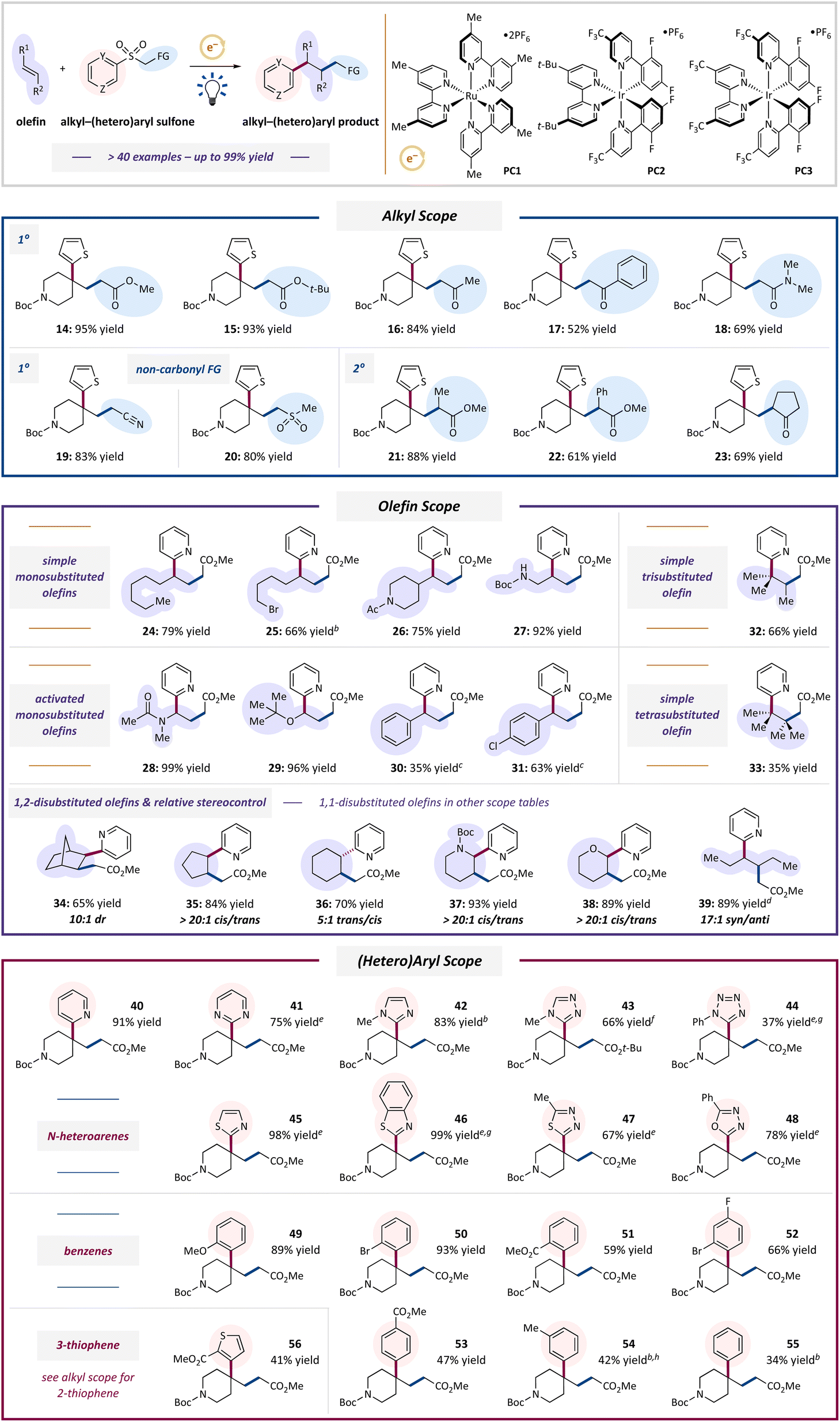
The scope of this transformation is detailed in Table 2. We sought to demonstrate clearly that this system could simultaneously address all the above-mentioned challenges: (1) using diversifiable and differently substituted alkyl groups, (2) engaging simple alkenes with any degree of substitution, (3) affording diastereocontrol, (4) adding benzenes and N-heteroarenes, and (5) generating tertiary and quaternary benzylic centers.
|
a Standard conditions follow Table 1, entry 1. Yields of isolated products. See ESI for experimental procedures.
b NMR yield.
c Olefin (1 equiv.) and green light (510–575 nm).
d Using trans-3-hexene. With cis-3-hexene, 39 was obtained in 88% yield and 15:1 syn/anti.
e Catalyst PC2 (1 mol%).
f Catalyst PC3 (1 mol%).
g DMSO as solvent or cosolvent.
h Mixture of regioisomers, see ESI.
|
|---|
|
Addressing challenge (1), a range of primary alkyl groups featuring carbonyls (esters, ketones, and an amide), nitrile, or sulfonyl functionalities were added to a simple olefin, accompanied by a 2-thienyl unit (14–20, 52–95% yields). Secondary alkyl groups were also added without complication, affording tertiary alkyl products 21–23 in 61–88% yields.
Addressing challenge (2), a full suite of olefin substitution patterns was tolerated. Monosubstituted olefins reacted well, including 1-octene, examples bearing an alkyl bromide or amide, and a Boc-protected allylic amine (24–27, 66–92% yields). Electron-rich olefins including an enamide and an enol ether reacted very efficiently, affording products 28–29 in 96–99% yields. Styrene (product 30, 35% yield) and p-chlorostyrene (product 31, 63% yield) were also viable alkenes. Notably, products 32 and 33 represented the first successful alkyl–arylations of simple tri- and tetrasubstituted olefins. Electron-deficient olefins such as acrylates were unproductive.
Next, 1,2-disubstituted alkenes underwent alkyl–(hetero)arylation diastereoselectively, addressing challenge (3). Products were obtained in good yields and syn-selectivities using rigid norbornene (34, 65% yield, 10:1 dr) and five-membered cyclopentene (35, 84% yield, >20:1 cis/trans), presumably because the putative cis-fused intermediates in these systems are the most-stable diastereomers.98,99 Six-membered cyclohexane, piperidine, and tetrahydropyran products 36–38 were also formed efficiently (70–93% yields). Interestingly, 6-membered heterocyclic alkenes underwent syn-alkyl–arylation in high distereoselectivity (>20:1 cis/trans for 37 and 38), whereas trans-selectivity was observed when using cyclohexene (36, 5:1 trans/cis). Lastly, internal, acyclic trans-3-hexene afforded 39 in 89% yield and 17:1 syn-selectivity (cis-3-hexene gave a nearly identical outcome: 88% yield, 15:1 syn/anti), representing the first stereoselective alkyl–arylation of a simple acyclic olefin. The stereochemical convergence observed for 39 likely arises from equilibration between alkyl-radical rotamers generated by addition of the olefin to the initial, electrophilic alkyl radical, but before (hetero)aryl migration (e.g., 8 in Fig. 2). This equilibrium is unaffected by the geometry of the olefin substrate.77 A preliminary explanation of all these stereochemical outcomes is included in the ESI.†
Addressing challenge (4), a range of useful (hetero)aryl groups was competent in this transformation. N-Heteroaromatic motifs with different ring sizes and even multiple Lewis basic nitrogen atoms reacted well, affording products with pyridine, pyrimidine, imidazole, triazole, tetrazole, thiazole, benzothiazole, thiadiazole, and oxadiazole groups (40–48, 37–99% yields, only 44 was below 66% yield). Benzene derivatives were also reliably obtained. Ortho-methoxy, bromo, and carbomethoxy substituents, as well the disubstituted o-bromo-p-fluoro pattern on the new phenyl ring gave products 49–52 in 59–96% yields. Substituents were also tolerated at the para- (p-CO2Me, 53, 47% yield) and meta- (m-Me, 54, 42% yield) positions. Unsubstituted phenyl product 55 (34% yield) and 3-thienyl product 56 (41% yield) were also generated in modest efficiencies.
Throughout these scope studies that afforded 42 products, 14 tertiary benzylic products and 28 quaternary products were obtained, successfully addressing challenge (5).
We then sought to demonstrate the utility of this alkyl–(hetero)arylation in a new synthesis of oliceridine, a novel painkiller approved by the FDA in 2020 (Scheme 1).100–103 To this end, olefin 57 (prepared in 3 steps from 3-buten-1-ol and cyclopentanone) underwent alkyl–(hetero)arylation with sulfone 12 (prepared in 2 steps from 2-mercaptopyridine and methyl bromoacetate). Using ideal 1:1 stoichiometry, alkyl–arylation product 58 was isolated in 95% yield. The methyl ester was then converted to protected amine 59 by hydrolysis and a modified Curtius rearrangement (83% yield over 2 steps), and oliceridine (61) was ultimately obtained by amine deprotection and reductive amination with aldehyde 60 (53% yield from 59; 44% yield from 58). Critically, the alkyl–(hetero)arylation of simple olefin 57 concurrently added a primary, synthetically diversifiable alkyl group and a Lewis basic 2-pyridyl group. Achieving either of these synthetic outcomes on their own has proven challenging in alkyl–arylations of simple olefins, and no previously reported systems have achieved them together.
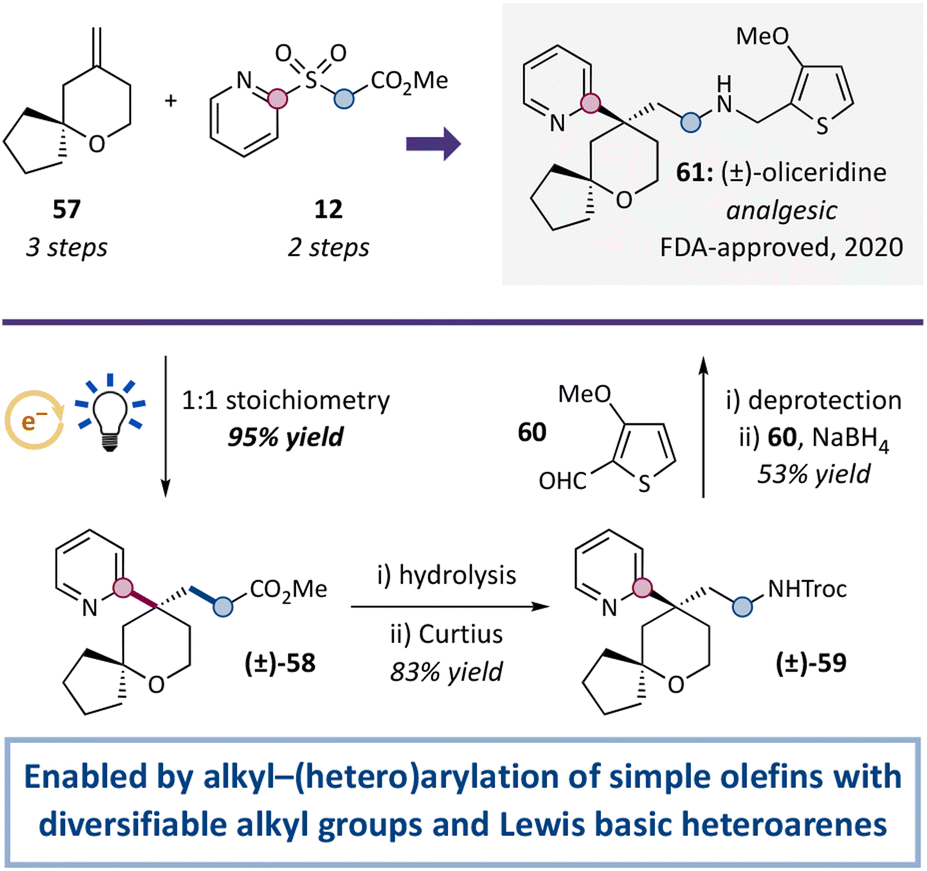 | ||
| Scheme 1 Concise synthesis of (±)-oliceridine featuring a key olefin alkyl–(hetero)arylation. Reaction of olefin 57 (1 equiv.) with alkyl–aryl sulfone 12 (1 equiv.) served as a key step in a new synthesis of (±)-oliceridine (61). See ESI† for detailed procedures. | ||
Since we proposed that many of this method's synthetic advantages result from its free-radical mechanism, we performed preliminary experiments to assess whether open-shell intermediates are indeed generated in this system. First, the formation of representative product 63 was completely shut down when a radical inhibitor (TEMPO, 3 equiv.) was added to the reaction (compared to 78% yield without TEMPO, Scheme 2a). Second, when vinylcyclopropane (64) was used as the olefin, none of desired product 65 was obtained. Instead, byproduct 68 was formed, which could arise from ring-opening of radical intermediate 66 (Scheme 2b). Only minimal consumption of the reactants occurred in this case, possibly because the formation of 68 from ring-opened radical 67 may not efficiently close the photoredox catalytic cycle, thereby deactivating the catalyst. Together, these outcomes are best explained if the alkyl–(hetero)arylation proceeded through a radical manifold. Lastly, we measured the quantum efficiency throughout the formation of product 63 (Scheme 2c). The relatively low value (2.7% quantum yield) suggests that a radical-chain mechanism is unlikely.
 | ||
| Scheme 2 Preliminary mechanistic experiments were consistent with the proposed radical mechanism (Fig. 2). (a) Addition of a persistent radical TEMPO completely inhibited the reaction. (b) Using vinylcyclopropane (64) as the olefin afforded only ring-opened byproduct 68 and none of desired product 65. (c) A low quantum yield (2.7%) was measured, which disfavors a chain mechanism. See ESI† for detailed procedures. | ||
Finally, fluorescence-quenching experiments confirmed that the excited-state photocatalyst activated the conjugate base of the sulfone as proposed in Fig. 2 (see ESI†).
Conclusions
A free-radical approach was leveraged to develop a synthetically versatile alkyl–(hetero)arylation of olefins. This transformation engaged a complete range of simple olefins, from mono- to tetrasubstituted. Further key outcomes included the use of synthetically diversifiable alkyl groups with different degrees of substitution, good stereocontrol in cyclic and acyclic systems, the introduction of heteroaryl groups with Lewis basic nitrogen atoms in addition to simple benzenes, and the efficient formation of either tertiary or quaternary benzylic centers. We are confident that a further suite of synthetically empowering transformations will be enabled by strategies featuring radical-mediated migrations.Data availability
The data supporting this article have been uploaded as part of the ESI.†Author contributions
D. J. B., A. J. W., J. L. B., S. M. S., and A. S. performed experiments. All authors analyzed data. D. J. B. and E. D. N. designed experiments. E. D. N. conceived of the project.Conflicts of interest
There are no conflicts to declare.Note added after first publication
This article replaces the version published on 2nd February 2024 which contained errors in Fig. 1a and 2. The RSC apologises for any confusion.Acknowledgements
The authors are grateful for financial support from the Pennsylvania State University Eberly College of Science (ECoS). S. M. S. was supported by a National Science Foundation REU Program (CHE-250927), and A. S. thanks ECoS for an Erickson Discovery Grant. David Iwig and the Booker Group (Penn State University) are thanked extensively for obtaining HRMS data. Christy George (Penn State University) provided support in the acquisition of NMR data.Notes and references
- Comprehensive Organic Synthesis, ed. P. Knochel, Elsevier, Amsterdam, 2nd edn, 2014 Search PubMed.
- J. Zhu, Q. Wang and M. Wang, Multicomponent Reactions in Organic Synthesis, Wiley-VCH, 2014 Search PubMed.
- M. J. C. M. Hulce, Tandem Vicinal Difunctionalization: β-Addition to α,β-Unsaturated Carbonyl Substrates Followed by α-Functionalization, Org. React., 1990, 38, 225–653 Search PubMed.
- R. I. McDonald, G. Liu and S. S. Stahl, Palladium(II)-catalyzed alkene functionalization via nucleopalladation: stereochemical pathways and enantioselective catalytic applications, Chem. Rev., 2011, 111, 2981–3019 CrossRef CAS PubMed.
- H. Egami and M. Sodeoka, Trifluoromethylation of alkenes with concomitant introduction of additional functional groups, Angew. Chem., Int. Ed., 2014, 53, 8294–8308 CrossRef CAS PubMed.
- G. Yin, X. Mu and G. Liu, Palladium(II)-Catalyzed Oxidative Difunctionalization of Alkenes: Bond Forming at a High-Valent Palladium Center, Acc. Chem. Res., 2016, 49, 2413–2423 CrossRef CAS PubMed.
- X.-W. Lan, N.-X. Wang and Y. Xing, Recent Advances in Radical Difunctionalization of Simple Alkenes, Eur. J. Org Chem., 2017, 2017, 5821–5851 CrossRef CAS.
- R. K. Dhungana, S. Kc, P. Basnet and R. Giri, Transition Metal-Catalyzed Dicarbofunctionalization of Unactivated Olefins, Chem. Rec., 2018, 18, 1314–1340 CrossRef CAS PubMed.
- J. Derosa, V. T. Tran, V. A. van der Puyl and K. M. Engle, Carbon–Carbon π Bonds as Conjunctive Reagents in Cross-Coupling, Aldrichimica Acta, 2018, 51, 21–32 Search PubMed.
- R. Giri and S. Kc, Strategies toward Dicarbofunctionalization of Unactivated Olefins by Combined Heck Carbometalation and Cross-Coupling, J. Org. Chem., 2018, 83, 3013–3022 CrossRef CAS PubMed.
- J. S. Zhang, L. Liu, T. Chen and L. B. Han, Transition-Metal-Catalyzed Three-Component Difunctionalizations of Alkenes, Chem.–Asian J., 2018, 13, 2277–2291 CrossRef CAS PubMed.
- J. Derosa, O. Apolinar, T. Kang, V. T. Tran and K. M. Engle, Recent developments in nickel-catalyzed intermolecular dicarbofunctionalization of alkenes, Chem. Sci., 2020, 11, 4287–4296 RSC.
- X. Qi and T. Diao, Nickel-Catalyzed Dicarbofunctionalization of Alkenes, ACS Catal., 2020, 10, 8542–8556 CrossRef CAS PubMed.
- H. Yao, W. Hu and W. Zhang, Difunctionalization of Alkenes and Alkynes via Intermolecular Radical and Nucleophilic Additions, Molecules, 2020, 26, 105 CrossRef PubMed.
- L. M. Wickham and R. Giri, Transition Metal (Ni, Cu, Pd)-Catalyzed Alkene Dicarbofunctionalization Reactions, Acc. Chem. Res., 2021, 54, 3415–3437 CrossRef CAS PubMed.
- S. Zhu, X. Zhao, H. Li and L. Chu, Catalytic three-component dicarbofunctionalization reactions involving radical capture by nickel, Chem. Soc. Rev., 2021, 50, 10836–10856 RSC.
- P. Gao, Y. J. Niu, F. Yang, L. N. Guo and X. H. Duan, Three-component 1,2-dicarbofunctionalization of alkenes involving alkyl radicals, Chem. Commun., 2022, 58, 730–746 RSC.
- M. Beller, J. Seayad, A. Tillack and H. Jiao, Catalytic Markovnikov and anti-Markovnikov functionalization of alkenes and alkynes: recent developments and trends, Angew. Chem., Int. Ed., 2004, 43, 3368–3398 CrossRef CAS PubMed.
- L. Liao, R. Jana, K. B. Urkalan and M. S. Sigman, A palladium-catalyzed three-component cross-coupling of conjugated dienes or terminal alkenes with vinyl triflates and boronic acids, J. Am. Chem. Soc., 2011, 133, 5784–5787 CrossRef CAS PubMed.
- B. J. Stokes, L. Liao, A. M. de Andrade, Q. Wang and M. S. Sigman, A palladium-catalyzed three-component-coupling strategy for the differential vicinal diarylation of terminal 1,3-dienes, Org. Lett., 2014, 16, 4666–4669 CrossRef CAS PubMed.
- X. Wu, H. C. Lin, M. L. Li, L. L. Li, Z. Y. Han and L. Z. Gong, Enantioselective 1,2-Difunctionalization of Dienes Enabled by Chiral Palladium Complex-Catalyzed Cascade Arylation/Allylic Alkylation Reaction, J. Am. Chem. Soc., 2015, 137, 13476–13479 CrossRef CAS PubMed.
- Z. Liu, T. Zeng, K. S. Yang and K. M. Engle, beta,gamma-Vicinal Dicarbofunctionalization of Alkenyl Carbonyl Compounds via Directed Nucleopalladation, J. Am. Chem. Soc., 2016, 138, 15122–15125 CrossRef CAS PubMed.
- B. Shrestha, P. Basnet, R. K. Dhungana, S. Kc, S. Thapa, J. M. Sears and R. Giri, Ni-Catalyzed Regioselective 1,2-Dicarbofunctionalization of Olefins by Intercepting Heck Intermediates as Imine-Stabilized Transient Metallacycles, J. Am. Chem. Soc., 2017, 139, 10653–10656 CrossRef CAS PubMed.
- P. Basnet, S. Kc, R. K. Dhungana, B. Shrestha, T. J. Boyle and R. Giri, Synergistic Bimetallic Ni/Ag and Ni/Cu Catalysis for Regioselective gamma,delta-Diarylation of Alkenyl Ketimines: Addressing beta-H Elimination by In situ Generation of Cationic Ni(II) Catalysts, J. Am. Chem. Soc., 2018, 140, 15586–15590 CrossRef CAS PubMed.
- J. Derosa, R. Kleinmans, V. T. Tran, M. K. Karunananda, S. R. Wisniewski, M. D. Eastgate and K. M. Engle, Nickel-Catalyzed 1,2-Diarylation of Simple Alkenyl Amides, J. Am. Chem. Soc., 2018, 140, 17878–17883 CrossRef CAS PubMed.
- P. Gao, L. A. Chen and M. K. Brown, Nickel-Catalyzed Stereoselective Diarylation of Alkenylarenes, J. Am. Chem. Soc., 2018, 140, 10653–10657 CrossRef CAS PubMed.
- S. Thapa, R. K. Dhungana, R. T. Magar, B. Shrestha, S. Kc and R. Giri, Ni-catalysed regioselective 1,2-diarylation of unactivated olefins by stabilizing Heck intermediates as pyridylsilyl-coordinated transient metallacycles, Chem. Sci., 2018, 9, 904–909 RSC.
- D. Anthony, Q. Lin, J. Baudet and T. Diao, Nickel-Catalyzed Asymmetric Reductive Diarylation of Vinylarenes, Angew. Chem., Int. Ed., 2019, 58, 3198–3202 CrossRef CAS PubMed.
- J. Derosa, T. Kang, V. T. Tran, S. R. Wisniewski, M. K. Karunananda, T. C. Jankins, K. L. Xu and K. M. Engle, Nickel-Catalyzed 1,2-Diarylation of Alkenyl Carboxylates: A Gateway to 1,2,3-Trifunctionalized Building Blocks, Angew. Chem., Int. Ed., 2020, 59, 1201–1205 CrossRef CAS PubMed.
- R. K. Dhungana, R. R. Sapkota, L. M. Wickham, D. Niroula and R. Giri, Ni-Catalyzed Regioselective 1,2-Dialkylation of Alkenes Enabled by the Formation of Two C(sp(3))-C(sp(3)) Bonds, J. Am. Chem. Soc., 2020, 142, 20930–20936 CrossRef CAS PubMed.
- T. Yang, Y. Jiang, Y. Luo, J. J. H. Lim, Y. Lan and M. J. Koh, Chemoselective Union of Olefins, Organohalides, and Redox-Active Esters Enables Regioselective Alkene Dialkylation, J. Am. Chem. Soc., 2020, 142, 21410–21419 CrossRef CAS PubMed.
- V. Aryal, L. J. Chesley, D. Niroula, R. R. Sapkota, R. K. Dhungana and R. Giri, Ni-Catalyzed Regio- and Stereoselective Alkylarylation of Unactivated Alkenes in γ,δ-Alkenylketimines, ACS Catal., 2022, 12, 7262–7268 CrossRef CAS PubMed.
- F. Lovering, J. Bikker and C. Humblet, Escape from flatland: increasing saturation as an approach to improving clinical success, J. Med. Chem., 2009, 52, 6752–6756 CrossRef CAS PubMed.
- E. Geist, A. Kirschning and T. Schmidt, sp3-sp3 Coupling reactions in the synthesis of natural products and biologically active molecules, Nat. Prod. Rep., 2014, 31, 441–448 RSC.
- T. Qin, J. Cornella, C. Li, L. R. Malins, J. T. Edwards, S. Kawamura, B. D. Maxwell, M. D. Eastgate and P. S. Baran, A general alkyl-alkyl cross-coupling enabled by redox-active esters and alkylzinc reagents, Science, 2016, 352, 801–805 CrossRef CAS PubMed.
- L. Zhang, G. J. Lovinger, E. K. Edelstein, A. A. Szymaniak, M. P. Chierchia and J. P. Morken, Catalytic conjunctive cross-coupling enabled by metal-induced metallate rearrangement, Science, 2016, 351, 70–74 CrossRef CAS PubMed.
- J. Derosa, V. T. Tran, M. N. Boulous, J. S. Chen and K. M. Engle, Nickel-Catalyzed beta,gamma-Dicarbofunctionalization of Alkenyl Carbonyl Compounds via Conjunctive Cross-Coupling, J. Am. Chem. Soc., 2017, 139, 10657–10660 CrossRef CAS PubMed.
- M. Kischkewitz, K. Okamoto, C. Muck-Lichtenfeld and A. Studer, Radical-polar crossover reactions of vinylboron ate complexes, Science, 2017, 355, 936–938 CrossRef CAS PubMed.
- S. Kc, R. K. Dhungana, B. Shrestha, S. Thapa, N. Khanal, P. Basnet, R. W. Lebrun and R. Giri, Ni-Catalyzed Regioselective Alkylarylation of Vinylarenes via C(sp(3))-C(sp(3))/C(sp(3))-C(sp(2)) Bond Formation and Mechanistic Studies, J. Am. Chem. Soc., 2018, 140, 9801–9805 CrossRef CAS PubMed.
- M. W. Campbell, J. S. Compton, C. B. Kelly and G. A. Molander, Three-Component Olefin Dicarbofunctionalization Enabled by Nickel/Photoredox Dual Catalysis, J. Am. Chem. Soc., 2019, 141, 20069–20078 CrossRef CAS PubMed.
- M. Chierchia, P. Xu, G. J. Lovinger and J. P. Morken, Enantioselective Radical Addition/Cross-Coupling of Organozinc Reagents, Alkyl Iodides, and Alkenyl Boron Reagents, Angew. Chem., Int. Ed., 2019, 58, 14245–14249 CrossRef CAS PubMed.
- S. Kc, R. K. Dhungana, N. Khanal and R. Giri, Nickel-Catalyzed alpha-Carbonylalkylarylation of Vinylarenes: Expedient Access to gamma,gamma-Diarylcarbonyl and Aryltetralone Derivatives, Angew. Chem., Int. Ed., 2020, 59, 8047–8051 CrossRef CAS PubMed.
- R. S. Mega, V. K. Duong, A. Noble and V. K. Aggarwal, Decarboxylative Conjunctive Cross-coupling of Vinyl Boronic Esters using Metallaphotoredox Catalysis, Angew. Chem., Int. Ed., 2020, 59, 4375–4379 CrossRef CAS PubMed.
- X. Wei, W. Shu, A. Garcia-Dominguez, E. Merino and C. Nevado, Asymmetric Ni-Catalyzed Radical Relayed Reductive Coupling, J. Am. Chem. Soc., 2020, 142, 13515–13522 CrossRef CAS PubMed.
- R. K. Dhungana, R. R. Sapkota, L. M. Wickham, D. Niroula, B. Shrestha and R. Giri, Ni-Catalyzed Arylbenzylation of Alkenylarenes: Kinetic Studies Reveal Autocatalysis by ZnX(2), Angew. Chem., Int. Ed., 2021, 60, 22977–22982 CrossRef CAS PubMed.
- M. J. Cabrera-Afonso, A. Sookezian, S. O. Badir, M. El Khatib and G. A. Molander, Photoinduced 1,2-dicarbofunctionalization of alkenes with organotrifluoroborate nucleophiles via radical/polar crossover, Chem. Sci., 2021, 12, 9189–9195 RSC.
- P. Dey, S. K. Jana, P. Rai and B. Maji, Dicarbofunctionalizations of an Unactivated Alkene via Photoredox/Nickel Dual Catalysis, Org. Lett., 2022, 24, 6261–6265 CrossRef CAS PubMed.
- F. Wang, D. Wang, X. Mu, P. Chen and G. Liu, Copper-catalyzed intermolecular trifluoromethylarylation of alkenes: mutual activation of arylboronic acid and CF3+ reagent, J. Am. Chem. Soc., 2014, 136, 10202–10205 CrossRef CAS PubMed.
- A. Garcia-Dominguez, Z. Li and C. Nevado, Nickel-Catalyzed Reductive Dicarbofunctionalization of Alkenes, J. Am. Chem. Soc., 2017, 139, 6835–6838 CrossRef CAS PubMed.
- A. Garcia-Dominguez, R. Mondal and C. Nevado, Dual Photoredox/Nickel-Catalyzed Three-Component Carbofunctionalization of Alkenes, Angew. Chem., Int. Ed., 2019, 58, 12286–12290 CrossRef CAS PubMed.
- L. Guo, H. Y. Tu, S. Zhu and L. Chu, Selective, Intermolecular Alkylarylation of Alkenes via Photoredox/Nickel Dual Catalysis, Org. Lett., 2019, 21, 4771–4776 CrossRef CAS PubMed.
- X. L. Lv, C. Wang, Q. L. Wang and W. Shu, Rapid Synthesis of gamma-Arylated Carbonyls Enabled by the Merge of Copper- and Photocatalytic Radical Relay Alkylarylation of Alkenes, Org. Lett., 2019, 21, 56–59 CrossRef CAS PubMed.
- W. Shu, A. Garcia-Dominguez, M. T. Quiros, R. Mondal, D. J. Cardenas and C. Nevado, Ni-Catalyzed Reductive Dicarbofunctionalization of Nonactivated Alkenes: Scope and Mechanistic Insights, J. Am. Chem. Soc., 2019, 141, 13812–13821 CrossRef CAS PubMed.
- L. Liu, W. Lee, C. R. Youshaw, M. Yuan, M. B. Geherty, P. Y. Zavalij and O. Gutierrez, Fe-catalyzed three-component dicarbofunctionalization of unactivated alkenes with alkyl halides and Grignard reagents, Chem. Sci., 2020, 11, 8301–8305 RSC.
- H. Wang, C. F. Liu, R. T. Martin, O. Gutierrez and M. J. Koh, Directing-group-free catalytic dicarbofunctionalization of unactivated alkenes, Nat. Chem., 2022, 14, 188–195 CrossRef CAS PubMed.
- J. R. Bour, N. M. Camasso and M. S. Sanford, Oxidation of Ni(II) to Ni(IV) with Aryl Electrophiles Enables Ni-Mediated Aryl-CF3 Coupling, J. Am. Chem. Soc., 2015, 137, 8034–8037 CrossRef CAS PubMed.
- M. Yuan, Z. Song, S. O. Badir, G. A. Molander and O. Gutierrez, On the Nature of C(sp(3))-C(sp(2)) Bond Formation in Nickel-Catalyzed Tertiary Radical Cross-Couplings: A Case Study of Ni/Photoredox Catalytic Cross-Coupling of Alkyl Radicals and Aryl Halides, J. Am. Chem. Soc., 2020, 142, 7225–7234 CrossRef CAS PubMed.
- W. Xue, X. Jia, X. Wang, X. Tao, Z. Yin and H. Gong, Nickel-catalyzed formation of quaternary carbon centers using tertiary alkyl electrophiles, Chem. Soc. Rev., 2021, 50, 4162–4184 RSC.
- Two further protocols (7f, h) feature single examples of 3-pyridyl products. These reactions may be special cases of N-heteroarenes, however, because (a) these isomers avoid catalyst deactivation by chelation that is often a problem for 2-azaheteroarenes, and (b) both examples feature an electronegative substituent on that lowers the basicity of the nitrogen atom that can be essential for biological activity and a problem for transition-metal catalysts.
- Open-shell approaches have also been developed featuring alkyl-radical addition to an alkene followed by Minisci addition of the resulting olefin-derived alkyl radical to a heteroarene, but these protocols are inherently restricted to electrophilic N-heteroaromatic substrates and products. For a leading example, see the reference immediately below.
- T. McCallum and L. Barriault, Direct alkylation of heteroarenes with unactivated bromoalkanes using photoredox gold catalysis, Chem. Sci., 2016, 7, 4754–4758 RSC.
- While we were completing this work, the report in the reference immediately below described nine examples of simple olefin alkyl–arylation with analogous sulfone reagents. The key synthetic challenges that plague transition-metal-catalyzed systems and that we sought to solve remained mostly unaddressed, however, as only monosubstituted alkenes were competent substrates (meaning that quaternary benzylic products and diastereocontrol were also absent), and only one example of any heteroaryl group (2-benzothiazole) appeared in the scope.
- S. Hong, M. Kim, K. Lee and S. Kim, Photoinduced Carboarylation of Alkenes by Using Bifunctional Reagents, Synlett, 2023, 34, 1437–1441 CrossRef.
- Bromoalkyl–aryl sulfones have been employed to a similar end via C–Br reduction using a stroichiometric thiol that is not commercially available. These sulfones also require 3–4 steps to access (4 steps whenever primary alkyl groups are added to the olefin), and no functional handles beyond halogens were tolerated near the reactive alkyl carbon, although they do permit the incorporation of alkyl groups devoid of functionality.
- J. Liu, S. Wu, J. Yu, C. Lu, Z. Wu, X. Wu, X. S. Xue and C. Zhu, Polarity Umpolung Strategy for the Radical Alkylation of Alkenes, Angew. Chem., Int. Ed., 2020, 59, 8195–8202 CrossRef CAS PubMed.
- M. H. Shaw, J. Twilton and D. W. MacMillan, Photoredox Catalysis in Organic Chemistry, J. Org. Chem., 2016, 81, 6898–6926 CrossRef CAS PubMed.
- C. S. Wang, P. H. Dixneuf and J. F. Soule, Photoredox Catalysis for Building C-C Bonds from C(sp(2))-H Bonds, Chem. Rev., 2018, 118, 7532–7585 CrossRef CAS PubMed.
- J. D. Bell and J. A. Murphy, Recent advances in visible light-activated radical coupling reactions triggered by (i) ruthenium, (ii) iridium and (iii) organic photoredox agents, Chem. Soc. Rev., 2021, 50, 9540–9685 RSC.
- A. Studer and M. Bossart, Radical aryl migration reactions, Tetrahedron, 2001, 57, 9649–9667 CrossRef CAS.
- Z. M. Chen, X. M. Zhang and Y. Q. Tu, Radical aryl migration reactions and synthetic applications, Chem. Soc. Rev., 2015, 44, 5220–5245 RSC.
- X. Wu and C. Zhu, Radical-Mediated Remote Functional Group Migration, Acc. Chem. Res., 2020, 53, 1620–1636 CrossRef CAS PubMed.
- X. Q. Chu, D. Ge, Y. Y. Cui, Z. L. Shen and C. J. Li, Desulfonylation via Radical Process: Recent Developments in Organic Synthesis, Chem. Rev., 2021, 121, 12548–12680 CrossRef CAS PubMed.
- X. Wu, Z. Ma, T. Feng and C. Zhu, Radical-mediated rearrangements: past, present, and future, Chem. Soc. Rev., 2021, 50, 11577–11613 RSC.
- A. R. Allen, E. A. Noten and C. R. J. Stephenson, Aryl Transfer Strategies Mediated by Photoinduced Electron Transfer, Chem. Rev., 2022, 122, 2695–2751 CrossRef CAS PubMed.
- R. Loven and W. N. Speckamp, A novel 1,4 arylradical rearrangement, Tetrahedron Lett., 1972, 13, 1567–1570 CrossRef.
- W. Kong, M. Casimiro, E. Merino and C. Nevado, Copper-catalyzed one-pot trifluoromethylation/aryl migration/desulfonylation and C(sp2)-N bond formation of conjugated tosyl amides, J. Am. Chem. Soc., 2013, 135, 14480–14483 CrossRef CAS PubMed.
- T. M. Monos, R. C. McAtee and C. R. J. Stephenson, Arylsulfonylacetamides as bifunctional reagents for alkene aminoarylation, Science, 2018, 361, 1369–1373 CrossRef CAS PubMed.
- J. Yu, Z. Wu and C. Zhu, Efficient Docking-Migration Strategy for Selective Radical Difluoromethylation of Alkenes, Angew. Chem., Int. Ed., 2018, 57, 17156–17160 CrossRef CAS PubMed.
- D. M. Whalley, H. A. Duong and M. F. Greaney, Alkene Carboarylation through Catalyst-Free, Visible Light-Mediated Smiles Rearrangement, Chemistry, 2019, 25, 1927–1930 CrossRef CAS PubMed.
- M. Wang, H. Zhang, J. Liu, X. Wu and C. Zhu, Radical Monofluoroalkylative Alkynylation of Olefins by a Docking-Migration Strategy, Angew. Chem., Int. Ed., 2019, 58, 17646–17650 CrossRef CAS PubMed.
- C. Hervieu, M. S. Kirillova, T. Suarez, M. Muller, E. Merino and C. Nevado, Asymmetric, visible light-mediated radical sulfinyl-Smiles rearrangement to access all-carbon quaternary stereocentres, Nat. Chem., 2021, 13, 327–334 CrossRef CAS PubMed.
- Y. Wei, H. Zhang, X. Wu and C. Zhu, Alkene Difunctionalization Triggered by a Stabilized Allenyl Radical: Concomitant Installation of Two Unsaturated C-C Bonds, Angew. Chem., Int. Ed., 2021, 60, 20215–20219 CrossRef CAS PubMed.
- A. R. Allen, J. F. Poon, R. C. McAtee, N. B. Watson, D. A. Pratt and C. R. J. Stephenson, Mechanism of Visible Light-Mediated Alkene Aminoarylation with Arylsulfonylacetamides, ACS Catal., 2022, 12, 8511–8526 CrossRef CAS PubMed.
- D. Wang, C. Muck-Lichtenfeld, C. G. Daniliuc and A. Studer, Radical Aryl Migration from Boron to Carbon, J. Am. Chem. Soc., 2021, 143, 9320–9326 CrossRef CAS PubMed.
- E. A. Noten, R. C. McAtee and C. R. J. Stephenson, Catalytic intramolecular aminoarylation of unactivated alkenes with aryl sulfonamides, Chem. Sci., 2022, 13, 6942–6949 RSC.
- N. Radhoff and A. Studer, 1,4-Aryl migration in ketene-derived enolates by a polar-radical-crossover cascade, Nat. Commun., 2022, 13, 3083 CrossRef CAS PubMed.
- C. He, K. Zhang, D. N. Wang, M. Wang, Y. Niu, X. H. Duan and L. Liu, Visible-Light-Induced Alkylarylation of Unactivated Alkenes via Radical Addition/Truce-Smiles Rearrangement Cascade, Org. Lett., 2022, 24, 2767–2771 CrossRef CAS PubMed.
- J. Yu, X. Zhang, X. Wu, T. Liu, Z.-Q. Zhang, J. Wu and C. Zhu, Metal-free radical difunctionalization of ethylene, Chem, 2023, 9, 472–482 CAS.
- F. Parsaee, M. C. Senarathna, P. B. Kannangara, S. N. Alexander, P. D. E. Arche and E. R. Welin, Radical philicity and its role in selective organic transformations, Nat. Rev. Chem, 2021, 5, 486–499 CrossRef CAS PubMed.
- A. Studer and M. Bossart, Stereoselective radical aryl migrations from sulfur to carbon, Chem. Commun., 1998, 1998, 2127–2128 RSC.
- S. Amrein, M. Bossart, T. Vasella and A. Studer, Stereoselective radical aryl migration from silicon to carbon, J. Org. Chem., 2000, 65, 4281–4288 CrossRef CAS PubMed.
- E. Vitaku, D. T. Smith and J. T. Njardarson, Analysis of the structural diversity, substitution patterns, and frequency of nitrogen heterocycles among U.S. FDA approved pharmaceuticals, J. Med. Chem., 2014, 57, 10257–10274 CrossRef CAS PubMed.
- M. W. Wong, A. Pross and L. Radom, Addition of tert-Butyl Radical to Substituted Alkenes: A Theoretical Study of the Reaction Mechanism, J. Am. Chem. Soc., 2002, 116, 11938–11943 CrossRef.
- F. G. Bordwell, J. A. Harrelson and X. Zhang, Homolytic bond dissociation energies of acidic carbon-hydrogen bonds activated by one or two electron acceptors, J. Org. Chem., 1991, 56, 4448–4450 CrossRef CAS.
- T. S. Teets, Y. Wu and D. Kim, Photophysical Properties and Redox Potentials of Photosensitizers for Organic Photoredox Transformations, Synlett, 2021, 33, 1154–1179 CrossRef.
- We also measured the oxidation potentials of some of the conjugate bases of the sulfones used in this study. These values ranged from +0.67 V to +0.91 V vs. SCE in MeCN (see ESI†).
- M. S. Alnajjar, X. M. Zhang, G. J. Gleicher, S. V. Truksa and J. A. Franz, Equilibrium acidities and homolytic bond dissociation energies of acidic C-H bonds in alpha-arylacetophenones and related compounds, J. Org. Chem., 2002, 67, 9016–9022 CrossRef CAS PubMed.
- S.-J. Chang, D. McNally, S. Shary-Tehrany, H. S. M. James and R. H. Boyd, Heats of combustion and strain energies of bicyclo[n.m.O]alkanes, J. Am. Chem. Soc., 1970, 92, 3109–3118 CrossRef CAS.
- N. L. Allinger, M. T. Tribble, M. A. Miller and D. H. Wertz, Conformational analysis. LXIX. Improved force field for the calculation of the structures and energies of hydrocarbons, J. Am. Chem. Soc., 1971, 93, 1637–1648 CrossRef CAS.
- X. T. Chen, P. Pitis, G. Liu, C. Yuan, D. Gotchev, C. L. Cowan, D. H. Rominger, M. Koblish, S. M. Dewire, A. L. Crombie, J. D. Violin and D. S. Yamashita, Structure-activity relationships and discovery of a G protein biased mu opioid receptor ligand, [(3-methoxythiophen-2-yl)methyl](2-[(9R)-9-(pyridin-2-yl)-6-oxaspiro-[4.5]decan-9-yl]ethyl)amine (TRV130), for the treatment of acute severe pain, J. Med. Chem., 2013, 56, 8019–8031 CrossRef CAS PubMed.
- A. Markham, Oliceridine: First Approval, Drugs, 2020, 80, 1739–1744 CrossRef CAS PubMed.
- H. S. Tan and A. S. Habib, Oliceridine: A Novel Drug for the Management of Moderate to Severe Acute Pain - A Review of Current Evidence, J. Pain Res., 2021, 14, 969–979 CrossRef PubMed.
- A. A. H. Azzam and D. G. Lambert, Preclinical discovery and development of oliceridine (Olinvyk(R)) for the treatment of post-operative pain, Expert Opin. Drug Discovery, 2022, 17, 215–223 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Materials, experimental details, stereochemical analysis and discussion, fluorescence-quenching data, cyclic voltammograms, and NMR spectra. See DOI: https://doi.org/10.1039/d3sc06476j |
| This journal is © The Royal Society of Chemistry 2024 |