
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
High-throughput assay exploiting disorder-to-order conformational switches: application to the proteasomal Rpn10:E6AP complex†
Christine S.
Muli
 a,
Sergey G.
Tarasov
b and
Kylie J.
Walters
*a
a,
Sergey G.
Tarasov
b and
Kylie J.
Walters
*a
aProtein Processing Section, Center for Structural Biology, Center for Cancer Research, National Cancer Institute, National Institutes of Health, Frederick, MD 21702, USA. E-mail: kylie.walters@nih.gov
bBiophysics Resource, Center for Structural Biology, Center for Cancer Research, National Cancer Institute, National Institutes of Health, Frederick, MD 21702, USA
First published on 6th February 2024
Abstract
Conformational switching is pervasively driven by protein interactions, particularly for intrinsically disordered binding partners. We developed a dually orthogonal fluorescence-based assay to monitor such events, exploiting environmentally sensitive fluorophores. This assay is applied to E3 ligase E6AP, as its AZUL domain induces a disorder-to-order switch in an intrinsically disordered region of the proteasome, the so-named Rpn10 AZUL-binding domain (RAZUL). By testing various fluorophores, we developed an assay appropriate for high-throughput screening of Rpn10:E6AP-disrupting ligands. We found distinct positions in RAZUL for fluorophore labeling with either acrylodan or Atto610, which had disparate spectral responses to E6AP binding. E6AP caused a hypsochromic shift with increased fluorescence of acrylodan-RAZUL while decreasing fluorescence intensity of Atto610-RAZUL. Combining RAZUL labeled with either acrylodan or Atto610 into a common sample achieved robust and orthogonal measurement of the E6AP-induced conformational switch. This approach is generally applicable to disorder-to-order (or vice versa) transitions mediated by molecular interactions.
Introduction
E3 ligases are critical for the transfer of ubiquitin to a protein substrate for ubiquitin-dependent proteasomal degradation.1 The 26S proteasome is a sophisticated multi-catalytic enzyme that recognizes ubiquitinated substrates with its 19S regulatory particle (RP), with subsequent degradation within its 20S core particle (CP).2 For targeted protein degradation, the activity of E3 ligases is therapeutically harnessed for development of proteolysis targeting chimeras (PROTACs), which act as heterobifunctional molecules that tether a substrate-of-interest to an E3, thereby inducing its ubiquitination.3–7 Akin to the PROTAC mechanism, E3 ligase E6AP/UBE3A is seized by the human papillomavirus (HPV) oncoprotein E6 to induce ubiquitination and subsequent degradation of tumor suppressor p53, driving HPV-associated cervical cancers.8–12 E6AP dysfunction is also implicated in a variety of other cancers, including prostate and breast cancer.13–16 Moreover, overexpression or gain-of-function of the UBE3A gene is linked to autism spectrum disorders,17–20 and loss or loss-of-function of UBE3A drives the neurological disorder Angelman syndrome.21–24 E6AP is present at the 26S proteasome18,25 by direct binding to the RP subunit Rpn10,26 which also regulates E6AP subcellular localization.27–29To date, E6AP is the only E3 ligase with a known proteasomal binding site. The E6AP AZUL (amino-terminal zinc-binding domain of ubiquitin E3a ligase) domain binds to an intrinsically disordered region in Rpn10 to induce a disorder-to-order transition in the so-named Rpn10 AZUL-binding domain (RAZUL).26 With its prevalent role in cancer and neurological disease and with the importance of E3s for targeted protein degradation, E6AP is an attractive therapeutic target. Chemical probes against the Rpn10:E6AP interaction would be useful for interrogating the physiological significance of the E6AP interaction with proteasomes and the contribution of this interaction to E6AP-associated diseases. Several E6AP-interacting small molecules have been identified that bind to domains other than the AZUL domain or at unknown sites.30–32 A ligand that blocks Rpn10 binding to K48-linked ubiquitin chains was also reported,33 but this outcome is independent of the RAZUL domain.
Recently, environmentally sensitive fluorophores have proven useful as probes against systems that undergo conformational changes, including conformational switching during enzymatic activity34 and between intrinsically disordered tau and tubulin dimers.35 To develop an assay for the discovery of ligands that disrupt or enhance the Rpn10:E6AP interaction, we used environmentally sensitive fluorophores as sensors of the RAZUL disorder-to-order switch, Fig. 1. Such exploitation of a disorder-to-order transition of an intrinsically disordered protein (IDP) or region (IDR) is of therapeutic interest,36–38 especially when induced by a native binding partner; for example, p27 with Cdk2/cyclin A,39,40 MAX with cMyc,41,42 and p53 with MDM2.43–45 Thus, the approach we describe herein for Rpn10:E6AP, whereby binding is monitored by using environmentally sensitive fluorophores as sensors for conformational switching, can be applied generally to other therapeutically relevant protein systems to either characterize dynamic behavior and/or to perform high-throughput (HTP) screening for therapeutic discovery.
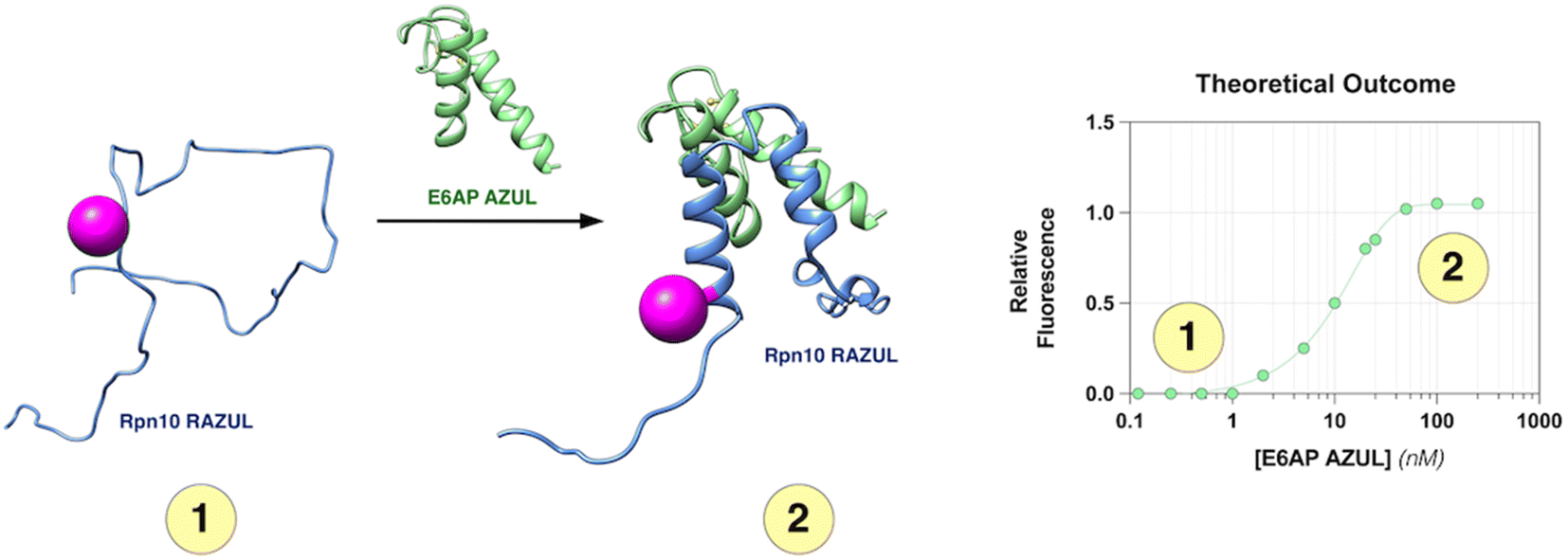 | ||
| Fig. 1 Assay design to monitor Rpn10 RAZUL binding to E6AP AZUL. RAZUL (blue) is intrinsically disordered (1) but becomes helical upon interaction with AZUL (green) to form an intermolecular 4-helix bundle (2). An environmentally sensitive fluorophore (magenta) is expected to have distinct signals for the (1) free and (2) bound states. PDB: 6U19. | ||
Results and discussion
Cysteine-substitutions in Rpn10 RAZUL do not disrupt E6AP binding or its disorder-to-order conformational switch
Rpn10 RAZUL spans amino acids 305–377 with no native cysteine. To allow for fluorogenic labeling, we rationally introduced a thiol by examining the Rpn10:E6AP structure26 for surface exposed serines. Three native serines (S337, S358, and S361) were identified in Rpn10 RAZUL proximal to but not within the E6AP AZUL interacting surface (Fig. S1A†). These serines are within regions that become helical by RAZUL binding to E6AP and are therefore likely to be sensitive to the RAZUL disorder-to-order transition. We tested and found that addition of the E6AP AZUL (24–87) domain induces helicity in RAZUL with cysteine substitution at S337, S358, or S361, with comparison to unmodified RAZUL. Specifically, experimentally measured circular dichroism (CD) spectra were compared to the theoretical sum of free RAZUL and AZUL to find induced helicity, with an enhanced effect for RAZUL S361C (Fig. S1B†). Based on these results, we concluded that AZUL induces helicity in all RAZUL mutants tested akin to wildtype RAZUL; however, we cannot preclude the possibility of the serine-to-cysteine mutations causing differences in the overall helical arrangements of mutant RAZUL when AZUL-bound. We therefore tested whether the binding affinity for AZUL is altered by these amino acid substitutions.Previously, the binding affinity of the RAZUL:AZUL interaction was characterized by surface plasmon resonance (SPR) and isothermal titration calorimetry (ITC), and binding dissociation constants (Kd) were agreeable at 11.6 ± 3.3 nM and 8.1 ± 1.4 nM, respectively.26 To test directly the impact of RAZUL cysteine substitution on AZUL binding affinity, Kd values were measured by ITC to find similarity with wildtype RAZUL and <30 nM affinity for all variants with AZUL (Fig. S1C†). The largest deviation was for RAZUL S361C at 6.90 ± 4.04 nM affinity. Altogether, these results indicate that cysteine substitution at codon 337, 358, or 361 does not affect Rpn10 RAZUL interaction with E6AP AZUL. Overall, the RAZUL cysteine mutants' retention of both the disorder-to-order transition by CD and binding interaction by ITC allowed for investigation of environmentally sensitive fluorophores with this system.
Acrylodan labeling at Rpn10 RAZUL S358C is optimal for detecting E6AP binding
To produce fluorescently labeled RAZUL constructs, we treated cysteine-substituted and wildtype (negative control) RAZUL with 2-fold equivalent acrylodan to achieve >99% labeling efficiency of the mutants, as indicated by intact protein mass spectrometry (Fig. S2†). Acrylodan labeling did not interfere with the RAZUL:AZUL interaction, as each labeled RAZUL variant bound AZUL with <35 nM affinity by ITC (Fig. 2A). Each acrylodan-labeled Rpn10 RAZUL (RAZULAcr) sample was analyzed by fluorescence spectroscopy to evaluate sensitivity to AZUL binding. In each case, the fluorescence of 500 nM Rpn10 RAZULAcr was measured with increasing quantities of E6AP AZUL up to 2 equivalences, recording with an excitation at 390 nm and emission scanning range of 410–600 nm. Relative fluorescent units (RFU) were plotted to find a dose-dependent hypsochromic shift for each RAZULAcr probe, with the maximum wavelength (λmax) of RAZUL S358CAcr shifted from 518 nm to 475 nm (Fig. 2B). More minor shifts of 4 nm and 8 nm were measured for RAZUL S337CAcr and S361CAcr, respectively (Fig. 2B). The RFU similarly increased dose-dependently with E6AP AZUL for each RAZULAcr mutant when the assay was translated to a 96-well format at 50 μL per well. The change in RFU (ΔRFU) was plotted with acrylodan-treated wildtype Rpn10 RAZUL (which was not labeled) used as a negative control (Fig. 2C). The affinity of RAZUL S358CAcr for AZUL was measured to be similar to RAZUL S337CAcr and S361CAcr by ITC (Fig. 2A), indicating that the more prominent spectral shift in RAZUL S358CAcr is driven by a greater change in the local chemical environment rather than by increased affinity. Since RAZUL S358CAcr elicited the largest response, it was chosen for further optimization. | ||
| Fig. 2 Acrylodan acts as an environmentally sensitive probe for Rpn10 RAZUL binding to E6AP AZUL, with greatest sensitivity for RAZUL S358CAcr. (A) Binding isotherm with raw ITC data (top left insert) for injections of E6AP AZUL into acrylodan-labeled Rpn10 RAZUL at 170 μM AZUL into 18 μM S337CAcr (left panel), 90 μM AZUL into 9 μM S358CAcr (middle panel), or 106 μM AZUL into 9 μM S361CAcr (right panel). Fitted thermodynamic values are included on each panel at the bottom right: n, stoichiometry; Kd, binding affinity; ΔH, change in enthalpy; ΔS, change in entropy. Measurements were made in 10 mM MOPS, pH 6.5, 50 mM NaCl, 2 mM TCEP, and 10 μM ZnSO4. (B) Fluorescence spectroscopy of 500 nM of RAZULAcr as indicated with 0–2.0 equivalences of E6AP AZUL in 10 mM MOPS, pH 6.5, 50 mM NaCl, 5 mM DTT, and 10 μM ZnSO4. All samples were excited at 390 nm on a FluoroMax-4C spectrofluorometer with emission scanning range from 410–600 nm. λmax refers to the wavelength (λ) at which the maximum absorption is observed. (C) Change in RFU (relative to RFUbackground) for 500 nM of acrylodan-treated WT or cysteine-substituted Rpn10 RAZUL mixed with 0 (red), 0.5 (yellow), 250 (blue), or 5000 (purple) nM of E6AP AZUL (n = 3). A 96-well plate was used with excitation of 390 nm and emission of 500 nm. ΔRFU = RFUsignal − RFUbackground. (D) Fluorescence measurements of 25 (red), 50 (green), 100 (blue), or 500 (purple) nM RAZUL S358CAcr with increasing quantities of E6AP AZUL normalized to RFUmax in a 384-well plate and buffer as in (B) but supplemented with 0.1% Tween 20 (n = 3). The relative signal was fit for specific binding to a Hill slope. (E) Fluorescence measurements of 100 nM RAZUL S358CAcr mixed with 0–1500 nM E6AP AZUL following storage at 4 °C for 0.25, 2, 4, or 30 hours. Buffer conditions of (D) were used but with 50 (red), 150 (green), or 300 (purple) mM NaCl (n = 3). Curves were fit as in (D), and the respective Kd values were plotted against time. For (C) through (E), each data point is represented as mean ± standard error of the mean (SEM). | ||
We next translated the acrylodan-based assay to a 384-well plate with 10–500 nM RAZUL S358CAcr and sequentially increased quantities of AZUL. The relative fluorescent signal was normalized to the maximum RFU value, namely at saturating AZUL concentrations (Fig. 2D). The assay stability and optimal buffer conditions were assessed to find that without detergent, a decrease in signal occurred after the saturation point from protein aggregation (Fig. S3A†). All binding curves could be fit with an R2 > 0.96 when Tween 20 was included, whereas without it, the R2 values ranged from 0.83 to 0.94. To ensure the fluorescence profile of RAZUL S358CAcr was not compromised by detergent, we evaluated free and bound RAZUL S358CAcr with 0–0.1% Tween 20 and found no significant dependence (Fig. S3B and C†). We next measured the reproducibility of the assay at 4 °C with 50, 150 or 300 mM NaCl to find stability over 30 hours based on Kd measurements, with best results at 150 mM NaCl for a consistent Kd of ∼18 nM (Fig. 2E). Therefore, we concluded that RAZUL S358CAcr conformational switching in response to AZUL binding could be measured in a 384-well plate format, with most stable signal when 150 mM NaCl and 0.1% Tween 20 are included in the buffer.
RAZUL S358CAcr detects competitive inhibition for E6AP AZUL in a HTP screening format
To interrogate the application of RAZULAcr for identifying molecules that inhibit the Rpn10:E6AP interaction, we aimed to compete the most sensitive RAZULAcr probe (S358CAcr) off AZUL to simulate competitive inhibition. With no available chemical tools for the Rpn10:E6AP interaction, a synthetic RAZUL322–366 peptide with 18.2 ± 4.4 nM affinity for AZUL26 was used. Equimolar concentrations at 10, 25, 50, and 100 nM of RAZUL S358CAcr:AZUL were incubated with 0–100 μM RAZUL322–366 and ΔRFU values calculated, plotted, and fit to obtain an inhibition constant (Ki). As expected, RAZUL322–366 competed with RAZUL S358CAcr for AZUL, reducing the relative RAZUL S358CAcr signal; greater quantities of RAZUL322–366 were required for competition when increasing concentration of RAZUL S358CAcr:AZUL (Fig. 3A). Ki values of 1.26 ± 0.28 μM and 18.2 ± 2.9 μM were measured for RAZUL322–366 at 25 nM and 100 nM RAZUL S358CAcr:AZUL, respectively. The slow off-rate of the RAZUL:AZUL interaction, which was previously found to be 2.11 ± 0.30 (×10−2 s−1),26 may cause the >800-fold increase in Ki compared to Kd at 25 nM RAZUL S358CAcr:AZUL. At 10–50 nM RAZUL S358CAcr:AZUL, the competition assay was stable over 4 hours at 4 °C, although less stability was detected at 100 nM RAZUL S358CAcr:AZUL (Fig. 3B).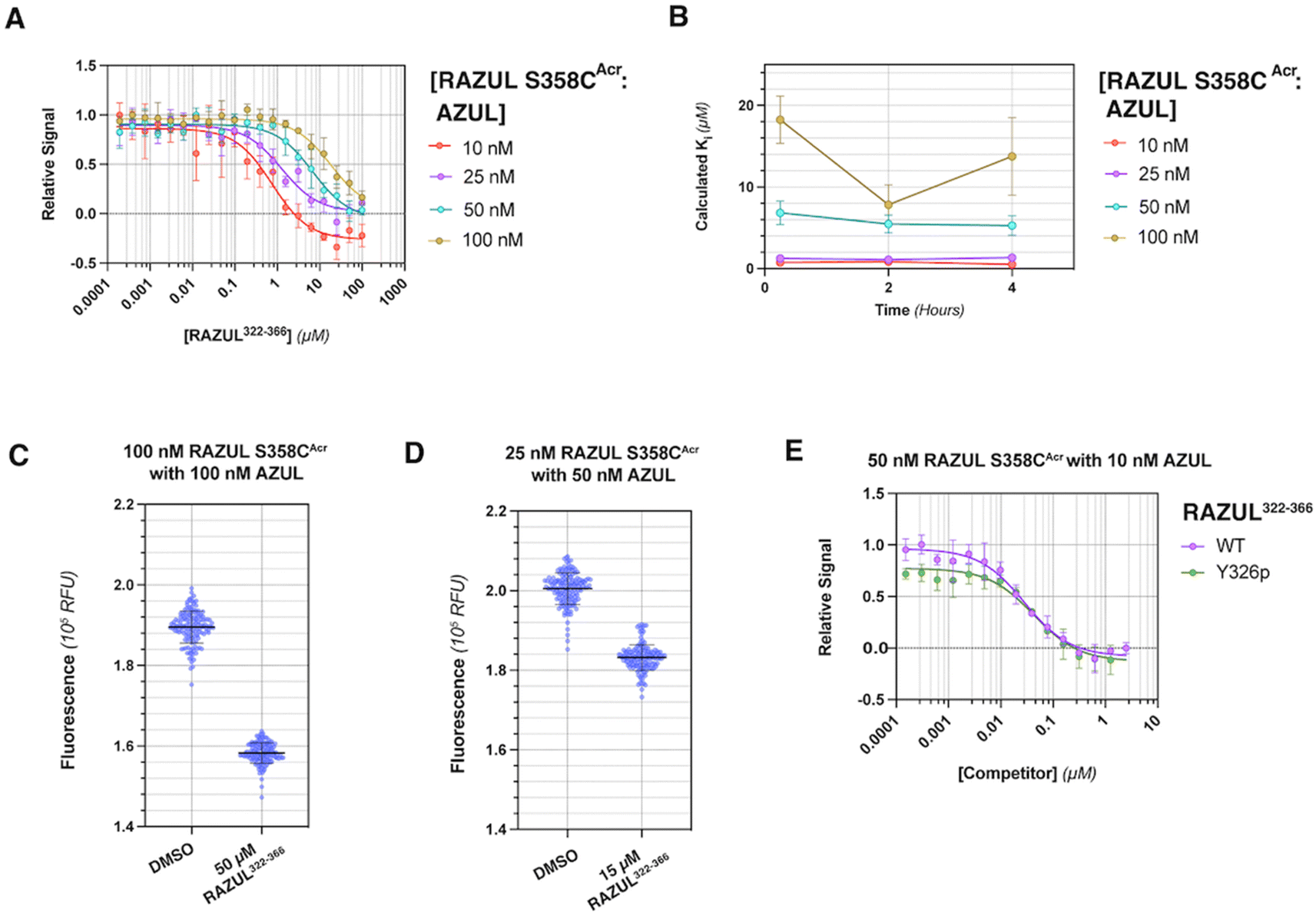 | ||
| Fig. 3 RAZUL S358CAcr is sensitive to competition for AZUL by RAZUL322–366. (A) Plot of RFU values normalized by RFUmax for 10 (red), 25 (purple), 50 (cyan), or 100 (gold) nM RAZUL S358CAcr:AZUL against concentration of RAZUL322–366 (n = 3). Measurements were in 10 mM MOPS, pH 6.5, 150 mM NaCl, 5 mM DTT, 10 μM ZnSO4, 0.1% Tween 20 supplemented with 5% DMSO. Curves were fit to dose response with variable slope on GraphPad Prism. (B) Plot of Ki values for RAZUL322–366 after 0.25, 2, or 4 hours storage at 4 °C, based on fluorescent measurements of 10 (red), 25 (purple), 50 (cyan), or 100 (gold) nM RAZUL S358CAcr:AZUL, with the 0.25 hour timepoint as displayed in (A). Competition curves from different timepoints were plotted and fitted for Ki values, which are plotted against storage time. (C) RFU of 100 nM RAZUL S358CAcr:AZUL with DMSO (vehicle control) or 50 μM RAZUL322–366 in buffer supplemented with 1% BSA (n = 154). (D) RFU of 25 nM RAZUL S358CAcr:50 nM AZUL with DMSO (vehicle control) or 15 μM RAZUL322–366 in buffer supplemented with 1% BSA (n = 154). (E) Plot of RFU values normalized by RFUmax for 10 nM AZUL mixed with 50 nM RAZUL S358CAcr against concentration of unmodified (purple) or Y326p (green) RAZUL322–366. Curves were fit to dose response with variable slope on GraphPad Prism. For all experiments, excitation and emission wavelengths of 390 nm and 475 nm were used, respectively. For (A), (B) and (E), each data point is represented as mean ± SEM. | ||
To evaluate the applicability of this acrylodan-based assay for large scale HTP screening of Rpn10:E6AP-disrupting ligands, the fluorescence of 100 nM RAZUL S358CAcr:AZUL with DMSO (vehicle control) or 50 μM RAZUL322–366 (positive control for inhibition) was measured (Fig. 3C). Within this experiment, 1% carrier protein bovine serum albumin (BSA) was included in the buffer to increase stability.46 Notably, no overlap be Tween the positive and vehicle control measurements was observed. A basic Z′ factor of 0.37 was calculated from the average mean and standard deviation47 of the controls. For nanomolar interactions, as is the case for RAZUL:AZUL, quality of HTP assays have relied on the robust Z′ factor 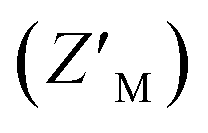 , which is calculated from the median and median absolute deviation.48,49 We calculated the
, which is calculated from the median and median absolute deviation.48,49 We calculated the  to be 0.63, indicating robustness of the RAZUL S358CAcr probe for competitive inhibition with AZUL binding. Decreasing amounts of material in the assay to 25 nM RAZUL S358CAcr:50 nM AZUL with or without 15 μM RAZUL322–366 reduces the
to be 0.63, indicating robustness of the RAZUL S358CAcr probe for competitive inhibition with AZUL binding. Decreasing amounts of material in the assay to 25 nM RAZUL S358CAcr:50 nM AZUL with or without 15 μM RAZUL322–366 reduces the 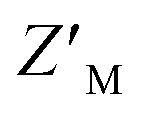 to 0.35 (Fig. 3D).
to 0.35 (Fig. 3D).
We tested how this competitive assay performs with unsaturating conditions of AZUL with RAZUL S358CAcr and whether it is sensitive to the 10-fold weaker affinity of phosphorylated RAZUL322–366 (Y326p) at 189.4 ± 43.8 nM.26 With 50 nM RAZUL S358CAcr and 10 nM AZUL, Ki values of 28.5 ± 4.6 nM and 42.6 ± 13.1 nM were measured for unmodified and Y326p RAZUL322–366, respectively (Fig. 3E). These reduced values signify a greater sensitivity with sub-stoichiometric AZUL but their similarity indicates limited sensitivity to affinity differences in the sub-micromolar range. Moreover, these conditions yielded a poor 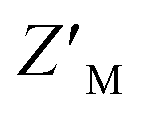 factor, similar to Fig. 3D. Altogether, 100 nM RAZUL S358CAcr:AZUL was found to be acceptable for HTP applications (Fig. 3C) and even lower quantities of material are suitable for non-HTP evaluation of competitors (Fig. 3A and B). These results indicate that acrylodan-labeled RAZUL can serve as an environmental probe for the E6AP-induced disorder-to-order transition.
factor, similar to Fig. 3D. Altogether, 100 nM RAZUL S358CAcr:AZUL was found to be acceptable for HTP applications (Fig. 3C) and even lower quantities of material are suitable for non-HTP evaluation of competitors (Fig. 3A and B). These results indicate that acrylodan-labeled RAZUL can serve as an environmental probe for the E6AP-induced disorder-to-order transition.
Detection of E6AP binding by Atto610 is optimal at Rpn10 RAZUL S337C
Based on its blue-shifted fluorescence, a major limitation for acrylodan as an environmental probe would be fluorescence overlap with potential compounds of interest.50 We therefore tested the red-shifted fluorophores Atto610 and DY647P1 by conjugating their maleimide-derivatives to the introduced cysteines of Rpn10 RAZUL. Conjugation conditions were optimized to >99% efficiency for 10 μM of cysteine-containing RAZUL mixed with 200 μM dye in 50 mM HEPES, 150 mM NaCl, pH 8, for 16–20 hours at 4 °C (Fig. S4 and S5†); insufficient and non-specific maleimide reactivity was measured at pH 7.4 and pH 8.5 respectively (Fig. S6†). The spectral profile of RAZUL S337CAtto or RAZUL S337CDY was measured without or with E6AP AZUL to find that the induced conformational switch causes an overall decrease in the emission scanning profile of RAZUL S337CAtto (excitation at 590 nm) (Fig. 4A, left panel) with only a minor reduction for RAZUL S337CDY (excitation at 630 nm) (Fig. 4A, right panel). In both cases, no change in λmax was detected.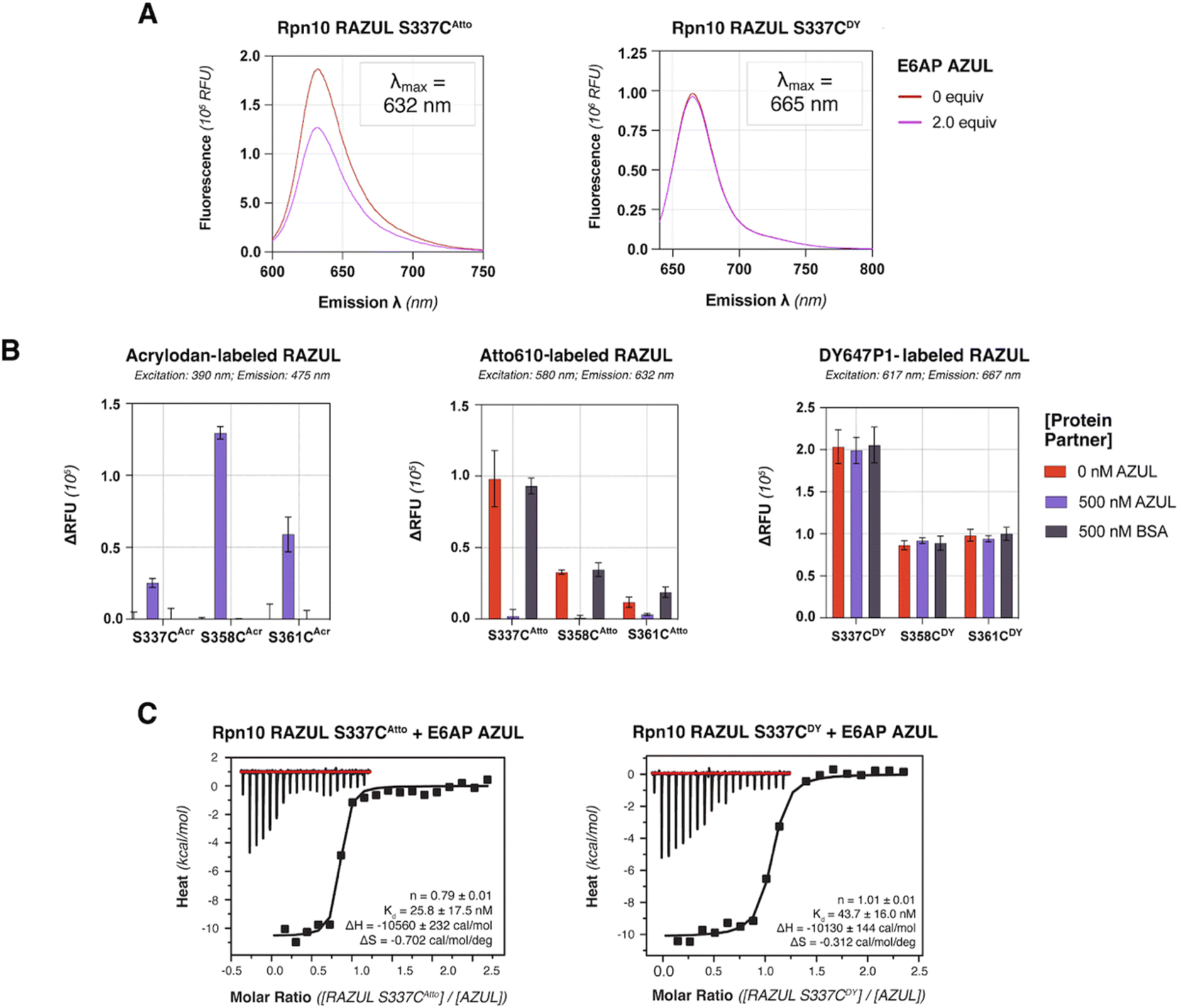 | ||
| Fig. 4 Evaluation of red-shifted fluorophores as probes for Rpn10 RAZUL binding to E6AP AZUL indicates greatest sensitivity for RAZUL S337CAtto. (A) Plot of RFU for 500 nM RAZUL S337CAtto (left) or RAZUL S337CDY (right) with 0 (red) or 2 (purple) equivalents of AZUL following excitation at 590 or 630 nm, respectively, with emission scanned from 600–750 nm or 640–800 nm, respectively, in 10 mM MOPS, pH 6.5, 50 mM NaCl, 5 mM DTT, and 10 μM ZnSO4. λmax values are indicated. (B) Change in RFU (relative to RFUbackground) for 500 nM of cysteine-substituted RAZUL labeled with acrylodan (left), Atto610 (middle), or DY647P1 (right) mixed with 0 (red) or 500 (purple) nM AZUL or 500 nM BSA (dark grey) (n = 3). Measurements were recorded in a 384-well plate in 10 mM MOPS, pH 6.5, 150 mM NaCl, 5 mM DTT, 10 μM ZnSO4, and 0.1% Tween 20. (C) Binding isotherms with raw ITC data (top insert) for injections of 90 μM E6AP AZUL into 9 μM RAZUL S337CAtto (left) or RAZUL S337CDY (right) in 10 mM MOPS, pH 6.5, 50 mM NaCl, 2 mM TCEP, and 10 μM ZnSO4. Fitted thermodynamic values are included in the bottom right of each panel: n, stoichiometry; Kd, binding affinity; ΔH, change in enthalpy; ΔS, change in entropy. | ||
We next evaluated how dye-labeled RAZUL S337C, S358C, or S361C compared in plate reader assays following addition of E6AP AZUL or BSA (negative control). Consistent with (Fig. 2C), AZUL caused an increase in the fluorescent intensity of RAZULAcr, with the largest increase for RAZUL S358CAcr (excitation: 390 nm; emission: 475 nm), Fig. 4B (left panel). As expected, no change was observed with BSA. AZUL caused no change for RAZULDY (excitation: 617 nm; emission: 667 nm), Fig. 4B (right panel), indicating that the minor decrease by fluorescence emission scanning was not detectable in 384-well plate reader assays. RAZULAtto fluorescence, however, was reduced by AZUL (excitation: 580 nm; emission: 632 nm), with greatest effect for S337CAtto, (Fig. 4B, middle panel). The lack of effect for RAZULDY was not caused by loss of affinity for AZUL, as the binding constant of RAZUL S337CDY and S337CAtto for AZUL was similar by ITC measurement (Fig. 4C and S7†). Notably, all cysteine-substituted positions on RAZULAcr and RAZULAtto were sensitive to AZUL binding and could be used as fluorescent probes to monitor the disorder-to-order conformational switch. For further HTP assay optimization, RAZUL S358C was chosen as the most sensitive position for E6AP binding with acrylodan-labeling, whereas Atto610-labeling was most sensitive at RAZUL S337C.
Evaluation of RFU with increasing quantities of AZUL for RAZUL S337CAtto (Fig. 5A) in comparison to RAZUL S358CAcr (Fig. 2D), with normalization to RFUmax, indicated similar sensitivity, but with a reversal in signal. Inflection points in plots of the normalized RFU signals for increasing concentration of AZUL ranged from 5–55 nM in assays with 25–500 nM RAZUL S337CAtto (Fig. 5A), resembling its binding affinity with E6AP AZUL (Fig. 4C, left panel). Addition of unlabeled RAZUL322–366 to 10 or 25 nM RAZUL S337CAtto:AZUL recovered the RFU signal (Fig. 5B) with a Ki of 0.664 ± 0.248 or 0.780 ± 0.199 μM, respectively. This Ki is less than the 1.26 ± 0.28 μM value for 25 nM RAZUL S358CAcr with AZUL, indicating that less RAZUL322–366 is required to compete with 25 nM RAZUL S337CAtto for AZUL compared to RAZUL S358CAcr. Moreover, binding (Fig. 5C) and competitive (Fig. 5D) assays were stable over time. We measured a 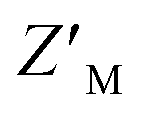 factor for DMSO and 50 μM RAZUL322–366 of 0.57 with 100 nM RAZUL S337CAtto:AZUL (Fig. 5E). Further reduction of material using 25 nM RAZUL S337CAtto:50 nM AZUL with and without 15 μM RAZUL322–366 maintained a similar
factor for DMSO and 50 μM RAZUL322–366 of 0.57 with 100 nM RAZUL S337CAtto:AZUL (Fig. 5E). Further reduction of material using 25 nM RAZUL S337CAtto:50 nM AZUL with and without 15 μM RAZUL322–366 maintained a similar 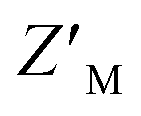 of 0.60 (Fig. 5F). Unlike RAZUL S358CAcr (Fig. 3D), the RAZUL S337CAtto response by the E6AP-induced conformational switch is sensitive enough to obtain robust
of 0.60 (Fig. 5F). Unlike RAZUL S358CAcr (Fig. 3D), the RAZUL S337CAtto response by the E6AP-induced conformational switch is sensitive enough to obtain robust 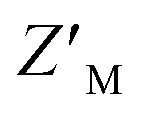 factors at lower quantities, which would be advantageous for HTP screening.
factors at lower quantities, which would be advantageous for HTP screening.
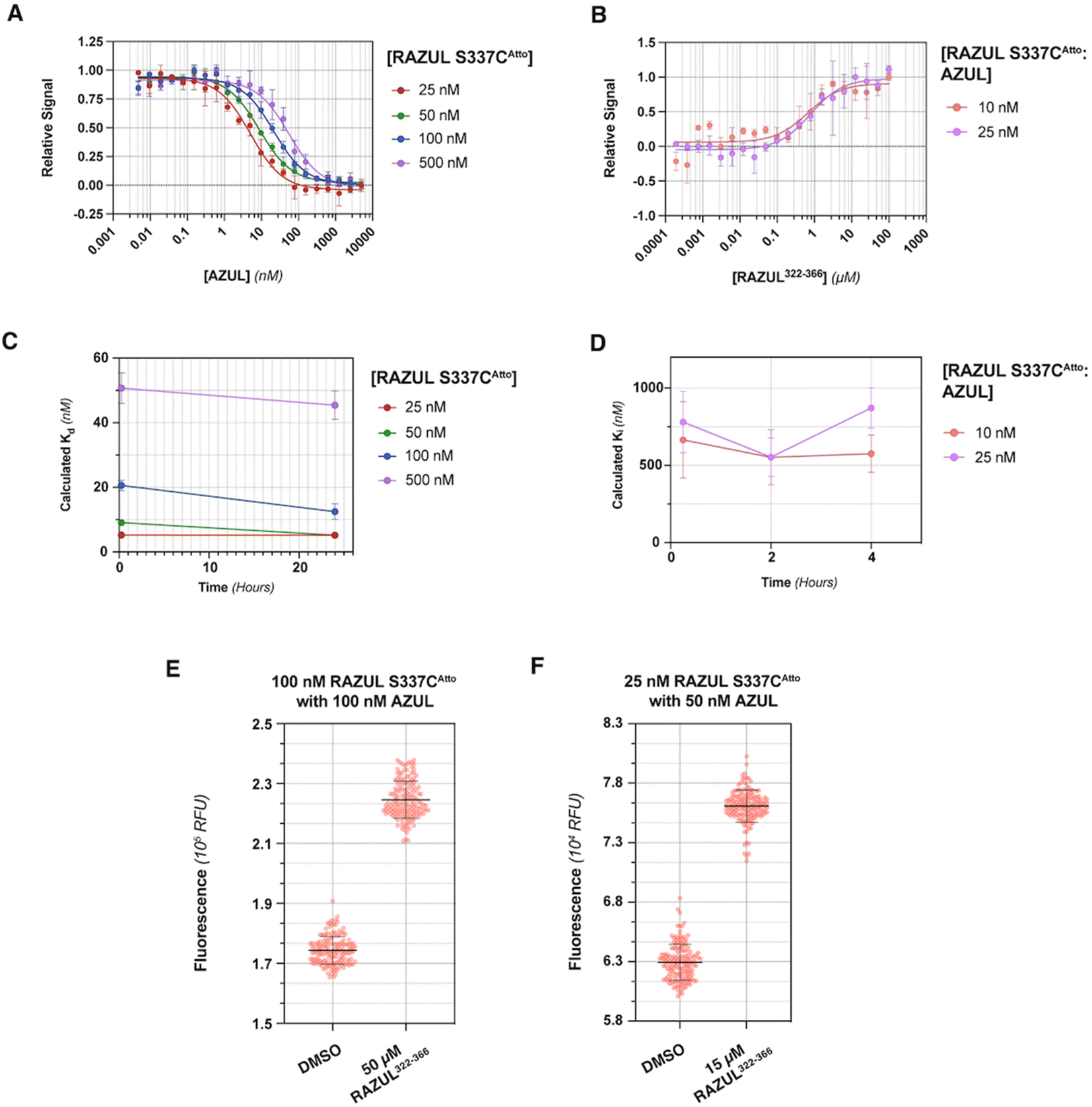 | ||
| Fig. 5 RAZUL S337CAtto shows similar sensitivity as RAZUL S358CAcr but with a reverse response. (A) Plot of RFU values normalized by RFUmax for 25 (red), 50 (green), 100 (blue), or 500 (purple) nM RAZUL S337CAtto with varying quantities of AZUL in a 384-well plate and 10 mM MOPS, pH 6.5, 150 mM NaCl, 5 mM DTT, 10 μM ZnSO4, and 0.1% Tween 20 (n = 3). (B) Plot of RFU values normalized by RFUmax for 10 (red) or 25 (purple) nM S358CAtto:AZUL against varying concentrations of RAZUL322–366 in the buffer of (A) supplemented with 5% DMSO (n = 3). (C) Plot of Kd values after 0.25 or 24 hours storage at 4 °C, with the 0.25 hour timepoint based on fluorescent measurements from (A). 25 (red), 50 (green), 100 (blue), or 500 (purple) nM RAZUL S337CAtto was prepared with 0–5000 nM E6AP AZUL in 10 mM MOPS, pH 6.5, 150 mM NaCl, 5 mM DTT, 10 μM ZnSO4, 0.1% Tween 20 (n = 3). After initial read of fluorescent values (shown in (A)), samples were stored at 4 °C and read by the CLARIOstar plate reader after 24 hours. (D) Plot of RAZUL322–366Ki values after 0.25, 2, or 4 hours storage at 4 °C based on fluorescence measurements of 10 (red) or 25 (purple) nM RAZUL S337CAtto:AZUL, with the 0.25 hour timepoint displayed in (B). The competition curves were plotted and fitted for Ki values. Those Ki values are plotted against storage time. In (A) through (D) the curves were fit in GraphPad Prism by dose response with variable slope and each data point is represented as mean ± SEM. (E) RFU for DMSO versus 50 μM RAZUL322–366 with 100 nM S337CAtto:AZUL (n = 154) in the buffer of (A) supplemented with 1% BSA and 5% DMSO. (F) RFU of 25 nM S337CAtto:50 nM AZUL with DMSO (vehicle control) or 15 μM RAZUL322–366 in buffer supplemented with 1% BSA and 5% DMSO (n = 154). For all experiments, excitation and emission wavelengths of 580 nm and 632 nm were used, respectively. | ||
Both RAZUL S337CAtto and RAZUL S358CAcr achieved adequate 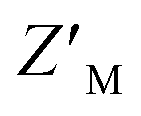 factors as single fluorescent conformational switch probes, but their differential spectral properties and responses upon AZUL binding led us to explore an assay that measured response with RAZUL S358CAcr and RAZUL S337CAtto present in the same well. We hypothesized that the incorporation of two fluorescent sensors that excite and emit at different wavelengths in the same well (Fig. 6A) could reduce false positives due to screening compound interference (quenching or autofluorescence).
factors as single fluorescent conformational switch probes, but their differential spectral properties and responses upon AZUL binding led us to explore an assay that measured response with RAZUL S358CAcr and RAZUL S337CAtto present in the same well. We hypothesized that the incorporation of two fluorescent sensors that excite and emit at different wavelengths in the same well (Fig. 6A) could reduce false positives due to screening compound interference (quenching or autofluorescence).
 | ||
| Fig. 6 Dual application of RAZUL S358CAcr and RAZUL S337CAtto yields an orthogonal assay with sensitivity appropriate for HTP screening. (A) Concept of an orthogonal fluorescent-based assay with RAZUL S358CAcr and RAZUL S337CAtto in a common well with E6AP AZUL yielding opposite RFU response to the RAZUL disorder-to-order conformational switch. With the introduction of a competitor (such as positive control RAZUL322–366), the fluorescent probes are expected to have reversed yet complimentary fluorescent responses. (B) Plot of RFU normalized by RFUmax for 25 nM RAZUL S358CAcr (blue) and 25 nM RAZUL S337CAtto (red) in a common well against varying amounts of AZUL in 10 mM MOPS, pH 6.5, 150 mM NaCl, 5 mM DTT, 10 μM ZnSO4, and 0.1% Tween 20. (C) Plot of Kd values of AZUL binding based on fluorescence measurements of RAZUL S358CAcr (blue) and RAZUL S337CAtto (red) after 0.25, 2, or 4 hours storage at 4 °C for a mixture of 25 nM RAZUL S358CAcr and 25 nM RAZUL S337CAtto, with the 0.25 hour timepoint displayed in (B). For (B) and (C), each data point is represented as mean ± SEM. (D) RFU for 100 nM S358CAcr (blue, left), S337CAtto (red, right), and E6AP AZUL following incubation with DMSO or 50 μM RAZUL322–366 in 10 mM MOPS, pH 6.5, 150 mM NaCl, 5 mM DTT, 10 μM ZnSO4, 0.1% Tween 20, 1% BSA, and 5% DMSO (n = 154). (E) Ratiometric RFU value with S358CAcr or S337CAtto in the numerator or denominator as indicated (n = 154). In (B) and (C), plots were generated by GraphPad Prism and fit by dose response with variable slope. In (B) through (E), Acr signal is the fluorescence measured with an excitation of 390 nm and emission of 475 nm, whereas Atto signal is measured with an excitation and emission of 580 nm and 632 nm, respectively. | ||
Dual monitoring of RAZUL S358CAcr and RAZUL S337CAtto achieves excellent robustness, enabling HTP screening
To determine whether RAZUL S358CAcr and RAZUL S337CAtto can detect the E6AP-induced conformational switch simultaneously, the RFU normalized to RFUmax for 25 nM of a mixture of each fluorescently labeled RAZUL domain was measured and plotted with varying concentrations of E6AP AZUL (Fig. 6B and C). In this dually orthogonal assay, RAZUL S358CAcr and RAZUL S337CAtto obtained inflection points of 2.97 ± 0.22 nM and 16.3 ± 1.2 nM at the 0.25 h timepoint, respectively. By ITC, RAZUL S358CAcr:AZUL (Fig. 2A) was measured to have a similar affinity to RAZUL S337CAtto:AZUL (Fig. 4C), but in a common sample, AZUL bound preferentially to RAZUL S358CAcr over RAZUL S337CAtto. The increase in the fluorescence of RAZUL S358CAcr with concomitant decrease for RAZUL S337CAtto following AZUL binding indicates that an inhibitory molecule would decrease S358CAcr fluorescence while increasing S337CAtto fluorescence at their respective excitation/emission wavelengths (Fig. 6A, bottom panel). With a positive control of 50 μM RAZUL322–366 and DMSO as a negative control, the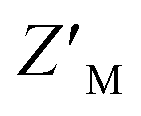 factor of RAZUL S358CAcr and RAZUL S337CAtto were measured to be 0.74 and 0.54, respectively (Fig. 6D). Comparison of how RAZUL S358CAcr (Fig. 3C) and S337CAtto (Fig. 5E) perform individually versus in a common assay (Fig. 6D) indicates the RAZUL S358CAcr
factor of RAZUL S358CAcr and RAZUL S337CAtto were measured to be 0.74 and 0.54, respectively (Fig. 6D). Comparison of how RAZUL S358CAcr (Fig. 3C) and S337CAtto (Fig. 5E) perform individually versus in a common assay (Fig. 6D) indicates the RAZUL S358CAcr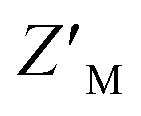 factor to increase from 0.63 to 0.74 while that of RAZUL S337CAtto slightly decreased from 0.57 to 0.54. Since these fluorophores monitor the RAZUL conformational switch in the same sample, a comparison within each well for RAZUL S358CAcr and RAZUL S337CAtto can be computed to obtain a ratiometric
factor to increase from 0.63 to 0.74 while that of RAZUL S337CAtto slightly decreased from 0.57 to 0.54. Since these fluorophores monitor the RAZUL conformational switch in the same sample, a comparison within each well for RAZUL S358CAcr and RAZUL S337CAtto can be computed to obtain a ratiometric  factor for RAZUL S337CAtto/S358CAcr or S358CAcr/S337CAtto of 0.73 (Fig. 6E, left panel) or 0.75 (Fig. 6E, right panel), respectively. These values indicate excellent robustness, as required for large scale HTP applications. Thus, this dually orthogonal assay is appropriate for HTP screening, and the combination of two fluorescent sensors will reduce false positives (Fig. 7) to aid in identification of true primary hits.
factor for RAZUL S337CAtto/S358CAcr or S358CAcr/S337CAtto of 0.73 (Fig. 6E, left panel) or 0.75 (Fig. 6E, right panel), respectively. These values indicate excellent robustness, as required for large scale HTP applications. Thus, this dually orthogonal assay is appropriate for HTP screening, and the combination of two fluorescent sensors will reduce false positives (Fig. 7) to aid in identification of true primary hits.
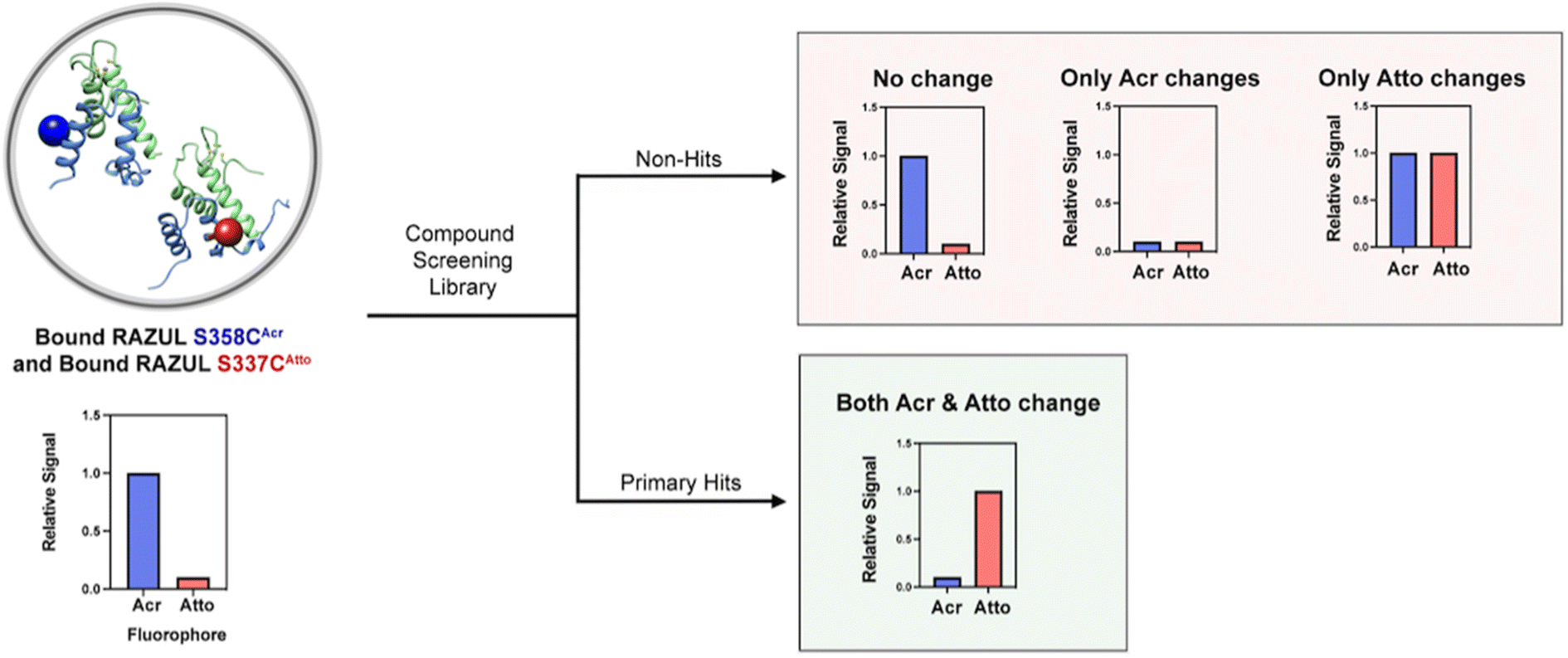 | ||
| Fig. 7 Model of a dually orthogonal assay with RAZUL S358CAcr/S337CAtto for elimination of false positives in a HTP screening assay that includes compounds with intrinsic fluorescent properties. Bound RAZUL S358CAcr/S337CAtto begins with high fluorescence in the acrylodan (ex. 390 nm; em. 475 nm) channel and low fluorescence in the Atto610 channel (ex. 580 nm; em. 632 nm). After introduction of a compound, a primary hit that inhibits the RAZUL:AZUL interaction will exhibit concomitant responses such that fluorescence of acrylodan decreases and Atto610 increases. Non-hits will elicit no change or only change one of the fluorescence channels. | ||
Conclusions
Here, we describe the design and development of a dually orthogonal fluorescent-based assay that can be used in a HTP screening campaign for Rpn10:E6AP chemical probes. Rpn10 RAZUL exhibits a disorder-to-order transition when associating with E6AP AZUL, and this conformational switch is captured with acrylodan- and Atto610-labeled Rpn10 RAZUL. Three RAZUL serines were cysteine-substituted to allow for nucleophilic addition to the acrylodan α,β-unsaturated ketone or Atto610 maleimide, with RAZUL S358C and S337C identified as the respective optimal positions. All acrylodan and Atto610 labeled RAZUL probes were sensitive to AZUL binding, but desirable placement for high signal-to-noise ratio was found to be fluorophore-dependent and affinity-independent, indicating local environmental reorientation dictates the extent of spectral change. Previous applications of acrylodan-based screening assays focused on the solvent-exposure of allosteric sites for kinases50–55 and phosphatases.56 Acrylodan-labeling of intrinsically disordered tau also creatively provided insight into how tau assembles into tubulin dimers, with major spectral hypsochromic shifts indicating that acrylodan was in a more hydrophobic environment.35 The greater hypsochromic shift of acrylodan at RAZUL S358C compared to S337CAcr and S361CAcr suggests that when at position 358, acrylodan is directed towards the RAZUL:AZUL interface. Similarly, the significant fluorescence change with Atto610 at RAZUL S337C and not when at S358C or S361C suggests that Atto610 is directed towards the RAZUL:AZUL interface when at position 337. The disparate location for optimal response to AZUL binding could originate from the longer linker region used with the Atto610 fluorophore compared to acrylodan (Fig. S2B and S4B,† respectively).Competition with unlabeled RAZUL322–366 indicated individual 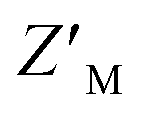 factors ranging from 0.35–0.63; however, the unique spectral signatures and response of RAZUL S358CAcr/S337CAtto binding to AZUL enabled their application in a common sample and a resulting ratiometric
factors ranging from 0.35–0.63; however, the unique spectral signatures and response of RAZUL S358CAcr/S337CAtto binding to AZUL enabled their application in a common sample and a resulting ratiometric 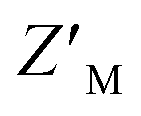 factor of 0.75. Molecules identified in future screening campaigns with this assay may bind to either RAZUL or AZUL. RAZUL-binding compounds could be rationally designed for direct 26S proteasome recruitment of a target substrate, akin to conjugating a 19S RP Rpn1-binding ligand to a potent BRD4 ligand for targeted protein degradation,57 whereas AZUL-binding compounds can be utilized to interrogate E6AP's function at Rpn10, the proteasome, or its other binding partners, such as UBQLN.58 Finally, our assay could be modified to identify molecular glues for Rpn10:E6AP, which could be therapeutically beneficial in diseases where mutations lessen their affinity.28
factor of 0.75. Molecules identified in future screening campaigns with this assay may bind to either RAZUL or AZUL. RAZUL-binding compounds could be rationally designed for direct 26S proteasome recruitment of a target substrate, akin to conjugating a 19S RP Rpn1-binding ligand to a potent BRD4 ligand for targeted protein degradation,57 whereas AZUL-binding compounds can be utilized to interrogate E6AP's function at Rpn10, the proteasome, or its other binding partners, such as UBQLN.58 Finally, our assay could be modified to identify molecular glues for Rpn10:E6AP, which could be therapeutically beneficial in diseases where mutations lessen their affinity.28
Despite the anticipated application of this dually orthogonal Rpn10:E6AP assay, there are inherent limitations, including the slow rate of dissociation (2.11 × 10−2 s−1)26 and <20 nM binding affinity of the native protein–protein interaction, making detection of inhibitors with micromolar affinity challenging. This limitation could be overcome however by making structure-based amino acid substitutions in either RAZUL or AZUL that weaken their binding affinity. Additionally, since this assay depends on the helical formation of Rpn10 RAZUL, screening molecules that non-specifically disrupt helicity would fall into the false positive pool. Thus, as with most screening assays, primary hits will require secondary validation by other biophysical or structural techniques.
To biochemically apply our approach to conformational switches, induced folding (or loss of folding) can be confirmed with the native and modified protein systems prior to fluorescent labeling through integrative biophysical/computational techniques such as CD,26,59 NMR,26,60,61 small X-ray light scattering (SAXS),62,63 single-molecule fluorescence resonance energy transfer (smFRET),64,65 and/or molecular modeling.66,67 Then, binding affinity retention with the unlabeled/fluorescently labeled proteins and their binding partner can be investigated through the suitable biophysical methods, such as NMR, ITC, SPR, and/or biolayer interferometry (BLI).68 If these methods are not accessible, however, acrylodan or Atto610 labeling of a disordered protein-of-interest with high efficiency, as described here, and monitoring fluorescent response under varied conditions can be investigated in 96-well and 384-well plate format. Like Rpn10 RAZUL, there are several therapeutically-relevant proteins that are intrinsically disordered, contain disordered domains, and/or exhibit extensive folding upon binding to a partner.69–72 An attractive cancer target that contains disordered regions is tumor suppressor p53,43,44 and we expect that environmentally sensitive fluorophores could be applied to discover p53-binding compounds and/or compounds that disrupt the E6-driven interaction of E6AP with p53. As we illustrate here with the RAZUL:AZUL high affinity interaction, the use of environmentally sensitive fluorophores presents screening opportunities for protein systems that exhibit such ubiquitous disorder-to-order (or vice versa) transitions.
Data availability
All data are available in the main text or the ESI.†Author contributions
C. S. M., S. G. T., and K. J. W. designed the studies. C. S. M. performed protein expression, purification, labeling, and plate reader assay experiments. C. S. M. and S. G. T. conducted and analyzed CD, fluorimeter, and isothermal titration calorimetry experiments. C. S. M. and K. J. W. wrote the initial draft of the manuscript, and all authors contributed to review and editing of the final manuscript.Conflicts of interest
The authors declare no competing interests.Acknowledgements
This work was supported by the Intramural Research Program through the Center for Cancer Research (CCR), National Cancer Institute, National Institutes of Health (1ZIABC011627), and the CCR FLEX Program. C. S. M. was supported by the Intramural Continuing Umbrella of Research Experiences (iCURE) program. We thank Marzena Dyba from the Biophysics Resource Core in the Center for Structural Biology, CCR, for assistance with LC-MS. We also thank Barry O'Keefe from the Molecular Targets Program (CCR, NCI, NIH) for scientific discussions and suggesting optimization conditions for the assay.References
- I. Dikic and B. A. Schulman, An expanded lexicon for the ubiquitin code, Nat. Rev. Mol. Cell Biol., 2023, 24(4), 273–287, DOI:10.1038/s41580-022-00543-1.
- V. Osei-Amponsa and K. J. Walters, Proteasome substrate receptors and their therapeutic potential, Trends Biochem. Sci., 2022, 47(11), 950–964, DOI:10.1016/j.tibs.2022.06.006.
- K. M. Sakamoto, K. B. Kim, A. Kumagai, F. Mercurio, C. M. Crews and R. J. Deshaies, Protacs: chimeric molecules that target proteins to the Skp1–Cullin–F box complex for ubiquitination and degradation, Proc. Natl. Acad. Sci. U. S. A., 2001, 98(15), 8554–8559, DOI:10.1073/pnas.141230798.
- Y. Wang, X. Jiang, F. Feng, W. Liu and H. Sun, Degradation of proteins by PROTACs and other strategies, Acta Pharm. Sin. B, 2020, 10(2), 207–238, DOI:10.1016/j.apsb.2019.08.001.
- S. Zeng, W. Huang, X. Zheng, L. Cheng, Z. Zhang, J. Wang and Z. Shen, Proteolysis targeting chimera (PROTAC) in drug discovery paradigm: recent progress and future challenges, Eur. J. Med. Chem., 2021, 210, 112981, DOI:10.1016/j.ejmech.2020.112981.
- M. Békés, D. R. Langley and C. M. Crews, PROTAC targeted protein degraders: the past is prologue, Nat. Rev. Drug Discovery, 2022, 21(3), 181–200, DOI:10.1038/s41573-021-00371-6.
- T. Ishida and A. Ciulli, E3 Ligase Ligands for PROTACs: How They Were Found and How to Discover New Ones, SLAS Discovery, 2021, 26(4), 484–502, DOI:10.1177/2472555220965528.
- M. Scheffner, J. M. Huibregtse, R. D. Vierstra and P. M. Howley, The HPV-16 E6 and E6-AP complex functions as a ubiquitin-protein ligase in the ubiquitination of p53, Cell, 1993, 75(3), 495–505, DOI:10.1016/0092-8674(93)90384-3.
- M. Traidej, L. Chen, D. Yu, S. Agrawal and J. Chen, The Roles of E6-AP and MDM2 in p53 Regulation in Human Papillomavirus-Positive Cervical Cancer Cells, Antisense Nucleic Acid Drug Dev., 2000, 10(1), 17–27, DOI:10.1089/oli.1.2000.10.17.
- S. Dymalla, M. Scheffner, E. Weber, P. Sehr, C. Lohrey, F. Hoppe-Seyler and K. Hoppe-Seyler, A novel peptide motif binding to and blocking the intracellular activity of the human papillomavirus E6 oncoprotein, J. Mol. Med., 2009, 87(3), 321–331, DOI:10.1007/s00109-008-0432-1.
- D. Martinez-Zapien, F. X. Ruiz, J. Poirson, A. Mitschler, J. Ramirez, A. Forster, A. Cousido-Siah, M. Masson, S. Vande Pol and A. Podjarny, et al., Structure of the E6/E6AP/p53 complex required for HPV-mediated degradation of p53, Nature, 2016, 529(7587), 541–545, DOI:10.1038/nature16481.
- C. Sailer, F. Offensperger, A. Julier, K.-M. Kammer, R. Walker-Gray, M. G. Gold, M. Scheffner and F. Stengel, Structural dynamics of the E6AP/UBE3A-E6-p53 enzyme-substrate complex, Nat. Commun., 2018, 9(1), 4441, DOI:10.1038/s41467-018-06953-0.
- S. Srinivasan and Z. Nawaz, E3 ubiquitin protein ligase, E6-associated protein (E6-AP) regulates PI3K-Akt signaling and prostate cell growth, Biochim. Biophys. Acta, 2011, 1809(2), 119–127, DOI:10.1016/j.bbagrm.2010.08.011.
- P. J. Paul, D. Raghu, A. L. Chan, T. Gulati, L. Lambeth, E. Takano, M. J. Herold, J. Hagekyriakou, R. L. Vessella and C. Fedele, et al., Restoration of tumor suppression in prostate cancer by targeting the E3 ligase E6AP, Oncogene, 2016, 35(48), 6235–6245, DOI:10.1038/onc.2016.159.
- L. Li, Z. Li, P. M. Howley and D. B. Sacks, E6AP and Calmodulin Reciprocally Regulate Estrogen Receptor Stability, J. Biol. Chem., 2006, 281(4), 1978–1985, DOI:10.1074/jbc.M508545200.
- A. Mani, A. S. Oh, E. T. Bowden, T. Lahusen, K. L. Lorick, A. M. Weissman, R. Schlegel, A. Wellstein and A. T. Riegel, E6AP Mediates Regulated Proteasomal Degradation of the Nuclear Receptor Coactivator Amplified in Breast Cancer 1 in Immortalized Cells, Cancer Res., 2006, 66(17), 8680–8686, DOI:10.1158/0008-5472.Can-06-0557 , acccessed 10/3/2023..
- R. C. Samaco, A. Hogart and J. M. LaSalle, Epigenetic overlap in autism-spectrum neurodevelopmental disorders: MECP2 deficiency causes reduced expression of UBE3A and GABRB3, Hum. Mol. Genet., 2005, 14(4), 483–492, DOI:10.1093/hmg/ddi045.
- S. Y. Lee, J. Ramirez, M. Franco, B. Lectez, M. Gonzalez, R. Barrio and U. Mayor, Ube3a, the E3 ubiquitin ligase causing Angelman syndrome and linked to autism, regulates protein homeostasis through the proteasomal shuttle Rpn10, Cell. Mol. Life Sci., 2014, 71(14), 2747–2758, DOI:10.1007/s00018-013-1526-7.
- X. Xu, C. Li, X. Gao, K. Xia, H. Guo, Y. Li, Z. Hao, L. Zhang, D. Gao and C. Xu, et al., Excessive UBE3A dosage impairs retinoic acid signaling and synaptic plasticity in autism spectrum disorders, Cell Research, 2018, 28(1), 48–68, DOI:10.1038/cr.2017.132.
- M. Elamin, A. Dumarchey, C. Stoddard, T. M. Robinson, C. Cowie, D. Gorka, S. J. Chamberlain and E. S. Levine, The role of UBE3A in the autism and epilepsy-related Dup15q syndrome using patient-derived, CRISPR-corrected neurons, Stem Cell Rep., 2023, 18(4), 884–898, DOI:10.1016/j.stemcr.2023.02.002.
- T. Kishino, M. Lalande and J. Wagstaff, UBE3A/E6-AP mutations cause Angelman syndrome, Nat. Genet., 1997, 15(1), 70–73, DOI:10.1038/ng0197-70.
- S. Kühnle, G. Martínez-Noël, F. Leclere, S. D. Hayes, J. W. Harper and P. M. Howley, Angelman syndrome-associated point mutations in the Zn2+-binding N-terminal (AZUL) domain of UBE3A ubiquitin ligase inhibit binding to the proteasome, J. Biol. Chem., 2018, 293(47), 18387–18399, DOI:10.1074/jbc.RA118.004653.
- T. Matsuura, J. S. Sutcliffe, P. Fang, R. J. Galjaard, Y. H. Jiang, C. S. Benton, J. M. Rommens and A. L. Beaudet, De novo truncating mutations in E6-AP ubiquitin-protein ligase gene (UBE3A) in Angelman syndrome, Nat. Genet., 1997, 15(1), 74–77, DOI:10.1038/ng0197-74.
- E. M. Cooper, A. W. Hudson, J. Amos, J. Wagstaff and P. M. Howley, Biochemical analysis of Angelman syndrome-associated mutations in the E3 ubiquitin ligase E6-associated protein, J. Biol. Chem., 2004, 279(39), 41208–41217, DOI:10.1074/jbc.M401302200.
- G. Martínez-Noël, J. T. Galligan, M. E. Sowa, V. Arndt, T. M. Overton, J. W. Harper and P. M. Howley, Identification and proteomic analysis of distinct UBE3A/E6AP protein complexes, Mol. Cell. Biol., 2012, 32(15), 3095–3106, DOI:10.1128/mcb.00201-12.
- G. R. Buel, X. Chen, R. Chari, M. J. O'Neill, D. L. Ebelle, C. Jenkins, V. Sridharan, S. G. Tarasov, N. I. Tarasova, T. Andresson and K. J. Walters, Structure of E3 ligase E6AP with a proteasome-binding site provided by substrate receptor hRpn10, Nat. Commun., 2020, 11(1), 1291, DOI:10.1038/s41467-020-15073-7.
- C. L. Sirois, J. E. Bloom, J. J. Fink, D. Gorka, S. Keller, N. D. Germain, E. S. Levine and S. J. Chamberlain, Abundance and localization of human UBE3A protein isoforms, Hum. Mol. Genet., 2020, 29(18), 3021–3031, DOI:10.1093/hmg/ddaa191.
- R. Avagliano Trezza, M. Sonzogni, S. N. V. Bossuyt, F. I. Zampeta, A. M. Punt, M. van den Berg, D. C. Rotaru, L. M. C. Koene, S. T. Munshi and J. Stedehouder, et al., Loss of nuclear UBE3A causes electrophysiological and behavioral deficits in mice and is associated with Angelman syndrome, Nat. Neurosci., 2019, 22(8), 1235–1247, DOI:10.1038/s41593-019-0425-0.
- F. I. Zampeta, M. Sonzogni, E. Niggl, B. Lendemeijer, H. Smeenk, F. M. S. de Vrij, S. A. Kushner, B. Distel and Y. Elgersma, Conserved UBE3A subcellular distribution between human and mice is facilitated by non-homologous isoforms, Hum. Mol. Genet., 2020, 29(18), 3032–3043, DOI:10.1093/hmg/ddaa194.
- F. Offensperger, F. Müller, J. Jansen, D. Hammler, K. H. Götz, A. Marx, C. L. Sirois, S. J. Chamberlain, F. Stengel and M. Scheffner, Identification of Small-Molecule Activators of the Ubiquitin Ligase E6AP/UBE3A and Angelman Syndrome-Derived E6AP/UBE3A Variants, Cell Chem. Biol., 2020, 27(12), 1510–1520, DOI:10.1016/j.chembiol.2020.08.017.
- B. Huang, L. Zhou, R. Liu, L. Wang, S. Xue, Y. Shi, G. H. Jeong, I. H. Jeong, S. Li, J. Yin and J. Cai, Activation of E6AP/UBE3A-Mediated Protein Ubiquitination and Degradation Pathways by a Cyclic γ-AA Peptide, J. Med. Chem., 2022, 65(3), 2497–2506, DOI:10.1021/acs.jmedchem.1c01922.
- S. Dengler, R. T. Howard, V. Morozov, C. Tsiamantas, W. E. Huang, Z. Liu, C. Dobrzanski, V. Pophristic, S. Brameyer and C. Douat, et al., Display Selection of a Hybrid Foldamer-Peptide Macrocycle, Angew Chem. Int. Ed. Engl., 2023, 62(46), e202308408, DOI:10.1002/anie.202308408.
- T. Du, Y. Song, A. Ray, X. Wan, Y. Yao, M. K. Samur, C. Shen, J. Penailillo, T. Sewastianik and Y.-T. Tai, et al., Ubiquitin receptor PSMD4/Rpn10 is a novel therapeutic target in multiple myeloma, Blood, 2023, 141(21), 2599–2614, DOI:10.1182/blood.2022017897.
- R. Hantani, S. Hanawa, S. Oie, K. Umetani, T. Sato and Y. Hantani, Identification of a New Inhibitor That Stabilizes Interleukin-2-Inducible T-Cell Kinase in Its Inactive Conformation, SLAS Discovery, 2019, 24(8), 854–862, DOI:10.1177/2472555219857542.
- X. H. Li, J. A. Culver and E. Rhoades, Tau Binds to Multiple Tubulin Dimers with Helical Structure, J. Am. Chem. Soc., 2015, 137(29), 9218–9221, DOI:10.1021/jacs.5b04561.
- S. J. Metallo, Intrinsically disordered proteins are potential drug targets, Curr. Opin. Chem. Biol., 2010, 14(4), 481–488, DOI:10.1016/j.cbpa.2010.06.169.
- P. Santofimia-Castano, B. Rizzuti, Y. Xia, O. Abian, L. Peng, A. Velazquez-Campoy, J. L. Neira and J. Iovanna, Targeting intrinsically disordered proteins involved in cancer, Cell. Mol. Life Sci., 2020, 77(9), 1695–1707, DOI:10.1007/s00018-019-03347-3.
- S. Saurabh, K. Nadendla, S. S. Purohit, P. M. Sivakumar and S. Cetinel, Fuzzy Drug Targets: Disordered Proteins in the Drug-Discovery Realm, ACS Omega, 2023, 8(11), 9729–9747, DOI:10.1021/acsomega.2c07708.
- C. A. Galea, A. Nourse, Y. Wang, S. G. Sivakolundu, W. T. Heller and R. W. Kriwacki, Role of intrinsic flexibility in signal transduction mediated by the cell cycle regulator, p27 Kip1, J. Mol. Biol., 2008, 376(3), 827–838, DOI:10.1016/j.jmb.2007.12.016.
- M. Tsytlonok, H. Sanabria, Y. Wang, S. Felekyan, K. Hemmen, A. H. Phillips, M. K. Yun, M. B. Waddell, C. G. Park and S. Vaithiyalingam, et al., Dynamic anticipation by Cdk2/Cyclin A-bound p27 mediates signal integration in cell cycle regulation, Nat. Commun., 2019, 10(1), 1676, DOI:10.1038/s41467-019-09446-w.
- E. M. Blackwood and R. N. Eisenman, Max: A Helix-Loop-Helix Zipper Protein That Forms a Sequence-Specific DNA-Binding Complex with Myc, Science, 1992, 251(4998), 1211–1217, DOI:10.1126/science.2006410.
- S. Schutz, C. Bergsdorf, B. Goretzki, A. Lingel, M. Renatus, A. D. Gossert and W. Jahnke, The Disordered MAX N-Terminus Modulates DNA Binding of the Transcription Factor MYC:MAX, J. Mol. Biol., 2022, 434(22), 167833, DOI:10.1016/j.jmb.2022.167833.
- A. C. Joerger and A. R. Fersht, The tumor suppressor p53: from structures to drug discovery, Cold Spring Harbor Perspect. Biol., 2010, 2(6), a000919 Search PubMed.
- M. M. Maslon and T. R. Hupp, Drug discovery and mutant p53, Trends Cell Biol., 2010, 20(9), 542–555 CrossRef CAS PubMed.
- H.-F. Chen and R. Luo, Binding Induced Folding in p53-MDM2 Complex, J. Am. Chem. Soc., 2007, 129(10), 2930–2937, DOI:10.1021/ja067877.
- K. E. D. Coan and B. K. Shoichet, Stability and equilibria of promiscuous aggregates in high protein milieus, Mol. BioSyst., 2007, 3(3), 208–213, 10.1039/B616314A.
- J. H. Zhang, T. D. Chung and K. R. Oldenburg, A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays, J. Biomol. Screening, 1999, 4(2), 67–73, DOI:10.1177/108705719900400206.
- R. Atmaramani, J. J. Pancrazio and B. J. Black, Adaptation of robust Z′ factor for assay quality assessment in microelectrode array based screening using adult dorsal root ganglion neurons, J. Neurosci. Methods, 2020, 339, 108699, DOI:10.1016/j.jneumeth.2020.108699.
- A. Birmingham, L. M. Selfors, T. Forster, D. Wrobel, C. J. Kennedy, E. Shanks, J. Santoyo-Lopez, D. J. Dunican, A. Long and D. Kelleher, et al., Statistical methods for analysis of high-throughput RNA interference screens, Nat. Methods, 2009, 6(8), 569–575, DOI:10.1038/nmeth.1351.
- R. Schneider, A. Gohla, J. R. Simard, D. B. Yadav, Z. Fang, W. A. van Otterlo and D. Rauh, Overcoming compound fluorescence in the FLiK screening assay with red-shifted fluorophores, J. Am. Chem. Soc., 2013, 135(22), 8400–8408, DOI:10.1021/ja403074j.
- M. Getlik, J. R. Simard, M. Termathe, C. Grutter, M. Rabiller, W. A. van Otterlo and D. Rauh, Fluorophore labeled kinase detects ligands that bind within the MAPK insert of p38alpha kinase, PLoS One, 2012, 7(7), e39713, DOI:10.1371/journal.pone.0039713.
- R. Schneider, C. Becker, J. R. Simard, M. Getlik, N. Bohlke, P. Janning and D. Rauh, Direct binding assay for the detection of type IV allosteric inhibitors of Abl, J. Am. Chem. Soc., 2012, 134(22), 9138–9141, DOI:10.1021/ja303858w.
- J. R. Simard, M. Getlik, C. Grütter, R. Schneider, S. Wulfert and D. Rauh, Fluorophore Labeling of the Glycine-Rich Loop as a Method of Identifying Inhibitors That Bind to Active and Inactive Kinase Conformations, J. Am. Chem. Soc., 2010, 132(12), 4152–4160, DOI:10.1021/ja908083e.
- J. R. Simard, C. Grütter, V. Pawar, B. Aust, A. Wolf, M. Rabiller, S. Wulfert, A. Robubi, S. Klüter, C. Ottmann and D. Rauh, High-Throughput Screening to Identify Inhibitors Which Stabilize Inactive Kinase Conformations in p38α, J. Am. Chem. Soc., 2009, 131(51), 18478–18488, DOI:10.1021/ja907795q.
- J. R. Simard, S. Kluter, C. Grutter, M. Getlik, M. Rabiller, H. B. Rode and D. Rauh, A new screening assay for allosteric inhibitors of cSrc, Nat. Chem. Biol., 2009, 5(6), 394–396, DOI:10.1038/nchembio.162.
- R. Schneider, C. Beumer, J. R. Simard, C. Grütter and D. Rauh, Selective Detection of Allosteric Phosphatase Inhibitors, J. Am. Chem. Soc., 2013, 135(18), 6838–6841, DOI:10.1021/ja4030484.
- C. Bashore, S. Prakash, M. C. Johnson, R. J. Conrad, I. A. Kekessie, S. J. Scales, N. Ishisoko, T. Kleinheinz, P. S. Liu and N. Popovych, et al., Targeted degradation via direct 26S proteasome recruitment, Nat. Chem. Biol., 2023, 19(1), 55–63, DOI:10.1038/s41589-022-01218-w.
- G. R. Buel, X. Chen, W. Myint, O. Kayode, V. Folimonova, A. Cruz, K. A. Skorupka, H. Matsuo and K. J. Walters, E6AP AZUL interaction with UBQLN1/2 in cells, condensates, and an AlphaFold-NMR integrated structure, Structure, 2023, 31(4), 395–410, DOI:10.1016/j.str.2023.01.012.
- B. A. Wallace, The role of circular dichroism spectroscopy in the era of integrative structural biology, Curr. Opin. Struct. Biol., 2019, 58, 191–196, DOI:10.1016/j.sbi.2019.04.001.
- C. Charlier, G. Bouvignies, P. Pelupessy, A. Walrant, R. Marquant, M. Kozlov, P. De Ioannes, N. Bolik-Coulon, S. Sagan and P. Cortes, et al., Structure and Dynamics of an Intrinsically Disordered Protein Region That Partially Folds upon Binding by Chemical-Exchange NMR, J. Am. Chem. Soc., 2017, 139(35), 12219–12227, DOI:10.1021/jacs.7b05823.
- W. Adamski, N. Salvi, D. Maurin, J. Magnat, S. Milles, M. R. Jensen, A. Abyzov, C. J. Moreau and M. Blackledge, A Unified Description of Intrinsically Disordered Protein Dynamics under Physiological Conditions Using NMR Spectroscopy, J. Am. Chem. Soc., 2019, 141(44), 17817–17829, DOI:10.1021/jacs.9b09002.
- A. G. Kikhney and D. I. Svergun, A practical guide to small angle X-ray scattering (SAXS) of flexible and intrinsically disordered proteins, FEBS Lett., 2015, 589(19 Pt A), 2570–2577, DOI:10.1016/j.febslet.2015.08.027.
- P. Bernado and D. I. Svergun, Structural analysis of intrinsically disordered proteins by small-angle X-ray scattering, Mol. BioSyst., 2012, 8(1), 151–167, 10.1039/c1mb05275f.
- S. J. LeBlanc, P. Kulkarni and K. R. Weninger, Single Molecule FRET: A Powerful Tool to Study Intrinsically Disordered Proteins, Biomolecules, 2018, 8(4), 140, DOI:10.3390/biom8040140.
- L. A. Metskas and E. Rhoades, Single-Molecule FRET of Intrinsically Disordered Proteins, Annu. Rev. Phys. Chem., 2020, 71, 391–414, DOI:10.1146/annurev-physchem-012420-104917.
- G. W. Gomes, M. Krzeminski, A. Namini, E. W. Martin, T. Mittag, T. Head-Gordon, J. D. Forman-Kay and C. C. Gradinaru, Conformational Ensembles of an Intrinsically Disordered Protein Consistent with NMR, SAXS, and Single-Molecule FRET, J. Am. Chem. Soc., 2020, 142(37), 15697–15710, DOI:10.1021/jacs.0c02088.
- P. Robustelli, S. Piana and D. E. Shaw, Mechanism of Coupled Folding-upon-Binding of an Intrinsically Disordered Protein, J. Am. Chem. Soc., 2020, 142(25), 11092–11101, DOI:10.1021/jacs.0c03217.
- N. B. Shah and T. M. Duncan, Bio-layer interferometry for measuring kinetics of protein-protein interactions and allosteric ligand effects, J. Visualized Exp., 2014, 84, e51383, DOI:10.3791/51383.
- C. Y.-C. Chen and W. I. Tou, How to design a drug for the disordered proteins?, Drug Discovery Today, 2013, 18(19), 910–915, DOI:10.1016/j.drudis.2013.04.008.
- N. Rezaei-Ghaleh, M. Blackledge and M. Zweckstetter, Intrinsically Disordered Proteins: From Sequence and Conformational Properties toward Drug Discovery, ChemBioChem, 2012, 13(7), 930–950, DOI:10.1002/cbic.201200093.
- J. Wang, Z. Cao, L. Zhao and S. Li, Novel Strategies for Drug Discovery Based on Intrinsically Disordered Proteins (IDPs), Int. J. Mol. Sci., 2011, 12(5), 3205–3219 CrossRef CAS PubMed.
- V. N. Uversky, C. J. Oldfield and A. K. Dunker, Showing your ID: intrinsic disorder as an ID for recognition, regulation and cell signaling, J. Mol. Recognit., 2005, 18(5), 343–384, DOI:10.1002/jmr.747.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc06370d |
| This journal is © The Royal Society of Chemistry 2024 |