
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA novel design of urea-assisted hydrogen production in electrochemical–chemical decoupled self-circulating systems†
Weihang Shu ,
Qi Sun* and
Mingrui Guo*
,
Qi Sun* and
Mingrui Guo*
College of Chemistry and Chemical Engineering, Institute for Sustainable Energy and Resources, Qingdao University, Qingdao 266071, Shandong, P. R. China. E-mail: gmrqddx@qdu.edu.cn
First published on 22nd August 2024
Abstract
In traditional water electrolysis processes, the oxidation and reduction reactions of water are coupled in both time and space, which presents significant challenges. Here, we propose an optimized design for an electrochemical–chemical self-circulating decoupled system. This system uses the continuous Ni2+/Ni3+ redox process on nickel hydroxide electrode sheets to stepwise couple the urea oxidation-assisted hydrogen production system, separating the hydrogen evolution reaction (HER) and urea oxidation reaction (UOR) into two distinct steps: electrochemical and chemical reactions. In the first electrochemical step, water is reduced at the cathode to produce hydrogen, while the single-electron electrochemical oxidation of Ni(OH)2 at the anode generates NiOOH. Then, in the second chemical reaction step, NiOOH spontaneously oxidizes urea, causing its decomposition and simultaneously reducing back to the Ni(OH)2 state. We concurrently investigated the effects of temperature and OH-concentration on the spontaneous oxidation of urea. At 80 °C and with a 1 M KOH concentration containing 50 mg of urea solution, the NiOOH electrode successfully catalyzed the spontaneous decomposition of urea, achieving conversion rate of 100% and faradaic efficiency of 98%.
1 Introduction
Hydrogen energy with its high energy density and zero harmful emissions is a crucial energy source for promoting sustainable societal development.1–4 Presently, water splitting for hydrogen and oxygen production is among the most promising methods for harnessing renewable energy and storing it as clean-burning and sustainable fuel.5–7 However, water splitting faces technical challenges related to improving efficiency, economic value, and global integration potential.8–11 In traditional water splitting, the oxidation and reduction reactions of water are coupled in time and space. The high overpotential and sluggish kinetics of the oxygen evolution reaction (OER) limit the overall energy efficiency of hydrogen production through water electrolysis.12–15 Simultaneously, researchers have found significant challenges in maintaining adequate separation between hydrogen and oxygen during water splitting processes driven by intermittent renewable energy sources.16In recent years, research exploring various approaches to decouple the oxidation and reduction reactions of water has garnered attention.17 Essentially, decoupling water splitting involves using a mediator with appropriate redox potentials to combine the OER with the mediator's reduction process, rather than directly generating hydrogen. Consequently, the hydrogen evolution reaction (HER) can proceed independently of the oxygen evolution reaction by coupling the HER to the mediator's re-oxidation, rather than water oxidation.18–20 This approach allows for the generation of oxygen and hydrogen at different times, rates, and even in entirely different electrochemical cells, offering solutions to these challenges. A suitable mediator requires properties that are stable in oxidized and reduced forms, and it should exhibit a reversible redox couple.21–23 Currently, several design approaches for decoupling systems have been primarily proposed based on the selection of mediator phases and standard potentials. Based on previous explorations, it has been found that decoupled water splitting systems can be classified into four categories depending on the potential of the decoupling mediator and whether input power is required for one or both steps. (1) Liquid mediator systems with two non-spontaneous steps. (2) Liquid mediator systems with a spontaneous second step. (3) Solid mediator systems with two non-spontaneous steps. (4) Solid mediator systems with a spontaneous second step.24–26 The last type of system is the least developed category, but it may represent a valuable technology because this system spontaneously drives half of the overall water splitting. The spontaneous half-reaction allows the system to pair directly with solar energy and perform spontaneous reactions at night. Cronin's group reported using silicotungstic acid (STA) solution as an intermediate medium to decouple the water splitting process.27 Similarly, Dotan et al. reported successfully decoupling the water splitting process through the reversible solid-state conversion of Ni(OH)2 to NiOOH.3 Evidently, by using an appropriately decoupling medium, only the energy consumption associated with the OER process needs to be provided, allowing the hydrogen production process to occur spontaneously. However, due to the high thermodynamic potential of the OER, suitable decoupling media are limited, and the low added value of oxygen restricts the industrial application of decoupled systems.
In previous studies, it has been reported that small molecule oxidation reaction is used to replace OER.28,29 The urea oxidation reaction (UOR) is a promising alternative to the OER. UOR has two main advantages: firstly, it has a low thermodynamic potential of 0.46 V (vs. RHE), and secondly, electrolyzing urea can help treat industrial urea-containing wastewater, which is beneficial for environmental protection.30–32 Moreover, during the UOR, nucleophilic groups with active hydrogen undergo dehydrogenation, losing protons and electrons to facilitate nucleophilic oxidation reaction (NOR).33–35 Research has found that the NOR of nickel-based hydroxides include both electrochemical oxidative dehydrogenation and a spontaneous non-electrochemical dehydrogenation process of the nucleophilic reagent. The two steps are the electro-oxidation process (Ni2+–OH + OH− = Ni3+–O + H2O + e−) and the spontaneous proton-coupled electron transfer (PCET) process (Ni3+–O + X–H = Ni2+–OH + X˙).36 Considering the modularity of decoupled water splitting systems and the compatibility between decoupling mediators and anodic oxidation reactions, along with advantageous performance characteristics in related electrochemical fields.37 We aim to explore the application of urea oxidation reactions in decoupled systems, designing optimized processes to overcome some of the challenges encountered in traditional electrolytic cells.
Here, we propose an optimized design of an electrochemical–chemical decoupled self-circulating system (ECC–UOR) using Ni(OH)2–NiOOH electrodes to decouple the urea-assisted hydrogen production system (Fig. S1†). The hydrogen evolution reduction process and urea oxidation process are divided into two distinct steps: an electrochemical reaction and a chemical reaction. In the first electrochemical step, HER process is paired with the oxidation of Ni(OH)2 to NiOOH, reducing water at the cathode and oxidizing at the anode. This single-electron reaction kinetics are significantly faster than the four-electron process of OER. The second step involves a spontaneous chemical process where the generated NiOOH electrophilic intermediate can capture protons and electrons from urea, spontaneously reverting to Ni(OH)2 while oxidizing urea. Simultaneously, we investigated the influence of temperature (25 °C–80 °C) and OH− concentration (0.1 M KOH–6 M KOH) on the spontaneous oxidation process of urea. The objective of this study is to produce hydrogen gas at low voltages within a simple cyclic process, demonstrating high efficiency, robustness, and safety potential. This research introduces new avenues for urea-assisted hydrogen production and provides greater flexibility for electrolytic hydrogen generation.
2 Experimental
2.1. Electrode materials preparation
The commercial Ni(OH)2 electrode was first pretreated by immersing it in ethanol solution and subjecting it to ultrasound for 30 minutes to remove surface contaminants. Subsequently, it was dried in an oven at 60 °C and kept aside for further use.2.2. Characterization of the electrode materials
The Ni(OH)2 electrode was tested using an X-ray diffractometer (XRD) on the Smart Lab 3KW instrument. The surface morphology of the electrode was captured using a scanning electron microscope (SEM) on the Thermo Scientific Apreo 2C instrument.2.3. Electrochemical analysis
The electrochemical workstation used for testing was the Shanghai Chenhua CHI760e. The OER performance was tested in 1 M KOH, while the UOR performance was tested in 1 M KOH + 0.33 M urea solution. Electrochemical testing was conducted under three-electrode conditions, with a mercury oxide electrode serving as the reference electrode, a platinum foil 1 × 1 cm2 as the counter electrode, and a Ni(OH)2 electrode 0.4 × 0.35 cm2 as the working electrode. In the two-electrode testing, a platinum-coated titanium mesh 3 × 3 cm2 was used as the counter electrode, and a Ni(OH)2 electrode 3 × 4 cm2 served as the working electrode. All electrochemical tests were conducted at room temperature. Using the Nernst equation, all potentials were converted to the reversible hydrogen electrode (RHE): ERHE = EHg/HgO + 0.098 + 0.059 V × pH. Cyclic voltammetry (CV) and linear sweep voltammetry (LSV) scan rates were set at 1 mV s−1 and 5 mV s−1, respectively. The constant potential method used potentials of 1.42 V and 1.12 V (vs. RHE). The stability of the electrode materials was tested using chronoamperometry with currents of 0.1 A and 0.25 A. Turnover frequency (TOF) values were calculated using the following equation: TOF = JA/4Fn. Where J stands for the current density in ampere, A is the electrode area, F represents the Faraday's constant (96![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 485.3 C mol−1), and n accounts for the total number of moles of metal that participate in the electrocatalytic reaction. For this work, it was assumed that all the Ni metal atoms present in the catalyst participated during UOR.
485.3 C mol−1), and n accounts for the total number of moles of metal that participate in the electrocatalytic reaction. For this work, it was assumed that all the Ni metal atoms present in the catalyst participated during UOR.
3 Results and discussion
3.1. Electrode performance
The phase composition of the Ni(OH)2 electrode was analyzed using X-ray diffraction (XRD) as shown in Fig. S2.† Sharp characteristic peaks were observed around 45.1°, 52.5°, and 76.9°, attributed to the nickel substrate. Additionally, distinct diffraction peaks appeared at 19.2°, 33.3°, 38.7°, 59.3°, 62.9°, 70.3°, and 73.1°, closely matching the β-Ni(OH)2 standard card (PDF# 73-1520), indicating high crystallinity. Furthermore, the surface morphology of the Ni(OH)2 electrode samples was examined using SEM. As shown in Fig. S3,† the electrode surface is predominantly characterized by a dense distribution of nanoscale particles adhering to the nickel substrate.The catalytic activities of the Ni(OH)2 electrode for the OER and UOR were evaluated using a three-electrode system in 1 M KOH and 1 M KOH containing 0.33 M urea. As shown in Fig. 1a, the CV curves of the Ni(OH)2 electrode exhibit an oxidation peak near 1.52 V and a reduction peak near 1.18 V, indicating the reversible transition between Ni(OH)2 and NiOOH. Additionally, the reduction potential of the Ni(OH)2 electrode in the urea-containing alkaline solution is significantly lower than in the urea-free alkaline solution (Fig. 1b). This indicates that the active material NiOOH is consumed and spontaneously gains electrons from urea, reducing back to the divalent nickel state. The UOR process differs from the OER process as it involves a stepwise electrochemical and chemical oxidation process.38 Fig. 1c shows the LSV curve of the Ni(OH)2 electrode, where it can be seen that in the urea-containing alkaline solution, the Ni(OH)2 electrode achieves a current density of 10 mA cm−2 at a voltage of 1.36 V. Compared to the OER process, the potential required to reach the same current density is lower, indicating that the urea oxidation reaction occurs more readily.
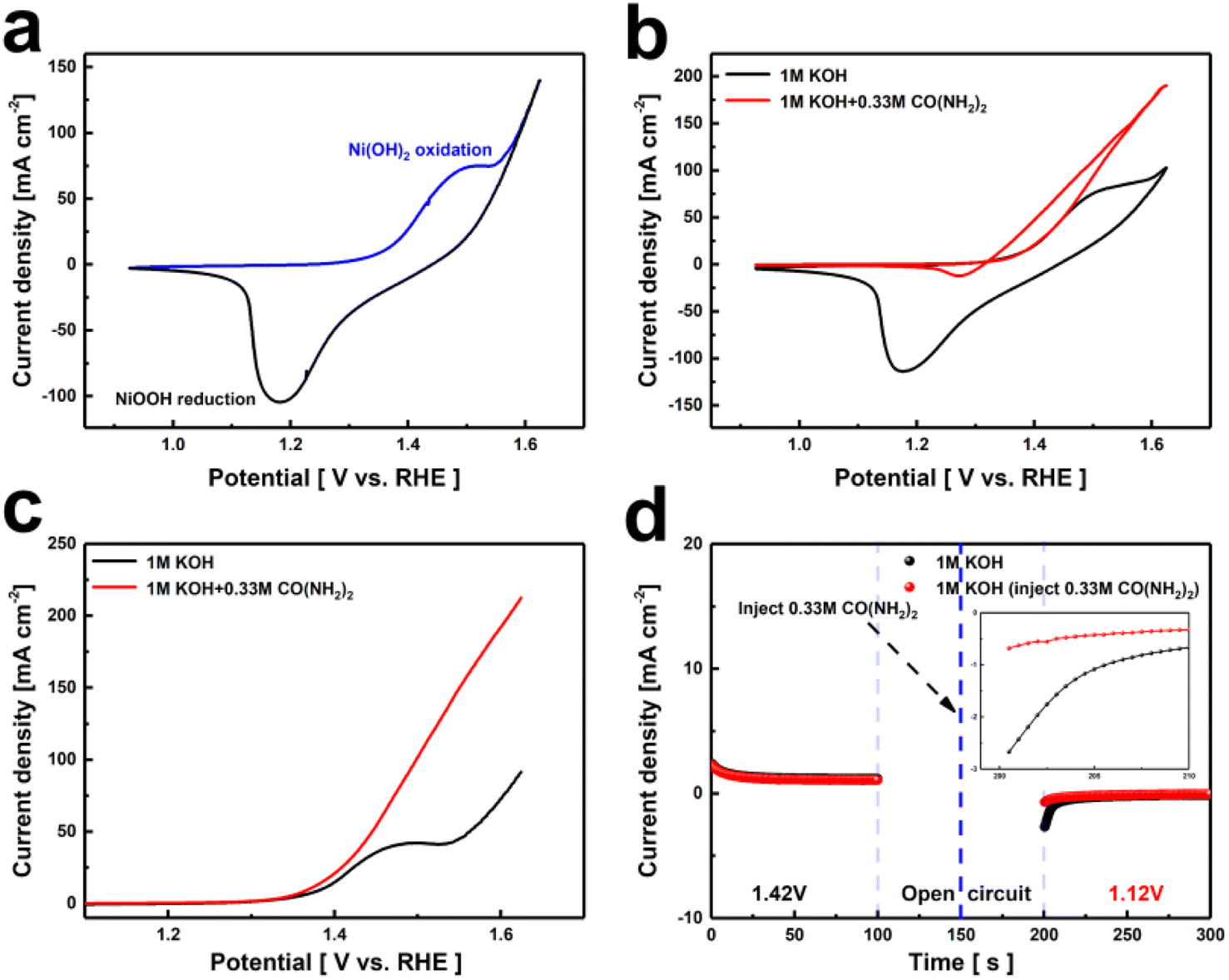 | ||
| Fig. 1 (a and b) CV curves, (c) LSV curves of the Ni(OH)2 electrode film in different media, (d) periodic chronocurrent measurement at 1.42 V (0–100 s), open-circuit state (100–200 s), and 1.12 V (200–300 s). | ||
Other electrochemical properties are shown in Fig. S4–S6,† the UOR catalytic performance of Ni(OH)2, compared with the reported catalyst, is shown in Table S1.† At 1.4 V, the turnover frequency (TOF) of the Ni(OH)2 is 0.0002 s−1. To investigate the spontaneity of NiOOH in the UOR reaction, as shown in Fig. 1d, two Ni(OH)2 electrodes were subjected to electro-oxidation at 1.42 V in a 1 M KOH solution (stage 1, 0–100 s). The reaction quickly reached a steady state, representing the formation of (oxy)hydroxide induced by the abundant OH− ions. The oxidized samples were then transferred to new solutions (1 M KOH + 0.33 M urea and 1 M KOH, respectively) and placed under open circuit potential conditions (stage 2, 100–200 s). Subsequently, in stage 3 (200–300 s), the sample exhibited a significant negative current (1.12 V) in the urea-free solution, indicating the electrochemical reduction of high-valence nickel species. No significant reduction current signal was detected in the urea solution, indicating that the active species, NiOOH, spontaneously reacts with urea, transferring electrons and protons from urea and reducing back to its original state in a spontaneous non-electrochemical process.36
Since OH− directly participates in the oxidation and dehydrogenation processes of nickel-based catalysts, leading to the formation of (oxy)hydroxide and the decomposition of urea, it may play a rate-controlling role.39 Given that the concentration of reactants in the solution may directly affect the catalytic performance, we systematically investigated the impact of different KOH concentrations on the catalytic activity. Fig. 2a and b show the CV and LSV curves of Ni(OH)2 electrodes in 0.1 M, 0.5 M, 1 M, 2 M, 3 M, and 6 M KOH concentrations, each containing 0.33 M urea. It is evident that the current density of UOR increases with the rise in KOH concentration from 0.5 M to 6 M, with the potential required to reach a current density of 10 mA cm−2 decreasing from 1.40 V to 1.30 V. This indicates that the kinetics of the UOR reaction are more strongly dependent on the OH− concentration. To understand the kinetic changes induced by different OH− concentrations, Tafel slopes were calculated from the polarization curves. As shown in Fig. 2c, the Tafel slope decreases with increasing OH− concentration, reaching a value of 57.86 mV dec−1 at 6 M KOH, demonstrating that higher OH− concentrations enhance the reaction kinetics. To better evaluate the catalytic activity of Ni(OH)2 for UOR, we also tested the performance of Ni(OH)2 electrodes at different temperatures. The CV and LSV curves (Fig. 2d and e) shows the catalytic performances of Ni(OH)2 electrode in 1 M KOH + 0.33 M urea at 25 °C, 50 °C, and 80 °C. The CV curves in Fig. 2d show a noticeable decrease in the reduction peak as the temperature increases, indicating that elevated temperatures facilitate the rapid reaction of highly oxidized Ni3+ with urea back to Ni2+. From the LSV curves in Fig. 2e, it can be seen that the current density corresponding to UOR increases with temperature. At 80 °C, a current density of 10 mA cm−2 is achieved at only 1.31 V. Furthermore, the Tafel slopes shown in Fig. 2f illustrate that the reaction kinetics of UOR is accelerated by increasing temperature.
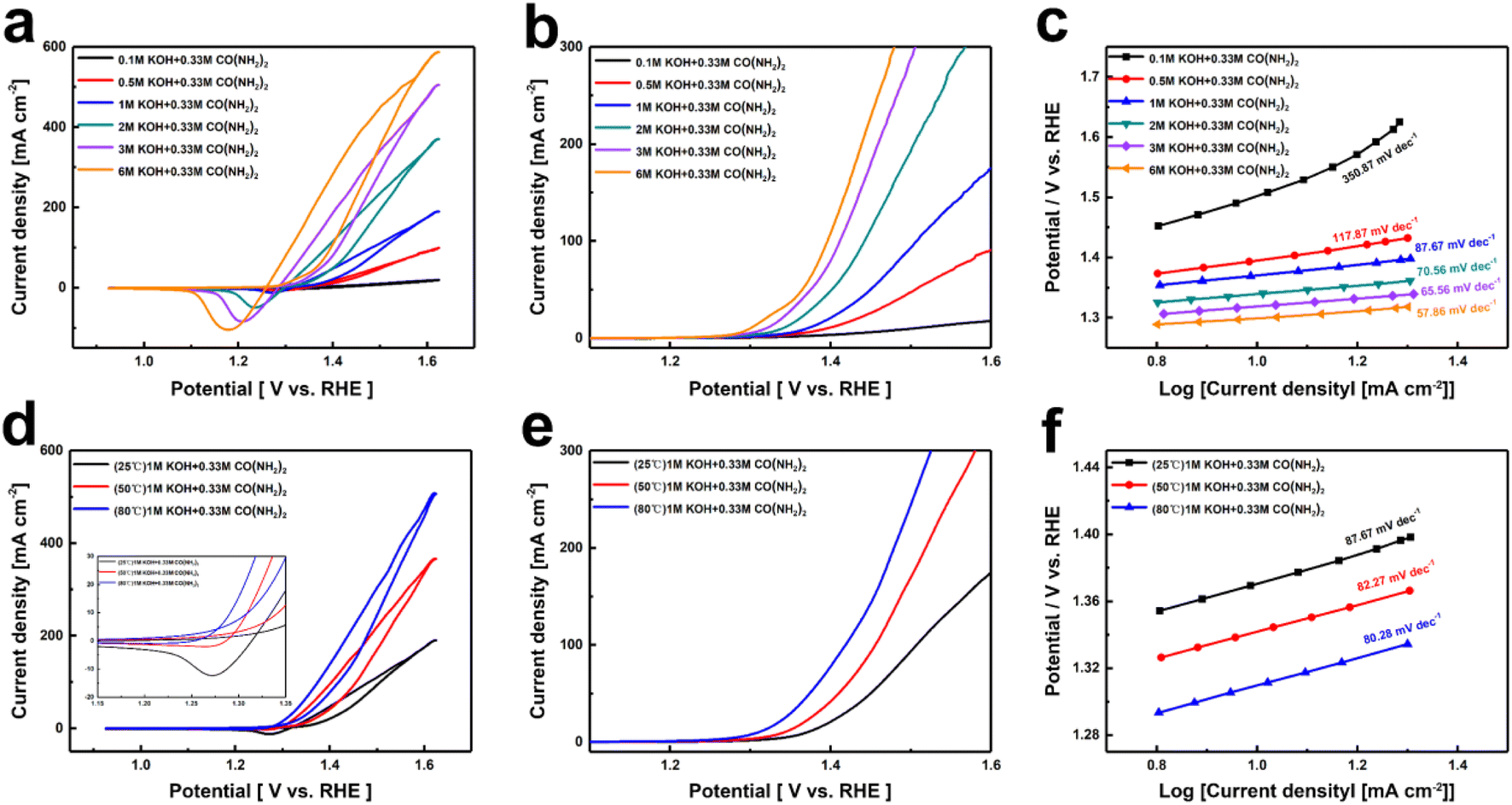 | ||
| Fig. 2 (a) CV curves, (b) LSV curves, (c) Tafel slopes of Ni(OH)2 electrode film for different OH−, (d) CV curves, (e) LSV curves, (f) Tafel slopes of Ni(OH)2 electrode film in the 1 M KOH and 0.33 M urea at different temperatures. | ||
Through previous electrochemical tests, we have determined that the nickel hydroxide electrode undergoes a two-step reaction during the UOR process. Initially, it is electrochemically oxidized to NiOOH, followed by the chemical oxidation of urea molecules at the NiOOH active sites involving dehydrogenation, hydration, and rearrangement.37 We further evaluated the spontaneous oxidation of urea and the reduction of NiOOH back to Ni(OH)2 under different KOH concentrations and temperatures using electrochemical methods. In 1 M KOH, NiOOH was accumulated through a 100 s electrochemical oxidation process and then subjected to constant current discharge under various KOH concentrations and temperatures to assess the rate capabilities of NiOOH. As shown in Fig. 3a and b, after charging to generate 100 C cm−2 of NiOOH, Ni(OH)2 was regenerated at 93.7, 65.9, 59.9, 50.3, 49.4, and 43.8 s in solutions of different KOH concentrations, producing 0.19, 2.05, 2.37, 3.01, 3.07, and 3.39 C cm−2 of Ni(OH)2, respectively. It is evident that at low concentrations, changes in OH− ion concentration significantly affect the rate capabilities of NiOOH. As the OH− ion concentration increases, the rate shows a gradual increase. High alkaline concentrations facilitate dehydrogenation, aiding subsequent N–N coupling and the formation of carbon oxides. In 1 M KOH with 0.33 M urea, changing the reaction temperature also significantly accelerated the product formation rate. Fig. 3c and d clearly show that as the temperature increases, the rates of NiOOH self-discharge and urea oxidation were accelerated. Based on the different discharge times of NiOOH after charging 100 C cm−2, the regenerated Ni(OH)2 coulombic amounts were 2.37, 3.73, and 4.51 C cm−2, demonstrating that increasing the temperature can effectively accelerate the spontaneous reduction of NiOOH back to Ni(OH)2 in urea-containing solutions.
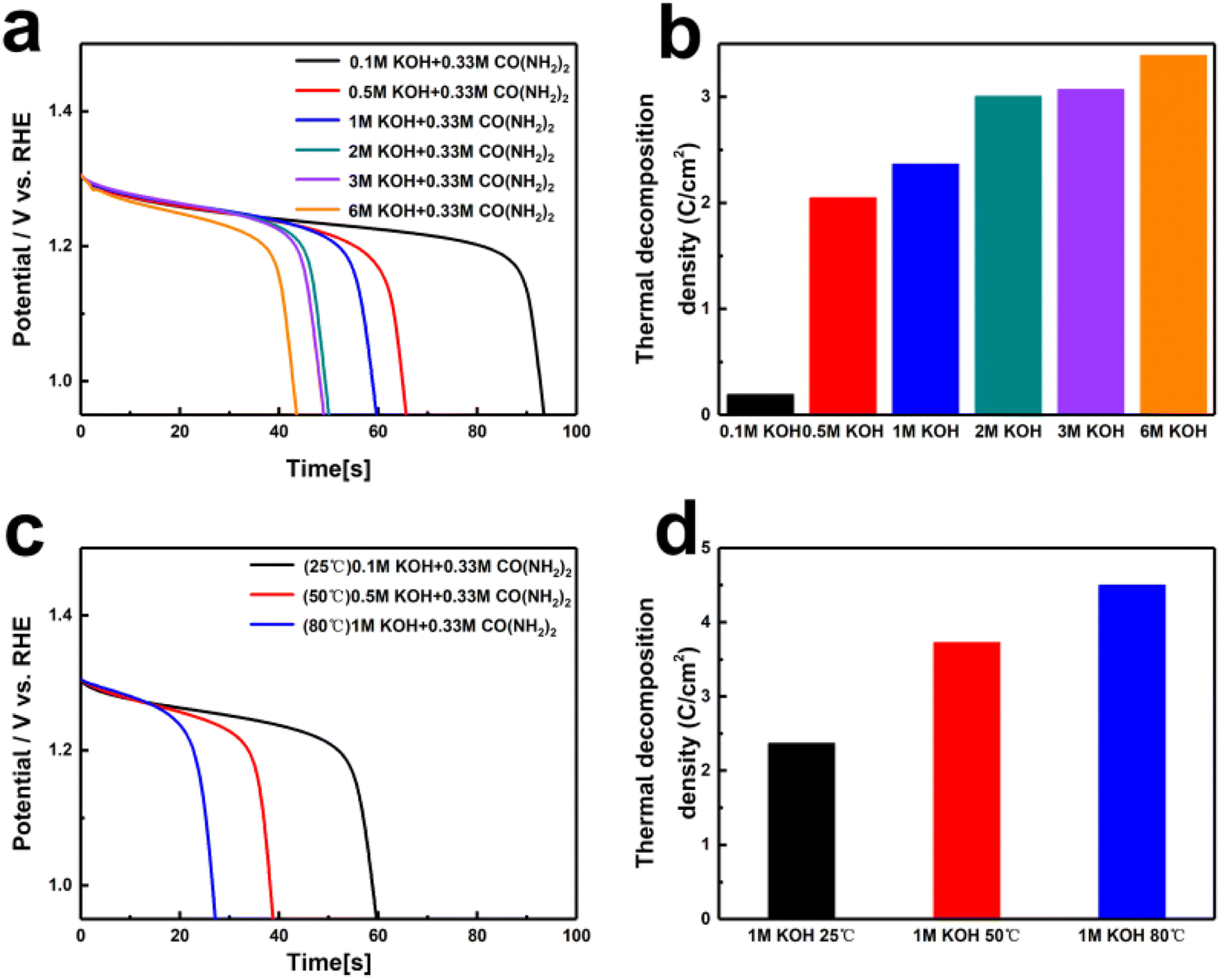 | ||
| Fig. 3 (a and c) The self-discharge time, and (b and d) rate capability of NiOOH electrode in different OH− and different temperature for KOH with 0.33 M urea. | ||
Moreover, the capacitance of the Ni(OH)2 electrode was evaluated through constant current charge–discharge testing. As shown in Fig. S7,† charge–discharge tests of the Ni(OH)2 electrode at a current of 100 mA reveal distinct plateaus during both the charging and discharging processes. The charging process corresponds to the transformation of Ni(OH)2 to NiOOH, while the discharging process indicates the reverse transformation of NiOOH to Ni(OH)2. The sudden drop in the curve signifies the complete conversion of NiOOH. Furthermore, to evaluate the cycling stability of the Ni(OH)2 electrode, charge–discharge tests were conducted for 100 cycles at current densities of 100 mA and 250 mA. The Ni(OH)2 electrode demonstrates excellent stability (Fig. S8†), with almost no loss in capacity after the testing cycles.
The above results show that the main UOR mechanism of NiOOH changes from the conventional electrochemical oxidation UOR process at room temperature to the ECC–UOR process at high temperatures, resulting in higher catalytic activity. In the ECC–UOR mechanism, we consider two steps, it is composed of reactions (1) and (2). The standard reversible potential (Ey) of reaction (1) relative to the RHE is 1.316 V, which is higher than that of the UOR (0.46 V).
Replacing the anodic reaction of water electrolysis with reaction (1) can reduce the overall voltage. However, reaction (1) is not sustainable because Ni(OH)2 will gradually be consumed and eventually depleted. NiOOH can be decomposed to Ni(OH)2 through reaction (2).
| Ni(OH)2 + OH− → NiOOH + H2O + e− | (1) |
| 6NiOOH + CO(NH2)2 + H2O → 6Ni(OH)2 + N2 + CO2 | (2) |
The standard Gibbs free energy change (ΔG) of reaction (2) is negative, indicating that NiOOH can spontaneously reduce. Since the sum of reactions (1) and (2) equals the urea oxidation reaction, the potential difference between reaction (1) and UOR (ΔEy = 1.316 − 0.46 = 0.856 V) provides the chemical driving force for reaction (2). Ni(OH)2 and NiOOH are interconversion by reactions (1) and (2).
3.2. Electrochemical–chemical self-cycling system
Thus, we designed an electrochemical–chemical decoupled self-circulating system. Fig. S9† shows optical photographs of the 3 × 3 cm2 Pt-coated Ti mesh and the 3 × 4 cm2 Ni(OH)2 electrode. In this system, a platinum-coated titanium mesh is used as the cathode, and the Ni(OH)2 electrode as the anode, forming a two-electrode setup. In the first electrochemical step, a current of 100 mA is applied for charging. As shown in Fig. S10,† hydrogen gas is generated by water reduction at the cathode, which is observed as numerous bubbles at the platinum-coated titanium mesh. Concurrently, at the anode, Ni(OH)2 is oxidized to NiOOH. In the second chemical urea oxidation step, the NiOOH electrode from the first step is transferred to an alkaline solution containing 0.33 M urea, numerous bubbles appear on the surface of the Ni(OH)2 electrode, indicating that in the urea-alkaline solution, NiOOH spontaneously oxidizes urea, producing CO2 and N2, reduces itself back to Ni(OH)2. To evaluate the urea conversion rate during the spontaneous chemical oxidation in the decoupled system, we employed high-performance liquid chromatography (HPLC). The standard curve for urea concentration was established due to its strong absorption at a wavelength of 190 nm in the UV-visible spectrum. The HPLC analysis was conducted under the following conditions: using an NH2 column with a mobile phase of 90% acetonitrile and 10% water, at a column temperature of 25 °C, and a UV detection wavelength of 190 nm. We prepared urea solutions with concentrations ranging from 0.01 to 1 g L−1 and measured corresponding HPLC peak areas (A). The linear relationship was determined to be A = 12265c + 240.3 (R2 = 0.9998). This standard curve allows accurate detection of urea concentrations within the range of 0.01 to 1 g L−1. Using a constant current method, we applied a current of 100 mA to the Ni(OH)2 electrode for 5200 s to accumulate NiOOH. The fully charged Ni(OH)2 electrodes were then transferred to 50 mL mixed solutions containing 50 mg urea (0.0167 M, 1 g L−1) and varying KOH concentrations (0.1 M to 6 M) for spontaneous urea oxidation process at room temperature in 180 minutes. As shown in Fig. 4a and b, the urea conversion rates were calculated based on the HPLC peak areas. The conversion rate increased with the OH− concentration. Comparing the results with the standard curve, the urea decomposition rate increased from 4.3% in 0.1 M KOH to 77.2% in 6 M KOH after 180 minutes. To determine the faradaic efficiency of the decoupled system for urea oxidation, we employed eqn (3)–(5).
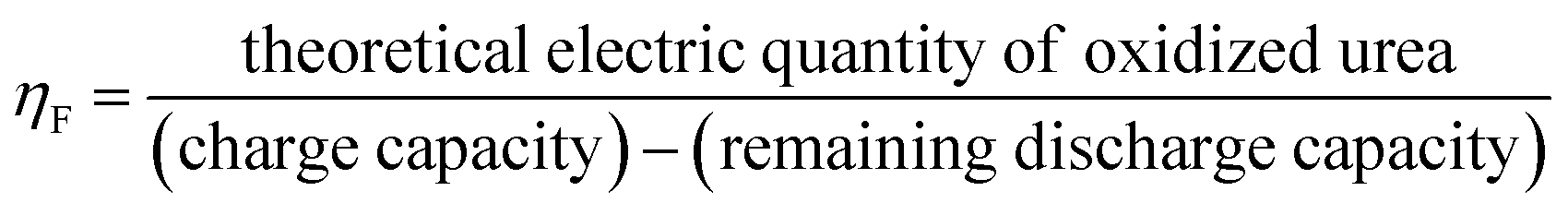 | (3) |
| Qcharging/discharge = I × t × ηF | (4) |
| Qurea = e × NA × 6 × n | (5) |
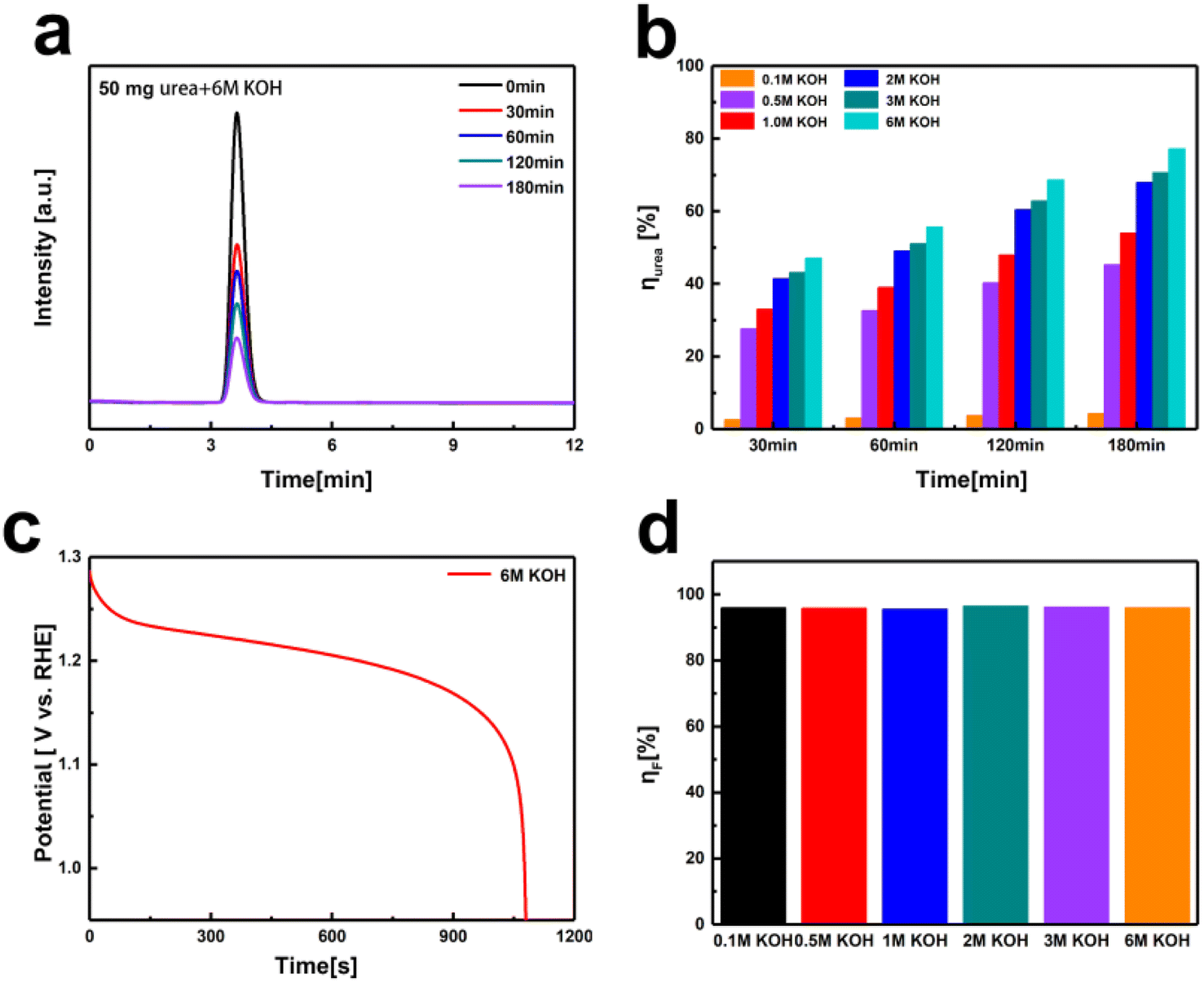 | ||
| Fig. 4 (a) HPLC of urea decomposition on Ni(OH)2 electrode in 6 M KOH concentration with the addition of 1 g L−1 urea over 0–180 minutes. (b) The urea conversion rate on Ni(OH)2 electrode at KOH concentrations ranging from 0.1 M to 6 M. (c) Discharge curve of Ni(OH)2 electrode after 180 minutes of urea decomposition in 6 M KOH concentration at 100 mA. (d) The corresponding faradaic efficiencies of urea decomposition at Ni(OH)2 electrodes under KOH concentrations ranging from 0.1 M to 6 M. | ||
ηF is the faradaic efficiency (%), I is the current (A), t is the discharge time (s), e is the elementary charge (C), NA is the Avogadro's constant (mol−1), n is the number of electrons for urea.
Taking the 6 M KOH concentration as an example, Qcharge = 0.1 A × 5200 s × 95% = 494 C, obtaining the discharge time for Fig. 4c through constant current discharge, Qdischarge = 0.1 A × 1085 s = 108.5 C, Qurea = 1.602 × 10−19 C × 6.022 × 1023 × 6 × 8.3 × 10−4 mol × 77% = 369.4 C, ηF = 96%. As shown in Fig. 4d and Table S1,† the faradaic efficiencies calculated for KOH concentrations ranging from 0.1 M to 6 M were 96%, 96%, 95%, 97%, 96%, and 96% respectively.
Due to the kinetic limitations of spontaneous urea oxidation at room temperature, the decomposition of urea on the NiOOH electrode is incomplete. Previous studies have shown that increasing the temperature can enhance the reaction kinetics and the decomposition conversion rate of urea.40 Consequently, we conducted decomposition tests at varying temperatures in a solution containing 50 mg of urea and 1 M KOH. As shown in Fig. 5a and b, the urea conversion rates at different temperatures in 180 minutes were determined by comparing the peak areas in the HPLC chromatograms with the standard calibration curve. The results clearly show a significant increase for urea conversion rate with rising temperature, from 54% (at 25 °C) to 100% (at 80 °C). Based on the discharge time of the NiOOH electrode (Fig. 5c), the faradaic efficiency for urea decomposition in urea–alkali solution at 80 °C was calculated to be 98%. The faradaic efficiencies at various temperatures were determined from the different discharge times (Fig. 5d and Table S2†), showing efficiencies of 95% at room temperature, 94% at 50 °C, and 98% at 80 °C. Increasing the temperature significantly enhances the decomposition reaction kinetics and the conversion rate in alkali solution of urea. At 80 °C, the urea conversion rate reached 100%, and the corresponding faradaic efficiency was 98%, indicating highly efficient urea oxidation under these conditions. This temperature dependency suggests that optimizing the reaction temperature is crucial for improving the efficiency of urea decomposition.
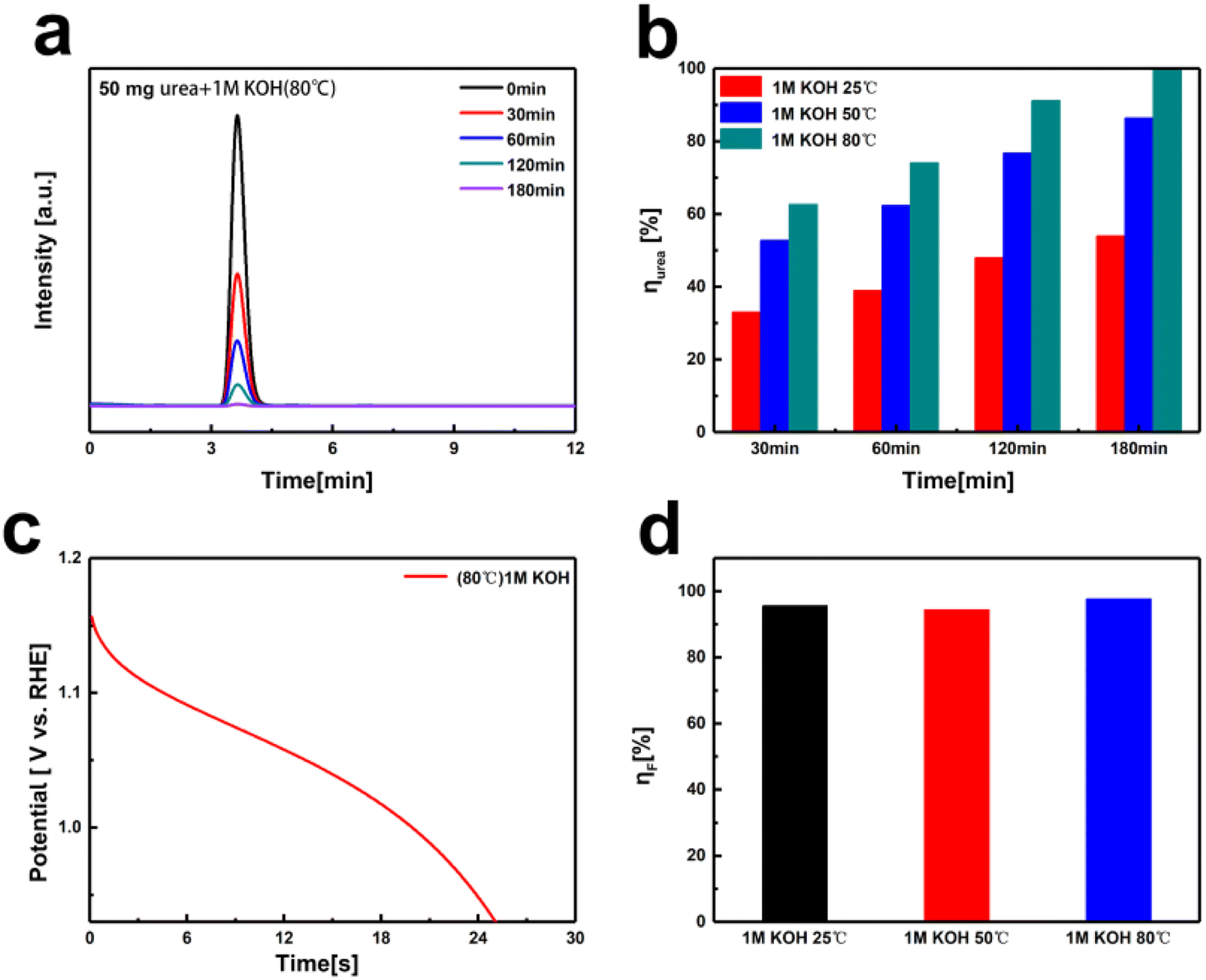 | ||
| Fig. 5 (a) HPLC of urea decomposition on Ni(OH)2 electrode in 1 M KOH concentration at 80 °C with the addition of 1 g L−1 urea over 0–180 minutes. (b) The urea conversion rates at 25 °C, 50 °C, and 80 °C. (c) Discharge curve of Ni(OH)2 electrode after 180 minutes of urea decomposition in 1 M KOH concentration at 80 °C at 100 mA. (d) The corresponding faradaic efficiencies of urea decomposition at Ni(OH)2 electrodes with 25 °C, 50 °C, and 80 °C. | ||
To evaluate the faradaic efficiency of hydrogen production in the decoupled electrochemical process, we measured the volume of hydrogen gas generated using the water displacement method. A current was applied to the Ni(OH)2 electrode to produce hydrogen gas, and the volume of the hydrogen produced was calculated using the water displacement method. As shown in Fig. S11,† at a current of 100 mA and an operation time of 1800 s, the volume of collected H2 gas was 925 μmol. Comparing the actual volume of hydrogen produced with the theoretically calculated volume, the faradaic efficiency was determined to be approximately 100%.
4 Conclusions
In this study, we propose an optimized design of an electrochemical–chemical self-cycling decoupled system that decouples the HER and UOR using a Ni(OH)2 electrode. The system operates in two distinct steps involving electrochemical and chemical processes. During the electrochemical step, the Ni(OH)2 anode undergoes oxidation to form NiOOH, while water reduction occurs at the cathode to produce hydrogen gas. In the subsequent chemical step, NiOOH spontaneously decomposes urea, reducing back to Ni(OH)2. Moreover, we investigated the effects of KOH concentration and temperature on the spontaneous urea oxidation process. In the first step of the electrochemical HER, the conversion of Ni(OH)2 to NiOOH is a single-electron process, which accelerates HER kinetics and promotes rapid and efficient hydrogen production. In the second step of spontaneous chemical urea oxidation, the NiOOH electrode successfully decomposes urea at 80 °C in a 1 M KOH solution containing 50 mg of urea, achieving a conversion rate of 100% and a faradaic efficiency of 98%. This work provides a novel approach to the modular production of hydrogen through a decoupled electrolysis system, offering higher flexibility and efficiency in the hydrogen production process.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (No. 22109077 and 22102078) and was also funded by the Shandong Provincial Natural Science Foundation, China (ZR2021QB190).Notes and references
- S. Cao, T. Sun, Q.-Z. Li, L. Piao and X. Chen, Trends Chem., 2023, 5, 947–960 CrossRef CAS.
- S. Chu and A. Majumdar, Nature, 2012, 488, 294–303 CrossRef CAS PubMed.
- H. Dotan, A. Landman, S. W. Sheehan, K. D. Malviya, G. E. Shter, D. A. Grave, Z. Arzi, N. Yehudai, M. Halabi, N. Gal, N. Hadari, C. Cohen, A. Rothschild and G. S. Grader, Nat. Energy, 2019, 4, 786–795 CrossRef CAS.
- X.-Y. Ji, Y.-Y. Wang and J. Tao, Mater. Chem. Front., 2023, 7, 5120–5139 RSC.
- L. Quan, H. Jiang, G. Mei, Y. Sun and B. You, Chem. Rev., 2024, 124, 3694–3812 CrossRef CAS PubMed.
- O. Schmidt, A. Gambhir, I. Staffell, A. Hawkes, J. Nelson and S. Few, Int. J. Hydrogen Energy, 2017, 42, 30470–30492 CrossRef CAS.
- T. Wu and J. Wang, IEEE Transactions on Sustainable Energy, 2023, 14, 920–934 Search PubMed.
- J. Du, F. Li and L. Sun, Chem. Soc. Rev., 2021, 50, 2663–2695 RSC.
- M. Guo, J. Zhan, Z. Wang, X. Wang, Z. Dai and T. Wang, Chin. Chem. Lett., 2023, 34, 107709 CrossRef CAS.
- B. Lu, Z. Li, J. Yin, K. Zhu and K. Ye, Appl. Catal., B, 2024, 350, 123940 CrossRef CAS.
- B. Lu, Y. Jing, J. Yin, K. Zhu, K. Ye and X. Li, J. Power Sources, 2024, 615, 235065 CrossRef CAS.
- H. N. Nong, T. Reier, H.-S. Oh, M. Gliech, P. Paciok, T. H. T. Vu, D. Teschner, M. Heggen, V. Petkov, R. Schlögl, T. Jones and P. Strasser, Nat. Catal., 2018, 1, 841–851 CrossRef CAS.
- Q. Qian, Y. Zhu, N. Ahmad, Y. Feng, H. Zhang, M. Cheng, H. Liu, C. Xiao, G. Zhang and Y. Xie, Adv. Mater., 2024, 36, e2306108 CrossRef PubMed.
- C. Deng, C. Y. Toe, X. Li, J. Tan, H. Yang, Q. Hu and C. He, Adv. Energy Mater., 2022, 12, 2201047 CrossRef CAS.
- B. Lu, C. Lv, Y. Xie, L. Gao, J. Yan, K. Zhu, G. Wang, D. Cao and K. Ye, Small, 2023, 19, e2302923 CrossRef PubMed.
- Z. Zhang, X. Li, C. Zhong, N. Zhao, Y. Deng, X. Han and W. Hu, Angew Chem. Int. Ed. Engl., 2020, 59, 7245–7250 CrossRef CAS PubMed.
- Z. P. Ifkovits, J. M. Evans, M. C. Meier, K. M. Papadantonakis and N. S. Lewis, Energy Environ. Sci., 2021, 14, 4740–4759 RSC.
- W. Ma, C. Xie, X. Wang, H. Wang, X. Jiang, H. Zhang, X. Guo, X. Zong, X. Li and C. Li, ACS Energy Lett., 2019, 5, 597–603 CrossRef.
- J. Lei, J. J. Yang, T. Liu, R. M. Yuan, D. R. Deng, M. S. Zheng, J. J. Chen, L. Cronin and Q. F. Dong, Chemistry, 2019, 25, 11432–11436 CrossRef CAS PubMed.
- S. Goodwin and D. A. Walsh, ACS Appl. Mater. Interfaces, 2017, 9, 23654–23661 CrossRef CAS PubMed.
- J. Piwek, C. R. Dennison, E. Frackowiak, H. Girault and A. Battistel, J. Power Sources, 2019, 439, 227075 CrossRef CAS.
- Z. Liu, G. Zhang, K. Zhang, H. Lan, H. Liu and J. Qu, J. Mater. Chem. A, 2020, 8, 4073–4082 RSC.
- X. Zheng, J. Yang, P. Li, Z. Jiang, P. Zhu, Q. Wang, J. Wu, E. Zhang, W. Sun, S. Dou, D. Wang and Y. Li, Angew Chem. Int. Ed. Engl., 2023, 62, e202217449 CrossRef CAS PubMed.
- D. Reynard, G. Bolik-Coulon, S. Maye and H. H. Girault, Chem. Eng. J., 2021, 407, 126721 CrossRef CAS.
- A. Landman, H. Dotan, G. E. Shter, M. Wullenkord, A. Houaijia, A. Maljusch, G. S. Grader and A. Rothschild, Nat. Mater., 2017, 16, 646–651 CrossRef CAS PubMed.
- L. Trotochaud, S. L. Young, J. K. Ranney and S. W. Boettcher, J. Am. Chem. Soc., 2014, 136, 6744–6753 CrossRef CAS PubMed.
- B. Rausch, M. D. Symes, G. Chisholm and L. Cronin, Science, 2014, 345, 1326–1330 CrossRef CAS PubMed.
- H. Y. Wang, L. Wang, J. T. Ren, W. Tian, M. Sun, Y. Feng and Z. Y. Yuan, ACS Nano, 2023, 17, 10965–10975 CrossRef CAS PubMed.
- W.-W. Tian, J.-T. Ren, H.-Y. Wang, L. Wang and Z.-Y. Yuan, Appl. Catal., B, 2024, 354, 124115 CrossRef CAS.
- K. Finlay, A. Patoine, D. B. Donald, M. J. Bogard and P. R. Leavitt, Limnol. Oceanogr., 2010, 55, 1213–1230 CrossRef CAS.
- K. Ye, G. Wang, D. Cao and G. Wang, Top. Curr. Chem., 2018, 376, 42 CrossRef PubMed.
- X. Gao, X. Bai, P. Wang, Y. Jiao, K. Davey, Y. Zheng and S. Z. Qiao, Nat. Commun., 2023, 14, 5842 CrossRef CAS PubMed.
- L. Liu, H.-m. Chang, H. Jameel, J.-Y. Park and S. Park, Ind. Eng. Chem. Res., 2017, 56, 14447–14453 CrossRef CAS.
- V. Vedharathinam and G. G. Botte, Electrochim. Acta, 2013, 108, 660–665 CrossRef CAS.
- Z. Ji, Y. Song, S. Zhao, Y. Li, J. Liu and W. Hu, ACS Catal., 2021, 12, 569–579 CrossRef.
- W. Chen, Y. Wang, B. Wu, J. Shi, Y. Li, L. Xu, C. Xie, W. Zhou, Y. C. Huang, T. Wang, S. Du, M. Song, D. Wang, C. Chen, J. Zheng, J. Liu, C. L. Dong, Y. Zou, J. Chen and S. Wang, Adv. Mater., 2022, 34, e2105320 CrossRef PubMed.
- W. Chen, C. Xie, Y. Wang, Y. Zou, C.-L. Dong, Y.-C. Huang, Z. Xiao, Z. Wei, S. Du, C. Chen, B. Zhou, J. Ma and S. Wang, Chem, 2020, 6, 2974–2993 CAS.
- W. Chen, L. Xu, X. Zhu, Y. C. Huang, W. Zhou, D. Wang, Y. Zhou, S. Du, Q. Li, C. Xie, L. Tao, C. L. Dong, J. Liu, Y. Wang, R. Chen, H. Su, C. Chen, Y. Zou, Y. Li, Q. Liu and S. Wang, Angew. Chem., 2021, 133, 7373–7383 CrossRef.
- D. Zhu, H. Zhang, J. Miao, F. Hu, L. Wang, Y. Tang, M. Qiao and C. Guo, J. Mater. Chem. A, 2022, 10, 3296–3313 RSC.
- B. Deng, G. Yu, W. Zhao, Y. Long, C. Yang, P. Du, X. He, Z. Zhang, K. Huang, X. Li and H. Wu, Energy Environ. Sci., 2023, 16, 5210–5219 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra04644g |
| This journal is © The Royal Society of Chemistry 2024 |