
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of novel pyrazole acetals of andrographolide and isoandrographolide as potent anticancer agents†
Siva Kumar Rokkam a,
Manohar Bhujela,
Dolly Jainc,
Lakshminath Sripadaa,
Srinivas Nandurib,
Avinash Bajajc and
Nageswara Rao Golakoti*a
a,
Manohar Bhujela,
Dolly Jainc,
Lakshminath Sripadaa,
Srinivas Nandurib,
Avinash Bajajc and
Nageswara Rao Golakoti*a
aDepartment of Chemistry, Sri Sathya Sai Institute of Higher Learning, Prasanthi Nilayam, Andhra Pradesh, India. E-mail: gnageswararao@sssihl.edu.in
bDepartment of Chemical Sciences, National Institute of Pharmaceutical Education and Research, Balanagar, Hyderabad, Telangana 500037, India
cLaboratory of Nanotechnology and Chemical Biology, Regional Centre for Biotechnology, NCR Biotech Science Cluster, 3rd Milestone, Faridabad-Gurgaon Expressway, Faridabad, Haryana 121001, India
First published on 22nd August 2024
Abstract
Globally, cancer is the most prevalent chronic disease-related cause of death. Although there are many anticancer drugs, some of them have adverse effects. Due to their limited side effects, natural products are preferred over synthetic drugs. Andrographolide and its derivatives are known to be potent anticancer agents. In this context, sixteen novel 3,19-(NH-3-aryl-pyrazole) acetals of andrographolide and isoandrographolide (1a–1h, 2a–2g, 2i) from 3-aryl-1-H-pyrazole-4-carboxaldehydes (a–i) were synthesized. All the synthesized compounds were characterized using 1H NMR, 13C NMR, HRMS, FT-IR, and UV-vis spectroscopy. All the compounds were evaluated against a panel of 60 different human cancer cell lines for their anticancer potential at NCI, USA. Four compounds, having promising GI50s (50% growth inhibitory activity) on all 60-cell lines were selected for further in vitro studies. Out of these four compounds, compound 1g exhibited the best IC50 (3.08 μM) against the colon cancer cell line, HCT-116. Cell cycle analysis, annexin V-FITC/PI, and ROS assays revealed that the apoptosis of HCT-116 cells induced by compound 1g could be mainly attributed to the elevated levels of intracellular ROS. Further, the structure–activity relationship revealed that the pyrazole moiety of andrographolide plays a key role in their anticancer properties. These compounds were further examined for in silico ADMET and Lipinski characteristics to assess their potential as lead compounds.
1. Introduction
Andrographis paniculata (A. paniculata) is an annual herb and a member of the family Acanthaceae that expands up to 1–2 meters in height mostly in shady places. Due to its exceptionally bitter flavor, it is often referred to as the “King of Bitters”. A. paniculata has a rich history in traditional medical practices in Southeast Asia, India, and China, and has been used for several generations.1,2 Traditionally, the leaves and roots of this plant have been utilized for a variety of therapeutic purposes in Asia and Europe. In China, this plant extract is used to eliminate toxins and cool the body.3,4 In India, it is used to cure diabetes, dysentery, herpes, jaundice, helminth infection, and ulcers, and is topically used to treat snake bites and skin infections.5,6Numerous research investigations have demonstrated that A. paniculata extracts exhibit a plethora of pharmacological activity, including anti-cancer, anti-inflammatory, anti-viral, anti-malarial, anti-oxidant, anti-diarrheal, hepatoprotective, and immunostimulatory activities.5–9 The observed activities of the methanolic extract are primarily attributed to the plant's major phytochemical constituent andrographolide.10,11 The recent research findings revealed that andrographolide and its derivatives exert their anticancer effects by cell cycle arrest via increasing the levels of tumor suppressor proteins p53, p21 and p16, inducing apoptosis.12–14 Andrographolide and its derivatives also suppress TNF-α, TGF-β1, IL-1β, IL-6 levels, angiogenic factor VEGF, NF-kB signaling pathway, and causes degradation of proto-oncogene tyrosine-protein kinase Src. Further, andrographolide acts as antiangiogenic and found to inhibit cell migration and invasion in A549, HT29, CT26, lovo, and cholangiocarcinoma cells by interfering with the production of proteins and cellular pathways that are crucial for the spread of cancer.15–18
Our previous studies on various andrographolide and isoandrographolide derivatives such as C-12-alkyl/benzyl/sulfamoyl-phenyl/amino-14-deoxyandrographolides, 3,19-benzylidene-andrographolide/isoandrographolide, 3,19-heteroaromatic acetals of andrographolide/isoandrographolide, 14-deoxy-12-hydroxy-andrographolide acetals displayed excellent anticancer activity on a panel of 60 different human cancer cell lines (at NCI).19–23 Further, the mechanism of action for the most potent derivatives was determined against breast cancer cell line MDA-MB-231. Since the acetals of andrographolide and isoandrographolide acted as very good anticancer agents, in our present work, we intended to link the pyrazole moiety to andrographolide and isoandrographolide and investigate their potential as anticancer agents. Pyrazoles were chosen because of their significant importance as pharmacophores in forming the basis for diverse pharmaceutical compounds.24–26
2. Experimental
2.1. Synthesis
All the chemicals were obtained from Sigma Aldrich, HiMedia, and Alfa Aesar, while the solvents were distilled before use.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :35 ratio as eluent.:acetone, 60:40).
:35 ratio as eluent.:acetone, 60:40).2.2. Instrumentation
The 1H and 13C-NMR spectra were recorded on BRUKER ASCEND 400 MHz and 100 MHz respectively. Tetramethylsilane (TMS) was used as an internal standard. Mass spectra were generated using AGILENT 6550 Q-TOF LC-MS (HRMS). Acetonitrile was used as a solvent to record UV-vis spectra in the range 200–600 nm using a Shimadzu 2450 spectrophotometer. Using KBr pellets, FT-IR spectra were acquired in the range 400 and 4000 cm−1 on AGILENT Cary 630 spectrophotometer. Agilent 1260 Infinity high-performance liquid chromatography system, equipped with a quaternary solvent delivery system, inline degasser, auto sampler, and photodiode array detector, was used. Samples were examined using water (10%) and acetonitrile (90%) as mobile phase on a Zorbax Extend-C18 (4.6 mm 250 mm, 5 μm) column at a flow rate of 1 mL min−1. At 25 °C, the separation was performed with an injection volume of 15 μL. A diode array detector was utilized to capture the chromatogram between 200 and 700 nm.2.3. In silico studies
All the active compounds were screened for their in silico Lipinski and ADMET properties using the pkCSM tool.273. Biological testing
3.1. Anticancer studies
4. Results and discussion
4.1. Chemistry
We intend to synthesize 1-isonicotinoyl-3-aryl-1H-pyrazole-4-carbaldehydes and further prepare their acetals with andrographolide and isoandrographolide. In step-1, we synthesized nine N′-(1-arylethylidene) isonicotinohydrazides (A–I), and in step-2, they were to be converted to 1-isonicotinoyl-3-aryl-1H-pyrazole-4-carbaldehydes (a–i) in the presence of DMF, POCl3 (1.2 equivalents) at 65 °C as reported by Beniwal et al.29 However, surprisingly, when we used 3 equivalents of POCl3, we observed the formation of 3-aryl-1H-pyrazole-4-carbaldehydes (a–i) instead of 1-isonicotinoyl-3-aryl-1H-pyrazole-4-carbaldehydes (Scheme 1). To investigate the effect of POCl3 in this reaction, we repeated the procedure using 2 equivalents of POCl3. However, we again observed the formation of 3-aryl-1H-pyrazole-4-carbaldehydes (a–i). This is the first report of the synthesis of 3-aryl-1H-pyrazole-4-carbaldehydes in a two-step reaction. Four of the nine synthesized carbaldehydes are novel (c, e, f, and g) in our work. Other five of these carbaldehydes were synthesized by You et al., following a three-step reaction and patented the work.30 In step-3, 3-aryl-1H-pyrazole-4-carbaldehydes (a–i) were reacted with andrographolide (ADG) (Scheme 2) and isoandrographolide (ISOADG) (Scheme 3) in the presence of mild acid catalyst pyridinium para-toluene sulfonate (PPTS) to obtain the desired products (1a–1h, 2a–2g, 2i). All the products were purified using column chromatography.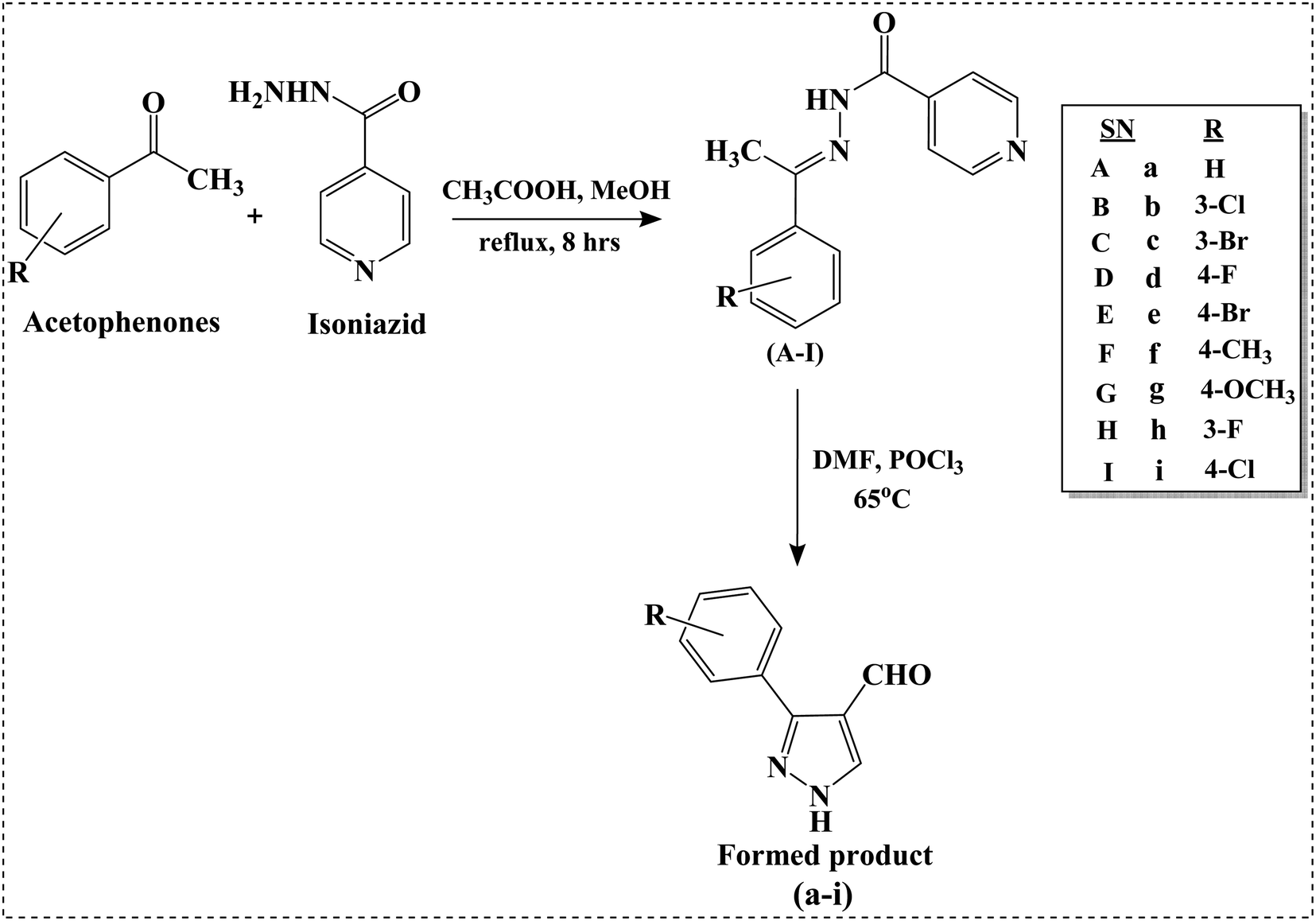 | ||
| Scheme 1 Synthesis of N′-(1-arylethylidene) isonicotinohydrazides (A–I) and 3-aryl-1H-pyrazole-4-carbaldehydes (a–i). | ||
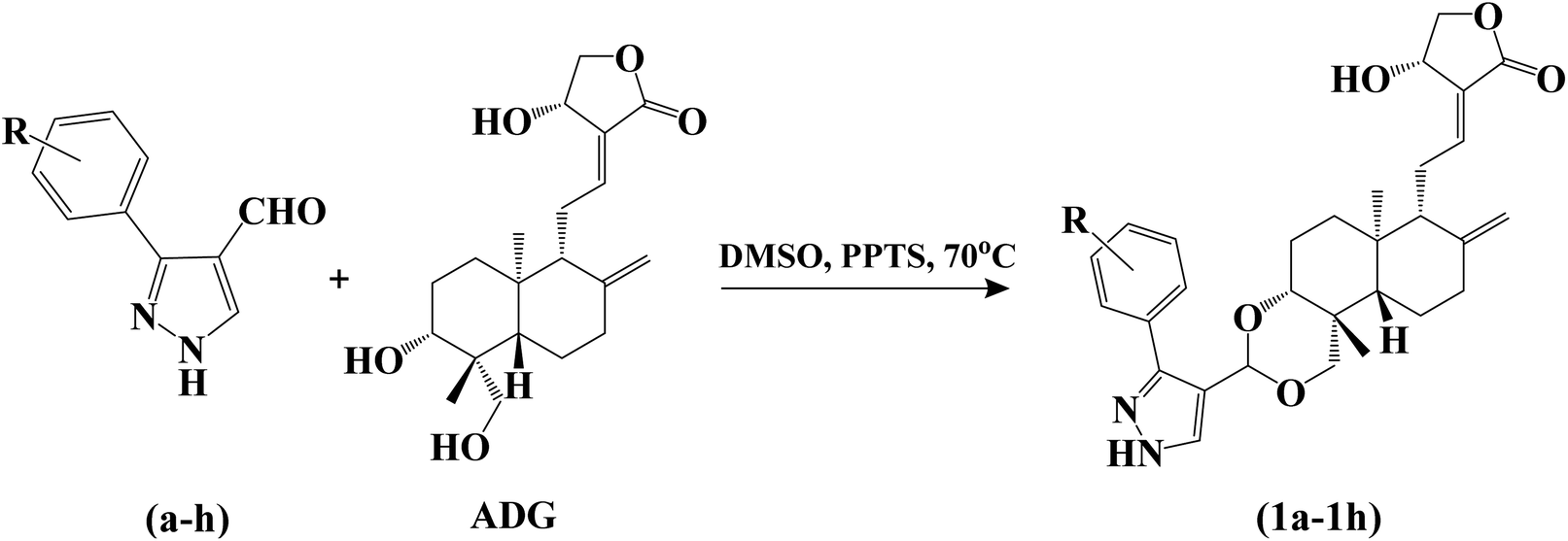 | ||
| Scheme 2 Synthesis of 3,19-(NH-3-aryl-pyrazole) acetals of andrographolide (1a–1h). | ||
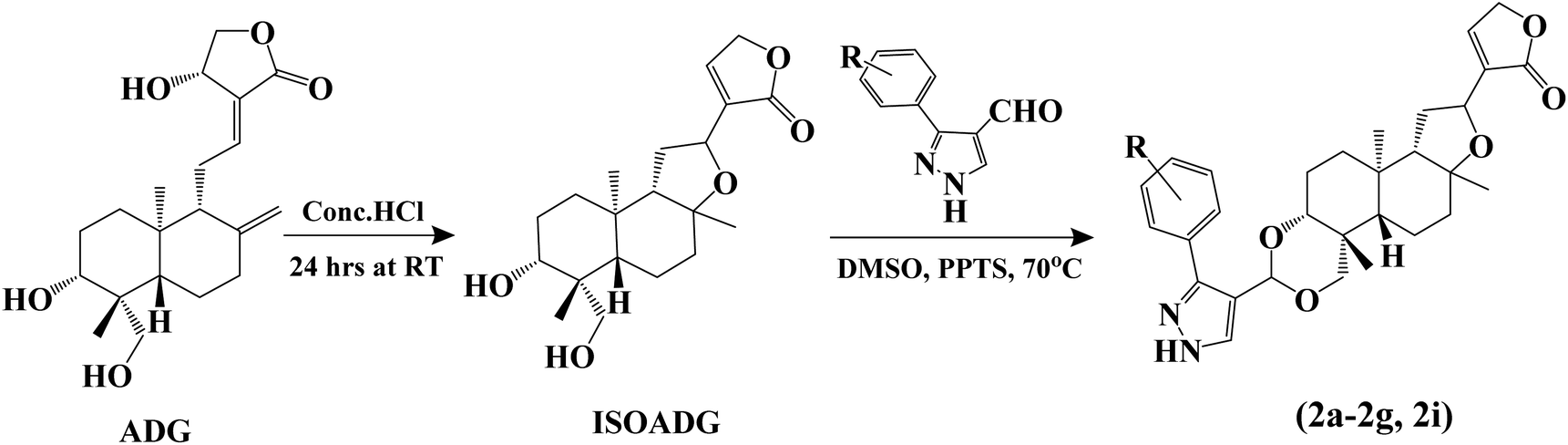 | ||
| Scheme 3 Synthesis of isoandrographolide and 3,19-(NH-3-aryl-pyrazole) acetals of isoandrographolide (2a–2g and 2i). | ||
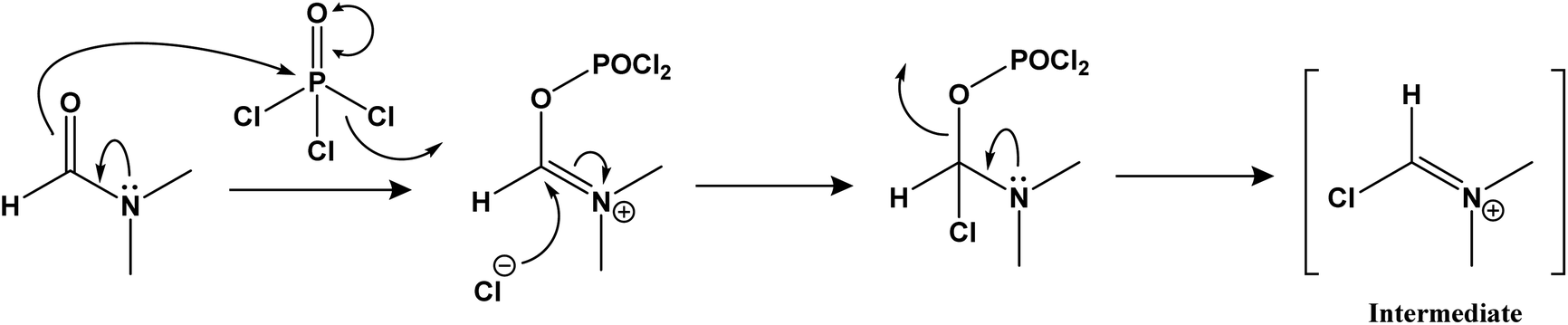
Step-2: reaction of N′-(1-arylethylidene) isonicotinohydrazides with iminium ion.
Step-3: nucleophile attack on the amide carbonyl to form 3-aryl-1H-pyrazole-4-carbaldehydes.

The amide carbonyl, as seen in step-3 above, is linked to two electron-withdrawing groups (pyridine and pyrazole-4-carboxaldehyde). This makes it more prone to react with nucleophiles. Hence, the possible mechanism for the formation of 3-aryl-1H-pyrazole-4-carbaldehydes (a–i) instead of 1-isonicotinoyl-3-aryl-1H-pyrazole-4-carbaldehydes could involve nucleophilic attack at the amide carbonyl, resulting in the hydrolysis of the amide bond.
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C groups. These peaks were absent for 3,19-(NH-3-aryl-pyrazole) acetals of isoandrographolide. –NH stretching peak in all the acetals appeared in the range 3321–3270 cm−1. The peak at ∼1755 cm−1 corresponds to the carbonyl group (CO) of the lactone moiety. Distinct absorptions at approximately 1200 to 1100 cm−1 can be attributed to the C–O stretching bands. Compounds with aryl chloride have bands at ∼1150 cm−1 (Ar–Cl stretch). Compounds with aryl fluorides showed a band around 1234 and 1219 cm−1 (Ar–F stretch), and compounds with aryl bromides have a band between 1018–1010 cm−1 (Ar–Br stretch).
C groups. These peaks were absent for 3,19-(NH-3-aryl-pyrazole) acetals of isoandrographolide. –NH stretching peak in all the acetals appeared in the range 3321–3270 cm−1. The peak at ∼1755 cm−1 corresponds to the carbonyl group (CO) of the lactone moiety. Distinct absorptions at approximately 1200 to 1100 cm−1 can be attributed to the C–O stretching bands. Compounds with aryl chloride have bands at ∼1150 cm−1 (Ar–Cl stretch). Compounds with aryl fluorides showed a band around 1234 and 1219 cm−1 (Ar–F stretch), and compounds with aryl bromides have a band between 1018–1010 cm−1 (Ar–Br stretch).In the 1H NMR spectra of all the acetals, the signal around δ 7.80 is attributed to the presence of H-5′, while peaks within the range of δ 7.90 to 6.95 correspond to the aromatic protons linked to the pyrazole ring (H-7′ to H-11′). The proton signal for the unsaturated lactone (H-14) in 3,19-(NH-3-aryl-pyrazole) acetals of isoandrographolide is observable at ∼δ 7.29. Additionally, the peak at around δ 6.95 corresponds to H-12 in 3,19-(NH-3-aryl-pyrazole) acetals of andrographolide. Across all compounds, a singlet peak at ∼δ 5.82 is for H-21.
In the 13C NMR spectra, signals approximately within the range of δ 173–170, confirm the presence of a carbonyl group (CO). The carbons in the pyrazole ring, aromatic ring, and olefin carbons were identified in the ∼δ 163 to 110 range. Notably, in the case of 3,19-(NH-3-aryl-pyrazole) acetals of isoandrographolide, the olefin carbons such as C-8 and C-17 are absent, leading to the absence of a signal around δ 110. A distinct signal around δ 90, associated with C-21 bonded to two oxygen atoms, strongly supports the formation of acetals. Carbon signals related to oxygen atoms (–O–C) were observed within the range of δ 82 to 66.31,32 The complete characterization and spectral data of the compounds (A–I, a–i, 1a–1h, 2a–2g, and 2i) is provided in the ESI.†
4.2. Biological evaluation
| Cancer | Sub panel | GI50 (μM) | |||
|---|---|---|---|---|---|
| 1c (R = 3-Br) | 1b (R = 3-Cl) | 1g (R = 4-OCH3) | 1e (R = 4-Br) | ||
| a NT – not tested. | |||||
| Leukemia | CCRF-CEM | NTa | 2.70 | 2.08 | 2.81 |
| HL-60(TB) | 1.87 | 2.91 | 2.69 | 2.30 | |
| K-562 | 1.93 | 2.41 | 2.71 | 2.24 | |
| MOLT-4 | 2.29 | 3.83 | 2.21 | 2.86 | |
| RPMI-8226 | 2.14 | 2.25 | 2.60 | 2.56 | |
| SR | NT | NT | 2.87 | NT | |
| Non-small cell lung | A549/ATCC | 3.37 | 9.73 | 3.32 | 11.10 |
| EKVX | 2.15 | 3.68 | 2.66 | 4.69 | |
| HOP-62 | 3.78 | 15.30 | 3.44 | 18.90 | |
| HOP-92 | 2.21 | 4.29 | 2.02 | 5.08 | |
| NCI-H226 | 1.62 | 1.66 | 3.58 | 1.93 | |
| NCI-H23 | 1.84 | 4.36 | 2.51 | 10.50 | |
| NCI-H322M | 5.09 | 10.40 | 4.71 | 12.50 | |
| NCI-H460 | 2.43 | 8.87 | 1.91 | 12.60 | |
| NCI-H522 | 1.90 | 1.75 | 2.49 | 2.08 | |
| Colon | COLO 205 | 2.18 | 4.85 | 1.95 | 4.45 |
| HCC-2998 | 1.87 | 3.49 | 2.87 | 9.11 | |
| HCT-116 | 1.83 | 1.84 | 1.86 | 1.94 | |
| HCT-15 | 1.66 | 1.79 | 2.53 | 1.97 | |
| HT29 | 2.10 | 2.70 | 2.86 | 2.80 | |
| KM12 | 2.44 | 3.29 | 2.38 | 7.17 | |
| SW-620 | 2.22 | 2.35 | 2.29 | 2.33 | |
| CNS | SF-268 | 12.60 | 17.30 | 3.25 | 16.50 |
| SF-295 | 1.83 | 4.27 | 2.13 | 12.10 | |
| SF-539 | 1.74 | 1.77 | 2.03 | 2.20 | |
| SNB-19 | 2.45 | 2.41 | 1.75 | 7.04 | |
| SNB-75 | NT | NT | 4.42 | NT | |
| U251 | 1.81 | 2.15 | 2.77 | 6.83 | |
| Melanoma | LOX IMVI | 1.39 | 1.44 | 1.73 | 1.37 |
| MALME-3M | 1.93 | 1.84 | 1.74 | 3.98 | |
| M14 | 2.52 | 2.19 | 3.14 | 3.40 | |
| MDA-MB-435 | 3.40 | 2.47 | 2.24 | 3.24 | |
| SK-MEL-2 | 2.55 | 2.71 | 3.97 | 5.85 | |
| SK-MEL-28 | 1.93 | 1.98 | 2.54 | 2.13 | |
| SK-MEL-5 | 1.75 | 2.27 | 1.52 | 1.04 | |
| UACC-257 | 2.85 | 11.20 | 2.46 | 11.8 | |
| UACC-62 | 1.55 | 1.69 | 1.68 | 3.44 | |
| Ovarian | IGROV1 | 2.72 | 4.15 | 3.98 | 13.80 |
| OVCAR-3 | 2.02 | 1.93 | 2.26 | 2.08 | |
| OVCAR-4 | 4.47 | 4.40 | 2.96 | 5.02 | |
| OVCAR-5 | 2.14 | 3.64 | 3.90 | 10.00 | |
| OVCAR-8 | 1.88 | 2.39 | 3.34 | 3.24 | |
| NCI/ADR-RES | 1.75 | 1.93 | NT | 2.20 | |
| SK-OV-3 | 5.80 | 16.00 | 10.50 | 16.80 | |
| Renal | 786–0 | 1.92 | 3.36 | 1.92 | 5.75 |
| A498 | 2.08 | 1.85 | 17.00 | 16.80 | |
| ACHN | 1.95 | 2.47 | 3.52 | 2.16 | |
| CAKI-1 | 2.77 | 4.00 | 2.96 | 8.18 | |
| RXF 393 | 1.81 | 1.73 | 1.74 | 1.90 | |
| SN12C | 2.03 | 2.94 | 3.54 | 7.51 | |
| TK-10 | 5.10 | 12.40 | 4.12 | 18.40 | |
| UO-31 | NT | NT | 1.70 | NT | |
| Prostate | PC-3 | 3.07 | 4.08 | 2.98 | 4.08 |
| DU-145 | 3.08 | 5.01 | 3.23 | 8.24 | |
| Breast | MCF7 | 1.28 | 2.06 | 2.48 | 2.13 |
| MDA-MB-231/ATCC | NT | NT | 2.39 | NT | |
| HS 578T | 5.27 | 9.04 | 2.20 | 17.70 | |
| BT-549 | 1.44 | 2.36 | 2.36 | 2.88 | |
| T-47D | NT | NT | 3.25 | NT | |
| MDA-MB-468 | 1.61 | 1.88 | 2.17 | 2.47 | |
Both meta and para substituted –Br derivatives 1c and 1e showed the highest activity with GI50s 1.28 μM and 2.13 μM respectively, against breast cancer cells MCF7, and compounds 1b and 1g demonstrated better activity on the MDA-MB-468 cell line with GI50s 1.88 μM and 2.17 μM.
All four compounds showed promising activity on four different leukemia cancer cell lines HL-60(TB) (1c, GI50: 1.87 μM), RPMI-8226 (1b, GI50: 2.25 μM), K-562 (1e, GI50: 2.24 μM) and CCRF-CEM (1g, GI50: 2.08 μM). Compounds 1c (GI50: 1.66 and 1.75 μM) and 1b (GI50: 1.79 and 1.93 μM) have exhibited potent activity against the colon and ovarian cancer cell lines HCT-15 and NCI/ADR-RES, respectively. However, compounds 1e and 1g showed their superior activity on HCT-116 (GI50: 1.93 and 1.86 μM) and OVCAR-3 (GI50: 2.08 and 2.26 μM) cell lines.
The compounds 1c, 1b, and 1e have the best activity on the melanoma cancer cell line LOX IMV (GI50s: 1.39, 1.44 and 1.37 μM respectively), the renal cancer cell line RXF 393 (GI50s: 1.81, 1.66 and 1.93 μM), non-small lung cancer cell line NCI-H226 (GI50s: 1.62, 1.73 and 1.90 μM) and the CNS cancer cell line SF-539 (GI50s: 1.74, 1.77 and 2.20 μM). In contrast, compound 1g displayed better activity against SK-MEL-5 (GI50: 1.52 μM), UO-31 (GI50: 1.70 μM), NCI-H460 (GI50: 1.91 μM) and SNB-75 (GI50: 1.75 μM) cell lines. All four compounds were more active on the prostate cancer cell line PC-3 with GI50s 3.07 μM (1c), 4.08 μM (1b), 2.13 μM (1e) and 2.98 μM (1g).
Total growth inhibition (TGI) and 50% lethal concentration (LC50) of these four compounds against the 60 human cancer cell lines are provided in the ESI (Tables S2 and S3).†
| Compound | IC50 (μM) |
|---|---|
| 1g | 3.08 |
| 1c | 3.16 |
| 1b | 3.40 |
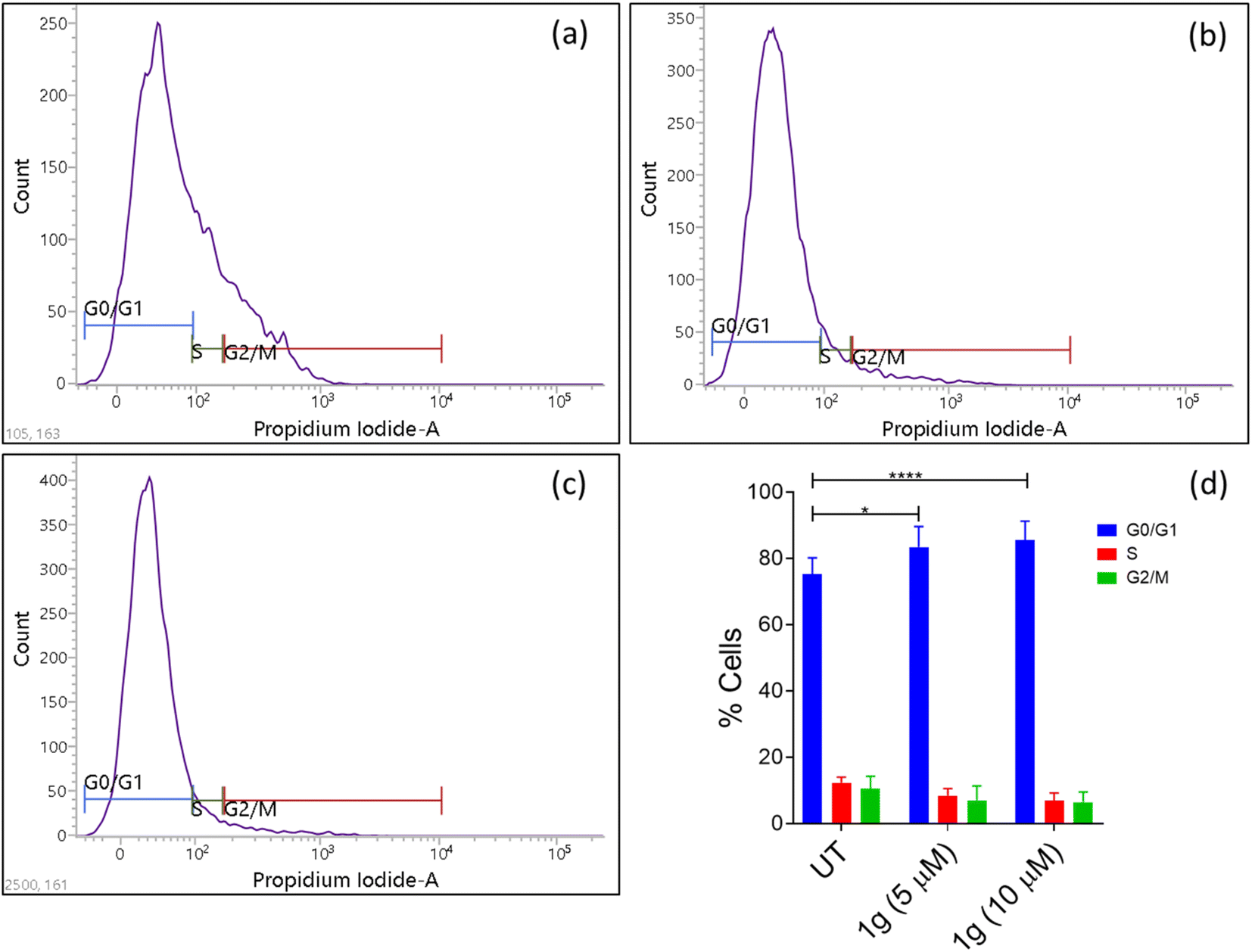 | ||
| Fig. 1 Effect of compound 1g on HCT-116 cell cycle arrest. (a) Untreated (control); (b) HCT-116 cells treated with 5 μM concentration of 1g; (c) HCT-116 cells treated with 10 μM concentration of 1g; (d) % cells in each phase after treatment with 1g. | ||
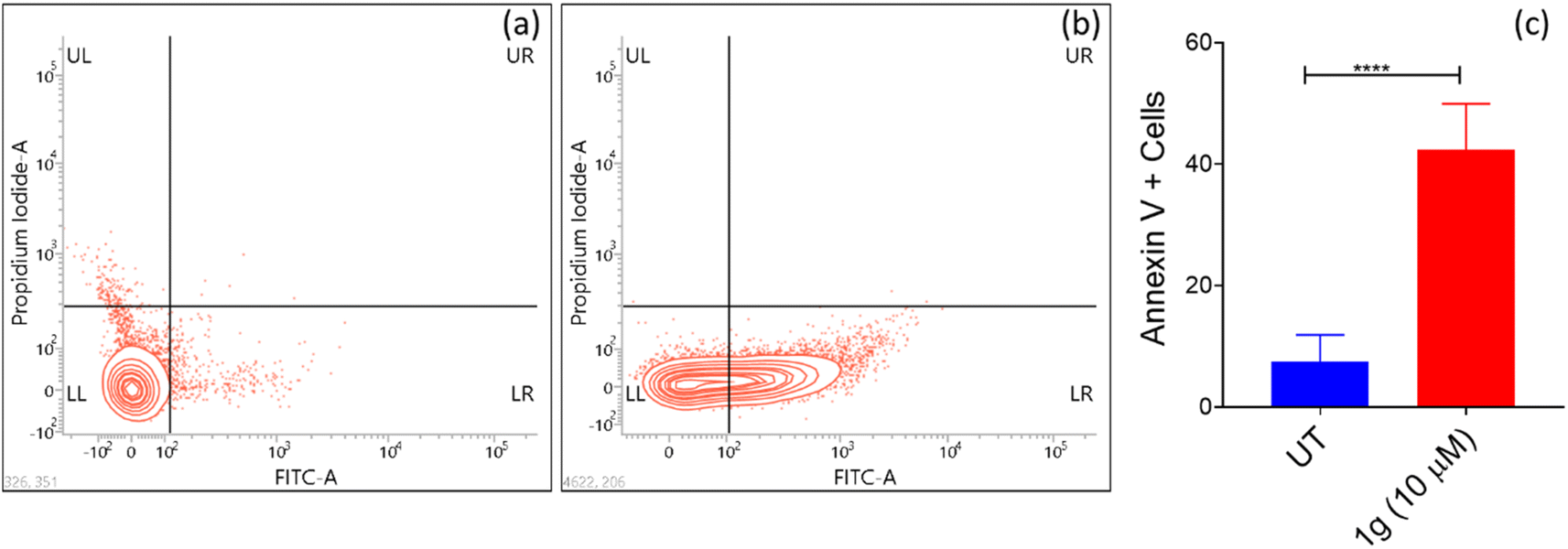 | ||
| Fig. 2 Flow cytometry plots using Annexin V-FITC/PI staining for apoptosis. HCT-116 cells were treated with (a) control (untreated cells) (b) 10 μM concentration of 1g. (c) % of apoptotic cells after treatment with 1g. | ||
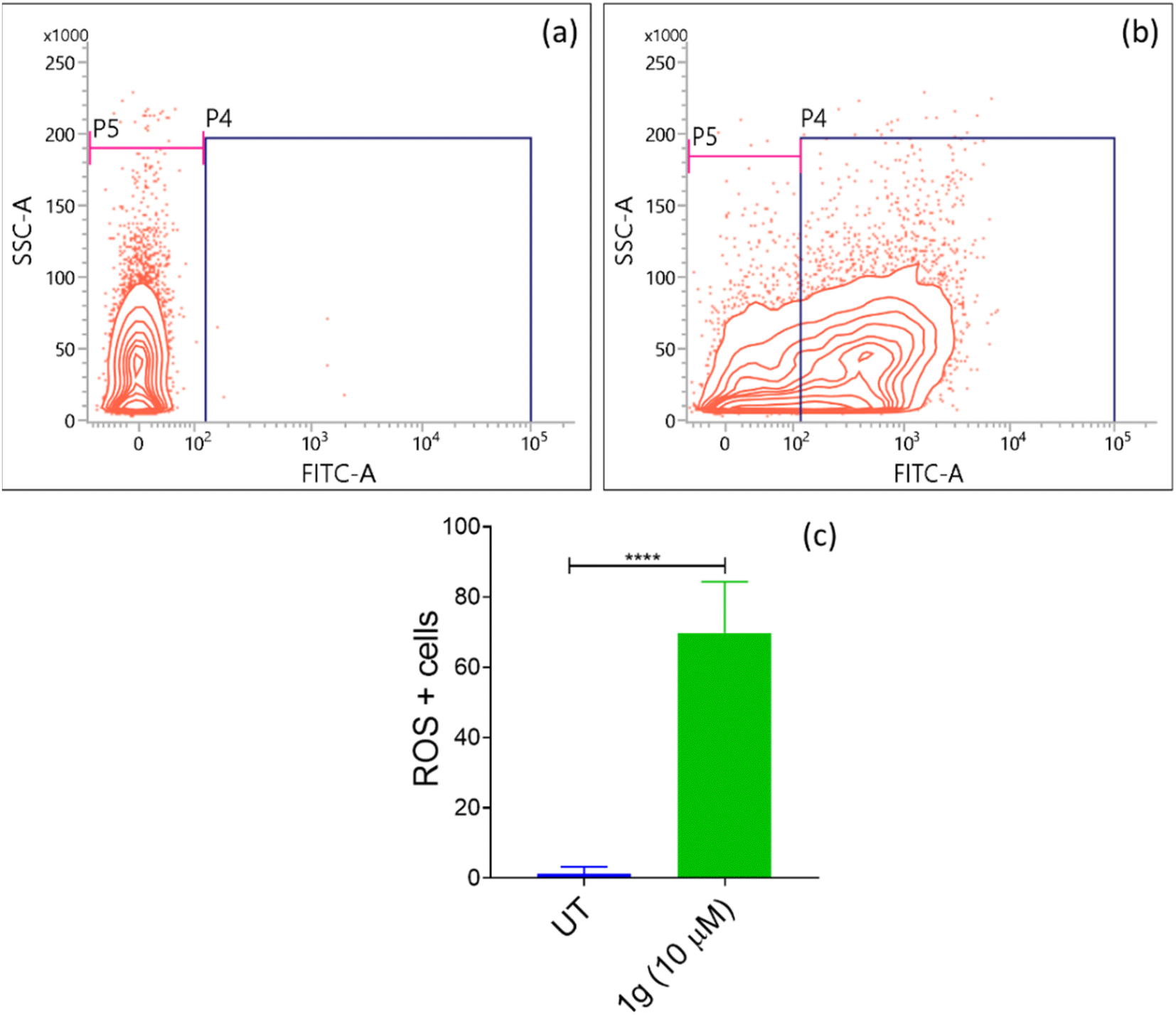 | ||
| Fig. 3 Determination of intracellular ROS levels in HCT-116 cells treated with (a) control (untreated cells), (b) 10 μM concentration of 1g; (c) % of ROS levels in the presence of 1g. | ||
4.3. Structural activity relationship based on NCI screening
As mentioned before, our group previously investigated the anticancer properties of 3,19-benzylidene/heteroaromatic acetals of andrographolide and isoandrographolide.22 These derivatives have displayed moderate to good anticancer activity. Therefore, from these series, we have selected and compared the most active compounds and the cell lines on which they showed good activity with the top three 3,19-(NH-3-aryl-pyrazole) acetals of andrographolide (1c, 1b and 1g) (% growth inhibition -Table 3, Fig. 4). Interestingly, on all the cell lines, the compounds 1c, 1b and 1g exhibited several-fold better % growth inhibition than the rest of the compounds. Indeed, these three compounds are lethal to most of the cancer cell lines. Here, the better anticancer activity could be attributed to the presence of pyrazole moiety in the acetals of andrographolide or isoandrographolide.| Cancer | Cell line | % Growth inhibition | |||||||
|---|---|---|---|---|---|---|---|---|---|
| ADG | Bz-ADG | Fur-ADG | Py-ADG | Thio-ADG | 1c | 1b | 1g | ||
| a n.a – not active; n.t – not tested, negative values represent lethality (example: −15.57% growth inhibition means 15.57% lethality). | |||||||||
| Leukaemia | CCRF-CEM | 76.51 | 90.97 | 89.84 | 86.21 | 66.78 | −15.57 | −14.87 | 99.40 |
| HL-60(TB) | n.aa | 51.12 | 1.86 | 13.79 | n.a | −16.26 | −15.08 | 98.41 | |
| K-562 | 34.47 | 56.31 | 55.44 | 47.29 | 23.54 | −2.18 | 95.00 | −31.21 | |
| RPMI-8226 | 16.71 | 71.77 | 82.88 | 58.08 | 29.46 | −6.49 | −11.69 | −27.00 | |
| SR | 20.46 | 63.12 | 70.99 | 83.82 | 23.59 | 97.32 | 84.29 | −29.91 | |
| NSCL | NCI-H522 | 82.86 | 36.48 | 36.90 | 34.87 | 15.89 | −55.96 | −59.98 | −18.40 |
| Colon | HCT-116 | 39.98 | 61.62 | 48.53 | 38.35 | 15.56 | −95.92 | −91.58 | −19.04 |
| HCT-15 | 38.11 | 34.48 | 40.82 | 28.68 | 13.59 | −67.53 | −59.20 | −35.29 | |
| HT29 | 72.99 | 45.20 | 36.91 | 30.12 | 15.95 | 98.48 | 99.69 | −26.28 | |
| SW-620 | 55.71 | 50.33 | 54.62 | 51.49 | 8.40 | −81.14 | −57.57 | n.t | |
| Melanoma | LOX IMVI | 78.16 | 66.18 | 77.96 | 77.02 | 27.00 | −100 | −100 | n.t |
| Ovarian | OVCAR-8 | 15.52 | 43.42 | 42.62 | 26.37 | 21.32 | −42.49 | −23.31 | −3.29 |
| NCI/ADR-RES | 16.25 | 42.98 | 23.28 | 12.07 | 6.23 | −50.15 | −56.43 | 93.33 | |
| Breast | MCF7 | 42.44 | 53.91 | 51.30 | 36.01 | 27.86 | −48.20 | −52.32 | 98.00 |
| BT-549 | 12.88 | 36.15 | n.a | n.a | n.a | −94.24 | −92.39 | −32.90 | |
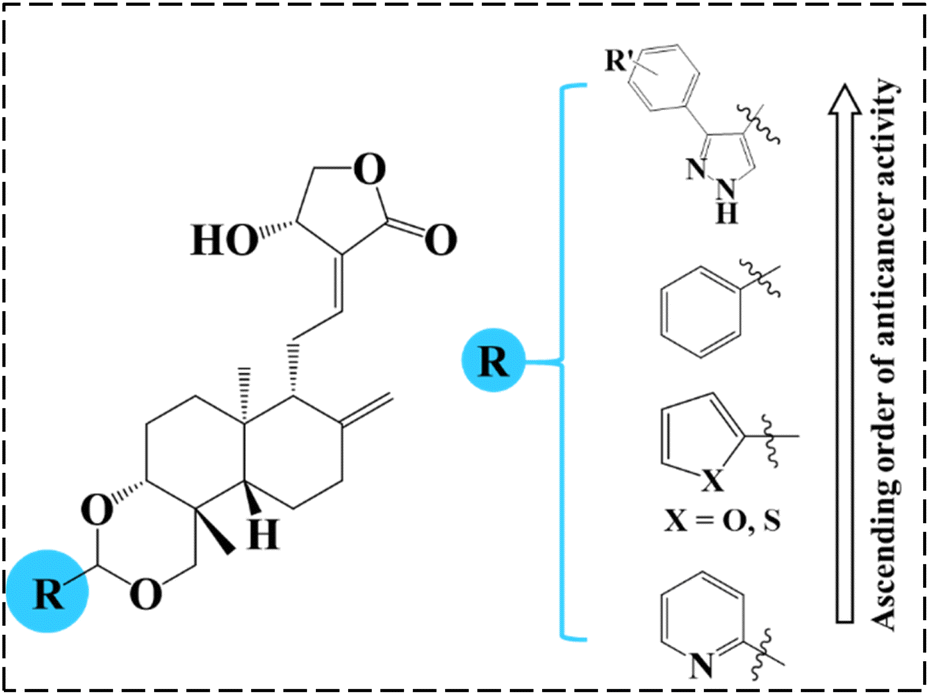 | ||
| Fig. 4 The order of anticancer activity of benzylidene/heteroaromatic/pyrazole acetals of andrographolide. | ||
4.4. Drug-likeness and ADMET prediction
Employing the pkCSM tool, in silico drug-likeness, pharmacokinetic, and toxicity studies (Table 4 and 5) were performed for the active compounds in order to determine their potential as drug candidates by analysing their Lipinski and ADMET parameters.| ADMET | Compound | ||
|---|---|---|---|
| 1b | 1c | 1g | |
| Water solubility (Logs) |
−5.24 | −5.27 | −5.01 |
| Skin permeability (LogKp) |
−2.84 | −2.83 | −2.83 |
| GI absorption | High | High | High |
| BBB permeability | Low | Low | Low |
| CYP1A2 inhibitor | No | No | No |
| Total clearance | 0.17 | 0.15 | 0.18 |
| Max. tolerated dose | −0.860 | −0.862 | −0.845 |
| AMES toxicity | No | No | No |
| hERG inhibitor | No | No | No |
| Skin sensitization | No | No | No |
| Minnow toxicity | −1.419 | −1.56 | −1.56 |
From the results, we note that the in silico molecular properties of the active compounds are in accordance with Lipinski's rule of five (RO5) barring molecular weights. The molecular weights of all the active compounds are in the range of 534–582 Daltons, slightly exceeding the acceptable limit (<500 Dalton). However, molecular weights of natural products are frequently considered an exception to Lipinski's rules.35 Molar refractivity of the compounds was found to be in the range 40–130 and LogP was observed to be <5. The total number of N–H and O–H hydrogen bond donors is <5 and the number of nitrogen and oxygen-hydrogen bond acceptors (HBA) is <10.
We also examined the adsorption, distribution, metabolism, excretion, and toxicity effects of the active compounds using pkCSM to obtain a comprehensive picture of their therapeutic efficacy.
Table 5 provides an overview of the ADMET properties of the various compounds with potential for use as drugs. The compounds showed moderate aqueous solubility, high gastrointestinal absorption, and low BBB permeability. In addition, logKp and maximum tolerated dose values were observed to be in the acceptable range, and all the compounds are non-inhibitors of CYP1A2 and hERG. Importantly, none of the compounds exhibited AMES toxicity, hERG inhibition, skin sensitization, and minnow toxicity. Compliance with Lipinski's parameters and the observed ADMET properties indicate that these molecules deserve to be investigated as potential anticancer agents.
5. Conclusion
In conclusion, nine 3-aryl-1H-pyrazole-4-carbaldehydes were synthesized, and among these four are novel. These pyrazole carbaldehydes were used as starting materials to synthesize eight 3,19-(NH-3-aryl-pyrazole) acetals of andrographolide and eight 3,19-(NH-3-aryl-pyrazole) acetals of isoandrographolide. All the synthesized compounds (1a–1h, 2a–2g, 2i) have been characterized extensively and were initially tested at the NCI (USA) for their anticancer properties on a panel of 60 human cancer cell lines. Interestingly, 3,19-(NH-3-aryl-pyrazole) acetals of andrographolide exhibited better anticancer activity than the acetals of isoandrographolide. Seven out of the eight 3,19-(NH-3-aryl-pyrazole) acetals of andrographolide (1a–1g) were further selected for five dose study to determine GI50s, total growth inhibition, and LC50s by NCI. The most potent among them (1b, 1c, and 1g) were further chosen to find out their IC50s against the colon cancer HCT-116 cell line. Based on the IC50 values, the most potent compound 1g (IC50 3.08 μM) was selected to investigate its mechanism of action on the HCT-116 cell line. The results revealed that 1g was responsible for the cell cycle arrest at the G0/G1 phase due to the early apoptosis of HCT-116 cells. In addition, ROS assay results indicated that the apoptosis of HCT-116 cells induced by compound 1g was mainly attributed to the elevated levels of intracellular ROS in HCT-116 cells. Further, the best three active compounds (1c, 1b and, 1g) were found to be more active than 3,19-benzylidene/heteroaromatic acetals of andrographolide and isoandrographolide, which were previously synthesized by our group. This comparison indicates that the pyrazole moiety played a crucial role in improving the anticancer activity. Further, in silico Lipinski and ADMET results of the most active compounds suggest that these can be modeled as potential therapeutic leads for the treatment of cancer.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Siva Kumar Rokkam carried out the synthesis, characterization, in silico studies, data analysis, and drafted the manuscript. Manohar Bhujel and Dolly Jain performed and analysed the in vitro anticancer studies: cell viability, cytotoxicity, cell cycle analysis, staining, and ROS assays. Avinash Bajaj supervised the anticancer studies. Nageswara Rao Golakoti, Lakshminath Sripada and Srinivas Nanduri conceived the study, participated in its design, analyzed the results, and contributed to drafting the manuscript. All authors read and approved the final manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors dedicate this work to the Founder Chancellor Bhagawan Sri Sathya Sai Baba. We thank the Central Research Instruments Facility-SSSIHL for providing the characterization facilities. Authors thank the Disease Biology lab, SSSIHL and Regional Center for Biotechnology (RCB) for providing lab facilities to carry out biological studies. GNR and NS are grateful to the Council of Scientific & Industrial Research (CSIR) for funding this project (no. 02(0304)/17/EMR-II).References
- C. Aromdee, Expert Opin. Ther. Pat., 2012, 22, 169–180 CrossRef CAS PubMed.
- S. G. S. Kandanur, N. Tamang, N. R. Golakoti and S. Nanduri, Eur. J. Med. Chem., 2019, 176, 513–533 CrossRef CAS PubMed.
- M. Jiang, F. Sheng, Z. Zhang, X. Ma, T. Gao, C. Fu and P. Li, J. Ethnopharmacol., 2021, 272, 113954 CrossRef CAS PubMed.
- P. Sharma, N. Kumar and R. Dudhe, Pharmacologyonline, 2011, 2, 11 Search PubMed.
- S. Kumar, B. Singh and V. Bajpai, J. Ethnopharmacol., 2021, 275, 114054 CrossRef CAS.
- A. Okhuarobo, J. Ehizogie Falodun, O. Erharuyi, V. Imieje, A. Falodun and P. Langer, Asian Pac. J. Trop. Dis., 2014, 4, 213–222 CrossRef CAS.
- L. S. Chua, Phyther. Res., 2014, 28, 1589–1598 CrossRef PubMed.
- A. Widyawaruyanti, M. Asrory, W. Ekasari, D. Setiawan, A. Radjaram, L. Tumewu and A. F. Hafid, Procedia Chem., 2014, 13, 101–104 CrossRef CAS.
- A. K. Jadhav and S. M. Karuppayil, Phyther. Res., 2021, 35, 5365–5373 CrossRef CAS.
- R. A. Kumar, K. Sridevi, N. V. Kumar, S. Nanduri and S. Rajagopal, J. Ethnopharmacol., 2004, 92, 291–295 CrossRef CAS.
- Y. Dai, S.-R. Chen, L. Chai, J. Zhao, Y. Wang and Y. Wang, Crit. Rev. Food Sci. Nutr., 2019, 59, S17–S29 CrossRef CAS PubMed.
- J. C. W. Lim, T. K. Chan, D. S. W. Ng, S. R. Sagineedu, J. Stanslas and W. S. F. Wong, Clin. Exp. Pharmacol. Physiol., 2012, 39, 300–310 CrossRef CAS.
- G. Liu and H. Chu, Oncol. Lett., 2018, 15, 5301–5305 Search PubMed.
- P. Udomwan, C. Pientong, P. Tongchai, A. Burassakarn, N. Sunthamala, S. Roytrakul, S. Suebsasana and T. Ekalaksananan, Int. J. Mol. Sci., 2021, 22, 6806 CrossRef CAS PubMed.
- Z. Malik, R. Parveen, B. Parveen, S. Zahiruddin, M. Aasif Khan, A. Khan, S. Massey, S. Ahmad and S. A. Husain, J. Ethnopharmacol., 2021, 272, 113936 CrossRef CAS PubMed.
- A. A. Farooqi, R. Attar, U. Y. Sabitaliyevich, N. Alaaeddine, D. P. de Sousa, B. Xu and W. C. Cho, Cancers, 2020, 12 Search PubMed.
- J. C. W. Lim, T. K. Chan, D. S. Ng, S. R. Sagineedu, J. Stanslas and W. F. Wong, Clin. Exp. Pharmacol. Physiol., 2012, 39, 300–310 CrossRef CAS PubMed.
- S. K. Mishra, S. Tripathi, A. Shukla, S. H. Oh and H. M. Kim, Front. Biosci., 2015, 7, 255–266 CrossRef.
- S. G. S. Kandanur, S. Nanduri and N. R. Golakoti, Bioorg. Med. Chem. Lett., 2017, 27, 2854–2862 CrossRef CAS PubMed.
- S. G. S. Kandanur, S. Kundu, C. Cadena, H. S. Juan, A. Bajaj, J. D. Guzman, S. Nanduri and N. R. Golakoti, Chem. Pap., 2019, 73, 1669–1675 CrossRef CAS.
- S. G. S. Kandanur, N. R. Golakoti and S. Nanduri, Bioorg. Med. Chem. Lett., 2015, 25, 5781–5786 CrossRef CAS PubMed.
- N. Tamang, C. Andrews, S. K. Mavileti, S. Nanduri, N. R. Golakoti and B. Karanam, New J. Chem., 2022, 46, 9745–9754 RSC.
- B. Karanam, N. Tamang, Y. Inabathina, S. K. Mavileti, L. Sripada, S. Nanduri and N. R. Golakoti, Cancer Res., 2023, 83, 3818 CrossRef.
- V. Kumar, K. Kaur, G. K. Gupta and A. K. Sharma, Eur. J. Med. Chem., 2013, 69, 735–753 CrossRef CAS.
- M. Faisal, A. Saeed, S. Hussain, P. Dar and F. A. Larik, J. Chem. Sci., 2019, 131, 70 CrossRef.
- A. Ansari, A. Ali, M. Asif and Shamsuzzaman, New J. Chem., 2016, 41, 16–41 RSC.
- D. E. V. Pires, T. L. Blundell and D. B. Ascher, J. Med. Chem., 2015, 58, 4066–4072 CrossRef CAS.
- A. Kar, D. Jain, S. Kumar, K. Rajput, S. Pal, K. Rana, R. Kar, S. K. Jha, N. Medatwal, P. S. Yavvari, N. Pandey, D. Mehta, H. Sharma, D. Bhattacharya, M. K. Pradhan, R. D. Sharma, A. Srivastava, U. Agrawal, A. Mukhopadhyay, S. Sengupta, V. S. Patil, A. Bajaj and U. Dasgupta, Sci. Adv., 2023, 9, 1–22 Search PubMed.
- M. Beniwal and N. Jain, J. Heterocycl. Chem., 2019, 56, 2508–2516 CrossRef CAS.
- L. You, Xu Qidong, Z. Xiaoli, Li Shiduo, Z. Hui, B. Wei Wu; X. Liu; L. Li; G. Xin; Z. Jiang and W. Xiaoke, China Pharmaceutical University, CN115960047A, 2023 Search PubMed.
- D. L. Pavia, G. M. Lampman and G. S. Kriz, Introduction to Spectroscopy-A Guide for Students of Organic Chemistry, Thomson Learning, Inc., 2001 Search PubMed.
- D. L. B. R. M. Silverstein, F. X. Webster and D. J. Kiemle, Spectrometric Identification of Organic Compounds, 8th edn, Wiley, 2014 Search PubMed.
- E. Miller, ed. S. P. Langdon, Humana Press, Totowa, NJ, 2004, pp. 191–202.
- G.-Y. Liou and P. Storz, Free Radical Res., 2010, 44, 479–496 CrossRef CAS PubMed.
- A. Ganesan, Curr. Opin. Chem. Biol., 2008, 12, 306–317 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Contains spectral characterization, 1H-NMR, 13C-NMR, HRMS spectra, and NCI in vitro results. See DOI: https://doi.org/10.1039/d4ra00547c |
| This journal is © The Royal Society of Chemistry 2024 |