
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Minor limonoid constituents from Swietenia macrophylla by simultaneous isolation using supercritical fluid chromatography and their biological activities†
Kathiravan Asokanab,
A. Zahir Hussainb,
Rajesh Kumar Gattua and
Andivelu Ilangovan *c
*c
aAragen Life Sciences Pvt Ltd, Bengaluru-562106, India
bDepartment of Chemistry, Jamal Mohamed College, Tiruchirappalli, Tamilnadu-620020, India
cSchool of Chemistry, Bharathidasan University, Tiruchirappalli, Tamilnadu-620024, India. E-mail: ilangovanbdu@yahoo.com
First published on 22nd August 2024
Abstract
This study reports simultaneous isolation of three new limonoids (1–3), six known regio isomers (6, 7, 9–12), and three more known limonoids (4, 5, 8) from Swietenia macrophylla (S. macrophylla) seeds. Structures of these compounds were determined via extensive study of their 1D/2D-NMR and mass spectral data. Known limonoids (4–12) were identified by comparing their physical and spectroscopic data with literature values. A novel environmentally friendly supercritical fluid chromatography (SFC) technique facilitated simultaneous and rapid separation of these compounds. The pharmacological activities of the new limonoids were investigated.
1 Introduction
Mahogany, or Swietenia macrophylla (S. macrophylla), is a significant medicinal plant cultivated in tropical and subtropical regions. Traditional medicine employs all parts of the S. macrophylla to cure a range of human ailments.1 The study of the phytochemical components found in S. macrophylla's various plant sections demonstrated the plant's abundance in triterpenes, referred to as limonoids, and their derivatives.2 These limonoids are prevalent in citrus fruits of S. macrophylla with a bitter taste and sweet or sour scent. With a furan ring attached at the C-17 position, they usually exhibit high degrees of oxidation and skeletal rearrangements. These carbon framework containing limonoids include andirobin, gedunin, mexicanolide, phragmalin, and D-ring-opened phragmalin (Fig. 1).2b Purified limonoids from S. macrophylla have been employed in several medical treatments such as hypertension,3 anti-diabetic,4,5 anti-bacterial,6 anti-inflammatory, antimicrobial, anti-malarial, anti-oxidant, anti-tumor, treatment of dengue virus7–10 and hypolipidemic activity.11,12 | ||
| Fig. 1 Selected biologically active limonoids (D-ring-opened phragmalin-type). | ||
The primary botanical families known to contain limonoids are Meliaceae, Rutaceae, and Simaroubaceae. Within these plant families, various species have been identified as rich sources of limonoids. These compounds are often found in the seeds, fruits, leaves, and bark of such plant species. More than 300 different limonoids have been identified from numerous plant sources,13,14 and there are undoubtedly many more limonoids yet to be found.
Medicinal plants possess complex matrices that allow them to produce compounds showing wide variety of biological activities. Purification, measurement, separation, and characterization of bioactive compounds from extracts of plant material have never been easy. Fortunately, several innovative techniques have emerged that provide compelling evidence for improving the method's sensitivity, selectivity, and run times in the evaluation of therapeutic herbs. Therefore, selecting a simple and appropriate separation technique for natural product separation has become increasingly crucial in recent years.
The development of new, supercritical technology instruments for a wide variety of column chemistries and fluid chromatography, along with innate technological qualities, have made supercritical fluid chromatography (SFC) a stand-in and renowned analytical platform for research on therapeutic plants. The SFC is a green technique, offers several advantages over Prep-HPLC such as easy to use, faster separations, and availability of wide variety of SFC stationary phases with diverging properties. Furthermore, SFC can assess substances that are insoluble in Prep-HPLC solvents, leading to more precise and well-resolved peaks. SFC is a desirable technique for the separation of non-polar natural components such as terpenes, fatty acids, vitamins, sterols, and also moderate-to-strong polar components using modifiers.15,16 This technique is particularly well-liked for the enantio separation of chiral compounds.17 Supercritical fluids have higher diffusivities and lower viscosities, which make them more efficient, easier to scale up, and need less time to analyse. It makes drying at lower temperatures easier and maintains the stability of the phytochemical elements because of the non-organic solvent systems.18 This perspective saw SFC as a promising substitute that safeguards the thermally labile compounds and gives a complimentary chromatography environment. Research to date indicates that the SFC technique has been used to analyze a wide range of phytochemical constituents, including vitamins,19–21 triterpenoids,22–26 alkaloids,27–31 and flavonoids.32–34 However, only a few instances SFC purification processes were used to isolate plant extracts such as the ergostane triterpenoids from Antrodia camphorata,35 polyphenols from Mangifera indica Linn,36 carsonic acid from rosemary extracts,37 and Piper kadsura.38
Phytochemicals from S. macrophylla were traditionally extracted and purified using energy-intensive methods like Prep-HPLC and repeated column chromatography,15,39 involving large volumes of petroleum hydrocarbon solvents, harmful to health and the environment.40–43 Isolating pure components was laborious, and many compounds degraded during drying. Due to the complexity and abundance, traditional isolation of limonoids from S. macrophylla posed challenges, due to their low UV absorbance (steroidal skeleton) and similarity in adsorption behavior.22 Availability of no report on the use of SFC for the study of phytochemicals from S. macrophylla set out the goal of the current work to use the SFC approach to separate the limonoids from the acetone and ethanol extracts of S. macrophylla seeds.
2 Results and discussion
Harvested fruits of S. macrophylla were collected from Salem, Tamilnadu, India, in the northern hemisphere of Asia, which is situated at latitude 11.65376 and longitude 78.15538. After shade drying and grinding the seeds to coarse powder, it was repeatedly defated using hexane at room temperature (Fig. 2) and successively extracted with chloroform, acetone, ethanol, methanol, and water.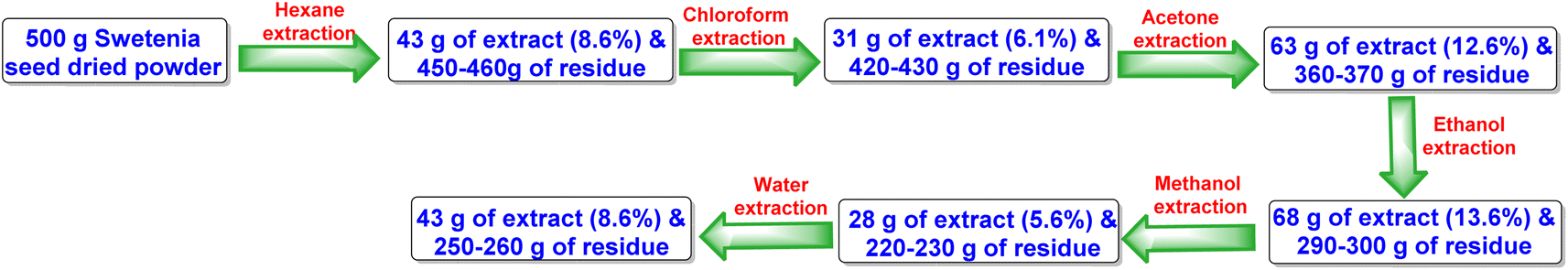 | ||
| Fig. 2 Room temperature extraction of S. macrophylla seeds using different solvents. | ||
Even though each extract had a variety of phytoconstituents, the acetone and ethanol extracts which provided higher quantity of crude phytoconstituents were selected for further study. Steps were taken to develop an effective, simultaneous, one-step SFC purification of several phytochemicals. To optimise SFC conditions, several factors encompassing the stationary phase, mobile phase, and other instrumental parameters were considered. Three achiral columns namely, Princeton 2-ethylpyridine, YMC Diol, and Daicel-P4VP, as well as nine chiral columns namely, LUX-i-Amylose-3, Chiralpak AD-H, LUX-Amylose-2, Chiralpak IG, Chiralpak AS-H, Chiral ART Amylose SA, Chiralpak IH, (R,R) Whelk-O1 and Chiralpak-IE, were examined. Due to the varying stereo-configurations of epimers, chiral columns typically exhibited significantly higher resolutions compared to achiral columns. Stationary phases primarily composed of polysaccharides offer enhanced resolution in separating regio-, E/Z and non-enantiomeric isomeric mixtures of compounds.44,45 Among these polysaccharide phases, the Amylose-based Chiralpak-IE column, specifically amylose tris(3,5-dichlorophenylcarbamate), was identified as providing superior separation of individual limonoid compounds while maintaining reasonable retention times (Fig. 3). After testing a variety of co-solvents, including ethanol, acetonitrile, isopropyl alcohol, the most effective co-solvent was identified as methanol and used along with supercritical carbon dioxide liquid as mobile phase for separating twelve different compounds from acetone and ethanol extracts. Structures of all compounds were determined by the interpretation of their 1D/2D-NMR, and additional spectroscopic data (Fig. 4).
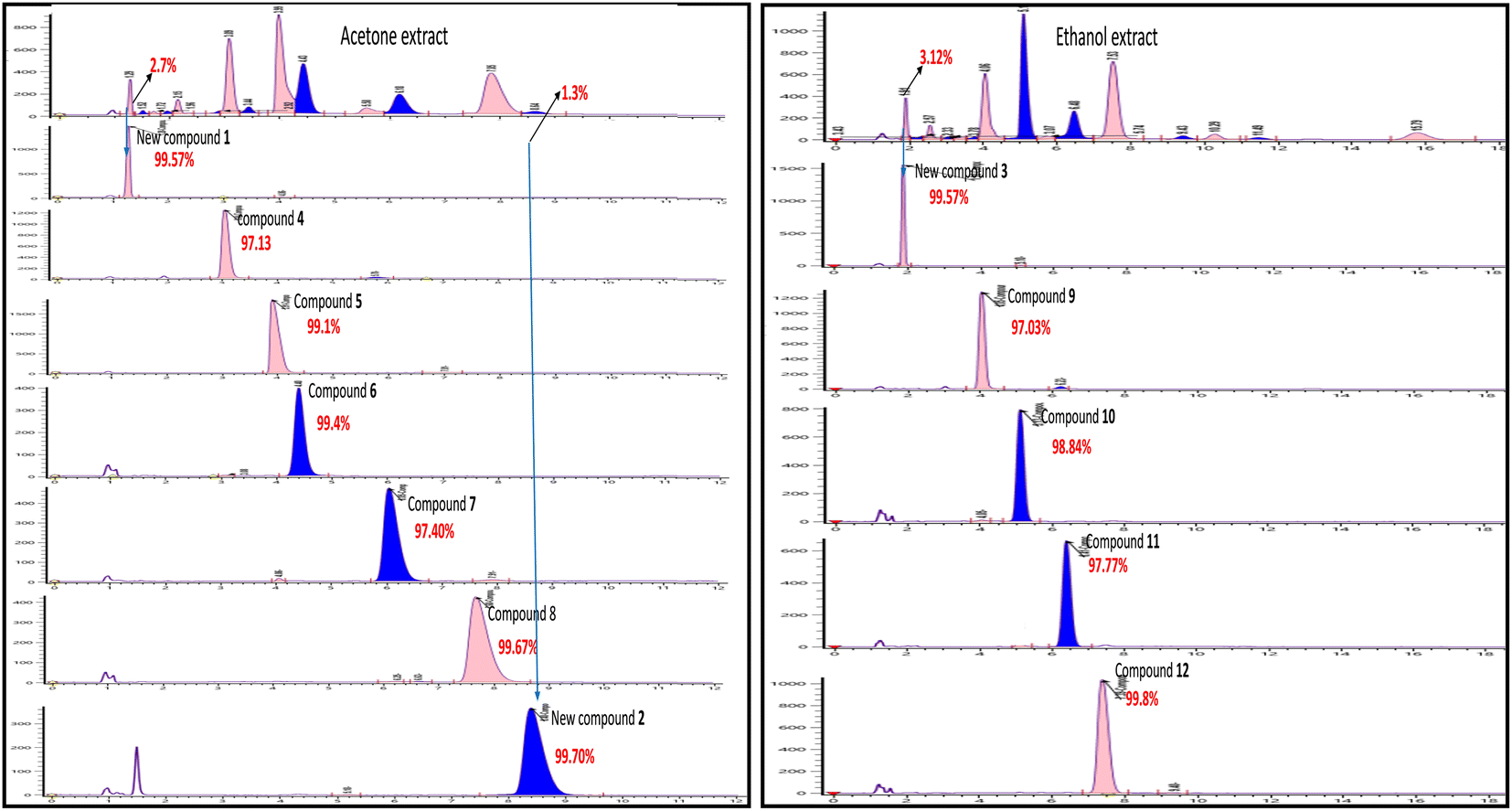 | ||
| Fig. 3 SFC chromatogram purity of compounds 1–12 isolated using Chiralpak IE (4.6 × 250 mm) 5μ column using methanol as co-solvent. | ||
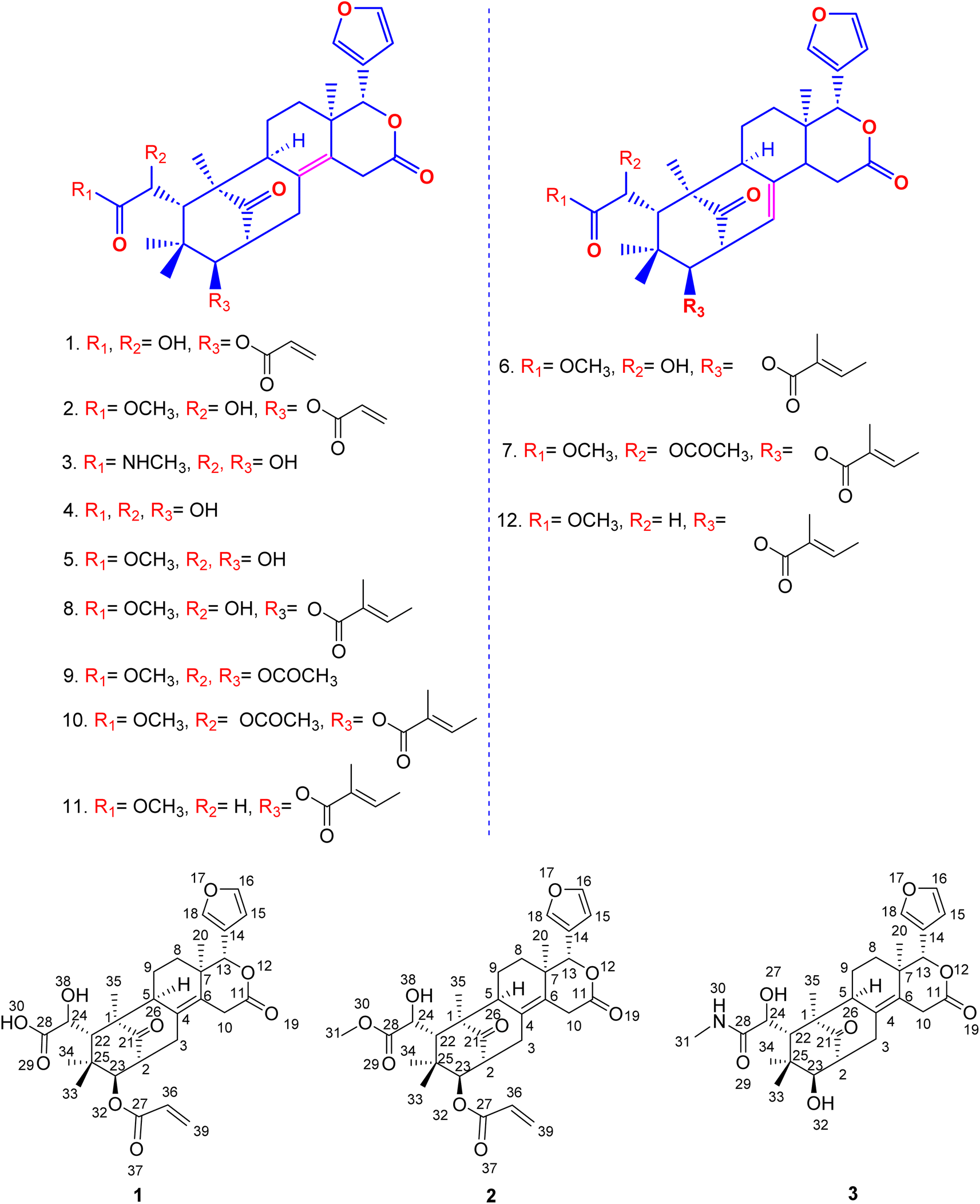 | ||
| Fig. 4 Molecular structure of new and known limonoids isolated from the seeds of Swietenia macrophylla. | ||
Compound 1 was obtained as an amorphous white solid having Mp 178–180 °C, [α]25D −180.8 and shows IR (KBr) absorptions at 3501 cm−1 (OH) and 1718 cm−1 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O). The molecular formula of C29H34O9 was determined from positive HR-ESI-MS value m/z 527.1511. Its structure was supported by 1H, 13C, HSQC, and HMBC NMR data, revealing features such as proton signals appearing at δH 5.87 (dd, J = 10.2, 1.8 Hz, 1H), 6.27 (dd, J = 17.2, 1.8 Hz, 1H), 6.07 (dd, J = 17.2, 10.2 Hz, 1H) due to the presence of substituted end alkene. Further, protons corresponding to the presence of a furan ring were observed at δH 6.49 (d, 1H), 7.65 (s, 1H), 7.67 (d, 1H), tertiary methyl groups at δH 0.89 (s, 3H), 1.22 (s, 3H), and oxygen-attached methine groups at δH 3.35 (d, 1H, J = 9.4 Hz), 4.40 (s, 1H), 5.47 (s, 1H) (Table 1). Notably, 29 peaks corresponding to different carbons, including methyls, methylenes, methines, aliphatic and aromatic quaternary carbons, and carbonyl groups, were observed in their expected δC values. Substituted end alkene group protons appearing at δH 5.87 (dd, J = 10.2, 1.8 Hz, 1H), 6.27 (dd, J = 17.2, 1.8 Hz, 1H), 6.07 (dd, J = 17.2, 10.2 Hz, 1H) were distinguished by HSQC and located at C-27 by the corresponding HMBC correlations of the alkene protons to C-27.
O). The molecular formula of C29H34O9 was determined from positive HR-ESI-MS value m/z 527.1511. Its structure was supported by 1H, 13C, HSQC, and HMBC NMR data, revealing features such as proton signals appearing at δH 5.87 (dd, J = 10.2, 1.8 Hz, 1H), 6.27 (dd, J = 17.2, 1.8 Hz, 1H), 6.07 (dd, J = 17.2, 10.2 Hz, 1H) due to the presence of substituted end alkene. Further, protons corresponding to the presence of a furan ring were observed at δH 6.49 (d, 1H), 7.65 (s, 1H), 7.67 (d, 1H), tertiary methyl groups at δH 0.89 (s, 3H), 1.22 (s, 3H), and oxygen-attached methine groups at δH 3.35 (d, 1H, J = 9.4 Hz), 4.40 (s, 1H), 5.47 (s, 1H) (Table 1). Notably, 29 peaks corresponding to different carbons, including methyls, methylenes, methines, aliphatic and aromatic quaternary carbons, and carbonyl groups, were observed in their expected δC values. Substituted end alkene group protons appearing at δH 5.87 (dd, J = 10.2, 1.8 Hz, 1H), 6.27 (dd, J = 17.2, 1.8 Hz, 1H), 6.07 (dd, J = 17.2, 10.2 Hz, 1H) were distinguished by HSQC and located at C-27 by the corresponding HMBC correlations of the alkene protons to C-27.
| Positions | Compound 1 | Compound 2 | Compound 3 | |||
|---|---|---|---|---|---|---|
| δH (J in Hz) | δC | δH (J in Hz) | δC | δH (J in Hz) | δC | |
| a m: multiplet, t: triplet d: doublet, s: singlet, dd: doublet of doublet, br: broad. | ||||||
| 1 | — | 53.5 | — | 53.5 | — | 53.3 |
| 2 | 2.80 (m) | 50.5 | 2.79 (m) | 50.5 | 2.80 (m) | 50.3 |
| 3 | 1.80, 3.06 (m) | 33.7 | 1.80, 3.06 (m) | 33.7 | 1.81, 3.05 (m) | 33.6 |
| 4 | — | 128.8 | — | 128.8 | — | 128.6 |
| 5 | 1.89 (m) | 52.8 | 1.88 (m) | 52.8 | 1.89 (m) | 52.7 |
| 6 | — | 129.9 | — | 129.9 | — | 129.8 |
| 7 | — | 37.6 | — | 37.6 | — | 37.1 |
| 8 | 0.95, 1.68 (m) | 28.9 | 0.93, 1.68 (m) | 28.9 | 0.95, 1.68 (m) | 28.8 |
| 9 | 1.66, 1.80 (m) | 18.4 | 1.66, 1.80 (m) | 18.4 | 1.66, 1.80 (m) | 18.2 |
| 10 | 3.41, 3.90 (m) | 33.1 | 3.29, 3.90 (m) | 33.1 | 3.43, 3.93 (m) | 32.9 |
| 11 | — | 170.3 | — | 170.3 | — | 170.0 |
| 12 | — | — | — | — | — | — |
| 13 | 5.47 (s) | 79.6 | 5.47 (s) | 79.6 | 5.48 (s) | 79.3 |
| 14 | — | 121.0 | — | 121.0 | — | 120.8 |
| 15 | 6.49 (d) | 110.2 | 6.49 (s) | 110.2 | 6.50 (s) | 110.0 |
| 16 | 7.67 (d) | 143.5 | 7.67 (s) | 143.5 | 7.66 (s) | 143.3 |
| 17 | — | — | — | — | — | — |
| 18 | 7.65 (s) | 141.3 | 7.64 (s) | 141.3 | 7.68 (s) | 141.2 |
| 19 | — | — | — | — | — | — |
| 20 | 0.90 (s) | 18.0 | 0.89 (s) | 18.0 | 0.90 (s) | 17.9 |
| 21 | — | 220.3 | — | 220.3 | — | 219.9 |
| 22 | 3.11 (s) | 43.6 | 3.10 (s) | 43.6 | 3.11 (s) | 43.4 |
| 23 | 3.36 (d, 9.4 Hz) | 77.1 | 3.35 (d, 9.4 Hz) | 77.1 | 3.36 (dd, 9.4, 4.8 Hz) | 76.9 |
| 24 | 4.42 (s) | 72.6 | 4.40 (s) | 72.6 | 4.41 (d, 4.5 Hz) | 72.4 |
| 25 | — | 39.6 | — | 39.6 | — | 39.4 |
| 26 | — | — | — | — | — | — |
| 27 | — | 167.1 | — | 167.1 | 5.28 (d, 4.5 Hz) | — |
| 28 | — | 177.2 | — | 176.6 | — | 172.8 |
| 29 | — | — | — | — | — | — |
| 30 | br (moisture) | — | — | — | 7.81 (q, 4.0 Hz) | — |
| 31 | — | — | 3.64 (s) | 52.1 | 2.56 (d, 4.8 Hz) | 28.2 |
| 32 | — | — | — | — | 5.17 (d, 4.8 Hz) | — |
| 33 | 0.84 (s) | 23.4 | 0.84 (s) | 23.4 | 0.84 (s) | 23.2 |
| 34 | 0.77 (s) | 24.0 | 0.77 (s) | 24.0 | 0.78 (s) | 23.8 |
| 35 | 1.22 (s) | 17.9 | 1.22 (s) | 17.9 | 1.22 (s) | 17.7 |
| 36 | 6.07 (dd, 17.20, 10.20 Hz) | 129.6 | 6.07 (dd, 17.20, 10.20 Hz) | 129.6 | ||
| 37 | — | — | — | — | ||
| 38 | br (moisture) | — | br (moisture) | — | ||
| 39 | 5.87 (dd, 10.20, 1.80 Hz) | 130.9 | 5.87 (dd, 10.20, 1.80 Hz) | 130.9 | ||
| 6.24 (dd, 17.20, 1.80 Hz) | 6.24 (dd, 17.20, 1.80 Hz) | |||||
The furan ring protons resonated at δH 6.49 (s, 1H), 7.64 (s, 1H), 7.67 (s, 1H), were distinguished by HSQC and were located at C-13 by the corresponding HMBC correlations of the furan ring group protons to C-13 (oxymethine group). The oxymethine groups at C-13 were assigned based on the HMBC correlations from H-15, H-18, H-20 to C-13, C-23 was assigned based on the HMBC correlations from H-3, H-22, H-33, H-34, and C-24 was assigned based on the HMBC correlations from H-22 to C-24 (Table 2). The HMBC cross-peaks of H-10, H-13, and H-20 were assigned two olefinic quaternary carbon atoms C-4 and C-6. The lactone carbonyl carbon at C-11 was assigned based on the HMBC correlations from H-13, and H-20, and one acid carbonyl carbon C-28 was assigned based on the HMBC correlations from H-22, H-24, while the HMBC cross-peaks of H-3, H-23, H-35 confirmed the most de-shielded keto carbon C-21. The positions of functional groups were discerned through HMBC correlations.
| Position | Compound 1 | Compound 2 | Compound 3 |
|---|---|---|---|
| HMBC | HMBC | HMBC | |
| 2 | C-21, C23 | C-21, C23 | C-21, C23 |
| 10 | C-4, C-6, C-7, C-11, C13 | C-4, C-6, C-7, C-11, C13 | C-4, C-6, C-7, C-11, C13 |
| 13 | C-6, C-11, C-15, C-18, C-20 | C-6, C-11, C-15, C-18, C-20 | C-6, C-11, C-15, C-18, C-20 |
| 15 | C-13, C-14, C-16, C-18 | C-13, C-14, C-16, C-18 | C-13, C-14, C-16, C-18 |
| 20 | C-6, C-7, C-8, C-13 | C-6, C-7, C-8, C-13 | C-6, C-7, C-8, C-13 |
| 22 | C-21, C-23, C-28, C-33 & 34 | C-21, C-23, C-28, C-33 & 34 | C-21, C-23, C-28, C33 & 34 |
| 23 | C-21, C-27, C-33 & 34 | C-21, C-27, C-33 & 34 | C-21, C-27, C-33 & 34 |
| 31 | C-28 | C-28 | |
| 39 | C-27, C-36 | C-27, C-36 |
Further, compound 2, also was obtained as an amorphous white solid with Mp 188–190 °C, [α]25D −119.40 and shows characteristic IR (KBr) spectral peaks at 3483 cm−1 (OH), 1733 cm−1 (CO), and 1251 cm−1 (CO–OCH3). The molecular formula of C30H36O9 was determined through HR-ESI-MS observed at m/z 541.2446 and was supported by the 1H, 13C, HSQC and HMBC NMR data. The 1H-NMR spectrum indicated the presence of a substituted end alkene through the proton peaks appearing at δH 5.87 (dd, J = 10.2, 1.8 Hz, 1H), 6.27 (dd, J = 17.2, 1.8 Hz, 1H), 6.07 (dd, J = 17.2, 10.2 Hz, 1H), a furan ring at δH 6.49 (s, 1H), 7.64 (s, 1H), 7.67 (s, 1H) and two tertiary methyl groups at δH 0.89 (s, 3H), 1.22 (s, 3H), one gem dimethyl group at δH 0.77 (s, 3H), 0.84 (s, 3H), three methine groups attached to oxygen appeared at δH 3.35 (d, 1H, J = 9.4 Hz), 4.40 (s, 1H), 5.47 (s, 1H). 13C NMR data displayed 30 different carbon resonances, which were resolved into five methyls, five methylenes, ten methines, three aliphatic quaternary carbons, three aromatic quaternary carbons, three ester carbonyl group and a keto carbonyl carbon. HSQC and HMBC correlations located functional groups: alkene (C-27), furan ring (C-13), methoxy (C-31), oxymethine (C-13), aromatic quaternary carbons (C-4, C-6), ester carbonyl (C-11) and keto carbon (C-21).
Similarly, the amide compound 3 was obtained as an amorphous white solid with Mp 195–197 °C, [α]25D −125.8, and IR (KBr) spectrum of compound 3 displayed peaks at 3426 cm−1 (OH), 1649 cm−1 (CO–NH). The molecular formula, C27H35NO7 was as established based on the HR-ESI-MS appearing at m/z 486.2492 and was supported by the 1H, 13C, and HSQC, HMBC NMR data. In the 1H-NMR spectrum, the secondary amide group protons appeared at δH 7.81 (q, 1H, NH), 2.56 (d, 3H), the presence of furan ring was detected based on the proton peaks appearing at δH 6.50 (s, 1H), 7.66 (s, 1H), 7.68 (s, 1H). Further, two tertiary methyl groups appeared at δH 0.90 (s, 3H), 1.22 (s, 3H), one gem dimethyl group resonated at δH 0.78 (s, 3H), 0.85 (s, 3H), three methine groups attached to oxygen appeared at δH 3.36 (d, 1H, J = 9.4 Hz), 4.41 (s, 1H), 5.48 (s, 1H) and two secondary alcohol group protons were found at δH 5.17 (d, 1H, J = 4.8 Hz), 5.28 (d, 1H, J = 4.5 Hz). Its 13C NMR data exhibited 27 carbons signals, which were resolved into five methyls, four methylenes, nine methines, three aliphatic quaternary carbons, three aromatic quaternary carbons, an acid carbonyl carbon, an ester carbonyl carbon and a keto carbonyl carbon by HSQC and HMBC data.
The secondary amide functional group appearing at δH 7.81 (q, 1H, NH), 2.56 (d, 3H) was distinguished by HSQC, nitrogen HSQC and were located at C-28 by the corresponding HMBC correlations of the secondary amide group protons to C-28. HSQC and HMBC correlations was used to locate the functional groups such as furan ring (C-13), oxymethine (C-13), olefinic quaternary carbons (C-4, C-6), ester carbonyl (C-11) and keto carbon (C-21). Many closely related compounds resembling the new compounds 1, 2, and 3, albeit with distinct substitutions at C-22 and C-23, have been previously documented in the literature from Swietenia macrophylla seeds. Therefore, the relative configuration of compounds 1, 2, and 3 at C-2, C-13, C-22, C-23, and C-24, as well as the ring junctions, were inferred based on biogenetic analogy with known swietenioides (Fig. 5).46–55
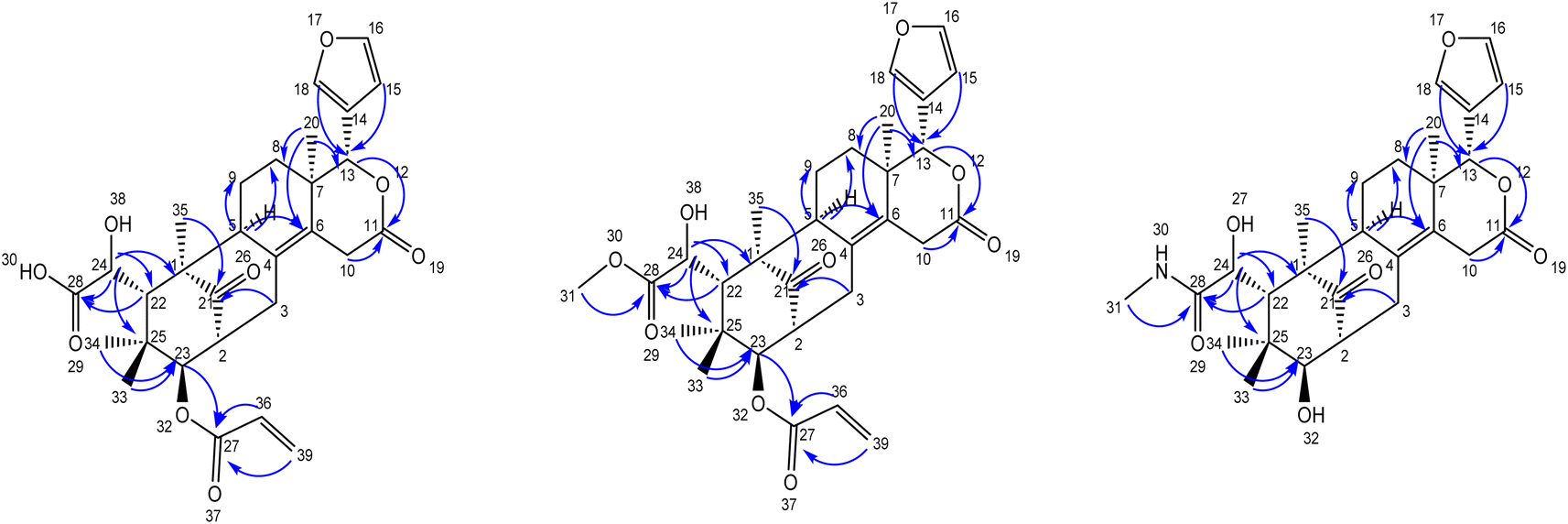 | ||
| Fig. 5 HMBC correlations for new limonoids 1, 2, and 3. | ||
2.1 Structural identification of the known isolates
The nine compounds demethylisoswietenolide (4),56,57 swietenolide (5),46,47 swietenine (6),48,49 swietenine acetate (7),49–51 3-O-tigloylswietenolide (8),48,49,52 diacetyl swietenolide (9),49,53 3-O-tigloyl-6-O-acetyl swietnolide (10),49,52,54 khayasin T (11),49,51,55 and febrifugine (12).49,51,52 isolated along with the unknown compounds were found to be known already in the literature and their structure was established and easily recognized by comparing their physical and spectroscopic data (LCMS, 1H-NMR, 13C and HMBC & HSQC and [α]D) with those of the matching authentic samples or literature values.Thus, overall SFC purification of acetone and ethanol extract resulted in simultaneous isolation and identification of three unknown, new limonoids 1–3, together with nine known compounds.
2.2 Biological activities
S. macrophylla exhibits a wide range of pharmacological benefits, including antibacterial, anti-inflammatory, antioxidant, antimutagenic, anticancer, antitumor, antidiabetic, anti-nociceptive, hypolipidemic, antidiarrheal, anti-infective, antiviral, antimalarial, acaricidal, and heavy metal phytoremediation activities.58 We examined the biological activities of newly discovered compounds 1, 2, and 3, in line with previously isolated compounds from S. macrophylla. Since several limonoids from S. macrophylla were the subject of thorough theoretical computations and computational analyses,59,60 we did not conduct experimental tests or empirical validation to support these theoretical conclusions.| Sl. no | Organism | Control fluconazole | Radius of the zone of inhibition (mm) | ||
|---|---|---|---|---|---|
| Compound 1 | Compound 2 | Compound 3 | |||
| a NI – no inhibition. | |||||
| 1 | Aspergillus niger | 25 mm | NI | NI | NI |
Nevertheless, against Aspergillus Niger, none of the test compounds 1, 2 and 3 demonstrated any inhibition at 25 μl (0.05 mg), 50 μl (0.1 mg), 75 μl (0.15 mg), or 100 μl (0.2 mg).61 This information suggests that while these compounds may exhibit antibacterial activity while they do not possess the any efficacy against Aspergillus Niger, highlighting the importance of considering the target organism when assessing the effectiveness of antimicrobial agents.
The anti-inflammatory effects of the newly isolated compounds 1, 2 and 3 (Table 4) revealed that while compounds 2 and 3 showed significantly higher efficacy in causing protein denaturation, compound 1 performed poorly. It was also evident that compound 3's ability to denaturize proteins rises in direct proportion to concentration.
| Sl no | Sample ID | Protein denaturation | Hemolytic activity | Heat-induced | |||
|---|---|---|---|---|---|---|---|
| Haemolytic activity | |||||||
| 25 μl(0.05 mg) | 50 μl(0.1 mg) | 25 μl (0.05 mg) | 50 μl (0.1 mg) | 25 μl (0.05 mg) | 50 μl (0.1 mg) | ||
| a NI – no inhibition. | |||||||
| 1 | Compound 1 | — | 7% | — | NI | 24.30% | 27.50% |
| 2 | Compound 2 | — | 14% | 34.46% | 65.30% | 45.62% | 52.30% |
| 3 | Compound 3 | 34% | 60% | NI | 23.43% | 48.12% | 62.30% |
Compound 2 exhibited increased hemolytic activity with increase compound concentration, while compound 3 exhibited hemolytic activity, compound 1 exhibited no activity at all. However, the same compounds studied against heat-induced hemolytic activity all the compounds exhibited increased hemolytic activity with increasing concentration.65,66
| S. no | Sample ID | Alpha amylase activity at 25 μl (0.05 mg) concentration | Alpha amylase activity at 50 μl (0.1 mg) concentration |
|---|---|---|---|
| a % inhibition= (absorbance-control − absorbance-test)absorbance-control ×100, where A-control = absorbance of the blank control and A-test = absorbance of the test sample. | |||
| 1 | Compound 1 | 20.53% | 26.78% |
| 2 | Compound 2 | 16.96% | 24.10% |
| 3 | Compound 3 | 28.57% | 41.96% |
Study of alpha-amylase inhibitory potency revealed that compounds 1, 2 and 3 possessed potential hypoglycemic activity. As the concentration of compounds 1, 2 and 3 rises, there is a corresponding increase in the percentage inhibition of alpha-amylase. This suggests that higher concentrations of the compounds lead to greater inhibition of the enzyme's activity.70,71 Inhibiting its activity can be desirable for various reasons, such as controlling blood sugar levels in diabetes management or preventing spoilage in food processing.
| Organism | Compound 1 | Compound 2 | Compound 3 | Control | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0.05 mg | 0.1 mg | 0.15 mg | 0.2 mg | 0.05 mg | 0.1 mg | 0.15 mg | 0.2 mg | 0.05 mg | 0.1 mg | 0.15 mg | 0.2 mg | 0.2 mg | |
| a Disc diffusion method (radius of the zone of inhibition mm). | |||||||||||||
| E. coli | NI | 6 | 7.5 | 8 | NI | NI | 6 | 7 | NI | NI | 4 | NI | 12.5 |
| B. cereus | NI | NI | 5 | 5 | NI | NI | 5 | 6 | NI | 4.5 | 6 | 6 | 15 |
| S. aureus | NI | NI | 5 | 5 | NI | NI | 5 | 5 | NI | NI | NI | NI | 13 |
| K. pneumoniae | 4 | NI | NI | NI | NI | NI | NI | 4 | NI | 5 | 6 | 6.5 | 15 |
The antibacterial activity of the compounds 1, 2 and 3 increases with the increase in concentration. This means there are more molecules available to target and inhibit the growth or kill the bacteria. As a result, the antibacterial effect becomes stronger because there's a higher likelihood of these compounds effectively neutralizing or eliminating the bacteria they encounter.
Compound 1 exhibited notable genotoxic effects on human lymphocytes (Table 7). This suggests that when exposed to compound 1, there was an observable damage to the genetic material of the lymphocytes. Such damage can have serious implications, including increased risk of cancer or other genetic disorders. On the other hand, compounds 2 and 3 show no discernible impacts on human lymphocytes in terms of genotoxicity. This implies that exposure to compounds 2 and 3 does not result in observable genetic damage or mutations in the lymphocytes.
| Sl. no | Sample ID | Sample concentration | Cell scored | Tail length ± SE |
|---|---|---|---|---|
| 1 | Untreated | — | 100 | 0.35 ± 0.02 |
| 2 | Compound 1 | 10 μl (0.02 mg) | 100 | 0.41 ± 0.05 |
| 20 μl (0.04 mg) | 100 | 0.45 ± 0.04 | ||
| 3 | Compound 2 | 10 μl (0.02 mg) | 100 | 0.59 ± 0.1 |
| 20 μl (0.04 mg) | 100 | 0.9 ± 0.15 | ||
| 4 | Compound 3 | 10 μl (0.02 mg) | 100 | 0.65 ± 0.015 |
| 20 μl (0.04 mg) | 100 | 01.1 ± 0.37 |
3 Experimental section
3.1 General experimental procedures
Using a digital polarimeter (JASCO P-2000), optical rotations were measured. Using a Shimadzu 2000 FT-IR spectrophotometer, IR spectra were acquired. NMR spectra were acquired using a 400 MHz Bruker Avance. A Waters ACQUITY UPLC H-Class coupled with SQ Detector-2 mass spectrometer was used to gather ESIMS data. ESIMS data with high resolution were acquired using an exploris 240 Thermo orbit trap. SFC purifications using Waters SFC-150 mgm equipment and SFC analysis performed on Waters SFC-Investigator instruments. Chiralpak-IE (4.6 × 250 mm; 5μ id) column from Diacel chiral technologies is used for purification, and Chiralpak-IE (4.6 × 250 mm; 5μ) for analytical development.3.2 Plant material
S. macrophylla fruits were collected from Salem located at latitude 11.65376 and longitude 78.15538. It is part of Asia and the northern hemisphere, Tamilnadu, India, and the seeds were removed by peeling them.3.3 Extraction and isolation
After being processed using an electronic grinder for a week to a coarse powder, the seeds were weighed, shade-dried, and stored in a dry place. 500 g of dry powder were continuously cold extracted using hexane residue, followed by chloroform, acetone, ethanol, methanol, and water three times each. A rotary evaporator was utilized to eliminate the solvents from every extract. The extracts were then kept at −70 °C for 48 h, and a freeze-dryer (Labconco Corporation, Denmark) was used to freeze-dry them under a vacuum for 24 h at −40 °C. Strictly sealed glass bottles containing each dried extract were kept at 4 °C. The extraction yield from S. macrophylla seeds was found to be highest in ethanol solvent (68 g with 13.6%) and lowest in aqueous solvent (28.0 g with 5.6%), according to a quantitative evaluation of the extracts observed from the seeds using different solvents.3.4 Optimizing SFC conditions for efficient phytochemical separation
We focused on developing an effective one-step SFC method for isolating phytochemicals, prioritizing compounds separable by acetone and ethanol. SFC offers superior separation efficiency and additional benefits such as online coupled processes, faster separations, reduced solvent consumption, wide applicability, and easy analyte recovery.SFC conditions were optimized, including stationary phase, mobile phase, and instrumental parameters, for separating limonoids from S. macrophylla seed extracts. Chiral columns showed higher resolution than achiral ones due to the presence of various positional isomers.
The Chiralpak-IE column was identified as optimal for limonoid separation using methanol as the most effective solvent. Key parameters included a column temperature of 35 °C, a 30% co-solvent composition, a sample volume of 10 μl, a flow rate of 4.0 ml min−1, and 100 bars of back pressure. These optimized conditions were applied in the analytical SFC for isolating compounds from ethanol and acetone extracts. Seven compounds 1, 2, 4, 5, 6, 7, and 8 were isolated from acetone extract, and five compounds 3, 9, 10, 11, and 12 from ethanol extract, even with extremely low abundances (2–3%), demonstrating the efficiency of the SFC approach.
The SFC purification was not performed for extracts of hexane, chloroform, methanol, and water due to their low solubility, poor peak shape, and retention behaviour. The purity of isolated compounds was confirmed using SFC investigation, and certain compounds were disregarded for further research as they were well-known and extensively reported in the literature. Initial LCMS analysis confirmed the presence of the same compounds in multiple extracts, leading to the selection of ethanol and acetone extracts for SFC purification.
4 Conclusion
In conclusion, we demonstrated for the first time that SFC could be used as a powerful technique for the simultaneous and superior separation of twelve compounds, in particular limonoids from S. macrophylla in a single-step. An optimum condition was arrived at based the study of different parameters such as column, pressure, temperature, and co-solvent composition. Compounds 1, 2, and 3 were previously unknown and challenging to isolate by conventional natural products separation methods. This study further shows that using the SFC isolation technique, even trace-level (2–3%) phyto constituents such as limonoids 1, 2, 3, 9, 10, 11 and 12 could be conveniently separated from complex mixtures. The use of recyclable carbon dioxide as the primary solvent, reduced use of organic solvent, efficient and simultaneous separation of minor natural product constituents by means SFC makes our separation technique sustainable, green and eco-friendly. These results are unattainable through traditional chromatographic methods, efficient alternative, and are unprecedented in the literature on S. macrophylla.Further, the new limonoids 1, 2 and 3 assessed for antimicrobial, anti-inflammatory, hemolytic, and genotoxic properties disclose novel biological activities. Compounds 1, 2 and 3 shows good hypoglycemic activity useful in controlling blood sugar levels and diabetes management, significant antibacterial activity and no anti-fungal activity. Compounds 2 and 3 showed significant anti-inflammatory activity and anti-mutagenic activity. Whereas compound 1 showed poor anti-inflammatory activity and genotoxic effects on human lymphocytes. Studying mechanism and therapeutic effects of the limonoids isolated from S. macrophylla, could facilitate the discovery of potential new drugs.
Data availability
All spectral data and other information supporting the contents of the main manuscript can be found in the ESI.†Conflicts of interest
The authors declared that they have no conflict of interest.Acknowledgements
The authors thank to Dr K. Muralidharan, Aragen life science pvt ltd for supporting to use of their analytical and synthesis facility. Prof. Andivelu Ilangovan thanks RUSA 2.0, and DST-FIST for providing facilities Bharathidasan University, Tiruchirappalli, India.References
- Y.-Y. Chen, X.-N. Wang, C.-Q. Fan and J.-M. Yue, Tetrahedron Lett., 2007, 48, 7480–7484, DOI:10.1016/j.tetlet.2007.08.066.
- (a) J. D. Connolly and C. Labbé, J. Chem. Soc., Perkin Trans. 1, 1980, 529–530, 10.1039/P19800000529; (b) B.-D. Lin, C.-R. Zhang, S.-P. Yang, S. Z. Y. Wu and J.-M. Yue, J. Nat. Prod., 2009, 72, 1305–1313, DOI:10.1021/np900139c.
- A. M. M. Eid, N. A. Elmarzugi and H. A. Enshasy, Int. J. Pharm. Sci., 2013, 5, 47 CAS.
- J.-Y. Duan, Y.-J. Wang, W. Chen, Y.-Q. Zhao, Z.-H. Bai, L.-L. He and C.-P. Zhang, J. Food Biochem., 2021, 45, e13668, DOI:10.1111/jfbc.13668.
- A. Maiti, S. Dewanjee, M. Kundu and S. C. Mandal, Pharm. Biol., 2009, 47, 132–136, DOI:10.1080/13880200802436703.
- A. K. M. S. Rahman, A. K. A. Chowdhury, H.-A. Ali, S. Z. Raihan, M. S. Ali, L. Nahar and S. D. Sarker, J. Nat. Med., 2009, 63, 41–45, DOI:10.1007/s11418-008-0287-3.
- V. Divakar, S. A. Fahath, A. Rishika, H. Prasad, M. Sivakumar and N. Deepa, Biochem. Cell. Arch., 2023, 23(2), 823–829, DOI:10.51470/bca.2023.23.2.823.
- Y.-P. Sun, Z. Xie, W.-F. Jin, Y.-W. Liu, L.-J. Sun, J.-S. Liu and G. K. Wang, Org. Biomol. Chem., 2024, 22, 2182–2186, 10.1039/D3OB02113K.
- K. Kalpana and K. V. Pugalendi, J. Basic Clin. Physiol. Pharmacol., 2011, 22(1–2), 11–21, DOI:10.1515/jbcpp.2011.001.
- Y.-B. Cheng, Y.-T. Chien, J.-C. Lee, C.-K. Tseng, H.-C. Wang, I.-W. Lo, Y.-H. Wu, S.-Y. Wang, Y.-C. Wu and F.-R. Chang, J. Nat. Prod., 2014, 77, 2367–2374, DOI:10.1021/np5002829.
- M. A. Hashim, M. F. Yam, S. Y. Hor, C. P. Lim, M. Z. Asmawi and A. Sadikun, Chin. Med., 2013, 8, 11, DOI:10.1186/1749-8546-8-11.
- S. Dewanjee, A. Maiti, A. K. Das, S. C. Mandal and S. P. Dey, Fitoterapia, 2009, 80, 249–251, DOI:10.1016/j.fitote.2009.02.004.
- K. Kojima, K. Isaka and Y. Ogihara, Chem. Pharm. Bull., 1998, 46(3), 523–525, DOI:10.1248/cpb.46.523.
- T. Yuan, C.-R. Zhang, S.-P. Yang and J.-M. Yue, J. Nat. Prod., 2010, 73, 669–674, DOI:10.1021/np1000158.
- D. Speybrouck and E. Lipka, J. Chromatogr. A, 2016, 1467, 33–55, DOI:10.1016/j.chroma.2016.07.050.
- E. Abbott, T. D. Veenstra, J. Haleem and H. Issaq, J. Sep. Sci., 2008, 31, 1223–1230, DOI:10.1002/jssc.200700579.
- K. L. Williams, L. C. Sander and S. A. Wise, J. Chromatogr. A, 1996, 746, 91–101, DOI:10.1016/0021-9673(96)00291-9.
- K. Tyśkiewicz, A. Dębczak, R. Gieysztor, T. Szymczak and E. Rój, J. Sep. Sci., 2018, 41, 336–350, DOI:10.1002/jssc.201700598.
- Q. Ningli, X. Gong, C. Feng, X. Wang, Y. Xu and L. Lin, Food Chem., 2016, 207, 157–161, DOI:10.1016/j.foodchem.2016.03.089.
- L. Nováková, M. Sejkorová and K. Smolková, Chromatographia, 2019, 82, 477–487, DOI:10.1007/s10337-018-3666-2.
- K. Tyśkiewicz, R. Gieysztor, I. Maziarczyk, P. Hodurek and E. Rój, RSC Adv., 2018, 8, 36792–36805, 10.1039/C8RA08289D.
- E. Lesellier, E. Destandau, C. Grigoras, L. Fougère and C. Elfakir, J. Chromatogr. A, 2012, 1268, 157–165, DOI:10.1016/j.chroma.2012.09.102.
- M. A. Hashim, M. F. Yam, S. Y. Hor, C. P. Lim, M. Z. Asmawi and A. Sadikun, J. Pharm. Biomed. Anal., 2015, 102, 400–408, DOI:10.1016/j.jpba.2014.10.013.
- X. G. Liu, L. W. Qi and Z. Y. Fan, J. Chromatogr. A, 2015, 1388, 251–258, DOI:10.1016/j.chroma.2015.02.031.
- X. T. Zhang, F. Ji, Y. Q. Li, Y. T. He, Y. Han and S. Z. Chen, Anal. Sci., 2018, 34, 407–413, DOI:10.2116/analsci.17P434.
- Y. Huang, T. Zhang and H. Zhou, J. Pharm. Biomed. Anal., 2016, 121, 22–29, DOI:10.1016/j.jpba.2015.12.056.
- M. Wang, E. J. Carrell and Z. Ali, J. Sep. Sci., 2014, 1–8, DOI:10.1002/jssc.201301389.
- A. Murauer and M. Ganzera, J. Chromatogr. A, 2018, 1554, 117–122, DOI:10.1016/j.chroma.2018.04.038.
- W. Z. Yang, Y. Zhang, H. Pan, C. Yao, J. Hou, S. Yao, L. Cai, R. Feng, W. Wu and D. Guo, J. Pharm. Biomed. Anal., 2016, 134, 352–360, DOI:10.1016/j.jpba.2016.10.021.
- Q. Fu, Z. Li, C. Sun, H. Xin, Y. Ke, Y. Jin and X. Liang, J. Supercrit. Fluids, 2015, 104, 85–93, DOI:10.1016/j.supflu.2015.05.006.
- K. Li, Q. Fu, H. Xin, Y. Ke, Y. Jin and X. Liang, Analyst, 2014, 139, 3577–3587, 10.1039/C4AN00438H.
- H. Yang, Y. Feng, G. Tang, M. Li, T. Zhang, M. Fillet, J. Crommen and Z. Jiang, J. Pharm. Biomed. Anal., 2017, 140, 384–391, DOI:10.1016/j.jpba.2017.03.012.
- M. G. Chini, N. Malafronte, C. M. Vaccaro, J. M. Gualtieri, A. Vassallo, M. Vasaturo, S. Castellano, C. Milite, A. Leone, G. Bifulco, N. D. Tommasi and F. Piaz, Chem.–Eur. J., 2016, 22, 1–16, DOI:10.1002/chem.201602242.
- M. Ganzera, J. Pharm. Biomed. Anal., 2015, 107, 364–369, DOI:10.1016/j.jpba.2015.01.013.
- X. Qiao, R. An and Y. Huang, J. Chromatogr. A, 2014, 1358, 252–260, DOI:10.1016/j.chroma.2014.06.074.
- T. F. Ponce, L. Casas, C. Mantell and E. M. Ossa, J. Supercrit. Fluids, 2014, 95, 444–456, DOI:10.1016/j.supflu.2014.10.005.
- G. Vicente, M. R. García-Risco and T. Fornari, J. Chromatogr. A, 2013, 1286, 208–215, DOI:10.1016/j.chroma.2013.02.044.
- H. Xin, Z. Dai, J. Cai, Y. Ke, J. Feng, Y. X. Feng, Q. Fu, Y. Jin and X. Liang, Chromatographia, 2018, 81, 1181–1187, DOI:10.1007/s10337-018-3544-y.
- Y.-P. Sun, L.-L. Zhu, J.-S. Liu, Y. Yu, Z.-Y. Zhou, G. Wang and G.-K. Wang, Fitoterapia, 2018, 125, 141–146, DOI:10.1016/j.fitote.2018.01.004.
- L. T. Taylor, Anal. Chem., 2010, 82, 4925–4935, DOI:10.1021/ac101194x.
- G. Guiochon and A. Tarafder, J. Chromatogr. A, 2011, 1218, 1037–1114, DOI:10.1016/j.chroma.2010.12.047.
- P. Ramírez, M. R. García-Risco, S. Santoyo, J. Señoráns, E. Ibáñez and G. Reglero, J. Pharm. Biomed. Anal., 2006, 41, 1606–1613, DOI:10.1016/j.jpba.2006.02.001.
- P. Ramírez, T. Fornari, F. J. Señoráns, E. Ibáñez and G. Reglero, J. Supercrit. Fluids, 2005, 35, 128–132, DOI:10.1016/j.supflu.2005.01.002.
- T. Ohji, A. Ohnishi and M. Ogasawara, ACS Omega, 2022, 7, 5146–5153, DOI:10.1021/acsomega.1c06187.
- M. Schaffrath, V. Weidmann and W. Maison, J. Chromatogr. A, 2014, 1363, 270–277, DOI:10.1016/j.chroma.2014.07.001.
- B. S. Mootoo, A. Ali, R. Motilal, R. Pingal, A. Ramlal, A. Khan, W. F. Reynolds and S. McLean, J. Nat. Prod., 1999, 62, 1514–1517, DOI:10.1021/np990199x.
- A. K. M. S. Rahman, A. K. A. Chowdhury, H.-A. Ali, S. Z. Raihan, M. S. Ali, L. Nahar and S. D. Sarker, J. Nat. Med., 2009, 63, 41–45, DOI:10.1007/s11418-008-0287-3.
- L.-C. Chen, H.-R. Liao, P.-Y. Chen, W.-L. Kuo, T.-H. Chang, P.-J. Sung, Z.-H. Wen and J.-J. Chen, Molecules, 2015, 20, 18551–18564, DOI:10.3390/molecules201018551.
- J.-J. Chen, S.-S. Huang, C.-H. Liao, D.-C. Wei, P.-J. Sung, T.-C. Wang and M.-J. Cheng, Food Chem., 2010, 120, 379–384, DOI:10.1016/j.foodchem.2009.09.093.
- A. H. Nour, M. B. Sulieman, M. Yousf, A. M. Abdurahman and A. M. Mazza, Aust. J. Basic Appl. Sci., 2016, 10, 55–62 Search PubMed.
- Y.-B. Cheng, Y.-T. Chien, J.-C. Lee, C.-K. Tseng, H.-C. Wang, I.-W. Lo, Y.-H. Wu, S.-Y. Wang, Y.-C. Wu and F.-R. Chang, J. Nat. Prod., 2014, 77, 2367–2374, DOI:10.1021/np5002829.
- Y.-Q. Ma, K. Jiang, Y. Deng, L. Guo, Y.-Q. Wan and C.-H. Tan, J. Asian Nat. Prod. Res., 2018, 20, 299–305, DOI:10.1080/10286020.2017.1335715.
- B. H. Goh, H. A. Kadir, S. N. A. Malek and S. W. Ng, Acta Crystallogr., 2010, 66, o1396, DOI:10.1107/S1600536810017733.
- C. K. Mahendra, H. L. Ser, S. Z. Abidin, S. U. Khan, P. Pusparajah, T. T. Htar, L. H. Chuah, S. Y. Tang, S. Y. Ming, L. C. Goh and K. W. Goh, Biomed. Pharmacother., 2023, 162, 114659, DOI:10.1016/j.biopha.2023.114659.
- J. Zhang, S.-X. Yang, X.-B. Yang, M.-Y. Li, G. Feng, J.-Y. Pan and T. Satyanandamurty, Chem. Pharm. Bull., 2010, 58, 552–555, DOI:10.1248/cpb.58.552.
- F. Robert, B. Mootoo, R. Ramsewak, A. Khan, A. Ramsubhag, W. Reynolds and M. Nair, Pest Manage. Sci., 2010, 66, 1298–1303, DOI:10.1002/ps.2013.
- G. R. Mootoo, S. Baldwin, R. S. Khan and A. Ramsewak, Pharm. Biol., 2012, 50, 264–267, DOI:10.3109/13880209.2011.581670.
- S. Z. Moghadamtousi, B. H. Goh, C. K. Chan, T. Shabab and H. A. Kadir, Molecules, 2013, 18, 10465–10483, DOI:10.3390/molecules180910465.
- P. T. Thuy, T. T. Hieu, D. X. Duc, H. V. Trung, N. H. Hung, W. N. Setzer, T. D. Thang and N. T. Son, J. Mol. Struct., 2023, 1283, 135264, DOI:10.1016/j.molstruc.2023.135264.
- L. S. W. F. Amrulloh, N. Harmastuti, A. Prasetiyo and A. R. Herowati, Jurnal Farmasi dan Ilmu Kefarmasian Indonesia, 2023, 10(3), 347–359, DOI:10.20473/jfiki.v10i32023.347-359.
- A. Guevara, A. Apilado, H. Sakurai, M. Kozuka and H. Takuda, Philipp. J. Sci., 1996, 125, 271–277 Search PubMed.
- A. Maiti, S. Dewanjee, S. C. Mandal and S. Annadurai, Iran. J. Pharmacol. Ther., 2007, 6, 99–102 Search PubMed.
- N. I. Osman, N. J. S. A. Awal, N. A. M. Adam and N. I. Rezali, J. Intercult. Ethnopharmacol., 2016, 5, 343, DOI:10.5455/jice.20160731025522.
- C. S. Kumari, N. Yasmin, M. R. Hussain and M. Babuselvam, Int. J. Pharm. Sci. Res., 2015, 6, 482–485 CAS.
- S. Chandra, P. Chatterjee, P. Dey and S. Bhattacharya, Asian Pac. J. Trop. Biomed., 2012, 2, S178–S180, DOI:10.1016/S2221-1691(12)60154-3.
- S. F. Wu, C. K. Lin, Y. S. Chuang, F. R. Chang, C. K. Tseng, Y. C. Wu and J. C. Lee, J. Viral Hepatitis, 2012, 19, 364–370, DOI:10.1111/j.1365-2893.2011.01558.x.
- R. Kusano, S. Ogawa, Y. Matsuo, T. Tanaka, Y. Yazaki and I. Kouno, J. Nat. Prod., 2011, 74, 119–128, DOI:10.1021/np100372t.
- R. Chakrabarti, B. Singh, V. N. Prakrith, L. Vanchhawng and K. Thirumurugan, Asian J. Pharm. Clin. Res, 2014, 7(4), 84–89 Search PubMed.
- K. Balan, P. Ratha, G. Prakash, P. Viswanathamurthi, S. Adisakwattana and T. Palvannan, Arabian J. Chem., 2014, 10, 732–738, DOI:10.1016/j.arabjc.2014.07.002.
- A. Maiti, S. Dewanjee, G. Jana and S. C. Mandal, Int. J. Green Pharm., 2008, 2, 224–227 CrossRef.
- K. Kalpana and K. V. Pugalendi, J. Basic Clin. Physiol. Pharmacol., 2011, 22, 11–21, DOI:10.1515/jbcpp.2011.001.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra03663h |
| This journal is © The Royal Society of Chemistry 2024 |