
Alkaline stability of pendant C2-protected poly(imidazolium)s†
Kate
Fraser‡
,
Anastasiia
Konovalova‡
 ,
Thomas
Skalski
,
Simon
Cassegrain
and
Steven
Holdcroft
*
,
Thomas
Skalski
,
Simon
Cassegrain
and
Steven
Holdcroft
*
Department of Chemistry, Simon Fraser University, 8888 University Drive, V5A 1S6 Burnaby, British Columbia, Canada. E-mail: holdcrof@sfu.ca
First published on 16th May 2024
Abstract
Non-fluorinated anion exchange polymers with sufficient ion exchange capacity, mechanical integrity and hydroxide-ion stability are required for the progression of alkaline electrochemical energy conversion devices. In this work, we present a new class of polycations, that possess C2-sterically protected imidazolium pendant groups which are incorporated via an olefinic backbone, and a synthetic route that requires no post-functionalization. The versatile 5-step synthetic route is a platform for future modifications of this family of C2-protected imidazolium-functionalised ion conducting polymers. Hydroxide-promoted degradation studies, along with DFT calculations, predict the robustness of polymers, with the benzyl-modified, C2-sterically protected imidazolium group showcasing a 8000 h half-life in 3 M NaOD/CD3OD/D2O at 80 °C. A humidity cycling degradation protocol, exposing the polymers to as low as 10 % relative humidity at 80 °C was implemented to reveal the relative chemical (in)stabilities of the polymers in extreme caustic conditions under accelerated timeframes.
Introduction
The versatility of ion conducting polymers with the ionic functionalities as pendant groups have gained attention in the fields of polymer and materials science.1,2 Pendant-like structures with the ionic groups bound as side chains to a polymer backbone enables tuneability of the desired material,1 and are often used in electrochemical conversion devices due to their high ionic conductivity.2,3In pursuit of a net-zero emissions goal, considerable effort has been invested into the development of renewable energy technologies such as water electrolysers, redox flow batteries, and fuel cells.4 Many of these electrochemical systems are dependent on ion conducting polymers for the transport of mobile charged species. Ion conducting polymers can be implemented within electrochemical devices as electrolytic membranes, providing ion transport between electrodes, whilst maintaining electrical insulation; and as an ionomer, binding components of catalyst layers, and improving ion transport between active sites, as well as between the membrane and electrodes. More recently, insoluble ion conducting polymer particles have been explored as additives within catalyst layers to promote ion conduction and improve the mass transport within the device.5–7
Proton exchange membrane fuel cells (PEMFCs) have been actively developed over the past few decades. However, the cost of platinum group metal (PGM) catalysts and corrosion-resistant components have proved to be significant roadblocks for widespread implementation of this technology.8,9 Moreover, the European Union recently proposed to ban per- and polyfluoroalkyl substances (PFAS), that are critical for the high performance of commercial PEMFC devices. Pendant group ion conducting polymers based on poly(phenylenes)10,11 and poly(aryl sulfones)12 have thus been incorporated into PEMFCs and shown to provide sufficient proton conductivity, but still require PGM catalysts.
Anion exchange membrane fuel cells (AEMFCs) offer distinct advantages over PEMFCs; namely, the ability to use non-PGM catalysts and alkaline, PFAS-free, hydroxide-conducting polymers.9 Due to the high pH local environment, ion conducting polymers in AEMFCs require different backbone chemistry and pendant groups than PEMs. Backbones with heteroatoms linkages, such as ethers and sulfones are shown to be unstable in alkaline conditions, especially when coupled with an electron-withdrawing cationic group.13–15 Polyolefinic backbones have been reported to possess enhanced stability to hydroxide attack, even with cationic pendant groups.13 Polyolefinic backbones are particularly interesting due to the versatile synthetic routes available, which include atom transfer radical polymerisation (ATRP),16 reversible addition–fragmentation chain transfer (RAFT),17 nitroxide-mediated polymerisation,18 Ziegler–Natta polymerisation19 and radical chain growth polymerisation. Pendant cations groups required for ion conductions are installed onto these polymers through post-functionalisation step, where the degree and location of such groups can be challenging to control.14,20
Hydroxide conducting cationic groups for AEMs have been thoroughly investigated. Cations include quaternary ammonium,21–23 cyclic ammoniums,24,25 phosphonium,26,27 guanidinium,28 benzimidazolium,29–31 and imidazolium.29,32–34 All of these cations have been studied with respect to their alkaline stability.35,36 Confined piperidinium cations have been identified as a group with enhanced alkaline stability,21,37 as have imidazolium cations with steric protection around the electrophilic C2 position.32–34 When incorporated into the polymer backbone in the form of an ionene,32,34 poly(imidazolium)s exhibit high conductivity and mechanical strength for integration into alkaline devices.38
Despite their incorporation into ionenes, highly stable C2-protected imidazoliums have yet to be employed as a pendant group on polyolefins: synthetic strategies have not been proven nor has the stability of pendant C2-protected imidazoliums been examined. In this work, we studied the synthesis of C2-protected imidazoliums bound to a polystyrene backbone. A free-radical initiated chain growth polymerisation route was designed to yield poly(vinyl C2-protected imidazoliums) without the requirement of additional a post-functionalisation. Through accelerated, hydroxide-promoted degradation studies, we identify the stability against caustic solutions for this new class of polymer, and describe future work required to render these materials suitable for alkaline electrochemical energy conversion devices.
Experimental
Materials
Ammonium acetate (crystals, ACS reagent), diethyl ether anhydrous (Et2O, ACS reagent), sodium chloride (NaCl, ACS reagent), and dimethylformamide (ACS reagent) were sourced from ACP Chemicals Inc., Canada and used as received. Acetone (ACS reagent), dichloromethane (DCM, ACS reagent, stabilized), methanol (MeOH, reagent grade), anhydrous magnesium sulfate (MgSO4, certified powder), n-butyl bromide (certified), iodomethane (99%, stabilized with copper), hydrogen bromide, 33% w/w (45% w/v) soln. in acetic acid, and paraformaldehyde (97%), mesitylene (98+%) were sourced from ThermoFisher Scientific, USA and used as received. Benzyl bromide was purchased from Mallinckrodt Inc., USA and used as received. Benzil (98%) was purchased from Combi-Blocks Inc., USA and used as received. 2,2′-Azobis(2-methylpropionitrile) (AIBN, 98%), 1-N-methyl-2-pyrrolidone (anhydrous, 99.5%), ethyl-bromide (98%, reagent grade), 4-chloromethyl styrene (90%), acetic acid (ACS reagent, >99.7%), aniline (Reagent plus® 99%), acetonitrile for HPLC, gradient grade (>99.9%), hexanes, mixture of isomers (ACS reagent, >98.5%), methanol-d4 (D, 99%), sodium deuteroxide (D, 99%), deuterium oxide (D, 99%) were purchased from Millipore Sigma Canada Co., Canada and were all used as received. Chloroform (CHCl3, ACS reagent), dimethyl sulfoxide (DMSO, ACS grade), and sodium hydroxide (NaOH, ACS grade) were purchased from VWR Chemicals BDH, USA. Potassium hydroxide (KOH, reagent grade, min 85%) and tetrahydrofuran (THF, ACS grade) were sourced from Caledon Laboratories, Canada. Dimethylsulfoxide-d6 (D, 99.9%), acetone-d6 (D, 99.9%), methylene chloride-d2 (D, 99.8%, CD2Cl2), and chloroform-d (CDCl3, 99%) were purchased from Cambridge Isotope Laboratories Inc., USA.Instruments
1H and 13C NMR spectra were recorded on a Bruker AVANCE III 500 MHz equipped with a 5 mm TXI Inverse probe at room temperature (T = 298 K) and Bruker 600 Hz QCI (Bruker Corp., USA). The following terms are defined: s for singlet, d for doublet, t for triplet, q for quadruplet and m for multiplet. Chemical shifts are reported in ppm and J-couplings are reported in Hz. Residual proton and carbon peaks for deuterated solvents were set at, respectively, 2.05 ppm and 29.84 ppm for d6-acetone, 2.50 ppm and 39.52 ppm for d6-DMSO, 5.32 ppm and 54.00 ppm for CD2Cl2, 7.26 ppm and 77.16 ppm for CDCl3.Mass spectra were recorded on an AB Sciex 4000 Q TRAP spectrometer (Danaher Corp., USA) and a Bruker MicrOTOF (Bruker Corp., USA) in both positive and negative electrospray ionization modes for ionic compounds. High accuracy mass measurements were obtained on an LC-TOF instrument (Agilent Technologies, Inc., USA) in positive atmospheric-pressure photoionization (APPI) mode. Molecular ions [M]+ was used for empirical formula confirmation.
Elemental analysis data were collected on an EA CHN 110 instrument (Thermo Fisher Scientific Inc., USA) under an O2 atmosphere up to 960 °C. Thermogravimetric Analysis was performed on a Shimadzu TGA-50 series (Shimadzu Corp., Japan). The samples were heated from ambient temperature to 450 °C at a heating rate of 10 °C min−1 under constant N2 flow. Dynamic Vapor Sorption (DVS) measurements were performed on a DVS Adventure humidity chamber and Ultrabalance (Surface Measurement Systems Ltd, UK).
Synthesis
1H NMR (500 MHz, CD2Cl2) δ (ppm): 7.58 (d, Jd = 7.59 Hz, 2H), 7.12–7.27 (m, 11H), 6.91 (d, Jd 7.64 = Hz, 2H), 6.83 (s, 2H), 2.25 (s, 3H), 2.11 (s, 6H).
13C NMR (125 MHz, CD2Cl2) δ (ppm): δ 147.05, 139.37, 138.84, 138.13, 137.11, 135.57, 131.65, 131.60, 131.57, 129.55, 128.97, 128.93, 128.68, 128.57, 128.34, 128.15, 127.76, 127.61, 126.85, 21.44, 20.52.
HRMS (m/z): [M + H]+ calculated for C30H26N2: 414.2096 found: 415.2284.
1H NMR (500 MHz, DMSO-d6) δ (ppm): 12.33 (s, 1H), 7.56 (d, Jd = 7.40 Hz, 2H), 7.47 (d, Jd = 7.82 Hz, 2H), 7.39 (t, Jt = 7.32 Hz, 2H), 7.29 (m, 3H), 7.20 (t, Jt = 7.20 Hz, 1H), 6.98 (s, 2H), 2.30 (s, 3H), 2.18 (s, 6H).
13C NMR (125 MHz, DMSO-d6) δ (ppm): 145.00, 137.81, 137.59, 136.08, 135.63, 131.33, 128.94, 128.62, 128.10, 127.89, 127.75, 127.26, 127.12, 126.47, 126.26, 20.75, 19.99.
HRMS (m/z): [M + H]+ calculated for C24H22N2: 338.4449, found: 339.1851.
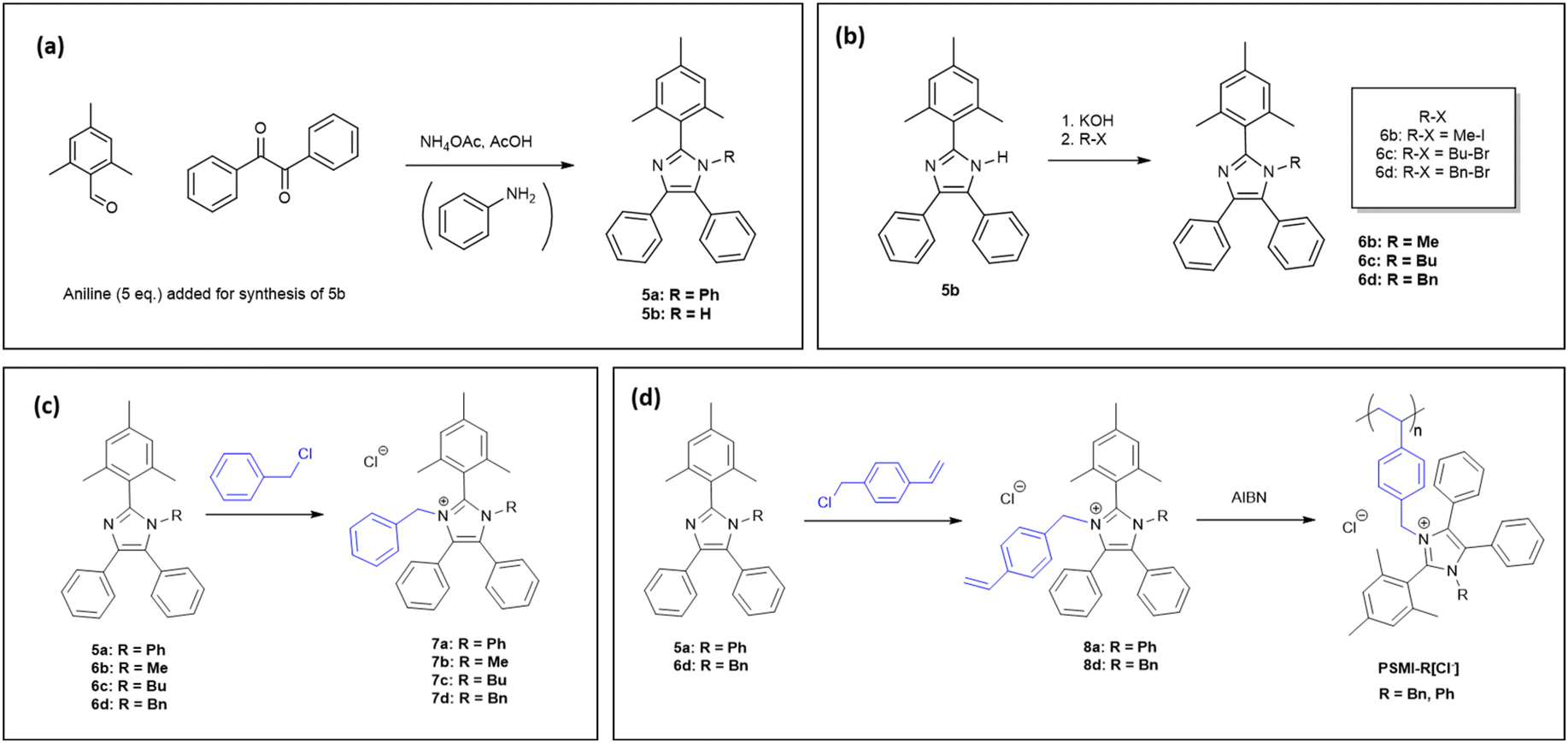 | ||
| Fig. 1 (a) Synthesis of imidazole monomer precursors (b) first N-functionalisation step (c) second N-functionalisation step for synthesis of imidazolium model compounds (d) second N-functionalisation for monomer synthesis followed by polymerisation. | ||
Characterisation
| Compound, M (g mol−1) | Molar concentration, mol l−1 |
|---|---|
| 7b, 443.61 | 0.077 |
| 7c, 485.69 | 0.070 |
| 7a, 505.68 | 0.067 |
| 7d, 519.71 | 0.065 |
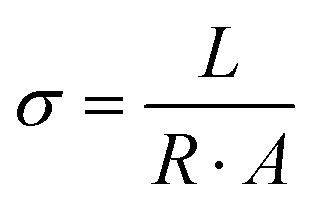 | (1) |
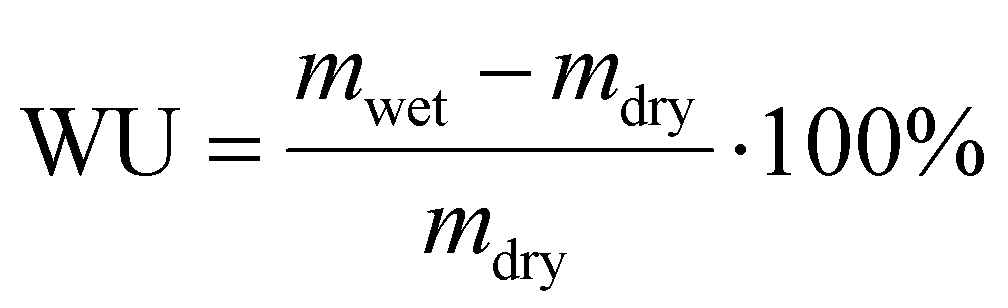 | (2) |
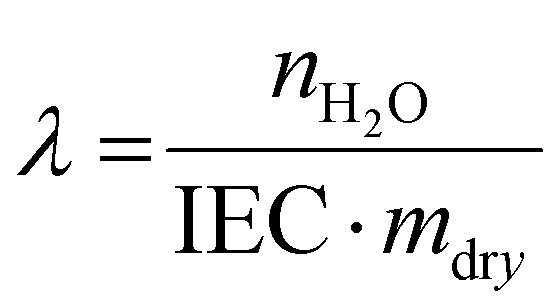 | (3) |
The theoretically derived IEC value was calculated using eqn (4), where M (PSMI-Bn) [mol g−1] is a molar mass of PSMI-Bn polymer and M (OH−) [mol g−1] is a molar mass of hydroxide ion and equals 1.7 mmol g−1.
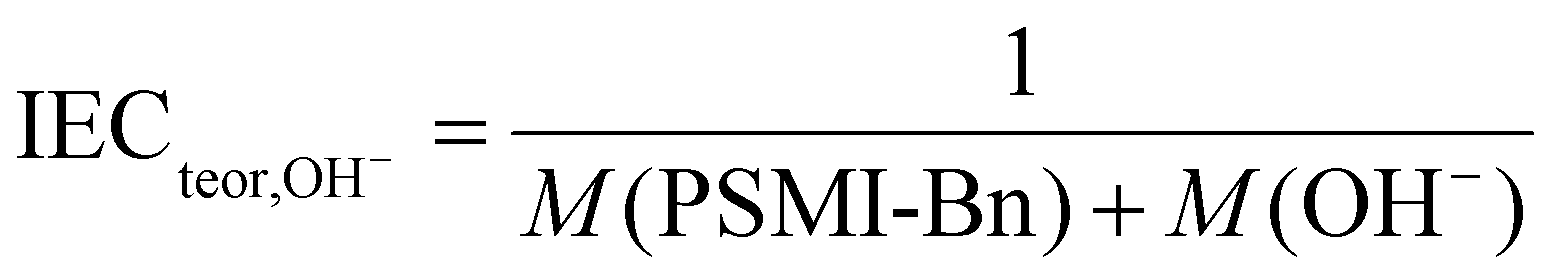 | (4) |
Results
Compound 5a was synthesised via a 4-component condensation route, using benzil, trimethylbenzaldehyde, ammonium acetate, and an excess of aniline (Fig. 1a). Compound 5b was synthesised via a 3-component (benzil, trimethylbenzaldehyde, and ammonium acetate) condensation reaction as per previously reported in the literature (Fig. 1a).325b was dissolved in DMSO, deprotonated with excess KOH and reacted with iodomethane, butyl-bromide or benzyl-bromide to yield the single N-alkylated imidazole (Fig. 1b). The N-functionalisation was varied to compare the impact of substituents on imidazolium stability, reactivity in the polymerisation step, and to tune the ion exchange capacity of the polymer. The second N-functionalisation of compounds 5a, and 6b–d proceeded with either benzylchloride (compounds 7a–d, Fig. 1c) or vinyl benzylchloride (compounds 8a–d, Fig. 1d). Fig. 2 depicts collected 1H NMR spectra of a representative synthetic route 5b–6d–7d–8d. All model compounds and monomers were characterized using 1H NMR, 13C NMR, and mass spectroscopy. Remaining spectral data can be found in the ESI (Fig. S2–S25†).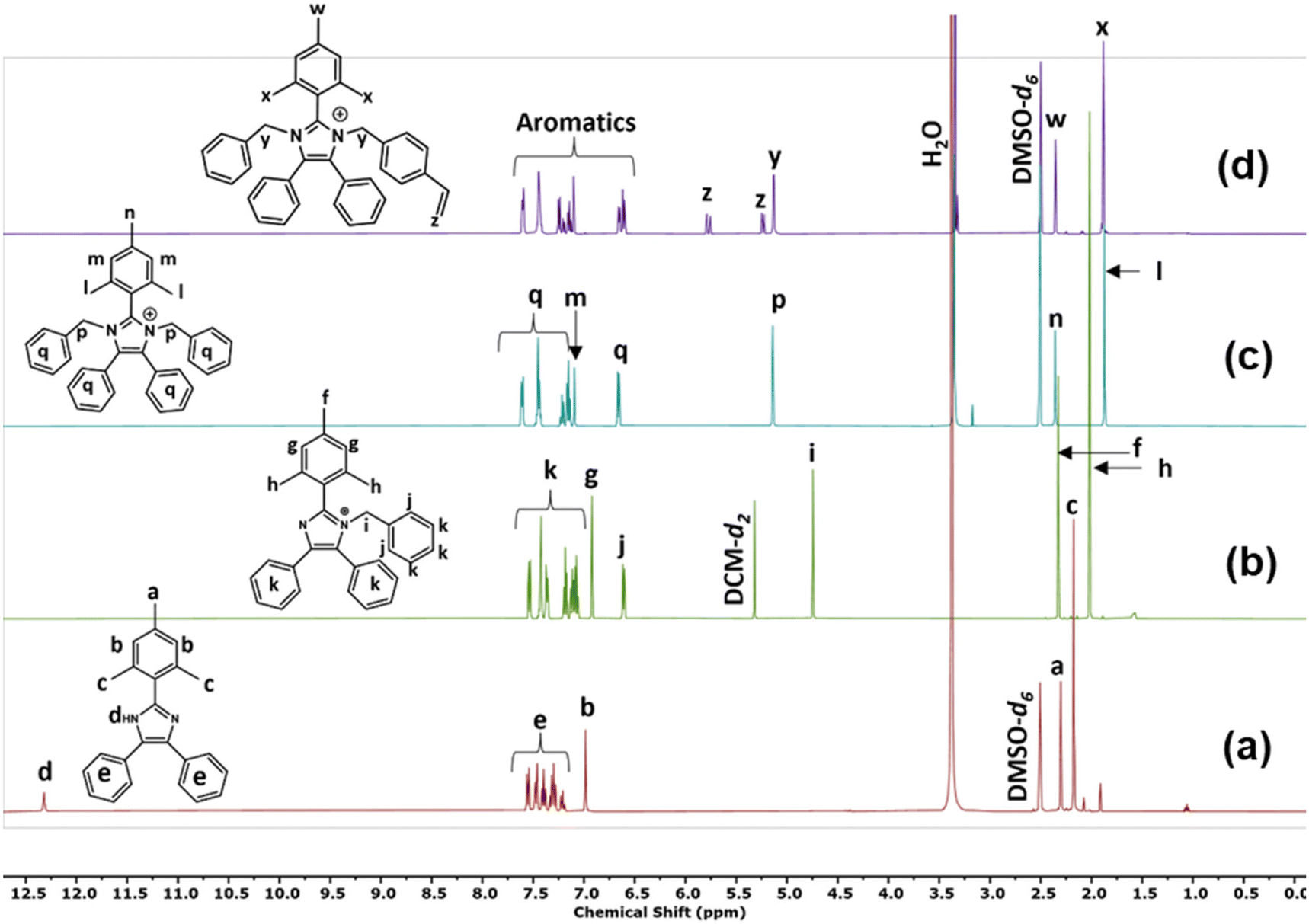 | ||
| Fig. 2 1H NMR spectra of compounds (a) 5b, (b) 6d, (c) 7d, and (d) 8d confirming the synthetic route progression. | ||
The dihedral angle was investigated via DFT calculations, using the optimized geometries of compounds 7a–d. The dihedral angles are measured between the mesityl groups and the imidazolium ring and consist of two values dependent on which side of the mesityl-imidazolium compound was measured. It was expected that the compounds with a dihedral angle closer to 90° would have the highest C2 steric protection and therefore the highest stability with respect to hydroxide attack at the C2 position.31 The N-functionalisation groups are known to have an impact on the mesityl-imidazolium dihedral angle.31 The variation of N-functionalisation for compounds 7a–d were expected to impact the dihedral angle and therefore the stability of the imidazoliums. Compound 7d has the closest dihedral angle to 90°, followed by 7c, then 7a and 7b as shown in Table 2. This suggests a stability trend as follows: 7d > 7c > 7a > 7b. However, this trend was not precisely observed experimentally, as illustrated below.
| Compound | Dihedral angle – DFT (°) | LUMO energy (eV) |
|---|---|---|
| 7a (Ph) | 76.5/78.6 | −1.51 |
| 7b (Me) | 75.8/77.2 | −1.45 |
| 7c (Bu) | 78.5/78.7 | −1.46 |
| 7d (Bn) | 82.3/86.8 | −1.49 |
The hydroxide stability of imidazoliums 7a–d were measured using the methods described by the Coates group.39 The compounds (0.02 M) were dissolved in a mixture of 3 M NaOD in CD3OD/D2O, stored at 80 °C and the changes in chemical structures were monitored using 1H NMR spectroscopy. Fig. 3a shows the evolution of the 1H NMR spectra of compound 7d, which was found to be the most stable molecule, over a period of 720 h. Significant deuterium exchange was observed for the –CH2 benzyl protons at the position M, 7.1 ppm and the –CH3 protons on the mesityl group position L, 1.75 ppm.40 The extent of degradation was quantified by comparing the change in integration of the benzyl –CH protons (Fig. 3a, protons q, 6.50, 7.12, and 7.21 ppm, respectively). There was no notable ring opening degradation from hydroxide attack at the C2-position, implying the benzyl groups provided sufficient C2 steric protection, as suggested by the dihedral angles. However, chemical degradation occurred through the de-functionalisation at the imidazolium N atoms as can be seen by the reduction of the peak P at 5.2 ppm. After 720 h, only 6% of compound 7d was degraded.
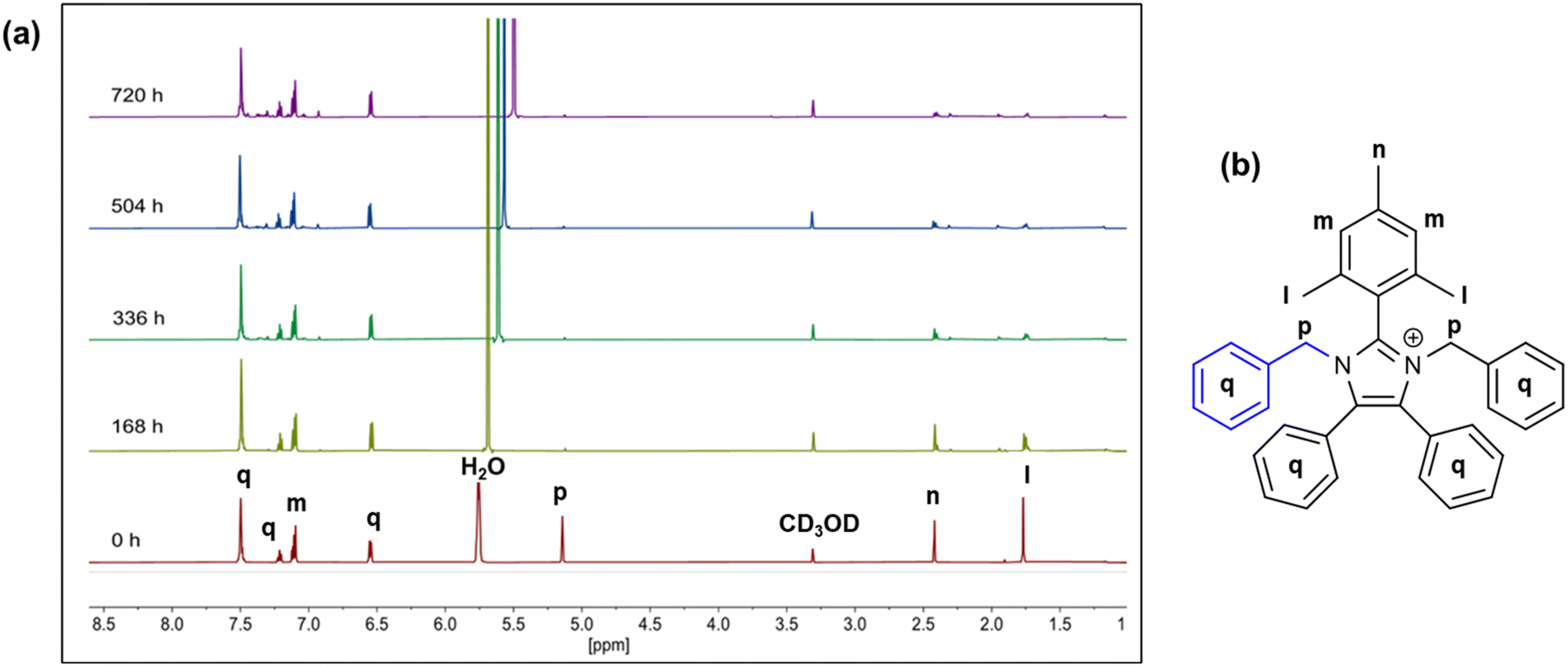 | ||
| Fig. 3 (a) 1H NMR of a representative compound 7d in 3 M NaOD/CD3OD/D2O at 80 °C at various intervals over 720 h. (b) Proton labeling of compound 7d. | ||
The degradation of compounds 7a–d, monitored by 1H NMR spectroscopy, was exponentially fitted to estimate the half-life (t1/2). The remaining degree of functionalization for each molecule is shown in Fig. 4. The steric encumberment provided by the N-benzyl and N-methyl groups restricted hydroxide attack at the C2-position but the methyl group was not sufficiently bulky to prevent de-alkylation, resulting in an estimated half-life of only 852 h and over 30% of degradation after 504 h for molecule 7b. The additional steric hindrance provided by the butyl (7c), phenyl (7a) and benzyl (7d) groups created a five-fold increase in stability for the molecules: half-lives of 3410, 3520 and 7730 h, respectively, with over 90% functionalization remaining after 504 h of degradation test for 7a and 7d.
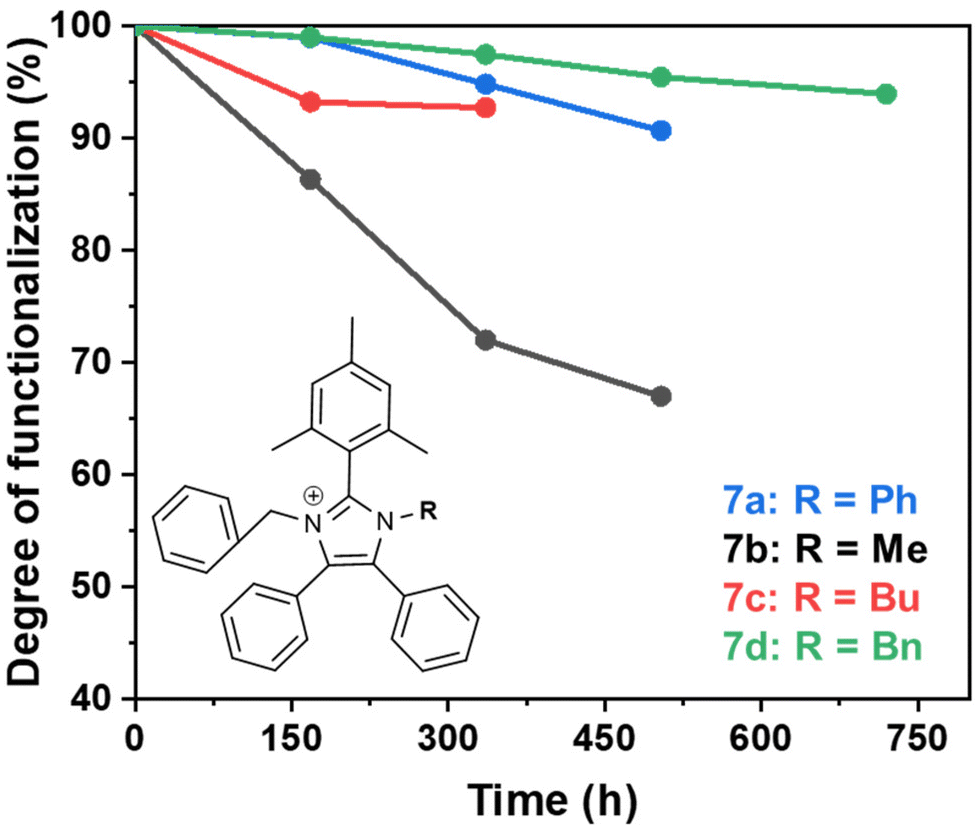 | ||
| Fig. 4 The degree of functionalization remaining after the alkaline degradation test performed on synthesised molecules in 3 M NaOD/CD3OD/D2O at 80 °C at various intervals over 720 h. | ||
Fan et al. compared half-lives of various imidazolium model compounds ranging from 1370 h to over 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 h. In this report, model compound 7d (Bn) with a half life of 7730 h compares to a previously reported imidazolium cation with 7790 h half life stability evaluated under similar conditions.32 Suggested degradation pathways of the model compounds are presented in Fig. 5 which depicts a general structure of synthesised molecules undergoing either de-alkylation via hydroxide attack on the benzyl methylene or a direct C2 attack followed by the ring opening.32,34
000 h. In this report, model compound 7d (Bn) with a half life of 7730 h compares to a previously reported imidazolium cation with 7790 h half life stability evaluated under similar conditions.32 Suggested degradation pathways of the model compounds are presented in Fig. 5 which depicts a general structure of synthesised molecules undergoing either de-alkylation via hydroxide attack on the benzyl methylene or a direct C2 attack followed by the ring opening.32,34
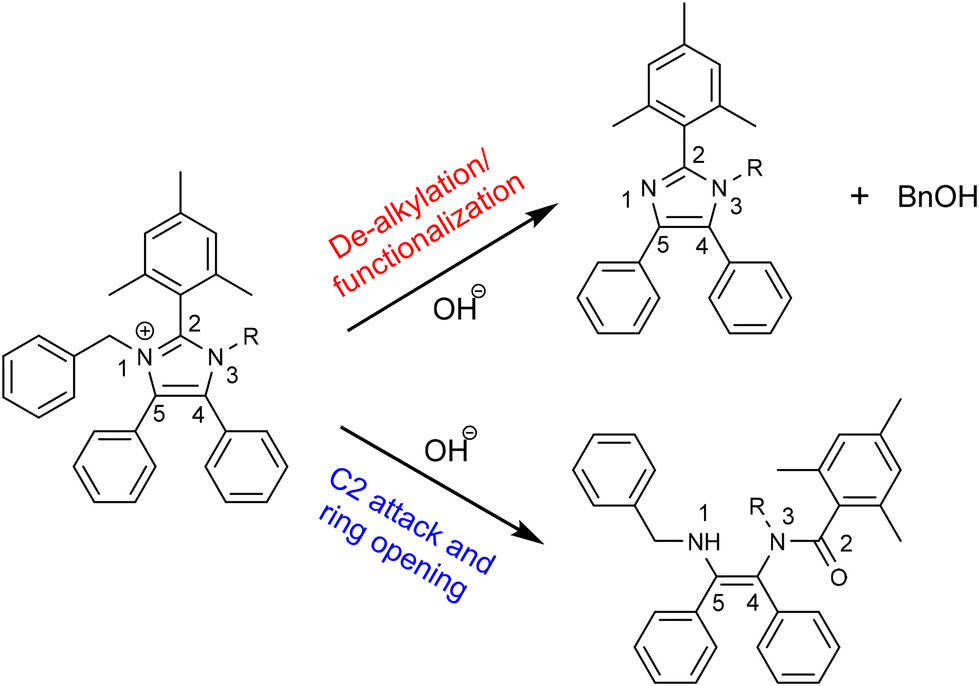 | ||
| Fig. 5 Possible hydroxide degradation scheme of the analysed model compounds 7a–d. | ||
The experimental and theoretical dihedral angles only partially agree with the degradation test results. The theoretical results give a stability trend of: 7d (Bn) > 7c (Bu) > 7a (Ph) > 7b (Me), in comparison to the experimental results which have a trend of: 7d (Bn) > 7a (Ph) > 7c (Bu) > 7b (Me).
A common finding for both theoretical and experimental trends is that the steric hindrance of the benzyl group, 7d exhibits an enhanced alkaline stability compared to the short alkyl chain functionalized polymer, 7b. There is an inconsistency between compounds 7c and 7a, where the theoretical results predict 7a having a lower stability than 7c, but the experimental results found this to be the opposite. The minor discrepancy between theoretical and experimental results could be related to the fact that the dihedral angles only consider steric encumberment around the C2 position and do not account for electronic effects that also impact stability. To investigate the electronic impact of the different N-functionalities, the lowest unoccupied molecular orbitals (LUMOs) of compounds 7a–d were calculated (ESI, Fig. S26 and Tables S1–S4†). The degradation reactions require the correct energy difference between the LUMO and the highest occupied molecular orbital (HOMO) of the hydroxide anion. Therefore, the lower the energy of the LUMO the more susceptible the compound is to hydroxide attack. Interestingly, the energies of the LUMOs were in the order 7b (Me) > 7c (Bu) > 7d (Bn) > 7a (Ph), which disagrees with the result obtained from the theoretical and experimental dihedral angles, and the degradation studies. However, there was very little difference in the values of the LUMOs, possibly suggesting there is a contribution from both the steric and electronic effects that impact the stability of the imidazoliums.
The high hydroxide stability of compound 7d (Bn) and 7a (Ph) inspired the incorporation of these C2-protected imidazoliums as potentially stable ionic groups on a polyolefinic backbone. Vinyl imidazolium monomers (8a, d) were polymerised using azobisisobutyronitrile (AIBN) in N-methyl-2-pyrrolidone (NMP) (Fig. 1d). The radical chain growth polymerisation mechanism created a polyolefin with C2-protected imidazolium pendant groups. The optimised synthesis of the PSMI-R[Cl−] polymers was conducted using 1.00 eq. of compound 8a, d, 0.05 eq. of AIBN, in NMP for 168 h. The polymers synthesised in these conditions were insoluble in most common organic solvents. Importantly, the design of the vinyl monomers and the polymerisation route ensured a homopolymer was synthesised with identical repeat units, each containing one imidazolium pendant group. No post-polymerisation functionalisation was required. The chemical composition of the polymers was investigated using elemental analysis and collected data is shown in Table 3.
| Sample | H (%) | N (%) | C (%) |
|---|---|---|---|
| PSMI-Ph (t) | 6.2 | 4.9 | 82.5 |
| PSMI-Ph (e) | 6.6 | 5.2 | 80.0 |
| PSMI-Bn (t) | 6.4 | 4.8 | 82.6 |
| PSMI-Bn (e) | 6.3 | 4.9 | 79.6 |
Thermogravimetric analysis was deployed to determine the thermal stability of the synthesised polymers. TGA curves of the polymers up to 450 °C under N2 shown in Fig. 6. Both curves display at least two distinctive steps, where the first step indicates the loss of the water trapped in the polymer and the second step reveals a thermal degradation of the olefinic polymer backbone which starts at about 350 °C.41 The TGA analysis of PSMI-Bn reveals an additional degradation that can be associated with a thermal decay of the pendant benzyl group. The temperature of 200 °C, at which degradation is initiated, is much above typical operational temperatures of polymer membrane electrochemical energy conversion devices.8,9
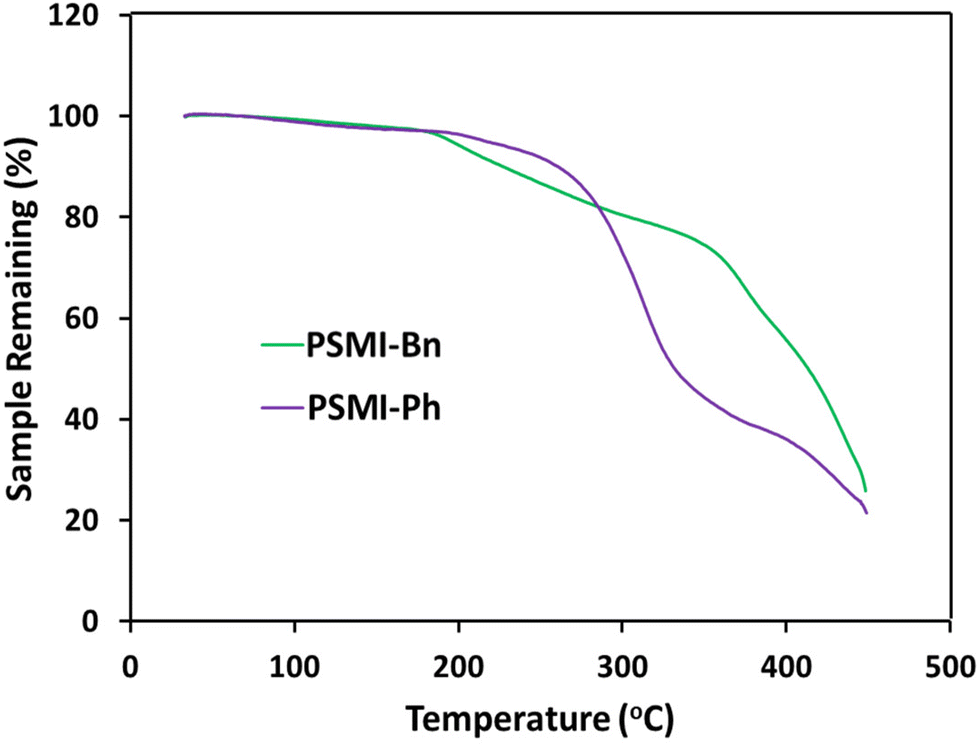 | ||
| Fig. 6 TGA curves of polymers under N2. | ||
Based on the liquid alkaline stability of model compounds, PSMI-Bn was expected to have the highest resistance to hydroxide attack and was therefore further investigated in polymer form by DVS technique to determine water uptake, hydration number and hydroxide stability under low hydration conditions. The water uptake of PSMI-Bn[Cl−] was determined to be 6.8 wt% at 70 %RH and 80 °C, yielding a hydration number of approximately 2. The chloride conductivity of this polymer in powder form was 1.18 mS cm−1 at 80 °C and 95 %RH evaluated via the method described by A.L.G. Biancolli et al.42 Previous research shows that poly(benzimidazolium) based polymers exhibit chloride conductivity of 3–4 mS cm−1 under similar conditions,43 while poly(imidazolium) based polymers in chloride-form have shown 10 mS cm−1.32 Named materials were evaluated in a free-standing film form, while ionic conductivity of PSMI-Bn was determined in its pelletized powder form, which contains a significant number of grain boundaries that reduces ionic conductivity. The stability of the polymer could not be investigated through a 1H NMR spectroscopic analysis due to the polymer's poor solubility, so the DVS degradation protocol described by Kreuer et al.,44 was employed. This method enables the stability of the polymer to be investigated in its powder form by measuring the change in water uptake whilst cycling through various levels of hydration. The hydration number (λ) of ionic polymer is correlated to its IEC at a given temperature and relative humidity (eqn (3)). Therefore, the degradation of the polymer can be investigated under controlled temperature and RH by measuring the mass loss of the sample. The mass loss of the sample occurs due to release of trapped water in the polymer structure, as the degree of functionalisation and therefore IEC of the sample is decreasing due to the degradation. The mass loss may also be exacerbated by loss of volatile degradation products, which are accounted for in this work.
Prior to the DVS measurement, the polymer sample was converted into its hydroxide form (as described in the Experimental section) and cycled from 70 %RH to 10 %RH. These conditions differ from the stability tests conducted in solvent, as described for the model compounds. The solution-based stability tests possess a much higher λ (H2O:OH−) ratio, than that under the low RH attained in this thermogravimetric method. The low hydration levels increase the reactivity of the hydroxide anion, rendering degradation much faster. The design of this method is more suitable for ion conducting polymers in AEMFC applications, since harsh hydroxide saturated conditions are mimicking drying of the cathode side of the cell caused by the electroosmotic drag of water, being especially degrading towards ionomer-binder incorporated into the catalyst layer.45,46
The test was conducted at 80 °C. Relative humidity of 70% was chosen as a probing humidity and it was held at intervals of 5 h, while a decomposition relative humidity of 10% was held for 10 h. As can be seen from the Fig. 7, after stabilizing the PSMI-Bn[OH−] polymer sample at 85 %RH and confirming its starting weight, the humidity was lowered to initial probing humidity of 70 %RH to begin the degradation cycling. The mass of the polymer was assumed to be stable during the probing periods of 70 %RH. The hydroxide degradation was identified from the decreasing difference in hydration number at both high and low humidities, as well as the recorded decrease in mass during decomposition periods. The fast initial degradation observed during first 20 h is attributed to the harsh conditions of the test. The IEC loss estimation was calculated from the first and the last decrease in hydration number as indicated on Fig. 7. According to the calculation, the IEC of PSMI-Bn[OH−] decreased by 75% of its initial IEC value over 90 h of harsh hydroxide-saturated conditions suggesting a comparable loss of the polymer's functionality.
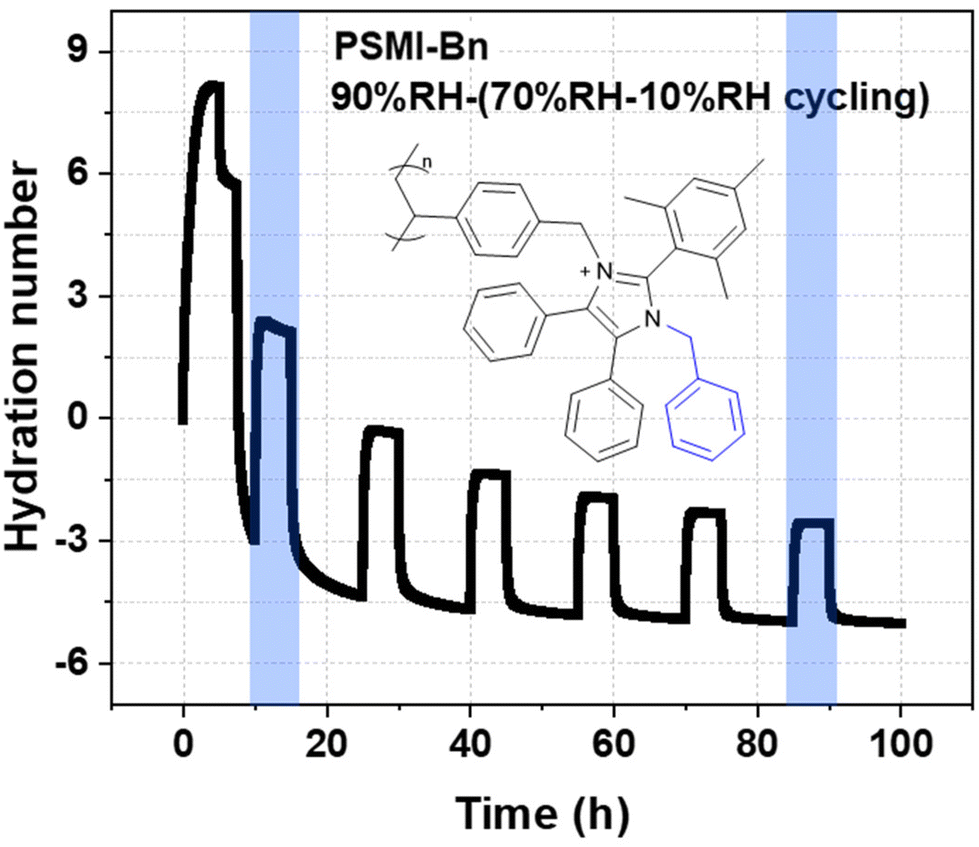 | ||
| Fig. 7 DVS curve collected at 80 °C. The change in weight is monitored via the change in hydration number assuming a constant theoretical IEC of 1.7 mmol g−1. Initial 5 h of experiment at 85 %RH was conducted to condition the polymer and collect a maximum accessible water uptake and hydration number value. | ||
Conclusions
For the first time, C2-protected imidazoliums were incorporated into a polyolefin backbone as pendant groups. N-functionalisation of the imidazoliums was used to probe the effect of steric bulk on hydroxide stability. The alkaline stability of the model compounds was assessed, using 1H NMR spectroscopy, dihedral angle analysis, and LUMO energies. DFT analyses showed the phenyl and benzyl N-functionalities caused the C-2 attached mesityl group to lie ∼90° with respect to the imidazolium ring. 1H NMR spectroscopic studies corroborated these results by finding the two benzyl N-functionalities provided the highest protection against hydroxide degradation. The benzyl substituted model compound, 7d, degraded only 6% after 720 h under the harsh alkaline conditions, yielding a projected half-life of 7730 h, which is at least 5 times higher than the corresponding model compound possessing a short methyl chain and 2 times higher than the rest of model compounds synthesized in this work.Polyolefins were synthesised using a radical chain growth polymerisation mechanism. In this method, the polymerisation reaction is the only synthetic step that removes the requirement for complex post-polymerisation functionalization reactions eliminating potential batch-to-batch inconsistencies and increasing reproducibility of the synthesis. The reaction yielded insoluble polymers that were characterised using elemental and thermogravimetric analysis for chemical structure. Elemental analysis confirmed the percentage composition of the product and TGA analyses revealed two-step temperature-induced changes for PSMI-Ph that suggests water removal and polyolefin backbone degradation at 350 °C, while PSMI-Bn sample had signs of functionalization loss at 200 °C. Water uptake, hydration number and stability of the polymer samples were evaluated by DVS. Humidity cycling between 70 %RH and 10 %RH of PSMI-Bn [OH−] revealed loss of polymer mass associated with loss of absorbed water (hydration number) that correlates with IEC. PSMI-Bn, while possessing a half-life of 7730 h in 3 M NaOD/CD3OD/D2O at 80 °C as a model compound, lost 75% of its initial IEC value over nearly 100 h when exposed to very dry conditions (10 %RH) at 80 °C under accelerated degradation environment where λ values can become as low as 2.
While PSMI-Bn is the most stable of the polyolefin cationic structures studied here, the observed instability under accelerated degradation conditions provides insight into the challenges surrounding the design and synthesis of stable hydroxide ion conducting polymers under extreme caustic exposure. Nonetheless, this work presents a synthetic platform for future structural modifications of either the polymer backbone or ionic group to improve the alkaline stability of C2-protected poly(imidazolium) materials.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by the Natural Sciences and Engineering Research Council of Canada (NSERC) through Discovery Grant RGPIN-2018-03698. The authors gratefully acknowledge Wen Zhou, Thomas Weissbach, and Carol Wu for useful discussions and technical help with the instruments.References
- W. Qian, J. Texter and F. Yan, Chem. Soc. Rev., 2017, 46, 1124–1159 RSC.
- P. Jannasch and E. A. Weiber, Macromol. Chem. Phys., 2016, 217, 1108–1118 CrossRef CAS.
- T. Ban, M. Guo, Y. Wang, Y. Zhang and X. Zhu, J. Membr. Sci., 2023, 668, 121255 CrossRef CAS.
- J. R. Varcoe, P. Atanassov, D. R. Dekel, A. M. Herring, M. A. Hickner, P. A. Kohl, A. R. Kucernak, W. E. Mustain, K. Nijmeijer, K. Scott, T. Xu and L. Zhuang, Energy Environ. Sci., 2014, 7, 3135–3191 RSC.
- E. Balogun, S. Cassegrain, P. Mardle, M. Adamski, T. Saatkamp and S. Holdcroft, ACS Energy Lett., 2022, 7, 2070–2078 CrossRef CAS.
- S. D. Poynton, R. C. T. Slade, T. J. Omasta, W. E. Mustain, R. Escudero-cid and J. R. Varcoe, J. Mater. Chem., 2014, 2, 5124–5130 RSC.
- C. He, A. C. Yang-Neyerlin and B. S. Pivovar, J. Electrochem. Soc., 2022, 169, 024507 CrossRef CAS.
- S. Gottesfeld, D. R. Dekel, M. Page, C. Bae, Y. Yan, P. Zelenay and Y. S. Kim, J. Power Sources, 2018, 375, 170–184 CrossRef CAS.
- D. R. Dekel, J. Power Sources, 2017, 2018, 158–169 Search PubMed.
- M. Adamski, T. J. G. Skalski, B. Britton, T. J. Peckham, L. Metzler and S. Holdcroft, Angew. Chem., Int. Ed., 2017, 6, 9186–9189 CrossRef.
- Z. Long, J. Miyake and K. Miyatake, ACS Appl. Energy Mater., 2019, 2, 7527–7534 CrossRef CAS.
- D. Yazili, E. Marini, T. Saatkamp, A. Münchinger, T. de Wild, L. Gubler, G. Titvinidze, M. Schuster, C. Schare, L. Jörissen and K. D. Kreuer, J. Power Sources, 2023, 563, 23791–23799 CrossRef.
- A. D. Mohanty, S. E. Tignor, J. A. Krause, Y. K. Choe and C. Bae, Macromolecules, 2016, 49, 3361–3372 CrossRef CAS.
- S. Noh, J. Y. Jeon, S. Adhikari, Y. S. Kim and C. Bae, Acc. Chem. Res., 2019, 52, 2745–2755 CrossRef CAS PubMed.
- E. J. Park and Y. S. Kim, J. Mater. Chem. A, 2018, 6, 15456–15477 RSC.
- J.-S. Wang and K. Matyjaszewski, Macromolecules, 1995, 28, 7901–7910 CrossRef CAS.
- J. Chiefari, Y. K. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1998, 31, 5559–5562 CrossRef CAS.
- G. Moad and E. Rizzardo, Macromolecules, 1995, 28, 8722–8728 CrossRef CAS.
- D. Breslow and N. Newburg, J. Am. Chem. Soc., 1057, 20, 5072–5073 Search PubMed.
- W. You, K. J. T. Noonan and G. W. Coates, Prog. Polym. Sci., 2020, 100, 101177 CrossRef CAS.
- M. G. Marino and K. D. Kreuer, ChemSusChem, 2015, 8, 513–523 CrossRef CAS PubMed.
- M. R. Sturgeon, C. S. Macomber, C. Engtrakul, H. Long and B. S. Pivovar, J. Electrochem. Soc., 2015, 162, 366–372 CrossRef.
- H. Long, K. Kim and B. S. Pivovar, J. Phys. Chem. C, 2012, 116, 9419–9426 CrossRef CAS.
- F. Gu, H. Dong, Y. Li, Z. Sun and F. Yan, Macromolecules, 2014, 47, 6740–6747 CrossRef CAS.
- L. Gu, H. Dong, Z. Sun, Y. Li and F. Yan, RSC Adv., 2016, 6, 94387–94398 RSC.
- B. Zhang, H. Long, R. B. Kaspar, J. Wang, S. Gu, Z. Zhuang, B. Pivovar and Y. Yan, RSC Adv., 2018, 8, 26640–26645 RSC.
- C. T. Womble, J. Kang, K. M. Hugar, W. Coates, S. Bernhard and K. J. T. Noonan, Organometallics, 2017, 36, 4038–4046 CrossRef CAS.
- B. Xue, X. Dong, Y. Li, J. Zheng, S. Li and S. Zhang, J. Membr. Sci., 2017, 537, 151–159 CrossRef CAS.
- H. Long and B. Pivovar, J. Phys. Chem. C, 2014, 118, 9880–9888 CrossRef CAS.
- A. G. Wright, T. Weissbach and S. Holdcroft, Angew. Chem., Int. Ed., 2016, 55, 4818–4821 CrossRef CAS PubMed.
- O. D. Thomas, K. J. W. Y. Soo, T. J. Peckham, M. P. Kulkarni and S. Holdcroft, J. Am. Chem. Soc., 2012, 134, 34 Search PubMed.
- J. Fan, A. G. Wright, B. Britton, T. Weissbach, T. J. G. Skalski, J. Ward, T. J. Peckham and S. Holdcroft, ACS Macro Lett., 2017, 6, 1089–1093 CrossRef CAS PubMed.
- K. M. Hugar, H. A. Kostalik and G. W. Coates, J. Am. Chem. Soc., 2015, 137, 8730–8737 CrossRef CAS PubMed.
- J. Fan, S. Willdorf-Cohen, E. M. Schibli, Z. Paula, W. Li, T. J. G. Skalski, A. T. Sergeenko, A. Hohenadel, B. J. Frisken, E. Magliocca, W. E. Mustain, C. E. Diesendruck, D. R. Dekel and S. Holdcroft, Nat. Commun., 2019, 10, 2306–2316 CrossRef PubMed.
- C. G. Arges, J. Parrondo, G. Johnson, A. Nadhan and V. Ramani, J. Mater. Chem., 2012, 22, 3733–3744 RSC.
- Z. Sun, B. Lin and F. Yan, ChemSusChem, 2018, 11, 58–70 CrossRef CAS PubMed.
- J. Wang, S. Gu, R. Kaspar, B. Zhang and Y. Yan, ChemSusChem, 2013, 6, 2079–2082 CrossRef CAS PubMed.
- M. Moreno-González, P. Mardle, S. Zhu, B. Gholamkhass, S. Jones, N. Chen, B. Britton and S. Holdcroft, J. Power Sources Adv., 2023, 19, 100109–100120 CrossRef.
- K. M. Hugar, W. You and G. W. Coates, ACS Energy Lett., 2019, 4, 1681–1686 CrossRef CAS.
- T. Weissbach, A. G. Wright, T. J. Peckham, A. Sadeghi Alavijeh, V. Pan, E. Kjeang and S. Holdcroft, Chem. Mater., 2016, 28, 8060–8070 CrossRef CAS.
- J. E. Mark, in The Physical Properties of Polymers Handbook, ed. J. E. Mark, SpringerNY, New York, 2nd edn, 2007 Search PubMed.
- A. L. G. Biancolli, A. Konovalova, E. I. Santiago and S. Holdcroft, Int. J. Electrochem. Sci., 2023, 18, 100288–100298 CrossRef.
- E. M. Schibli, J. C. Stewart, A. A. Wright, B. Chen, S. Holdcroft and B. J. Frisken, Macromolecules, 2020, 53, 4908–4916 CrossRef CAS.
- K. Kreuer and P. Jannasch, J. Power Sources, 2018, 375, 361–366 CrossRef CAS.
- X. Wang, J. P. McClure and P. S. Fedkiw, Electrochim. Acta, 2012, 79, 126–132 CrossRef CAS.
- Y. S. Li, T. S. Zhao and W. W. Yang, Int. J. Hydrogen Energy, 2010, 35, 5656–5665 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4py00235k |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |