
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnprecedented associative exchange in CO2-sourced cyclic S,O-acetal-based covalent adaptable networks†
Stephan
Maes‡
 a,
Thomas
Habets‡
b,
Susanne M.
Fischer
a,
Bruno
Grignard
bc,
Christophe
Detrembleur
*bd and
Filip E.
Du Prez
*a
a,
Thomas
Habets‡
b,
Susanne M.
Fischer
a,
Bruno
Grignard
bc,
Christophe
Detrembleur
*bd and
Filip E.
Du Prez
*a
aPolymer Chemistry Research group, Centre of Macromolecular Chemistry (CMaC), Faculty of Sciences, Ghent University, Krijgslaan 281-S4, Ghent, 9000, Belgium. E-mail: Filip.DuPrez@UGent.be
bCenter for Education and Research on Macromolecules (CERM), CESAM Research Unit, University of Liege, Sart-Tilman B6a, 4000 Liege, Belgium. E-mail: Christophe.Detrembleur@ULiege.be
cFRITCO2T Platform, University of Liege, Sart-Tilman B6a, 4000 Liege, Belgium
dWEL Research Institute, Avenue Pasteur 6, 1300 Wavre, Belgium
First published on 10th May 2024
Abstract
New dynamic chemistry is nowadays sought to widen the library of accessible covalent adaptable networks (CANs). Here, we investigate the dynamic nature of CO2-sourced cyclic S,O-acetal bonds under unexplored conditions. Model molecule studies were conducted on various compounds and supported by extensive DFT calculations to understand the required conditions for triggering exchange and the underlying reaction mechanisms. This is the first study to report dynamic S,O-acetal bonds with an unprecedented associative exchange mechanism occurring through nucleophilic attack onto a remote function from the exchanged site. Our findings were translated to macromolecular engineering with the successful production of CO2-sourced CANs embedding cyclic S,O-acetal bonds from bifunctional alkylidene cyclic carbonates and polythiols. The polymer properties were tuned by the use of structurally divergent monomers, affording materials with distinct thermal and mechanical properties (e.g. Tg ranging from 2 to 51 °C). Complex relaxation behaviour was recorded by rheology experiments, suggesting concurrent exchange reactions to take place at elevated temperatures. The materials dynamics was leveraged through recycling by compression molding for over five cycles. Furthermore, a proof-of-concept coating application was developed, showcasing damage healing at high temperatures.
Introduction
Regardless the excellent potential of polymer materials, issues regarding environmental pollution have become increasingly prominent in today's society.1,2 Therefore, research has been intensified in the last few years with the aim of producing plastics from bio-renewable resources and/or waste (such as CO2) while enabling their recycling or upcycling.3During the last decade, covalent adaptable networks (CANs) have emerged as a new class of polymeric materials that bridge the gap between thermoplastics and thermosets in terms of (re-) processability and mechanical superiority.4,5 Interestingly, while these materials are effectively cross-linked under standard conditions, an appropriate trigger – most commonly heat – enables the reshuffling of bonds and subsequently allows for recycling of polymeric networks. CANs are generally divided into two categories depending on their bond exchange mechanism: (i) associative exchange involving new bond creation followed by bond breaking, and (ii) dissociative exchange where bond creation is preceded by bond breaking. Meanwhile, well-established dynamic functionalities have been widely used for the preparation of CANs,6–8 for example esters,9–16 carbamates,17–19 dicarboximides,20 Diels–Alder adducts,21,22 or sulfur-based chemistries23 (e.g. disulfides, thioesters, or sulfonium salts).24–31 While modifying the architectural features of CANs (e.g. cross-link density, backbone stiffness and polarity) allows for fine-tuning their dynamic properties,32 there remains a need to further explore new dynamic chemistry platforms. These investigations aim to expand the library of recyclable CANs, with a focus on identifying the most promising candidates for various applications.
X,X-acetal bonds (X for heteroatom) have attracted significant attention because of their versatility and accessibility through multiple synthetic pathways from various precursors to provide dynamic networks (Scheme 1A).33–37 The most straightforward acetal system was initially developed by Zhu et al., producing O,O-acetal-based thermosets from alcohols and vinyl ethers.33,34,38 The same group reported S,O-acetal derivatives by switching the nucleophile to thiols.39 Well-known as protecting groups in organic synthesis, five- and six-membered cyclic O,O-acetal groups were also reported as efficient reversible bonds.40,41 Although these systems were reported to exchange under catalyst-free conditions, experiments with proton traps confirmed that the reactions were likely activated by residual water providing acidic protons at high temperature. Recently, Detrembleur and co-workers demonstrated that N,S-acetal bonds within alkylidene oxazolidone moieties showed reversibility in the presence of an acid catalyst.42 In 2019, Du Prez and co-workers studied the base-catalyzed exchange of S,S-acetal-based CANs that were prepared through reversible Michael additions on electron-poor alkenes.35 It must be noted that all aforementioned X,X-acetal bonds were of dissociative nature. Interestingly, in 2018, Kalow's group reported the first example of catalyst-free associative exchangeable S,S-acetal bonds using dithioalkylidene cross-linkers.36,43,44
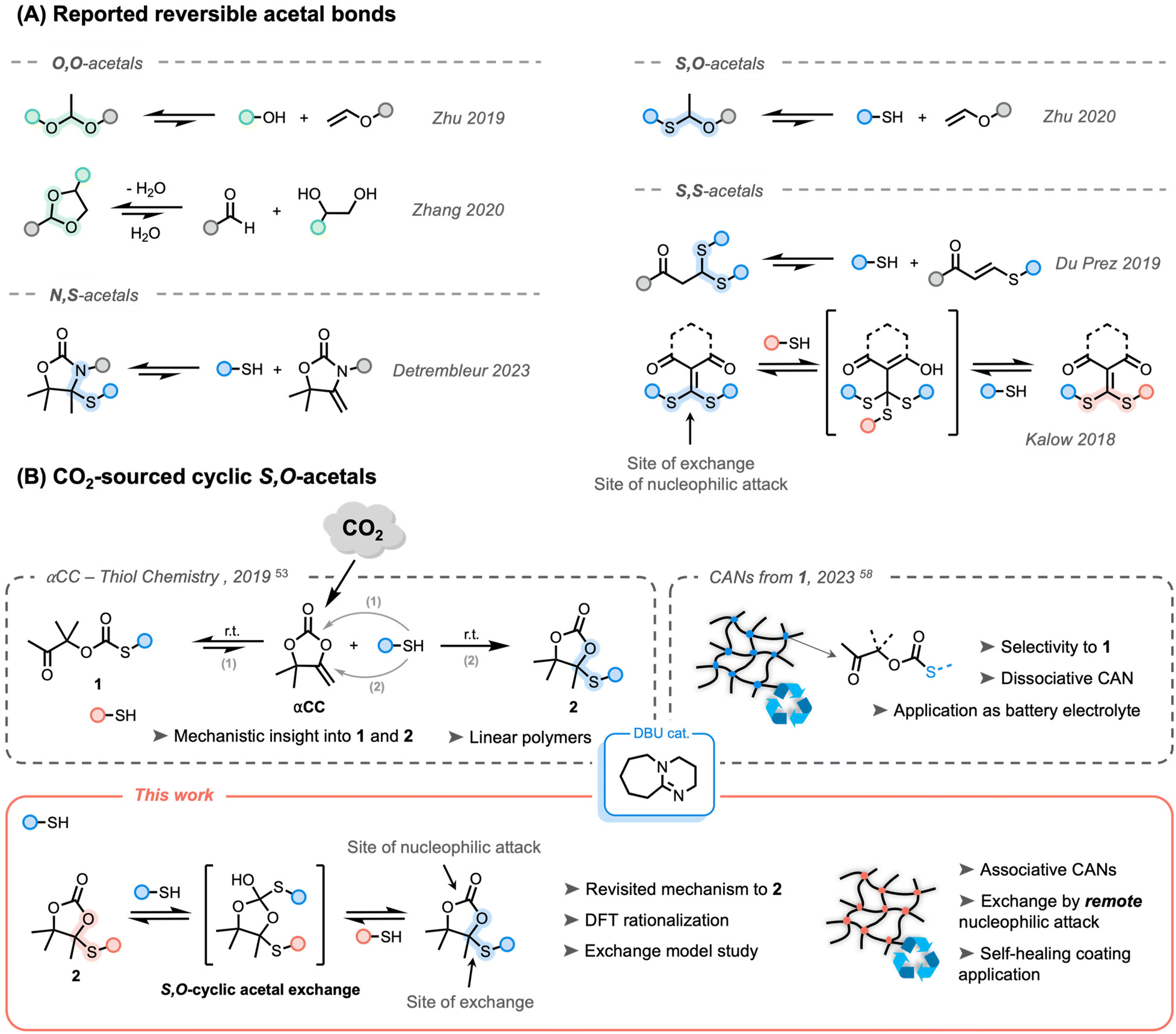 | ||
| Scheme 1 (A) Reported acetal-derived bonds used in covalent adaptable networks. (B) Previous work on αCC-thiol chemistry and a new mechanism proposed in this work for producing 2 toward covalent adaptable networks. | ||
While the design of recyclable polymer networks is an appealing strategy to bypass product disposal at the end of service, the valorization of waste in the manufacturing process is an effective way to produce more sustainable materials. Carbon dioxide (CO2) is a prevalent waste product on the Earth with well-known dramatic consequences for the environment. Its use as a C1 building block is, therefore, of great interest in chemistry and polymer science, leading to the design of many CO2-sourced polymers.45–47 Recently, some of us reported the synthesis of α-alkylidene cyclic carbonates (αCCs) from CO2. These unconventional cyclic carbonates have shown to benefit from high reactivity through increased ring strain provided by the exocyclic alkene.48,49 These compounds can thus easily undergo copolymerization by reaction with various nucleophiles under mild (catalytic) conditions, i.e. with amines,50,51 with alcohols52 and thiols.53 The scope of the obtained polymers was further expanded by different groups to bring more diversification, tunability, functionality, and emerging applications54–58 without omitting recyclability.42,59,60
The organobase-catalyzed reaction of αCCs with thiols can provide two different products. If a hard base (e.g. DBU) is used, the oxo-thiocarbonate 1 is rapidly formed, followed by a rearrangement into a more stable cyclic carbonate embedding an S,O-acetal function 2 (Scheme 1B).53 Using a soft organobase (e.g. DBU![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :acetate), high selectivity toward 1 can be obtained. Interestingly, it was previously determined experimentally and computationally that the formation of 1 was fast and reversible. By taking advantage of the dissociative nature of the bond in the presence of a soft base catalyst, dissociative poly(oxo-thiocarbonate) CANs were produced that could be processed into membranes for solid polymer electrolytes.58
:acetate), high selectivity toward 1 can be obtained. Interestingly, it was previously determined experimentally and computationally that the formation of 1 was fast and reversible. By taking advantage of the dissociative nature of the bond in the presence of a soft base catalyst, dissociative poly(oxo-thiocarbonate) CANs were produced that could be processed into membranes for solid polymer electrolytes.58
Although it was claimed in an earlier work (Scheme 1B, top left)53 that the formation of 2 was irreversible, the dynamic nature of acetal derivative bonds made us wonder whether the reversibility of these cyclic S,O-acetal bonds could be triggered under previously unexplored conditions.
In this work, we show that the previously claimed irreversible cyclic S,O-acetal linkages within cyclic carbonates 2 can undergo an associative exchange with free thiolates by an unprecedented mechanism that involves a remote nucleophilic attack at a different position than the exchange location of the molecule (Scheme 1B, bottom). First, a model study was conducted to demonstrate the associative nature of the exchange reaction and suitable conditions were evaluated by kinetics at elevated temperatures. By means of DFT calculations, a new mechanism rationalizing the formation of 2 was determined to be more favourable than the previously reported one. Next, cross-linked polymer networks embedding cyclic S,O-acetal linkages were prepared from bifunctional CO2-sourced α-alkylidene cyclic carbonates and polythiols. Their dynamics were carefully characterized by rheological measurements and a proof-of-concept applicability was highlighted for their use as healable coatings.
Results and discussion
While the dissociative behaviour of linear oxo-thiocarbonate 1 motifs was recently discussed for CANs,58 the potential reversibility of the S,O-acetal bond in 2 remained unexplored. Therefore, we first conducted initial model studies to assess the bond stability and reversibility before translating the process to the material level through the synthesis of cyclic S,O-acetal-containing CANs. Although the formation of 2 was previously claimed to be irreversible at room temperature, we envisaged that heat might trigger reactivity.Synthesis of model compounds and evaluation of the exchange mechanism by model studies
First, model cyclic carbonates embedding a cyclic S,O-acetal bond 2a–d were prepared in a straightforward fashion from CO2-sourced αCCs and thiols using DBU as the catalyst (Fig. 1, see the ESI† for details). We initiated our model studies by heating a solution of 2a in DMF with a catalytic amount of DBU (Fig. 2A). Analysis by 1H-NMR spectroscopy of the reaction medium (quenched with formic acid) after 2 h up to 120 °C did not reveal any significant change in the spectrum, demonstrating the thermal stability of 2a with no identifiable dissociation occurring at this temperature (Fig. S1†). Furthermore, when equimolar amounts of 2b and 2d were combined with DBU in DMF and heated to 85 °C, crossover products were not observed from LC-MS analysis, suggesting that dissociation did not occur (Fig. 2B and Fig. S2†), even after prolonged heating. This result confirmed that the S,O-acetal bonds in 2 did not present the usual dissociative nature of common acetals reported in CANs.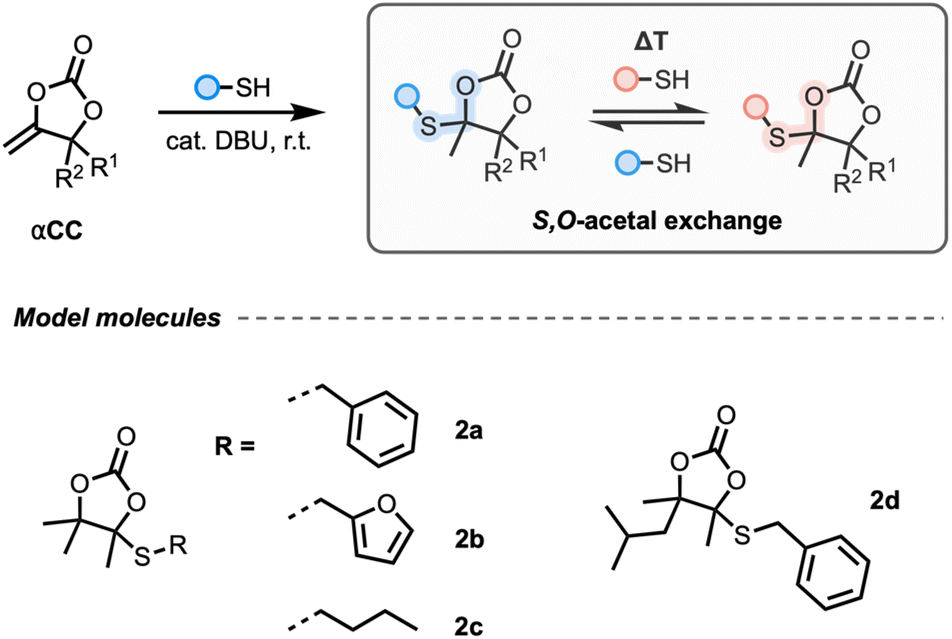 | ||
| Fig. 1 Synthesis of 2a–d from αCCs and a hypothesized exchange reaction. | ||
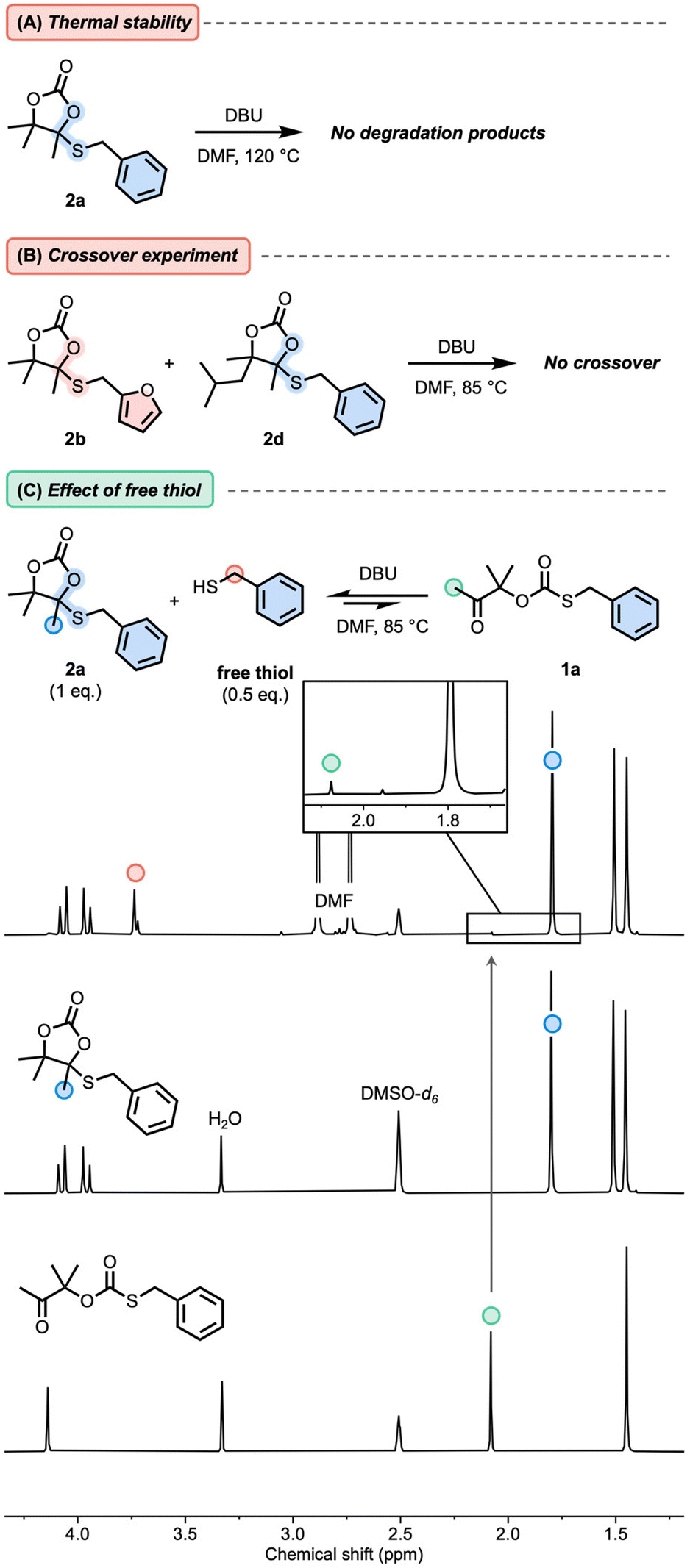 | ||
| Fig. 2 (A) Effect of temperature and catalyst (DBU, 0.01 eq.) on the stability of 2a. (B) Cross-over experiments between 2b and 2d. (C) Observed reaction between 2a and 0.5 equivalents of benzyl mercaptan in the presence of DBU (0.01 eq.) at 85 °C as observed by 1H-NMR spectroscopy (400 MHz, DMSO-d6). Pure NMR spectra of 2a and 1a were stacked to the crude reaction spectrum. | ||
Interestingly, when 0.5 equivalents of benzyl mercaptan were introduced in the reaction mixture containing 2a and DBU at 85 °C, the corresponding oxo-thiocarbonate 1a (1 mol% compared to 2a) was observed via NMR spectroscopy after 1 h (Fig. 2C and Fig. S3†). This indicates that the stability of 2 might be altered in the presence of free thiol and DBU.
Motivated by these preliminary results, pointing towards reactivity triggered by the presence of free thiols, we started investigating the DBU-catalyzed exchange reaction kinetics of 2b with a large excess of free thiol (5 equivalents of benzylmercaptan) to produce 2a (Fig. 3A). Under these conditions, a pseudo first-order reaction law might be used as an approximation to extract reaction rates at different temperatures. Because of inconvenient overlap in NMR, the reaction was followed by means of LC-MS analysis using trimethoxybenzene (TMB) as the internal standard in the temperature range from 70 to 95 °C (Fig. 3B and Fig. S4†). The reaction has shown to be active at 70 °C, with a nearly 10-fold increase in reaction rate when increasing the temperature to 95 °C (0.03 min−1 at 70 °C and 0.26 min−1 at 95 °C, Table S1†). From the related Arrhenius plot (Fig. 3C), constructed from the extracted reaction rate constants at the respective different temperatures, an activation energy of 83.5 ± 6.6 kJ mol−1 was determined. This is slightly higher than previously reported model systems (a range of 66.2–76.8 kJ mol−1 for S,S-acetals and 64.9 kJ mol−1 for N,S-acetals).35,42 To ensure that reactivity is not only triggered by the benzyl or furfuryl nature of the substituent in 2a and 2b, an additional reaction was carried out from an aliphatic-derived 2c (from n-butanethiol) at 70 °C with dodecanethiol, showing successful exchange with a similar reaction rate to that of 2b at the same temperature (0.04 min−1) (Fig. S5†).
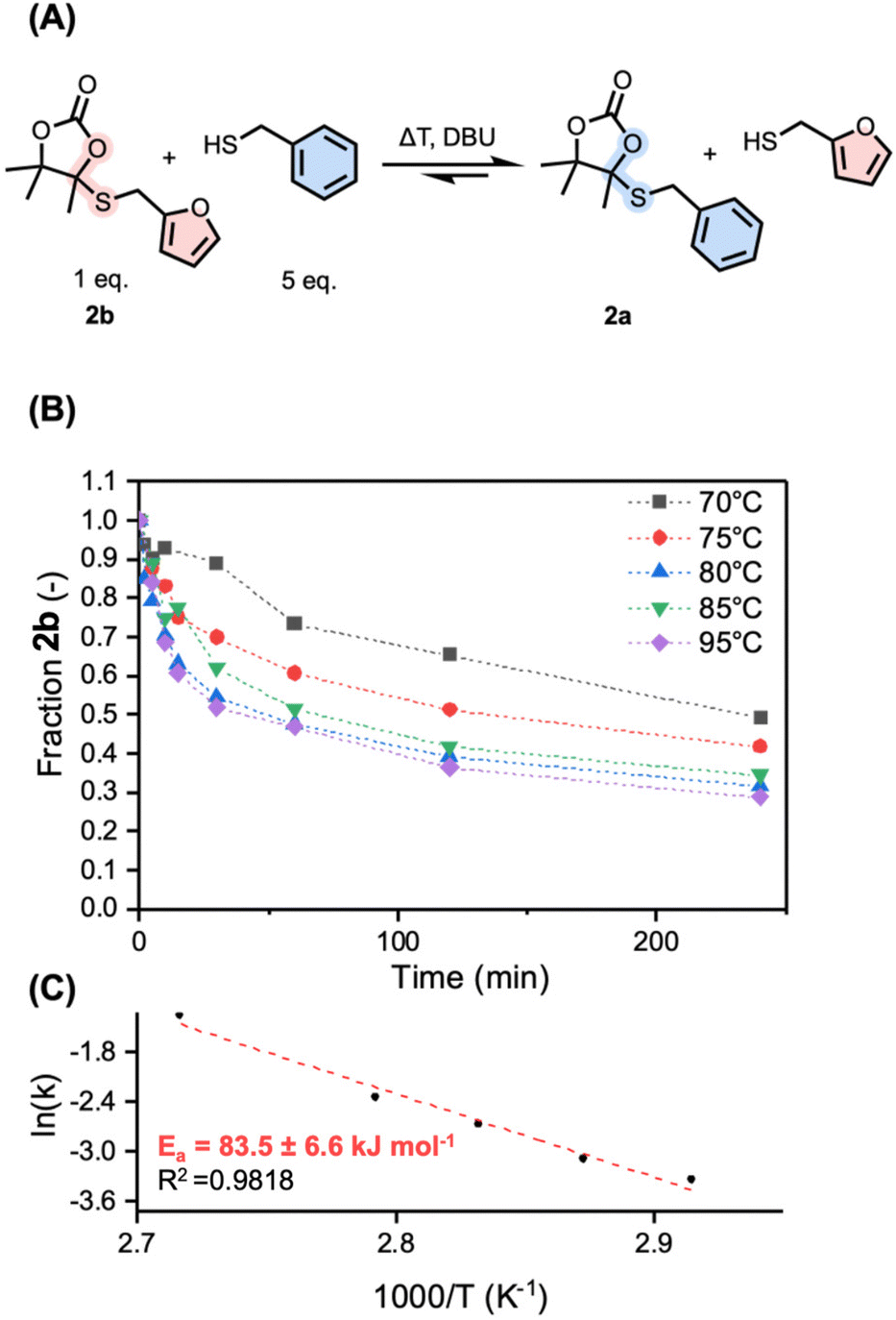 | ||
| Fig. 3 (A) Dynamic exchange reaction between 2b and 5 equivalents of benzyl mercaptan at different temperatures catalyzed by DBU. (B) Evolution of the fraction of 2b over time as followed by LC-MS with 1,3,5-trimethoxybenzene as an internal standard detected at 280 nm. (C) The Arrhenius plot fitted to a linear fit (dashed line). | ||
After successfully demonstrating the temperature dependence of the S,O-acetal exchange in 2, the influence of the concentration of the nucleophile was studied by mixing 2b with 10 equivalents of benzyl mercaptan (instead of 5 eq.). The reaction was followed over time at 75 °C and displayed a significant increase in reaction rate constant (from 0.046 to 0.57 min−1) (Fig. S6†). Both the requirement of free thiolates to trigger exchange and the acceleration of reaction rate by the presence of an increased amount of free thiols demonstrate that the exchange mechanism is of associative nature.
Mechanistic insight into involved reactions
To shed light on this unreported exchange reaction, density functional theory (DFT) calculations were performed at the ωB97-XD/6-311++G(d,p) level of theory on model molecules. In a previous work, the mechanism of αCC thiolation was studied through experimental model reactions and DFT calculations.53 This study concluded that an oxo-thiocarbonate 1 was first obtained by the reversible DBU-catalyzed attack of the thiolate onto the carbonyl group of αCC. The formation of 2 was rationalized by two different mechanisms that are rather close in energy, both involving the attack of the thiolate onto the internal carbon of the exocyclic alkene function of αCC. However, this previous study only provided calculations involving benzyl-derived thiols and some reactions were modelled without including DBU. In the same study, it was, however, demonstrated that the choice of the DBU catalyst was critical to the selective formation of 2 when other superbases (such as DBN, TBD or MTBD) failed at this task. For a systematic comparison between all herein studied pathways and to eliminate any possible effect of benzyl groups on the energetics of the reactions, our model involves DBU in all steps and methanethiol was selected as a simple aliphatic thiol substrate. All modelled pathways and the relative Gibbs free energies of all studied molecules can be found in the ESI (Fig. S7–S14†) including a more thorough discussion.We started our investigation by exploring a new pathway, coined pathway 1, where 2 undergoes thiolation on the electrophilic carbonate function to provide a tetrahedral intermediate int1 stabilized by DBUH+ (Fig. 4B). Subsequently, this intermediate can ring-open to form a hemithioacetal intermediate int2. The final step involves the ejection of the thiolate to yield the oxo-thiocarbonate 1. The rate-determining step of this mechanism involving TS2 is characterized by an energy barrier of 104.0 kJ mol−1. Interestingly, the reverse reaction, from 1 to 2, exhibits a similar energy barrier of 91.7 kJ mol−1, suggesting reversibility. However, the five-membered cyclic carbonate 2 is thermodynamically slightly favored, with an extra stabilization of 12.3 kJ mol−1 compared to the linear thiocarbonate 1. Apart from the experimental observation that 1 is observed when free thiols are added to pure 2 and DBU (see Fig. 2), DFT calculations successfully manage to rationalize the stepwise associative exchange through (i) ring-opening of 2 and (ii) ring-closing of 1.
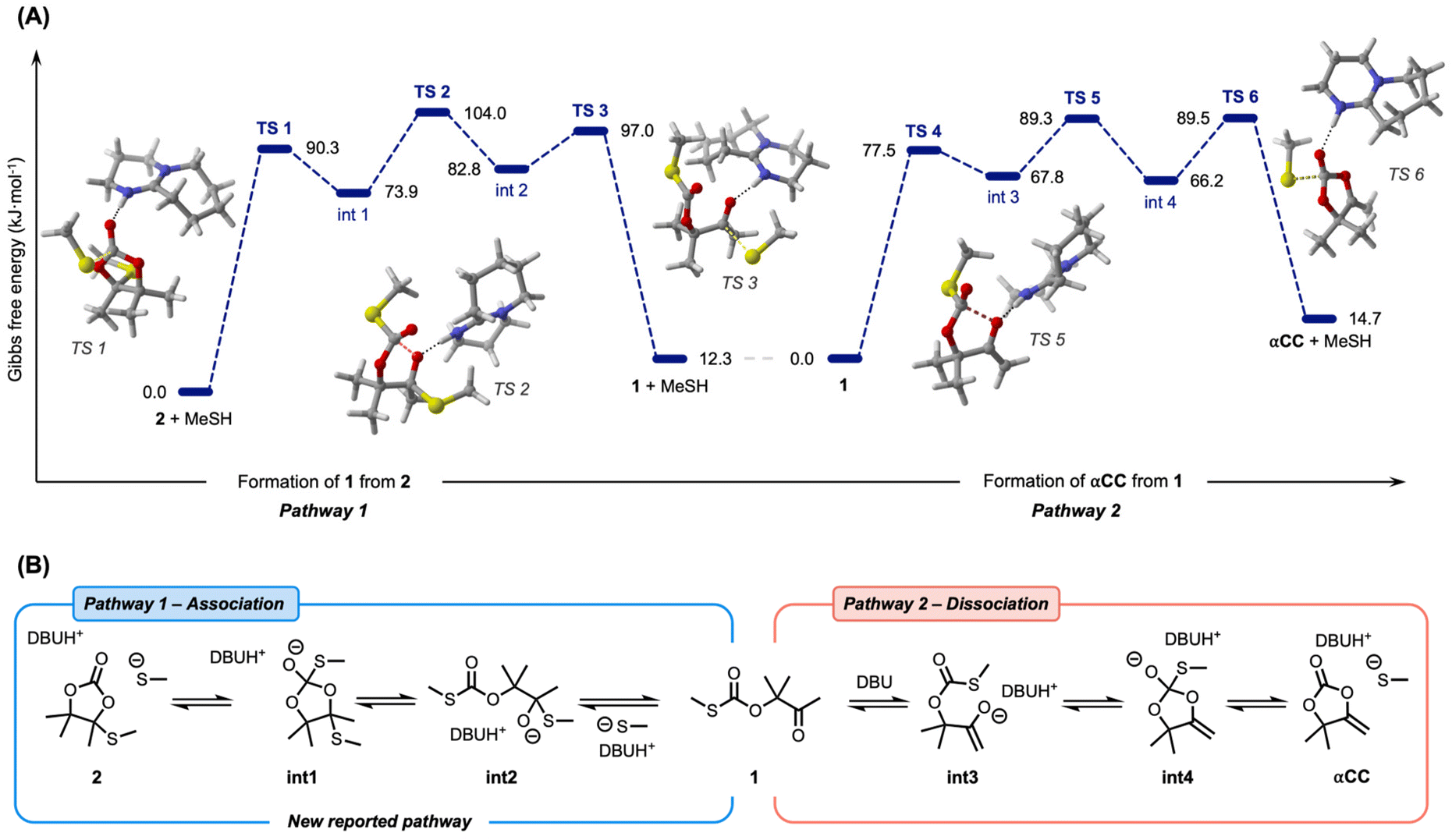 | ||
| Fig. 4 (A) Gibbs’ free energies in kJ mol−1 of the studied reaction pathways and representative structures of the transition state geometries. (B) Schematic representation of associative and dissociative pathways. | ||
Compound 1 is a dynamic molecule prone to dissociation into αCC and a free thiol (pathway 2). Therefore, the formation of 1 as an intermediate opens the door to an additional dissociation process within the system, as this compound is prone to dissociation into αCC and a free thiol. In the presence of DBU, the ketone in 1 can rearrange into an enolate intermediate int3. This intermediate can undergo ring-closure through the attack of the enolate onto the thiocarbonate function to provide a tetrahedral intermediate stabilized by DBUH+int4. The third step of the reaction involves ejection of the thiolate and re-formation of the carbonate to yield αCC. This reaction is characterized by an activation barrier of 89.5 kJ mol−1 and a backward reaction barrier of only 74.8 kJ mol−1, suggesting reversibility and fast reaction kinetics from αCC to 1. Furthermore, the formation of 1 is thermodynamically favored by 14.7 kJ mol−1.
Additionally, we reproduced previously reported pathways toward 2 through direct thiol attack onto αCC (in a stepwise or concerted fashion), labelled as pathway 3 in our study (see the ESI† for details). The most favorable stepwise mechanism showed a high energy barrier of 150.7 kJ mol−1, in close agreement with previously determined values (155.6 kJ mol−1).53 Our newly introduced pathway 1, providing 2 from 1 instead of αCC, thus exhibited a much lower energy barrier of 91.7 kJ mol−1 (59.0 kJ mol−1 less). This huge difference suggests this new path to be much more favorable than the previously reported pathway 3 to explain the formation of 2. Since the backward reaction barriers are even greater (177.7 kJ mol−1), it is unlikely that pathway 3 is the source of dissociation from 2 to αCC and a thiol. This is in line with experiments displaying no dissociation or crossover of 2 at high temperatures in the absence of free thiols.
Overall, these DFT calculations suggest an unprecedented mechanism of acetal exchange via thiol association onto a remote carbonyl function of 2, yielding 1 and a free thiol as intermediates. Compound 1 can then undergo thiolation to provide an exchanged cyclic S,O-acetal 2via a newly proposed mechanism which might be more feasible than the previously reported mechanism starting from αCC. Once 1 is formed, both association toward 2 or dissociation to αCC might concurrently occur.
Preparation and characterization of CANs
After successfully establishing the reaction mechanism, cross-linked materials could be prepared in a straightforward manner from bis(α-alkylidene cyclic carbonate)s (obtained from propargylic alcohols and CO2) (5a–c, see the ESI† for full experimental details and characterisation) and multifunctional thiols (Fig. 5) to exploit this dynamic exchange at the material level. According to the mechanistic model study, polymeric matrices were produced with an excess of thiol (10 mol% excess of thiol groups with respect to the exocyclic double bonds) to enable associative exchange and, by consequence, reprocessing of the thermoset material.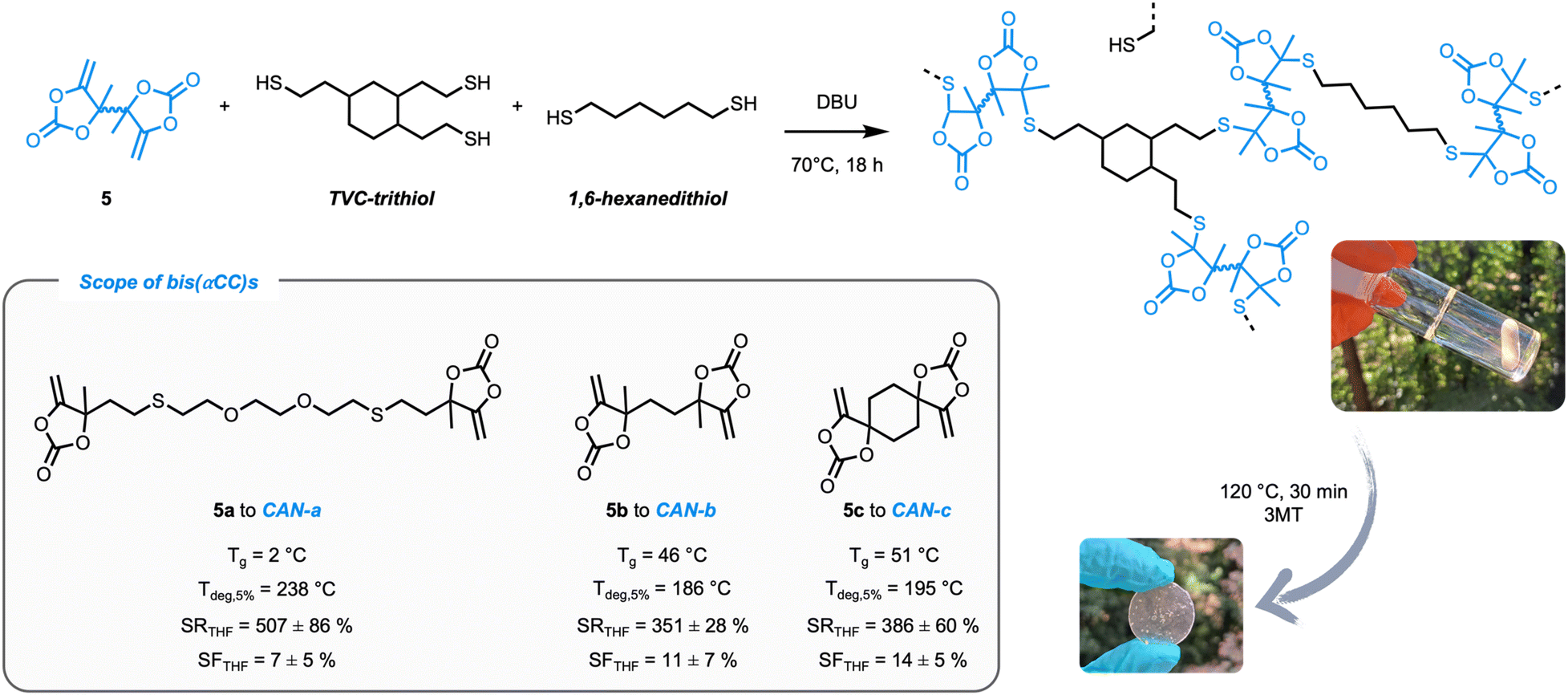 | ||
| Fig. 5 Preparation of covalent adaptable networks starting from bis(α-alkylidene cyclic carbonate)s 5a–c with multifunctional thiols (TVC-trithiol and 1,6-hexanedithiol) using DBU as the catalyst (1 mol% vs. thiol functions). Tg (glass transition temperature) was determined by DSC; Tdeg,5% (temperature at 5% of degradation) was determined by TGA; SR (swelling ratio) and SF (soluble fraction) after immersion of the networks for 24 h in THF. | ||
Initially, a commercially available multifunctional thiol (pentaerythritol tetrakis(3-mercaptoacetate)) was considered. However, this crosslinker could possibly induce issues under the conditions needed for the exchange (high-temperature, basic DBU as the catalyst) due to e.g. transesterification reactions.35 In order to obtain the best understanding about the dynamics of the S,O-acetals, an aliphatic trithiol obtained from trivinylcyclohexane (TVC-trithiol) devoid of other functional groups was therefore synthesized and used as the crosslinker. Networks (CAN-a–c) were subsequently prepared using compounds 5a–c together with TVC-trithiol and 1,6-hexanedithiol as the chain extender (in a 2.25:1:1 molar ratio) and a catalytic amount of DBU (1 mol% with respect to the thiol functionalities). Conversion of bis(αCC)s was conveniently monitored using ATR-FTIR spectroscopy as the disappearance of the signal attributed to the exovinylene function at 1675 cm−1 provided evidence of the αCC monomer consumption (Fig. 6). A small shoulder at 1718 cm−1 corresponding to the linear thiocarbonate intermediate can be observed. Nevertheless, a strong band at 1797 cm−1, corresponding to the stretching vibration of the carbonyl from the cyclic carbonate, confirmed the successful preparation of CANs containing cyclic S,O-acetal 2 linkages.
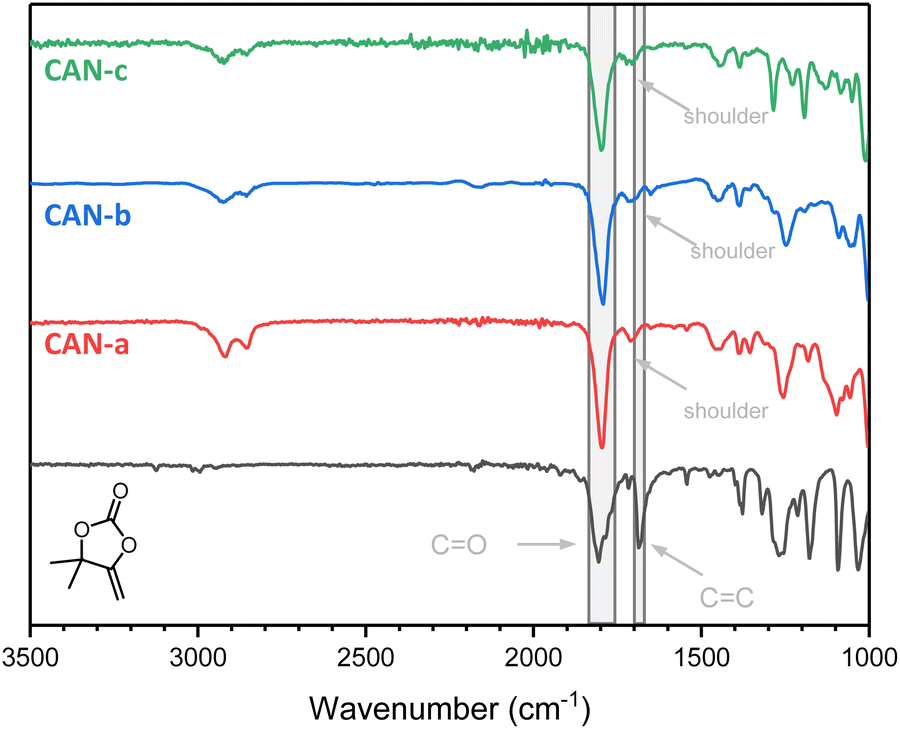 | ||
| Fig. 6 FTIR spectra of αCC and CANa–c. | ||
Homogeneous samples for all CANs were subsequently obtained by compression molding of the pristine material at 120 °C for 30 min under 3 metric tons of pressure. By DSC analysis, glass transition temperatures (Tg) of 2, 46 and 51 °C were determined for CAN-a–c, respectively (Fig. S15†). As expected, the lowest Tg is obtained for CAN-a embedding the most flexible bis(α-alkylidene cyclic carbonate) monomer. The thermal stability of the materials was assessed using TGA and degradation temperatures (Tdeg5%) around 200 °C were measured for all polymers (Fig. S16†). Furthermore, isothermal TGA measurements at 130 °C for 2 h validated the thermal stability of the materials during processing conditions (Fig. S17†). Finally, adequate network formation was confirmed by acceptable swelling ratios (between 300 and 600%) and soluble fractions of approximately 10% in THF at room temperature.
In the next step of this study, the rheological behavior of the materials was investigated via temperature sweep experiments. While the cross-over of the storage and loss modulus was not observed for all materials, a rather complex profile is observed (Fig. S18†). The dynamic behavior of the networks was characterized by stress-relaxation experiments in the viscoelastic region of the materials within a temperature range of 100–130 °C (Fig. 7). Initial experiments were performed on low Tg material CAN-a, displaying full and fast relaxation (less than 100 s) at 130 °C (Fig. 7A). However, consecutive experiments at lower temperatures showed an unanticipated increase in relaxation modulus. When the temperature was cycled back to 130 °C for a new experiment, a different relaxation pattern was observed with an increased storage modulus and relaxation time (Fig. S19a†). To examine the material behavior at a high temperature for a prolonged amount of time, a time sweep experiment was performed at 130 °C for 6 h and showed an increase in G′ over time, likely attributed to additional crosslinking (Fig. S19b†). As this undesired behavior was unique to CAN-a, we hypothesized some side reactions to occur from thioether and/or ether functions; however, we did not further investigate this phenomenon. We, therefore, focused our efforts on CAN-b and CAN-c that are characterized by fully aliphatic chemical structures.
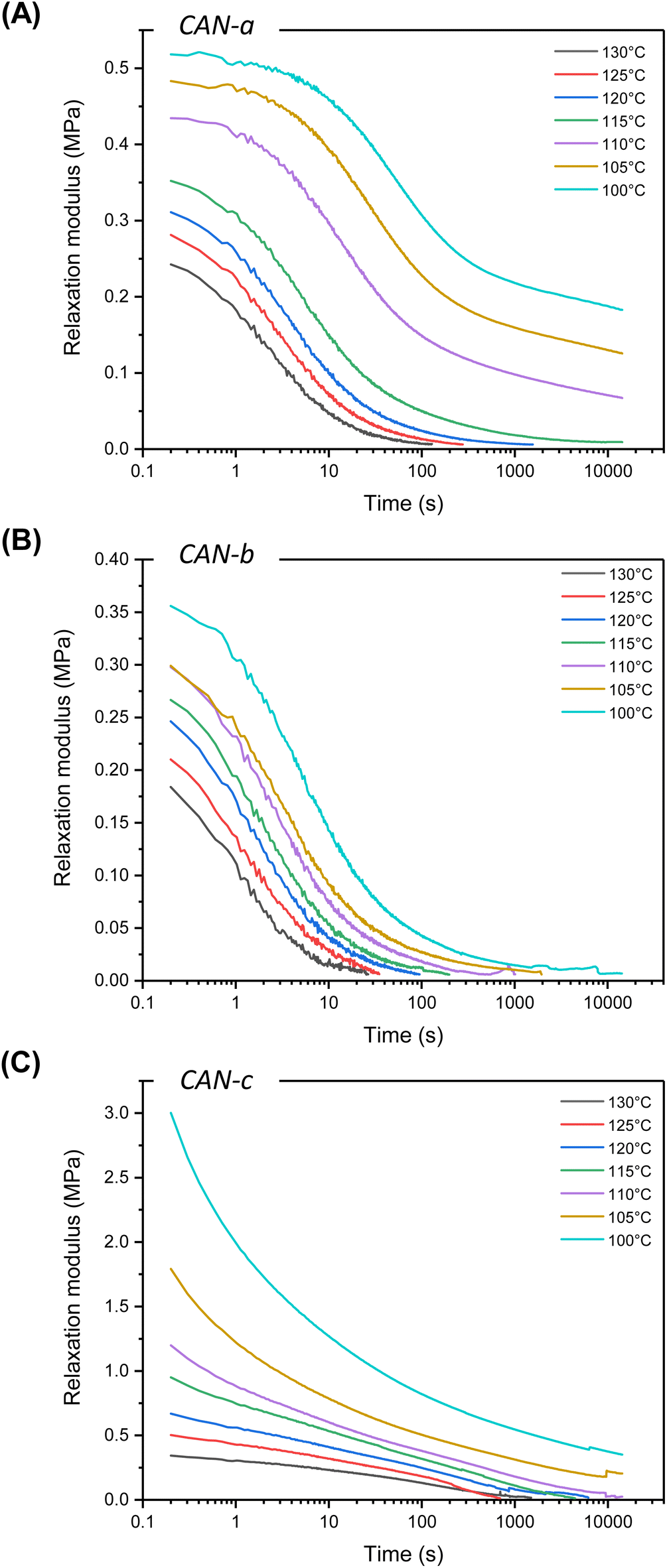 | ||
| Fig. 7 Non-normalized stress-relaxation plots of (A) CAN-a, (B) CAN-b and (C) CAN-c from 130 to 100 °C. | ||
CAN-b and CAN-c were both able to fully relax in the temperature range of 100–130 °C, highlighting the recyclability of the cyclic S,O-acetal materials (Fig. 7B and C). Surprisingly, despite their closely related chemical structures and similar Tgs, the stress-relaxation profiles of CAN-b and CAN-c exhibited divergent trends.
It is well-established that – while starting with a single Maxwell approximation to describe relaxation processes is generally a good starting point – reaching more accurate fitting of experimental data generally requires more sophisticated models such as the stretched exponential or the multi-element Maxwell models.42,61–65 For CAN-b, a single element Maxwell model poorly described the rheological data. (Fig. S20†) Applying a stretched single Maxwell element already enhanced the fitting quality. However, incorporating more elements (i.e. going to a description with two or, even better, three Maxwell elements) improved the fitting quality significantly (Fig. S20†). On the other hand, CAN-c presented a very particular relaxation profile, which did not seem to follow a Maxwell behavior. We were indeed unable to fit to any of the cited models, indicating that far more complex relaxation mechanisms might occur (Fig. S21†). As opposed to CAN-b, an abrupt loss in relaxation modulus was observed at low relaxation times and full relaxation was reached at much longer times. Nevertheless, a common feature to both systems was the striking temperature dependence of the relaxation modulus, readily decreasing at higher temperature. Non-normalized stress relaxation profiles indeed showed a decrease of initial modulus with temperature, indicating a decrease in cross-linking density. This behavior might be explained by dissociation events, occurring when the intermediate oxo-thiocarbonate 1 is formed. Once formed, 1 can either react with a free thiol to provide the cyclic S,O-acetal 2 or dissociate into αCC and a free thiol (Fig. 4). Although our calculations have shown the pathway to be of a much higher energy barrier, a direct dissociation of the S,O-cyclic acetal cannot be fully excluded in the high-temperature regime. A set of different associative and dissociative reactions, all leading to material relaxation, are thus expected to happen simultaneously and lead to stress relaxation profiles of high complexity.
We hypothesized that the unexpected divergence in stress relaxation profiles between CAN-b and CAN-c could originate from a complex set of reactions characterized by different kinetics over the temperature range. The incorporation of a spirocyclic cyclohexyl ring into CAN-c could indeed potentially influence the global energetics of different reactions compared to CAN-b (containing a simple linear linker). To investigate this hypothesis, we performed again DFT calculations on model molecules and compared all pathways from already studied αCC (with two methyl groups) to a spirocyclic derivative (with a cyclohexyl group). Although some changes in energetics were observed for elemental steps of both pathway 1 (association reaction) and pathway 2 (dissociation reaction), the change of the substituent did not seem to affect the overall calculated energy barrier in both cases and, consequently, the reaction rates.
Recycling and proof-of-concept coating application
To demonstrate the recyclability of cyclic S,O-acetal CAN materials, CAN-c was subjected to multiple shredding and compression molding cycles at 120 °C for 30 min under 3 metric tons of pressure. While Tg over the different reprocessing cycles was varying, the degradation temperatures only slightly altered over the cycles (Fig. S22 and S23 and Table S2†). Moreover, the FTIR spectra show no significant change in signals (Fig. 8).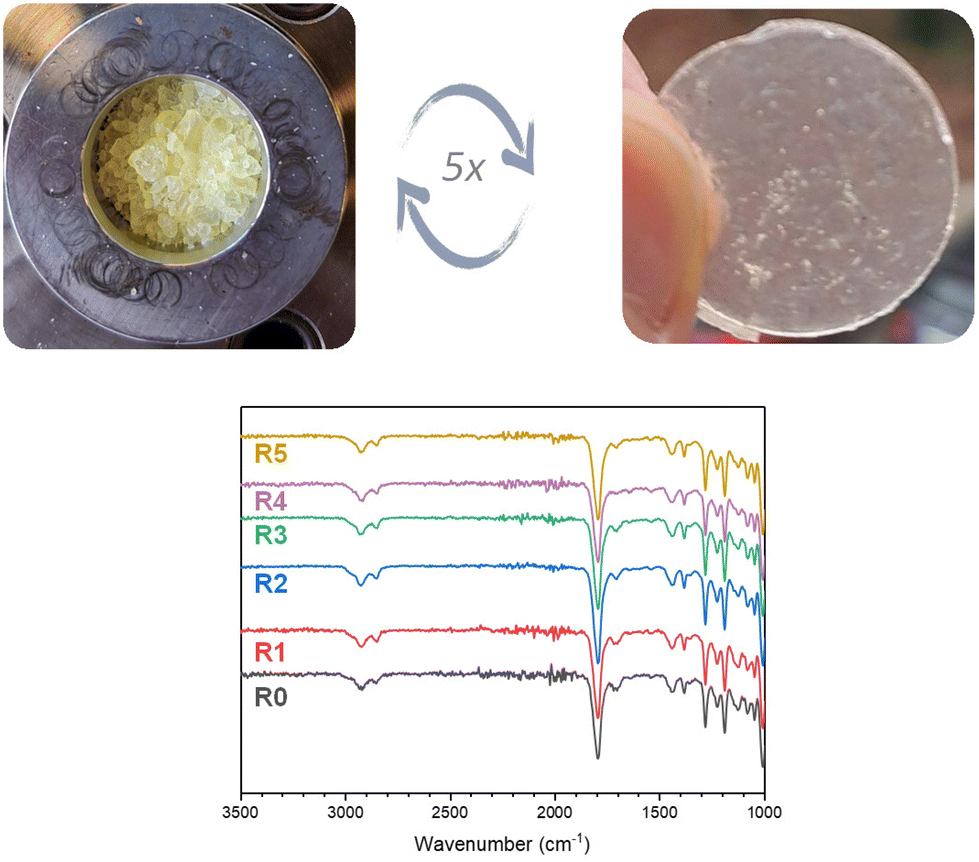 | ||
| Fig. 8 Physical appearance and FTIR spectra of the recycled CAN-c after reprocessing at 120 °C under a pressure of 3 metric ton for 30 min for multiple cycles. | ||
In the final part of this study, the focus was shifted towards an application involving coatings. The most important function of a coating is to protect a substrate and preferably consists of more resilient thermosetting materials. However, when damage occurs to the coating, its protective capacity is typically compromised, necessitating the application of a new coating. By integrating dynamic bonds in the coating, the inflicted damage could in principle be healed upon applying an appropriate trigger, thereby restoring the protecting capacity.66 To enable the dynamics in the exchange chemistry of S,O-acetals, free thiolates are essential. Interestingly, thiol groups also find utility in photochemical thiol–ene reactions, which are commonly used in coating applications.67 Moreover, besides the spatio-temporal control, the thiol–ene reaction is rather insensitive to oxygen and because of the mild orthogonal reaction conditions, this reaction can be carried out even with sensitive molecules.67,68
The dynamic S,O-acetal groups were introduced into the coating materials starting from a prepolymer, following a similar approach to that previously described for vinylogous urethane coatings.66 Polymer synthesis using bisαCC was earlier described;53 however, by slightly adjusting the stoichiometry (i.e. using a small excess of thiols, see the ESI† for details), a thiol end-capped prepolymer was obtained and characterized by NMR spectroscopy (Fig. S24–S26†). Subsequently, the thiol end groups were reacted photochemically with TVC using DMPA as the photoinitiator (Fig. 9A).
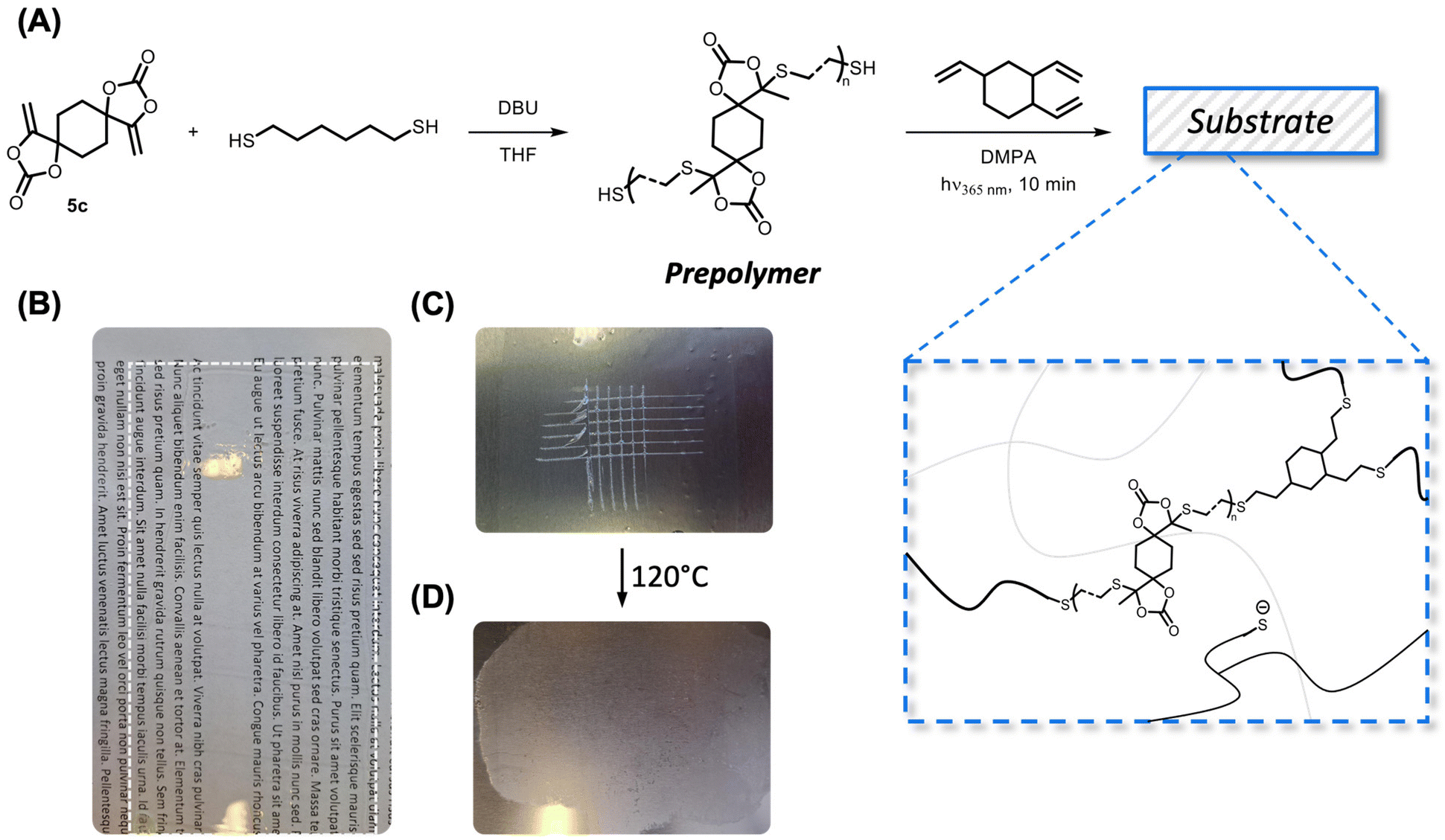 | ||
| Fig. 9 (A) General scheme for the preparation of S,O-acetal containing coatings starting from a prepolymer that was reacted photochemically in a thiol–ene reaction. (B) Excellent transparency is observed when the coating is applied on a glass substrate. (C) The film exhibits good adhesive properties when scratched on a metal surface and (D) scratches could be mended after the application of heat. | ||
Crosslinker 5c was selected to produce a coating with the desired Tg value of approximately 50 °C (Fig. S27†). FTIR spectra were recorded and compared between compound 5c, 1,6-hexanedithiol, the prepolymer, TVC and the coating (Fig. S28†). No significant differences between the FTIR spectrum from the coating and the spectrum from CAN-c were observed. The amount of TVC was calculated to ensure the availability of free thiolates in the final material. A transparent layer was obtained when coated on a glass substrate (Fig. 9B). The adhesive properties were tested on a metal surface, following the ASTM D3359-02 standard, which yielded a value of 3B (Fig. 9C). The scratched surface was thereafter subjected to heat (120 °C) for 30 min, which enabled the healing of the coating through the dynamic S,O-acetal exchange (Fig. 9D). While the scratches were visibly healed, the coating also seemed to flow excessively. This preliminary scratch healing test showed that the dynamics of S,O-acetals is indeed operational in the coatings but more formulations should be investigated to optimize the coating properties, which is beyond the scope of the current study.
Conclusion
In summary, we have demonstrated an unprecedented associative exchange in CO2-sourced cyclic S,O-acetals. The dynamic nature of these cyclic S,O-acetals was investigated through model molecule studies and reaction kinetics at different temperatures. Additionally, DFT calculations were performed to support the findings from the model studies and to elucidate the newly proposed reaction mechanism.CO2-sourced bifunctional alkylidene cyclic carbonates were combined with an excess of polythiols to provide covalent adaptable networks of the associative type. The dynamic nature of these materials was studied by rheology through stress relaxation experiments, revealing complex relaxation profiles that might originate from a set of concurrent exchange mechanisms. Nevertheless, compression molding allowed for the reprocessing of the materials, with good retention of their properties. The potential of these materials was finally assessed in a coating application with excellent transparency, adhesion to the substrates, and healing of scratches at elevated temperature.
Author contributions
The manuscript was written through the contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
S. M. acknowledges Ghent University for funding of his doctoral research (DOCT/009009). The authors from the University of Liège thank FNRS for the financial support in the frame of the CO2 Switch project under grant T.0075.20. C. D. is a F.R.S.-FNRS Research Director. Computational resources have been provided by the Consortium des Équipements de Calcul Intensif (CÉCI), funded by the Fonds de la Recherche Scientifique de Belgique (F.R.S.-FNRS) under grant no. 2.5020.11 and by the Walloon Region. Computational resources (Stevin Supercomputer Infrastructure) and services used in this work were provided by the VSC (Flemish Supercomputer Center), funded by Ghent University, FWO and the Flemish Government- department EWI. The NMR expertise centre (Ghent University) is also acknowledged for providing support and access to NMR infrastructure. 400 MHz used in this work has been funded by a grant/project of the Research Foundation Flanders (FWO I006920N) and the Bijzonder Onderzoeksfonds (BOF.BAS.2022.0023.01). Bernhard De Meyer and Jan Goeman are acknowledged for technical support. Vincent Scholiers is acknowledged for his help during the synthesis of the TVC-trithiol.References
- C. Jehanno, J. W. Alty, M. Roosen, S. De Meester, A. P. Dove, E. Y. X. Chen, F. A. Leibfarth and H. Sardon, Critical Advances and Future Opportunities in Upcycling Commodity Polymers, Nature, 2022, 603(7903), 803–814 CrossRef CAS PubMed.
- R. Geyer, J. R. Jambeck and K. L. Law, Production, Use, and Fate of All Plastics Ever Made, Sci. Adv., 2017, 3(7), 25–29 Search PubMed.
- F. Vidal, E. R. van der Marel, R. W. F. Kerr, C. McElroy, N. Schroeder, C. Mitchell, G. Rosetto, T. T. D. Chen, R. M. Bailey, C. Hepburn, C. Redgwell and C. K. Williams, Designing a Circular Carbon and Plastics Economy for a Sustainable Future, Nature, 2024, 626(7997), 45–57 CrossRef CAS PubMed.
- W. Denissen, J. M. Winne and F. E. Du Prez, Vitrimers: Permanent Organic Networks with Glass-like Fluidity, Chem. Sci., 2016, 7(1), 30–38 RSC.
- J. M. Winne, L. Leibler and F. E. Du Prez, Dynamic Covalent Chemistry in Polymer Networks: A Mechanistic Perspective, Polym. Chem., 2019, 10(45), 6091–6108 RSC.
- W. Zou, J. Dong, Y. Luo, Q. Zhao and T. Xie, Dynamic Covalent Polymer Networks: From Old Chemistry to Modern Day Innovations, Adv. Mater., 2017, 29(14), 1606100 CrossRef PubMed.
- P. Chakma and D. Konkolewicz, Dynamic Covalent Bonds in Polymeric Materials, Angew. Chem., Int. Ed., 2019, 58(29), 9682–9695 CrossRef CAS PubMed.
- S. Huang, X. Kong, Y. Xiong, X. Zhang, H. Chen, W. Jiang, Y. Niu, W. Xu and C. Ren, An Overview of Dynamic Covalent Bonds in Polymer Material and Their Applications, Eur. Polym. J., 2020, 141, 110094 CrossRef CAS.
- D. Montarnal, M. Capelot, F. Tournilhac and L. Leibler, Silica-Like Malleable Materials from Permanent Organic Networks, Science, 2011, 334(6058), 965–968 CrossRef CAS PubMed.
- M. Capelot, D. Montarnal, F. Tournilhac and L. Leibler, Metal-Catalyzed Transesterification for Healing and Assembling of Thermosets, J. Am. Chem. Soc., 2012, 134(18), 7664–7667 CrossRef CAS PubMed.
- M. Wrighton, The Photochemistry of Metal Carbonyls, 1974, vol. 74 Search PubMed.
- J. L. Self, N. D. Dolinski, M. S. Zayas, J. Read de Alaniz and C. M. Bates, Brønsted-Acid-Catalyzed Exchange in Polyester Dynamic Covalent Networks, ACS Macro Lett., 2018, 7(7), 817–821 CrossRef CAS PubMed.
- S. Chappuis, P. Edera, M. Cloitre and F. Tournilhac, Enriching an Exchangeable Network with One of Its Components: The Key to High- TgEpoxy Vitrimers with Accelerated Relaxation, Macromolecules, 2022, 55(16), 6982–6991 CrossRef CAS.
- M. Delahaye, J. M. Winne and F. E. Du Prez, Internal Catalysis in Covalent Adaptable Networks: Phthalate Monoester Transesterification As a Versatile Dynamic Cross-Linking Chemistry, J. Am. Chem. Soc., 2019, 141(38), 15277–15287 CrossRef CAS PubMed.
- M. Delahaye, F. Tanini, J. O. Holloway, J. M. Winne and F. E. Du Prez, Double Neighbouring Group Participation for Ultrafast Exchange in Phthalate Monoester Networks, Polym. Chem., 2020, 11(32), 5207–5215 RSC.
- M. L. Bender, General Acid-Base Catalysis in the Intramolecular Hydrolysis of Phthalamic Acid, J. Am. Chem. Soc., 1957, 79 (5), 1258–1259 CrossRef.
- E. Delebecq, J. Pascault, B. Boutevin and F. Ganachaud, On the Versatility of Urethane/Urea Bonds: Reversibility, Blocked Isocyanate, and Non-Isocyanate Polyurethane, Chem. Rev., 2013, 113(1), 80–118 CrossRef CAS PubMed.
- P. C. Colodny and A. V. Tobolsky, Chemorheological Study of Polyurethan Elastomers 1, J. Am. Chem. Soc., 1957, 79(16), 4320–4323 CrossRef CAS.
- J. A. Offenbach and A. V. Tobolsky, Chemical Relaxation of Stress in Polyurethane Elastomers, J. Colloid Sci., 1956, 11(1), 39–47 CrossRef CAS.
- F. Van Lijsebetten, Y. Spiesschaert, J. M. Winne and F. E. Du Prez, Reprocessing of Covalent Adaptable Polyamide Networks through Internal Catalysis and Ring-Size Effects, J. Am. Chem. Soc., 2021, 143(38), 15834–15844 CrossRef CAS PubMed.
- J. M. Craven, Cross-Linked Thermally Reversible Polymers Produced from Condensation Polymers with Pendant Furan Groups Cross-Linked with, Maleimides, 1969 Search PubMed.
- F. Orozco, J. Li, U. Ezekiel, Z. Niyazov, L. Floyd, G. M. R. Lima, J. G. M. Winkelman, I. Moreno-Villoslada, F. Picchioni and R. K. Bose, Diels-Alder-Based Thermo-Reversibly Crosslinked Polymers: Interplay of Crosslinking Density, Network Mobility, Kinetics and Stereoisomerism, Eur. Polym. J., 2020, 135, 109882 CrossRef CAS.
- A. G. Orrillo and R. L. E. Furlan, Sulfur in Dynamic Covalent Chemistry, Angew. Chem., Int. Ed., 2022, 61(26), e202201168 CrossRef CAS PubMed.
- R. W. Clarke, T. Sandmeier, K. A. Franklin, D. Reich, X. Zhang, N. Vengallur, T. K. Patra, R. J. Tannenbaum, S. Adhikari, S. K. Kumar, T. Rovis and E. Y. X. Chen, Dynamic Crosslinking Compatibilizes Immiscible Mixed Plastics, Nature, 2023, 616(7958), 731–739 CrossRef CAS PubMed.
- A. Ruiz De Luzuriaga, R. Martin, N. Markaide, A. Rekondo, G. Cabañero, J. Rodríguez and I. Odriozola, Epoxy Resin with Exchangeable Disulfide Crosslinks to Obtain Reprocessable, Repairable and Recyclable Fiber-Reinforced Thermoset Composites, Mater. Horiz., 2016, 3(3), 241–247 RSC.
- J. Canadell, H. Goossens and B. Klumperman, Self-Healing Materials Based on Disulfide Links, Macromolecules, 2011, 44(8), 2536–2541 CrossRef CAS.
- L. Imbernon, E. K. Oikonomou, S. Norvez and L. Leibler, Chemically Crosslinked yet Reprocessable Epoxidized Natural Rubber via Thermo-Activated Disulfide Rearrangements, Polym. Chem., 2015, 6(23), 4271–4278 RSC.
- M. Podgórski, S. Mavila, S. Huang, N. Spurgin, J. Sinha and C. N. Bowman, Thiol–Anhydride Dynamic Reversible Networks, Angew. Chem., Int. Ed., 2020, 59(24), 9345–9349 CrossRef PubMed.
- D. Van Ooteghem, R. Deveux and E. J. Goethals, Study of the Reaction of Sulfides and Sulfonium Salts, J. Sulphur Chem., 1973, 8(1), 31–35 CAS.
- B. Hendriks, J. Waelkens, J. M. Winne and F. E. Du Prez, Poly(Thioether) Vitrimers via Transalkylation of Trialkylsulfonium Salts, ACS Macro Lett., 2017, 6(9), 930–934 CrossRef CAS PubMed.
- V. Scholiers, B. Hendriks, S. Maes, T. Debsharma, J. M. Winne and F. E. Du Prez, Trialkylsulfonium-Based Reprocessable Polyurethane Thermosets, Macromolecules, 2023, 56(23), 9559–9569 CrossRef CAS.
- N. De Alwis Watuthanthrige, P. Chakma and D. Konkolewicz, Designing Dynamic Materials from Dynamic Bonds to Macromolecular Architecture, Trends Chem., 2021, 3(3), 231–247 CrossRef CAS.
- Q. Li, S. Ma, S. Wang, Y. Liu, M. A. Taher, B. Wang, K. Huang, X. Xu, Y. Han and J. Zhu, Green and Facile Preparation of Readily Dual-Recyclable Thermosetting Polymers with Superior Stability Based on Asymmetric Acetal, Macromolecules, 2020, 53(4), 1474–1485 CrossRef CAS.
- Q. Li, S. Ma, P. Li, B. Wang, Z. Yu, H. Feng, Y. Liu and J. Zhu, Fast Reprocessing of Acetal Covalent Adaptable Networks with High Performance Enabled by Neighboring Group Participation, Macromolecules, 2021, 54(18), 8423–8434 CrossRef CAS.
- N. Van Herck, D. Maes, K. Unal, M. Guerre, J. M. Winne and F. E. Du Prez, Covalent Adaptable Networks with Tunable Exchange Rates Based on Reversible Thiol–Yne Cross–Linking, Angew. Chem., Int. Ed., 2020, 59(9), 3609–3617 CrossRef CAS PubMed.
- J. S. A. Ishibashi and J. A. Kalow, Vitrimeric Silicone Elastomers Enabled by Dynamic Meldrum's Acid-Derived Cross-Links, ACS Macro Lett., 2018, 7(4), 482–486 CrossRef CAS PubMed.
- H. Zeng, Z. Tang, Y. Duan, S. Wu and B. Guo, Recyclable Crosslinked Elastomer Based on Dynamic Dithioacetals, Polymer, 2021, 229, 124007 CrossRef CAS.
- Q. Li, S. Ma, S. Wang, W. Yuan, X. Xu, B. Wang, K. Huang and J. Zhu, Facile Catalyst-Free Synthesis, Exchanging, and Hydrolysis of an Acetal Motif for Dynamic Covalent Networks, J. Mater. Chem. A, 2019, 7(30), 18039–18049 RSC.
- Q. Li, S. Ma, N. Lu, J. Qiu, J. Ye, Y. Liu, S. Wang, Y. Han, B. Wang, X. Xu, H. Feng and J. Zhu, Concurrent Thiol–Ene Competitive Reactions Provide Reprocessable, Degradable and Creep-Resistant Dynamic–Permanent Hybrid Covalent Networks, Green Chem., 2020, 22(22), 7769–7777 RSC.
- S. Yu, S. Wu, C. Zhang, Z. Tang, Y. Luo, B. Guo and L. Zhang, Catalyst-Free Metathesis of Cyclic Acetals and Spirocyclic Acetal Covalent Adaptable Networks, ACS Macro Lett., 2020, 9(8), 1143–1148 CrossRef CAS PubMed.
- Z. M. Png, J. Zheng, S. Kamarulzaman, S. Wang, Z. Li and S. S. Goh, Fully Biomass-Derived Vitrimeric Material with Water-Mediated Recyclability and Monomer Recovery, Green Chem., 2022, 24(15), 5978–5986 RSC.
- T. Habets, G. Seychal, M. Caliari, J.-M. Raquez, H. Sardon, B. Grignard and C. Detrembleur, Covalent Adaptable Networks through Dynamic N, S -Acetal Chemistry: Toward Recyclable CO 2 -Based Thermosets, J. Am. Chem. Soc., 2023, 145(46), 25450–25462 CrossRef CAS PubMed.
- B. M. El-Zaatari, J. S. A. Ishibashi and J. A. Kalow, Cross-Linker Control of Vitrimer Flow, Polym. Chem., 2020, 11(33), 5339–5345 RSC.
- V. Zhang, J. V. Accardo, I. Kevlishvili, E. F. Woods, S. J. Chapman, C. T. Eckdahl, C. L. Stern, H. J. Kulik and J. A. Kalow, Tailoring Dynamic Hydrogels by Controlling Associative Exchange Rates, Chem, 2023, 9(8), 2298–2317 CAS.
- C. Hepburn, E. Adlen, J. Beddington, E. A. Carter, S. Fuss, N. Mac Dowell, J. C. Minx, P. Smith and C. K. Williams, The Technological and Economic Prospects for CO2 Utilization and Removal, Nature, 2019, 575(7781), 87–97 CrossRef CAS PubMed.
- B. Song, A. Qin and B. Z. Tang, Syntheses, Properties, and Applications of CO2-Based Functional Polymers, Cell Rep. Phys. Sci., 2022, 3(2), 100719 CrossRef CAS.
- B. Grignard, S. Gennen, C. Jérôme, A. W. Kleij and C. Detrembleur, Advances in the Use of CO2 as a Renewable Feedstock for the Synthesis of Polymers, Chem. Soc. Rev., 2019, 48 (16), 4466–4514 RSC.
- S. Gennen, B. Grignard, T. Tassaing, C. Jérôme and C. Detrembleur, CO2-Sourced α-Alkylidene Cyclic Carbonates: A Step Forward in the Quest for Functional Regioregular Poly(Urethane)s and Poly(Carbonate)S, Angew. Chem., Int. Ed., 2017, 56(35), 10394–10398 CrossRef CAS PubMed.
- C. Ngassam Tounzoua, B. Grignard and C. Detrembleur, Exovinylene Cyclic Carbonates: Multifaceted CO2-Based Building Blocks for Modern Chemistry and Polymer Science, Angew. Chem., Int. Ed., 2022, 61(22), e202116066 CrossRef CAS PubMed.
- T. Habets, F. Siragusa, B. Grignard and C. Detrembleur, Advancing the Synthesis of Isocyanate-Free Poly(Oxazolidones)s: Scope and Limitations, Macromolecules, 2020, 53(15), 6396–6408 CrossRef CAS.
- A. R. Wong, M. Barrera, A. Pal and J. R. Lamb, Improved Characterization of Polyoxazolidinones by Incorporating Solubilizing Side Chains, Macromolecules, 2022, 55(24), 11006–11012 CrossRef CAS.
- F. Siragusa, E. Van Den Broeck, C. Ocando, A. J. Müller, G. De Smet, B. U. W. Maes, J. De Winter, V. Van Speybroeck, B. Grignard and C. Detrembleur, Access to Biorenewable and CO 2 -Based Polycarbonates from Exovinylene Cyclic Carbonates, ACS Sustainable Chem. Eng., 2021, 9(4), 1714–1728 CrossRef CAS.
- F. Ouhib, B. Grignard, E. Van Den Broeck, A. Luxen, K. Robeyns, V. Van Speybroeck, C. Jerome and C. Detrembleur, A Switchable Domino Process for the Construction of Novel CO 2 –Sourced Sulfur–Containing Building Blocks and Polymers, Angew. Chem., Int. Ed., 2019, 58(34), 11768–11773 CrossRef CAS PubMed.
- S. Dabral, U. Licht, P. Rudolf, G. Bollmann, A. S. K. Hashmi and T. Schaub, Synthesis and Polymerisation of α-Alkylidene Cyclic Carbonates from Carbon Dioxide, Epoxides and the Primary Propargylic Alcohol 1,4-Butynediol, Green Chem., 2020, 22(5), 1553–1558 RSC.
- Y. F. Zhang, W. M. Lai, S. Xie, H. Zhou and X. B. Lu, Facile Synthesis Structure and Properties of CO2-Sourced Poly(Thioether-: Co -Carbonate)s Containing Acetyl Pendants via Thio-Ene Click Polymerization, Polym. Chem., 2022, 13(2), 201–208 RSC.
- T. Habets, F. Siragusa, A. J. Müller, Q. Grossman, D. Ruffoni, B. Grignard and C. Detrembleur, Facile Construction of Functional Poly(Monothiocarbonate) Copolymers under Mild Operating Conditions, Polym. Chem., 2022, 13(21), 3076–3090 RSC.
- F. Ouhib, L. Meabe, A. Mahmoud, N. Eshraghi, B. Grignard, J. M. Thomassin, A. Aqil, F. Boschini, C. Jérôme, D. Mecerreyes and C. Detrembleur, CO2-Sourced Polycarbonates as Solid Electrolytes for Room Temperature Operating Lithium Batteries, J. Mater. Chem. A, 2019, 7(16), 9844–9853 RSC.
- T. Habets, J. L. Olmedo-Martínez, R. del Olmo, B. Grignard, D. Mecerreyes and C. Detrembleur, Facile Access to CO 2 –Sourced Polythiocarbonate Dynamic Networks And Their Potential As Solid–State Electrolytes For Lithium Metal Batteries, ChemSusChem, 2023, 16(14), e202300225 CrossRef CAS PubMed.
- F. Siragusa, T. Habets, R. Méreau, G. Evano, B. Grignard and C. Detrembleur, Catalyst-Free Approach for the Degradation of Bio- and CO2-Sourced Polycarbonates: A Step toward a Circular Plastic Economy, ACS Sustainable Chem. Eng., 2022, 10(27), 8863–8875 CrossRef CAS.
- F. Siragusa, J. Demarteau, T. Habets, I. Olazabal, K. Robeyns, G. Evano, R. Mereau, T. Tassaing, B. Grignard, H. Sardon and C. Detrembleur, Unifying Step-Growth Polymerization and On-Demand Cascade Ring-Closure Depolymerization via Polymer Skeletal Editing, Macromolecules, 2022, 55(11), 4637–4646 CrossRef CAS.
- F. Van Lijsebetten, K. De Bruycker, E. Van Ruymbeke, J. M. Winne and F. E. Du Prez, Characterising Different Molecular Landscapes in Dynamic Covalent Networks, Chem. Sci., 2022, 13(43), 12865–12875 RSC.
- S. Maes, F. Van Lijsebetten, J. M. Winne and F. E. Du Prez, N -Sulfonyl Urethanes to Design Polyurethane Networks with Temperature-Controlled Dynamicity, Macromolecules, 2023, 56(5), 1934–1944 CrossRef CAS.
- S. K. Schoustra, T. Groeneveld and M. M. J. Smulders, The Effect of Polarity on the Molecular Exchange Dynamics in Imine-Based Covalent Adaptable Networks, Polym. Chem., 2021, 12(11), 1635–1642 RSC.
- M. L. Martins, X. Zhao, Z. Demchuk, J. Luo, G. P. Carden, G. Toleutay and A. P. Sokolov, Viscoelasticity of Polymers with Dynamic Covalent Bonds, Concepts and Misconceptions, Macromolecules, 2023, 56(21), 8688–8696 CrossRef CAS.
- V. Zhang, B. Kang, J. V. Accardo and J. A. Kalow, Structure–Reactivity–Property Relationships in Covalent Adaptable Networks, J. Am. Chem. Soc., 2022, 144(49), 22358–22377 CrossRef CAS PubMed.
- F. Van Lijsebetten, S. Engelen, E. Bauters, W. Van Vooren, M. M. J. Smulders and F. E. Du Prez, Recyclable Vitrimer Epoxy Coatings for Durable Protection, Eur. Polym. J., 2022, 176, 111426 CrossRef CAS.
- C. Resetco, B. Hendriks, N. Badi and F. Du Prez, Thiol-Ene Chemistry for Polymer Coatings and Surface Modification-Building in Sustainability and Performance, Mater. Horiz., 2017, 4(6), 1041–1053 RSC.
- B. Hendriks, O. van den Berg and F. E. Du Prez, Urethane Polythioether Self-Crosslinking Resins, Prog. Org. Coat., 2019, 136, 105215 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Details about instrumentation, synthesis and experimental procedures, 1H- and 13C NMR spectra, computational details, IR spectra, DSC and TGA thermograms, stress-relaxation data. See DOI: https://doi.org/10.1039/d4py00359d |
| ‡ These authors contributed equally to the work. |
| This journal is © The Royal Society of Chemistry 2024 |