
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBridging of poly(acetylene)s and PEG-modified poly(olefin)s through ring-opening metathesis copolymerization (ROMCP)†
Santhosh
Kumar Podiyanachari
a,
Maciej
Barłóg
b,
Mohammed
Al-Hashimi
 *b and
Hassan S.
Bazzi
*ac
*b and
Hassan S.
Bazzi
*ac
aDivision of Arts and Science, Texas A&M University at Qatar, P.O. Box 23874, Doha, Qatar. E-mail: hassan.bazzi@qatar.tamu.edu
bDepartment of Chemical Engineering, Texas A&M University at Qatar, P.O. Box 23874, Doha, Qatar. E-mail: mohammed.al-hashimi@qatar.tamu.edu
cDepartment of Materials Science & Engineering, Texas A&M University, 209 Reed MacDonald Building, College Station, TX 77843-3003, USA
First published on 9th May 2024
Abstract
Amphiphilic copolymers of highly conjugated poly(acetylene)s and poly(ethylene glycol)-functionalized perylene diimide (PEG750–PDI)-incorporating poly(olefin)s have been synthesized via tandem cyclopolymerization and ring-opening metathesis copolymerization (ROMCP) methodologies. Both di- and tri-block copolymers were prepared from 1,6-heptadiyne and oxanorbornene imide-based cycloolefin monomers using ruthenium-based alkylidene initiators. The relative atomic weight percentages of both di- and tri-block copolymers were estimated using X-ray photoelectron spectroscopy (XPS) analysis. Photophysical properties of both copolymers have been explained based on both UV-Vis and fluorescence spectroscopic analysis shedding more light on their different stages of organization as well as the π–π stacking interactions of the PEG750-incorporating perylene cores in aqueous solutions. Furthermore, these investigations elucidate the modulation of the photophysical properties and H-type aggregation processes of polymers in aqueous solutions that lead to the formation of PEG750–PDI-derived amphiphiles. Copolymeric surface analysis, segmental patterns and film morphology were examined by atomic force microscopy (AFM) revealing globular or spherical morphological features enhanced by various functional groups present in the polymer bulk. In addition, the formation of these spherical morphologies was further visualized in the thin film cross-sections of both di-and tri-block polymers by scanning electron microscopy (SEM) to confirm the surface morphologies determined by AFM analysis of both polymeric materials.
Introduction
Metathesis polymerization is a well-established technique used for synthesizing conjugated poly(acetylene)s and poly(ene)s from terminal dialkyne monomers.1,2 Conjugated poly(acetylene)s acquired through cyclopolymerization of functionalized 1,6-heptadiyne monomers using metathesis catalysts exhibit cyclic structures along the conjugated backbone, particularly those derived from oxidative polymerization. This unique structural feature provides enhanced stability and excellent processability of the resulting polymers.3,4 The repeat units of these polymers can consist of either five-membered rings (via α-addition) or six-membered rings (via β-addition), or a combination of both, depending on the alkyne unit's insertion mode into the metal–alkylidene initiator.1,2 This versatility in ring formation imparts a tailored and tunable aspect to the resulting polymer structure, enabling fine control over its properties. Among various catalytic systems, Schrock and Buchmeiser reported pioneering results in the cyclopolymerization of various terminal dialkynes, such as 1,6-heptadiynes, using well-defined [Mo]- or [Ru]-based metathesis initiators.2,3,5–7 Later, many cyclopolymerization-derived conjugated poly(acetylene)s were synthesized and have been well utilised for various conducting applications.8–11Choi and co-workers also explored different Grubbs’ catalysts for the cyclopolymerization of different functionalized terminal di(alkyne) monomers. Their investigation led to the synthesis of the first example of β-selective cyclopolymerization-derived poly(ene)s, characterized by exclusive incorporation of six-membered ring structures along the polymer backbone.12–20 Those conjugated polymers obtained via cyclopolymerization have found extensive use in the fabrication of nanosheets, nanoparticles, and various self-assembly applications.21–26 Furthermore, Choi has extended the application of cyclopolymerization, by reporting the synthesis of diblock copolymers, combining cyclopolymerization with ring-opening metathesis (ROM). Copolymers obtained via cyclopolymerization and ROMCP have been specifically employed in the preparation of nanostructures and the synthesis of polyolefin-linked conjugated poly(ene)s under controlled and living polymerization.16,27–29 In addition, such random and diblock copolymers containing conjugated polymer backbones with various functionalities have been well-studied in various self-assembly applications.30
However, there are a limited number of reports on the synthesis of such copolymers based on poly(ene)s and poly(olefin)s via tandem cyclopolymerization and ROMCP in a unified, one-step metathesis polymerization process. Our research group has actively contributed to this field, investigating the alkyne-insertion metathesis polymerization31 and ROMP reactivities of diverse functional and nonfunctional cycloolefin monomers using Grubbs or Hoveyda–Grubbs type [Ru]–alkylidene initiators. This approach has led to the development of a new class of structurally well-defined poly(olefin)s.32–37 Our efforts have yielded a series of functional poly(olefin)s based on polypentenamers,36,38 poly(vinyl alcohol)s,34,38 poly(norbornene)s32,33,39,40 and rhodamine-based poly(olefin)s showcasing versatility in material design for various biological applications.41,42 We recently reported the synthesis of highly conjugated bay-functional perylene diimide chromophore incorporating poly(oxanorbornene imide)s via ROMP,43 by investigating their hyperbranched propensity behavior.44 Based upon our findings, ROMP-derived poly(olefin)s containing PDI-grafted alkoxy-functionalized poly(oxanorbornene imide)s are promising candidates for the preparation of a diverse range of novel polymeric materials tailored for applications in organic electronics.43,44
In this work, we describe a tandem approach for the synthesis of amphiphilic copolymers, combining conjugated poly(acetylene)s and poly(ethylene glycol)-incorporating perylene diimide (PEG750–PDI)-grafted poly(olefin)s via cyclopolymerization and ROMCP. The copolymers derived from PEG–PDI have attracted considerable attention, particularly due to their unique structural and dynamic characteristics in diluted aqueous solutions.45–49 Investigations by various research groups have revealed the amphiphilic nature of these compounds and their self-assembly behavior, driven by a delicate interplay between hydrophobic and π–π interactions. The capacity to manipulate their aggregation behavior through molecular engineering, temperature variation, and solvent composition has paved the way for the incorporation of these systems into a diverse range of applications, including catalysis, membranes, nanotubes, sensing and bioimaging agents.50–54 Furthermore, the photoluminescence properties of this class of materials, such as the quantum yield (ϕPL) of PDI derivatives, often face limitations, like aggregation caused quenching (ACQ) at high concentration in solution or in the solid film state. Tuning the fluorescent properties of the PDI unit from ACQ to aggregation induced emission (AIE) has been typically achieved via molecular engineering of both the amide and bay functionality with bulky groups designed to control the π–π stacking and aggregation behavior.55–57 Our tandem approach offers a versatile platform for accessing a range of tunable PEG750-incorporating chromophores within the non-conjugated polymer matrix, serving as a copolymer counterpart of the cyclopolymerization-derived conjugated poly(acetylene). Our findings demonstrate that tandem cyclopolymerization, followed by ROMP, yields copolymers with diverse properties and applications including electrical conductivity, bioimaging and photoluminescence. This methodology provides a promising route for the preparation of novel polymeric materials with tailored properties.
To the best of our knowledge, this work constitutes the first example of incorporating perylene cores into a non-conjugated polymer, with optical properties modulated by the poly(ene) block composition and the polarity of the medium. This innovative approach to control PDI aggregation through the strategic integration of perylene cores into non-conjugated polymers, coupled with the tunable and reversible nature of the interactions, introduces new avenues for the advancement of this class of systems. The modular composition of the copolymer, allowing for tailored adjustments, presents a versatile platform with potential applications across various fields.
Results and discussion
Polymer synthesis
The molecular structures of all the monomers (M1–M3) and the ruthenium–alkylidene initiator [Ru] are outlined in Scheme 1A. The dialkyne-terminal monomer dipropargyl diethyl malonate (DEDPM) M1 was synthesized following a previously reported method.58 The cycloolefin monomers PEG750–PDI-based oxanorbornene imide monomer M259,60 (Fig. S1 and S2, ESI†) and N-alkyl substituted oxa-norbornene imide monomer M361 were synthesized following a modified procedure based on prior reports.59 Poly(acetylene) P1 was derived from cyclopolymerization of 1,6-hexadiyne M1 using a trifluoroacetate modified Hoveyda–Grubbs type 2nd generation catalyst,62 resulting in polymer P1 as a dark-red solid.62 Additionally, PDI-based oxa-norbornene imide M2 underwent polymerization using the same initiator, yielding the ROMP-derived P2 as a dark red solid. To extend the cyclopolymerization by incorporating both hydrophilic and hydrophobic units into the polymers, diblock copolymer P3 was synthesized via cyclopolymerization of M1, followed by the addition of PEG750-derived PDI oxa-norbornene imide monomer M2via ROMCP. Copolymerization was carried out with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of both monomers using a 2 mol% catalyst loading, and the monomer conversion was monitored by 1H NMR spectroscopy. After terminating the polymerization using ethyl vinyl ether, the resulting polymer was purified via precipitation of the concentrated polymer solution in n-pentane. After multiple reprecipitations, the pure copolymer P3 was isolated as a dark-red solid in excellent yield (98%) (Scheme 1B). Similarly, triblock copolymer P4 was synthesized in a 1:1:1 ratio via cyclopolymerization with M1, followed by ROMCPs of M2 and M3 to yield a dark-red solid in excellent yield (96%) (Scheme 1B). In addition, random versions of diblock polymer P5 and triblock polymer P6 were synthesized using the same monomer ratio and catalyst loading (Scheme 1C). Both P5 and P6 were isolated as dark red solids in excellent yields (95% isolated yield for P5 and 97% for P6).
:1 ratio of both monomers using a 2 mol% catalyst loading, and the monomer conversion was monitored by 1H NMR spectroscopy. After terminating the polymerization using ethyl vinyl ether, the resulting polymer was purified via precipitation of the concentrated polymer solution in n-pentane. After multiple reprecipitations, the pure copolymer P3 was isolated as a dark-red solid in excellent yield (98%) (Scheme 1B). Similarly, triblock copolymer P4 was synthesized in a 1:1:1 ratio via cyclopolymerization with M1, followed by ROMCPs of M2 and M3 to yield a dark-red solid in excellent yield (96%) (Scheme 1B). In addition, random versions of diblock polymer P5 and triblock polymer P6 were synthesized using the same monomer ratio and catalyst loading (Scheme 1C). Both P5 and P6 were isolated as dark red solids in excellent yields (95% isolated yield for P5 and 97% for P6).
 | ||
| Scheme 1 (A) Structure of monomers M1–M3 and trifluoroacetate-modified Hoveyda–Grubbs type 2nd generation catalyst [Ru], (B) synthesis of diblock copolymer P3, and triblock copolymer P4, (C) synthesis of random binary copolymer P5 and ternary copolymer P6. | ||
The copolymer composition and structural features of copolymers P3–P6 were characterized by 1H NMR spectroscopy. The 1H NMR spectrum of copolymer P3 displays a broad multiplet together with a singlet signal ranging from δ = 6.6–6.9 ppm, indicating olefinic protons within the conjugated backbone of poly(acetylene) (P1), resulting from the cyclopolymerization of the dialkyne monomer M1 and the olefinic protons of the poly(oxanorbornene imide) copolymer counterpart derived from PEG-containing oxanorbornene imide monomer M2. Triblock copolymer P4 shows an additional olefinic proton signal along with the abovementioned peaks for P3 as a broad singlet signal at 6.0 ppm representing the poly(oxanorbornene imide) copolymer counterpart derived from N-alkyl substituted oxa-norbornene imide monomer M3 (Fig. S3–S6, ESI†). In the 1H NMR spectra of random diblock P5 and triblock P6 copolymers, olefinic proton signals appeared as heterodyad peaks around 6.9 ppm and 5.9 ppm indicating olefinic proton signals of conjugated backbones and poly(oxanorbornene imide)s (Fig. S7–S10, ESI†).
The relative atomic weight percentages of the constituent elements in copolymers P3 and P4 were further characterized by XPS analysis. In the XPS spectra, characteristic signals corresponding to carbon, nitrogen and oxygen atoms within the copolymer composition were observed, with corresponding binding energies (BE) at 282 eV for carbon, 397 eV for nitrogen, and 529 eV for oxygen, respectively (Fig. S11†). These binding energy values were consistent for carbon, nitrogen, and oxygen in both diblock P3 and triblock P4 polymers. The relative atomic weight percentages estimated from the peak areas of the carbon, nitrogen and oxygen signals were determined for diblock copolymer P3 as 74.79% for carbon, 1.19% for nitrogen, and 20.68% for oxygen. In the case of the triblock copolymer P4, the relative atomic weight percentages were estimated as 77.33%, 1.99%, and 24.02%, respectively, for carbon, nitrogen, and oxygen. The slight excess in the relative weight percentages of carbon, nitrogen, and oxygen in the case of the triblock copolymer further indicates the presence of different C, N, and O proportions in the triblock copolymer P4. This XPS analysis provides additional confirmation of the distinct atomic weight percentages of the di- and tri-block copolymers.
Molecular weights and thermal stability of the polymers
The number average molecular weights (Mn) of the polymers P3–P6 were measured using size-exclusion chromatography (SEC) in chlorobenzene. By SEC analysis, we aimed to investigate Mn as well as the amount of diblock and triblock copolymers obtained via tandem metathesis polymerization. The obtained copolymers displayed relatively high molecular weights with slightly elevated dispersities (Đ), i.e., Mn = 62.0 kDa with Đ of 1.5 for P3, Mn = 79.0 kDa with Đ of 2.1 for P4, Mn = 74.3 kDa with Đ of 1.9 for P5 and Mn = 88.3 kDa with Đ of 2.4 for P6 (Fig. S12, ESI†). These high Mn values demonstrate the formation of copolymers via combining both conjugated poly(acetylene) backbones and poly(olefin)s in the case of both di- and tri-block polymers. The SEC traces of the polymers displayed a slightly bimodal distribution, and this is possibly due to partial catalyst deactivation that should lead to copolymer randomization during the formation of the di- and tri-block polymers. However, the larger values of Đ observed for these polymers can be attributed to chain coupling, leading to the formation of significant chain transfer during the secondary metathesis polymerization.63,64 These results contribute to a comprehensive understanding of the structural characteristics and macromolecular compositions of the synthesized copolymers.The stability of copolymers P3 and P4 was investigated by thermal gravimetric analysis (TGA), conducted in a temperature range from 30 to 800 °C at a heating rate of 10 °C min−1 under a nitrogen atmosphere. The thermal decomposition temperatures (Td) corresponding to 5% weight loss were determined, resulting in Td values of 219 °C and 377 °C for P3, and 234 °C, 391 °C, 484 °C for P4 (Fig. S13, ESI†). The high thermal degradation temperature for diblock copolymer P3 can be attributed to the presence of the conjugated poly(ene) in the main chain and the incorporation of the bulky PEG750–PDI-based cycloolefin as the copolymeric counterpart. Similarly, P4 containing a bulky polyolefinic counterpart of PEG750–PDI and alkyl-functionalized oxanorbornene imide units along with the conjugated poly(acetylene) led to the three different degradation temperatures mentioned above. These molecular and structural features play an important role in enhancing the thermal stability of the copolymers. Differential scanning calorimetry (DSC) was used to determine the glass transition temperature (Tg). The samples were initially heated from 30 to 400 °C to complete the first cycle of the calorimetry experiment, then cooled to room temperature, and re-heated again from 30 to 400 °C at a heating/cooling rate of 10 °C min−1. There was no significant Tg value recorded for the copolymer samples in the temperature range from 40 to 400 °C. This absence of a glass transition temperature aligns with observations in previously reported PDI-derived polymers.44,59
Photophysical properties
In order to study changes in the aggregation modes across the series of copolymers, full photophysical characterization was performed. As depicted in Fig. 1a, the UV-Vis spectra of PEG750–PDI-derived random copolymers P5 and P6, recorded in chloroform, display characteristic perylene S0–0 transition signals around 573 nm and S0–1 dependent maxima at 534 nm as well as a low intensity S0–2 signal at 400 nm. The first absorption band in this range is commonly referred to as the “fingerprint” region for PDI derivatives, and it highly depends on the presence of π–π interactions between perylene cores.65 Both absorption maxima are significantly red-shifted compared to the unsubstituted PDI moiety.66 This shift is attributed to the presence of electron-donating substituents in the bay position, which expands the system's conjugation, and therefore narrows the band gap.67–70 The absorption profiles of the PEG750–PDI-derived copolymers P5 and P6 (Fig. 1a) in solution are virtually identical and display similar spectroscopic changes when measured in the thin film state (Fig. 1a). The films of copolymers P5 and P6 show a clear blueshift of the absorption to 513 nm with complete disappearance of the secondary peak. This indicates the presence of strong π–π interactions accompanied by the formation of H-aggregates of the PDI moieties.71,72 For the random copolymers P5 and P6, the absorption profiles depend on the average interactions between perylene cores, while polyacetylene does not show interference in the absorption profile. | ||
| Fig. 1 UV-Vis absorption spectra of (a) random binary P5 and ternary P6 copolymers, (b) diblock P3 and triblock P4 copolymers, and (c) PEG750–PDI-derived homopolymer P2 in chloroform (solid lines) and their thin film state cast from the same solvent (dotted lines). | ||
The UV-Vis absorption spectra of the tandem cyclopolymerization- and ROMP-derived block copolymers are displayed in Fig. 1b and c revealing specific characteristic features. In solution polymers P2, P3 and P4 show virtually identical profiles with a characteristic double maximum at 527 and 563 nm. Additionally, all 3 polymer solutions exhibit a shoulder around 500 nm, which was assigned to the presence of the polyacetylene fragment with high absorption in this region.6 In the solid film state, polymers P2, P3, and P4 exhibit substantially different absorption profiles, despite no changes to the chromophore core. Polymer P2 in the solid film exhibits an almost complete absence of the S0–0 transition signal and a significant blueshift of the S0–1 maxima to 514 nm. This phenomenon was attributed to strong π–π interactions between perylene cores, further supported by regular distances between them, enforced by the homopolymeric structure of the backbone. The film of the diblock polymer P3 shows an absorption profile like that of polymer P2 with a single absorption peak blue shifted to 512 nm indicating similar π–π interactions and formation of H-aggregates. On the other hand, the triblock polymer P4 exhibits its maximum absorption redshifted to 556 nm as a broad band with a shoulder around 500 nm. These differences underscore the significant variations in perylene aggregation behavior influenced by the polymer structure. We assume that changes in the stacking of the perylene cores and thus changes in the absorption spectra are the result of the interplay of several factors such as π–π stacking and hydrophilic and lipophilic fragment interactions combined with steric effects exerted by the polymer backbone. Moreover, the reduction of the intensity of the 0–0 and 0–1 absorption peaks and the drop in fluorescence efficiency indicate H-type aggregation rather than J-type aggregation in the THF and THF/water solutions.73
The modular structure of block copolymers P3 and P4 allows for the introduction of both hydrophobic hydrocarbon and aromatic groups, as well as hydrophilic PEG750. This composition enables precise control of the polymer aggregation based on the medium's polarity. The amphiphilic nature of these PEG750–PDI-based polymers allows them to be dissolved in typical hydrophobic solvents like chloroform or chlorobenzene, as well as highly polar solvents such as DMF and methanol, which has been also reported for other PDI derivatives.74 These features allow amphiphiles to self-assemble in aqueous solutions making them suitable for use as markers in biological systems.54 This self-assembly behaviour results in significant changes in both their absorption and emission spectra, which are influenced by the polymer composition and polarity of the solvent. The absorption profile in THF/water solutions shows a decrease in the intensity of the vibronic progression peak around 530 nm, illustrating the effect of the aqueous fraction on the aggregation behaviour of the polymers (Fig. 2a–c).
 | ||
| Fig. 2 UV-Vis absorption spectra of (a) homopolymer P2 in THF and combinations of THF and water, (b) diblock copolymer P3 in THF and combinations of THF and water, and (c) triblock copolymer P4 in THF and combinations of THF and water. | ||
This effect has been widely reported in the literature and is typically assigned to the formation of aggregates with strong π–π stacking interactions among the PDI cores in polar solvents.54,65,75–77 Moreover, the intensity ratio of S0–1/S0–0 can be used to qualitatively evaluate the proximity of the PDI units in solution and the degree of aggregation in a given system.78 This effect is further amplified by raising the aqueous concentration in the solvent, subsequently increasing the polarity of the medium and allowing for additional hydrogen bonding interactions. Raising the aqueous concentration up to 100% water results in a significant decrease in absorption in the visible region for copolymers P3 and P4 and the observable formation of micelles. This change can be attributed to the presence of an additional alkylated monomer, which significantly alters the polymer's hydrophobic properties. Interestingly, homopolymer P2, containing the highest content of PEG750 chains, exhibits a clear absorption peak at 530 nm even with 100% water content. This can be rationalized as the effect of the PEG chains providing solubility in highly polar solvents, countering their aggregating impact on the PDI cores. When the solvent was replaced with a higher polarity one such as methanol or a MeOH/water mixture copolymers P3 and P4 exhibited broad maxima around 530 nm with decreasing intensity as the water content increased. A similar trend was observed for P2; however, in solution the PDI absorption profile was retained, confirming the ability of P2 to retain partial solubility in highly polar solvents (Fig. S14–S16, ESI†).
To further understand the aggregation modes of the polymers and their structure dependence, the emission spectra of polymers P2, P3 and P4 were analysed (Fig. 3). In a chloroform solution, the polymers exhibit an emission profile that closely matches the one reported in the literature for phenoxy bay functionalized PDI.66 This spectrum shows strong peaks with a maximum at 593 nm and a characteristic shoulder in the emission spectrum at around 640 nm. In the thin film state, the emission spectra exhibit a singular strong band around the 700 nm region, which is consistent with previous reports on PDI-based aggregates and supramolecular structures.74,79–81 Interestingly this emission is clearly visible in the solution of homopolymer P2 and random copolymers P3 and P4, which can be assigned to enhanced π–π interactions and the directing effect of the specific polymeric backbone structure (Fig. 3). In addition, analysis of the emission of the chloroform solutions and thin films of diblock polymers P3vs. P5 (Fig. S17, ESI†) and triblock polymers P4vs.P6 (Fig. S18, ESI†) indicated identical PDI-chromophore cores and degrees of aggregation, and thus the photophysical properties can be altered by changing the backbone composition. The films of polymers P3–P6 exhibit consistent emission at 700 nm indicating that strong π–π stacking is consistent with the 0–2 transition becoming predominant over the lower-energy processes in every scenario. Interestingly even in chloroform solution a significant emission band can be observed at 700 nm indicating that consistent distances between the PDI cores enforced by the polymer backbone support AEI, even in highly diluted media.
 | ||
| Fig. 3 Emission spectra of homopolymer P2 (pink solid line), diblock copolymer P3 (red solid line) and triblock copolymer P4 (blue solid line) in chloroform and the thin film state of P3 (red dotted line), P4 (blue dotted line) and P2 (pink dotted line) cast from the same solvent. | ||
Due to their modular structure and amphiphilic properties, the polymers studied here exhibit solubility in a wide range of solvents and all three polymers display similar emissions in THF. However, the introduction of water results in significant and dramatic changes in their fluorescence profiles indicating different π–π stacking aggregation of the PDI cores. Depending on the composition of the polymers and the solvent, the emissions from these polymers exhibit typical characteristic features of both solution and solid states. The homopolymer P2 (Fig. 4a), which contains the highest concentration of both PEG750 and PDI fragments, shows a significant decrease in emission at 586 nm when 5% water is added. However, when the aqueous concentration is increased up to 10%, there is only a minor further change in emission. This is accompanied by a red shift and a gradual enhancement of the 0–2 emission signal. This phenomenon can be estimated as reaching an equilibrium point where π–π stacking interactions are balanced by the increasing impact of the PEG moieties in a more polar medium. In the case of the diblock polymer P3 (Fig. 4b), which contains additional polar diester groups, there is a slower drop in the intensity of the main emission band at 587 nm upon the addition of water. However, there is a significant increase in the solid-state emission band when the aqueous concentration reaches up to 10%. We assume that the 10% aqueous concentration in solution is the point at which the ester groups are no longer polar enough to keep the polymer in solution, leading to rapid aggregation. A similar scenario was observed for the triblock copolymer P4 (Fig. 4c), where the presence of additional aliphatic fragments caused an even more pronounced increase in the solid-state emission at 700 nm, becoming the main band for this material. A schematic representation of the aggregation of both polymers P3 and P4 is outlined in Scheme 2 displaying self-assembling factors such as π-stacking and the formation of hydrophobic and hydrophilic domains as well as the directing effect of the backbone composition. When the solvent was replaced with the more polar MeOH the predominant peak at 700 nm was observed for all the polymers, with additional absorption at 600 nm observed for P3 and P4. When the water content was increased from 0–100% all the polymers exhibited solely 700 nm maxima with various intensities (ESI†).
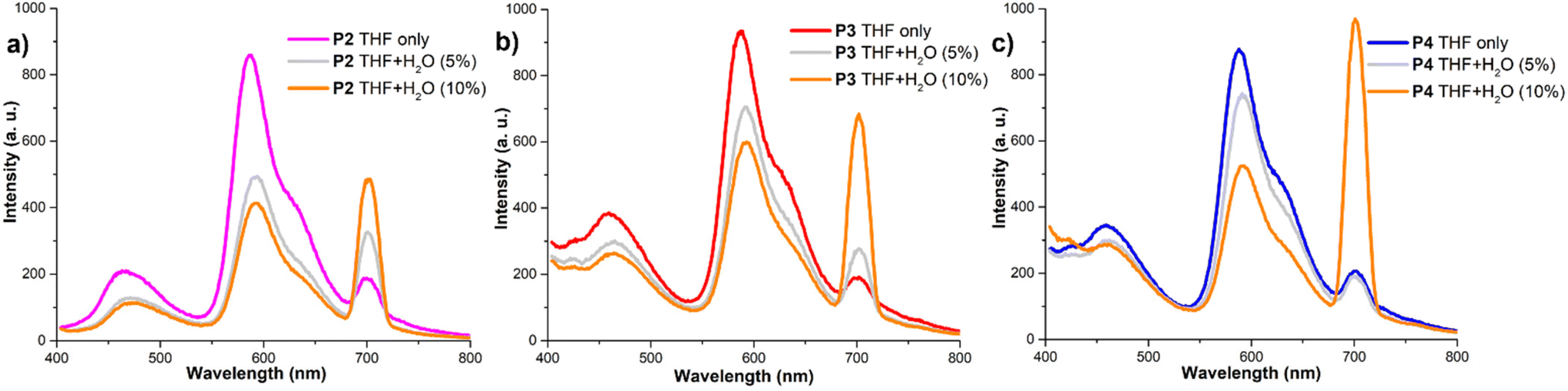 | ||
| Fig. 4 Emission spectra of (a) homopolymer P2 in THF and combinations of THF and water, (b) diblock copolymer P3 in THF and combinations of THF and water, and (c) triblock copolymer P4 in THF and combinations of THF and water. | ||
 | ||
| Scheme 2 A schematic representation of aggregation of both polymers P3 and P4 displaying self-assembly factors such as (a) π-stacking, (b) and (c) formation of hydrophobic and hydrophilic domains and (d) the directing effect of backbone composition. | ||
Morphological properties of the block copolymers
To further confirm the copolymer sizes and self-assembly behaviors of P3 and P4, their morphological properties were investigated using atomic force microscopy (AFM) and scanning electron microscopy (SEM) analysis. Samples for AFM and SEM analysis were prepared by casting drops of their chloroform solutions on a clean glass slide, followed by drying at room temperature. AFM was employed to investigate the morphology of solid films of the diblock copolymer P3 (A) and triblock polymer P4 (B) as shown in Fig. 5. The AFM images of polymers P3 and P4 clearly demonstrate the presence of globular and nano-ring like morphologies with clearly visible internal empty cavities between them. Similar morphological features have been reported and assigned to π–π interactions between the PDI aromatic rings and H-bond driven aggregation.82 As shown in Fig. 5, the AFM image of copolymer P3 displayed a slightly more homogeneous texture in comparison to the triblock copolymer P4, which exhibited significant uniformity albeit with various globular deformations on the surface having spherical morphologies.82 Furthermore, the AFM images show similar surface values of root-mean-square (RMS) roughness of 38.6 nm for P3 and 40.6 nm for P4, which results in the grain size being comparable. These differences are indicated by the 3D-height topography presented on the same scale for both polymers. Similar thin film formations for both P3 and P4 were observed, which are smoother in general, but with some defects seen for P3, while P4 is slightly rough but has smaller size irregularities on the order of nanometers in both cases.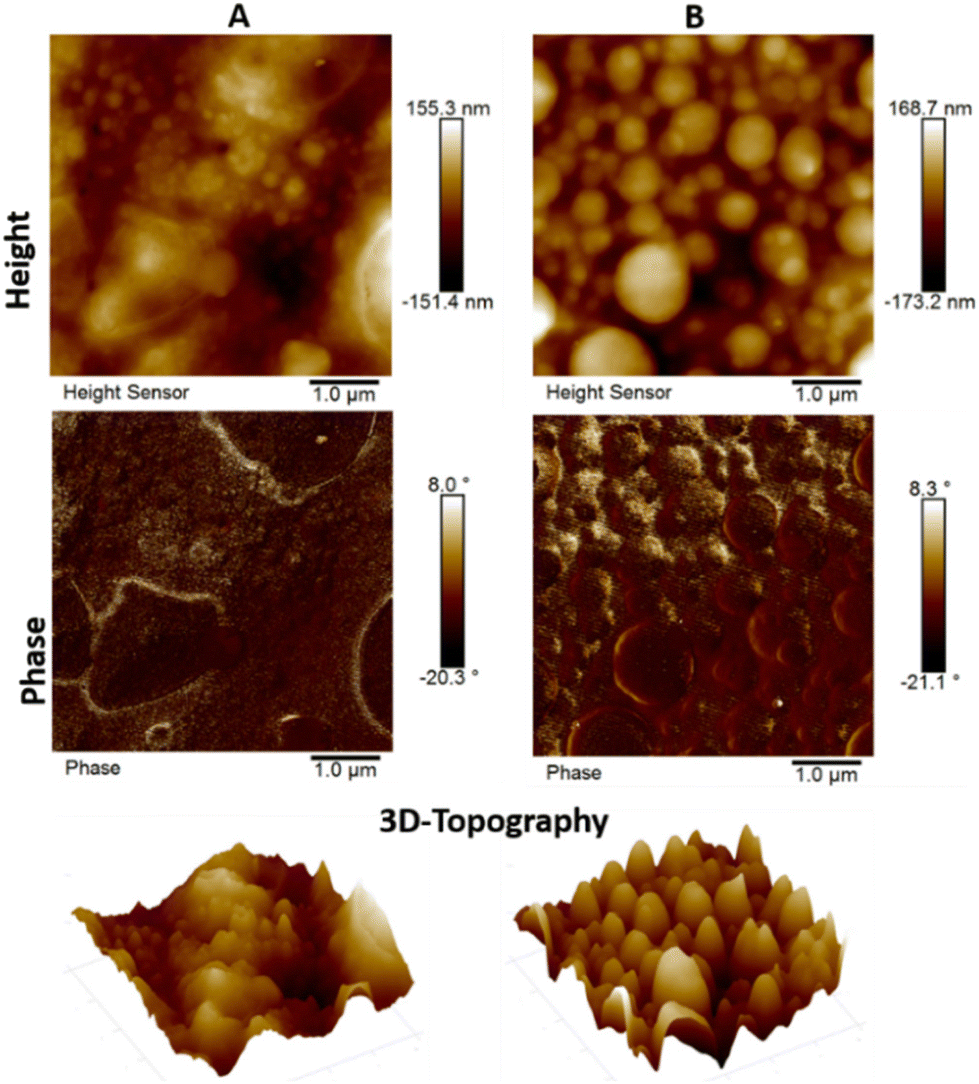 | ||
| Fig. 5 AFM images and 3D-topographical plots of P3 (A) and P4 (B). | ||
Nearly spherical morphologies were also evidenced, as expected, by SEM analysis of the cross-sections of copolymers P3 and P4 confirming the surface morphologies determined by AFM analysis. SEM images of the copolymers also showed various sizes of spherical morphologies and aggregation of cores dispersed within the poly(ene) and poly(oxanorbornene) copolymer matrix (Fig. 6A and B). It is well known that the dispersion of these polymer blocks in a polymer matrix plays a dominant role in influencing the morphological properties of the polymer.83 However, the type of aggregation of PEG-grafted PDI groups and the phase separation in these two copolymers have not been well addressed by SEM analysis.84
 | ||
| Fig. 6 SEM images of cross-sections of diblock copolymer P3 (A) and triblock copolymer P4 (B) films prepared by controlled evaporation from CHCl3. | ||
Conclusions
In summary, we successfully synthesized copolymers of highly conjugated poly(acetylene)s and PEG750–PDI-incorporating or N-alkyl-derived poly(olefin)s in a single polymer chain via one-pot tandem cyclopolymerization and ROMP methodologies. Both di- and tri-block copolymers and their random versions were prepared from 1,6-heptadiynes and oxanorbornene imide-based cycloolefin monomers using a trifluoroacetate-modified [Ru]–alkylidene-based metathesis initiator. The relative weight percentages of both di- and tri-block copolymers determined by XPS analysis revealed a slight excess of the percentages of atomic concentrations in the case of the triblock copolymers. The photophysical properties examined by UV-Vis and fluorescence spectroscopic analysis demonstrated different stages of the organization of the polymers in solution as well as in the formation of micelles upon diluting polymer solutions. Furthermore, these investigations provided more insight into the tuning and self-organization of PEG750–PDI derivatives of block copolymers in aqueous solution at various concentrations. In addition, AFM and SEM analysis elucidated copolymer surface and morphological information of both di-and tri-block polymers in their solid film state. In brief, our approach gives access to a variety of tunable PEG750-incorporating chromophores held by non-conjugated polymers as a copolymer counterpart of the cyclopolymerization-derived poly(acetylene). Based on our findings, tandem cyclopolymerization and subsequent ROMP-derived copolymers can be potentially used for the preparation of a variety of novel polymeric materials for electrical conductivity, bioimaging and photoluminescence applications.Author contributions
All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Texas A&M University at Qatar.References
- H. H. Fox and R. R. Schrock, Organometallics, 1992, 11, 2763–2765 CrossRef CAS.
- H. H. Fox, M. O. Wolf, R. O'Dell, B. L. Lin, R. R. Schrock and M. S. Wrighton, J. Am. Chem. Soc., 1994, 116, 2827–2843 CrossRef CAS.
- F. J. Schattenmann and R. R. Schrock, Macromolecules, 1996, 29, 8990–8991 CrossRef CAS.
- P. S. Kumar, K. Wurst and M. R. Buchmeiser, J. Am. Chem. Soc., 2009, 131, 387–395 CrossRef CAS PubMed.
- U. Anders, O. Nuyken, M. R. Buchmeiser and K. Wurst, Angew. Chem., Int. Ed., 2002, 41, 4044–4047 CrossRef CAS PubMed.
- J. O. Krause, M. T. Zarka, U. Anders, R. Weberskirch, O. Nuyken and M. R. Buchmeiser, Angew. Chem., Int. Ed., 2003, 42, 5965–5969 CrossRef CAS PubMed.
- S. K. Podiyanachari, H. S. Bazzi and M. Al-Hashimi, Curr. Org. Chem., 2021, 25, 2791–2805 CrossRef CAS.
- E. B. Anderson, P. S. Kumar, D. Schawaller, S. Mavila, M. Voss, A. Freyer, W. Knolle, F. Hermanutz and M. R. Buchmeiser, Macromol. Chem. Phys., 2013, 214, 1047–1051 CrossRef CAS.
- H. Li, J. Wang, H. Han, J. Wu and M. Xie, React. Funct. Polym., 2018, 127, 20–28 CrossRef CAS.
- J. Wang, H. Li, X. Liao, M. Xie and R. Sun, Polym. Chem., 2016, 7, 4912–4923 RSC.
- M. R. Buchmeiser, Polym. Rev., 2017, 57, 15–30 CrossRef CAS.
- K. Jung, E.-H. Kang, J.-H. Sohn and T.-L. Choi, J. Am. Chem. Soc., 2016, 138, 11227–11233 CrossRef CAS PubMed.
- H. Jung, K. Jung, M. Hong, S. Kwon, K. Kim, S. H. Hong, T.-L. Choi and M.-H. Baik, J. Am. Chem. Soc., 2018, 140, 834–841 CrossRef CAS PubMed.
- K. Jung, T. S. Ahmed, J. Lee, J.-C. Sung, H. Keum, R. H. Grubbs and T.-L. Choi, Chem. Sci., 2019, 10, 8955–8963 RSC.
- G. I. Peterson, S. Yang and T.-L. Choi, Acc. Chem. Res., 2019, 52, 994–1005 CrossRef CAS PubMed.
- C. Kang, K. Jung, S. Ahn and T.-L. Choi, J. Am. Chem. Soc., 2020, 142, 17140–17146 CrossRef CAS PubMed.
- E.-H. Kang and T.-L. Choi, ACS Macro Lett., 2013, 2, 780–784 CrossRef CAS PubMed.
- E.-H. Kang, I. S. Lee and T.-L. Choi, J. Am. Chem. Soc., 2011, 133, 11904–11907 CrossRef CAS PubMed.
- E.-H. Kang, S. Y. Yu, I. S. Lee, S. E. Park and T.-L. Choi, J. Am. Chem. Soc., 2014, 136, 10508–10514 CrossRef CAS PubMed.
- C. Kang, E.-H. Kang and T.-L. Choi, Macromolecules, 2017, 50, 3153–3163 CrossRef CAS.
- S. Yang, S.-Y. Kang and T.-L. Choi, J. Am. Chem. Soc., 2019, 141, 19138–19143 CrossRef CAS PubMed.
- K.-Y. Yoon, I.-H. Lee and T.-L. Choi, RSC Adv., 2014, 4, 49180–49185 RSC.
- E. H. Kang, S. Yang, S. Y. Yu, J. Kim and T. L. Choi, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 3058–3066 CrossRef CAS.
- S. Yang, S.-Y. Kang and T.-L. Choi, Nat. Commun., 2021, 12, 2602 CrossRef CAS PubMed.
- S. Yang and T.-L. Choi, Chem. Sci., 2020, 11, 8416–8424 RSC.
- S. Yang, S. Shin, I. Choi, J. Lee and T.-L. Choi, J. Am. Chem. Soc., 2017, 139, 3082–3088 CrossRef CAS PubMed.
- H. Park, H.-K. Lee and T.-L. Choi, Polym. Chem., 2013, 4, 4676–4681 RSC.
- C. Kang, H. Park, J.-K. Lee and T.-L. Choi, J. Am. Chem. Soc., 2017, 139, 11309–11312 CrossRef CAS PubMed.
- H. Park and T.-L. Choi, J. Am. Chem. Soc., 2012, 134, 7270–7273 CrossRef CAS PubMed.
- J. Kim, E.-H. Kang and T.-L. Choi, ACS Macro Lett., 2012, 1, 1090–1093 CrossRef CAS PubMed.
- S. K. Podiyanachari, S. Moncho, E. N. Brothers, S. Al-Meer, M. Al-Hashimi and H. S. Bazzi, Macromolecules, 2020, 53, 4330–4337 CrossRef CAS.
- M. Al-Hashimi, C. Hongfa, B. George, H. S. Bazzi and D. E. Bergbreiter, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 3954–3959 CrossRef CAS.
- R. Tuba, R. Corrêa da Costa, H. S. Bazzi and J. A. Gladysz, ACS Catal., 2012, 2, 155–162 CrossRef CAS.
- R. Tuba, M. Al-Hashimi, H. S. Bazzi and R. H. Grubbs, Macromolecules, 2014, 47, 8190–8195 CrossRef CAS.
- M. Al-Hashimi, M. A. Bakar, K. Elsaid, D. E. Bergbreiter and H. S. Bazzi, RSC Adv., 2014, 4, 43766–43771 RSC.
- M. Al-Hashimi, R. Tuba, H. S. Bazzi and R. H. Grubbs, ChemCatChem, 2016, 8, 228–233 CrossRef CAS.
- A. R. Hlil, J. Balogh, S. Moncho, H. L. Su, R. Tuba, E. N. Brothers, M. Al-Hashimi and H. S. Bazzi, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 3137–3145 CrossRef CAS.
- R. Tuba, J. Balogh, A. Hlil, M. Barłóg, M. Al-Hashimi and H. Bazzi, Engineering, 2016, 4, 6090–6094 CAS.
- J. Suriboot, C. E. Hobbs, W. Guzman, H. S. Bazzi and D. E. Bergbreiter, Macromolecules, 2015, 48, 5511–5516 CrossRef CAS.
- J. Suriboot, Y. Hu, T. J. Malinski, H. S. Bazzi and D. E. Bergbreiter, ACS Omega, 2016, 1, 714–721 CrossRef CAS PubMed.
- U. R. Gandra, A. Sinopoli, S. Moncho, M. NandaKumar, D. B. Ninković, S. D. Zaric, M. Sohail, S. Al-Meer, E. N. Brothers and N. A. Mazloum, ACS Appl. Mater. Interfaces, 2019, 11, 34376–34384 CrossRef CAS PubMed.
- U. R. Gandra, R. Courjaret, K. Machaca, M. Al-Hashimi and H. S. Bazzi, Sci. Rep., 2020, 10, 19519 CrossRef CAS PubMed.
- S. K. Podiyanachari, M. Barłóg, M. Comí, S. Attar, S. Al-Meer, M. Al-Hashimi and H. S. Bazzi, J. Polym. Sci., 2021, 59, 3150–3160 CrossRef CAS.
- M. Barłóg, S. K. Podiyanachari, S. Attar, D. N. Sredojević, H. S. Bazzi and M. Al-Hashimi, Polym. Chem., 2022, 13, 5912–5922 RSC.
- N. Jouault, Y. Xiang, E. Moulin, G. Fuks, N. Giuseppone and E. Buhler, Phys. Chem. Chem. Phys., 2012, 14, 5718–5728 RSC.
- E. Zhang, L. Liu, F. Lv and S. Wang, ACS Omega, 2018, 3, 8691–8696 CrossRef CAS PubMed.
- Z. Liang, R. A. Cormier, A. M. Nardes and B. A. Gregg, Synth. Met., 2011, 161, 1014–1021 CrossRef CAS.
- K. Trofymchuk, A. Reisch, I. Shulov, Y. Mély and A. S. Klymchenko, Nanoscale, 2014, 6, 12934–12942 RSC.
- X. Zhang, Z. Chen and F. Würthner, J. Am. Chem. Soc., 2007, 129, 4886–4887 CrossRef CAS PubMed.
- Z. Yang and X. Chen, Acc. Chem. Res., 2019, 52, 1245–1254 CrossRef CAS PubMed.
- E. Cohen, H. Weissman, I. Pinkas, E. Shimoni, P. Rehak, P. Král and B. Rybtchinski, ACS Nano, 2018, 12, 317–326 CrossRef CAS PubMed.
- H. Li and O. S. Wenger, Angew. Chem., Int. Ed., 2022, 61, e202110491 CrossRef CAS PubMed.
- P. Singh, A. Hirsch and S. Kumar, TrAC, Trends Anal. Chem., 2021, 138, 116237 CrossRef CAS.
- O. Krupka and P. Hudhomme, Int. J. Mol. Sci., 2023, 24, 6308 CrossRef CAS PubMed.
- L. Zong, H. Zhang, Y. Li, Y. Gong, D. Li, J. Wang, Z. Wang, Y. Xie, M. Han and Q. Peng, ACS Nano, 2018, 12, 9532–9540 CrossRef CAS PubMed.
- Y. Hu, K. Wang, Y. Wang and L. Ma, Dyes Pigm., 2023, 210, 110948 CrossRef CAS.
- B. Zhang, I. Lyskov, L. J. Wilson, R. P. Sabatini, A. Manian, H. Soleimaninejad, J. M. White, T. A. Smith, G. Lakhwani and D. J. Jones, J. Mater. Chem. C, 2020, 8, 8953–8961 RSC.
- M. S. Ryoo, W. C. Lee and S. K. Choi, Macromolecules, 1990, 23, 3029–3031 CrossRef CAS.
- S. K. Podiyanachari, M. Barłóg, M. Comi, S. Attar, S. Al-Meer, M. Al-Hashimi and H. S. Bazzi, J. Polym. Sci., 2021, 59, 3150–3160 CrossRef CAS.
- X. Wang, T. Zeng, M. Nourrein, B.-H. Lai, K. Shen, C.-L. Wang, B. Sun and M. Zhu, RSC Adv., 2017, 7, 26074–26081 RSC.
- R. Chen, S. J. Benware, S. D. Cawthern, J. P. Cole, J. J. Lessard, I. M. Crawford-Eng, R. Saxena and E. B. Berda, Eur. Polym. J., 2019, 112, 206–213 CrossRef CAS.
- J. O. Krause, M. T. Zarka, U. Anders, R. Weberskirch, O. Nuyken and M. R. Buchmeiser, Angew. Chem., Int. Ed., 2003, 42, 5965–5969 CrossRef CAS PubMed.
- H. Park and T.-L. Choi, J. Am. Chem. Soc., 2012, 134, 7270–7273 CrossRef CAS PubMed.
- R. T. Mathers, K. Damodaran, M. G. Rendos and M. S. Lavrich, Macromolecules, 2009, 42, 1512–1518 CrossRef CAS.
- F. Spreitler, M. Sommer, M. Thelakkat and J. Köhler, Phys. Chem. Chem. Phys., 2012, 14, 7971–7980 RSC.
- Y. Chen, Y. Feng, J. Gao and M. Bouvet, J. Colloid Interface Sci., 2012, 368, 387–394 CrossRef CAS PubMed.
- W. S. Shin, H.-H. Jeong, M.-K. Kim, S.-H. Jin, M.-R. Kim, J.-K. Lee, J. W. Lee and Y.-S. Gal, J. Mater. Chem., 2006, 16, 384–390 RSC.
- L. Rocard, A. Goujon and P. Hudhomme, Molecules, 2020, 25, 1402 CrossRef CAS PubMed.
- E. E. Beauvilliers, M. R. Topka and P. H. Dinolfo, RSC Adv., 2014, 4, 32866–32875 RSC.
- G. S. Perez, S. Dasgupta, W. Żuraw, R. F. Pineda, K. Wojciechowski, L. K. Jagadamma, I. Samuel and N. Robertson, J. Mater. Chem. A, 2022, 10, 11046–11053 RSC.
- Y. Zhang, X. Guo, W. Su, B. Guo, Z. Xu, M. Zhang and Y. Li, Org. Electron., 2017, 41, 49–55 CrossRef CAS.
- H. Chang, Z. Chen, X. Yang, Q. Yin, J. Zhang, L. Ying, X.-F. Jiang, B. Xu, F. Huang and Y. Cao, Org. Electron., 2017, 45, 227–233 CrossRef CAS.
- H. Liu, L. Shen, Z. Cao and X. Li, Phys. Chem. Chem. Phys., 2014, 16, 16399–16406 RSC.
- J. Liu, Y. Zhang, C. Zhang, P. Zhang, R. Zeng, J. Cui and J. Chen, Mater. Adv., 2020, 1, 1330–1336 RSC.
- C. Fuller and C. Finlayson, Phys. Chem. Chem. Phys., 2017, 19, 31781–31787 RSC.
- P. Su, G. Ran, H. Wang, J. Yue, Q. Kong, Z. Bo and W. Zhang, Molecules, 2023, 28, 3003 CrossRef CAS PubMed.
- Z. Chen, B. Fimmel and F. Würthner, Org. Biomol. Chem., 2012, 10, 5845–5855 RSC.
- B. Fimmel, M. Son, Y. M. Sung, M. Grüne, B. Engels, D. Kim and F. Würthner, Chem. – Eur. J., 2015, 21, 615–630 CrossRef CAS PubMed.
- D. Sriramulu, E. L. Reed, M. Annamalai, T. V. Venkatesan and S. Valiyaveettil, Sci. Rep., 2016, 6, 35993 CrossRef CAS PubMed.
- J. Das, R. B. K. Siram, D. Cahen, B. Rybtchinski and G. Hodes, J. Mater. Chem. A, 2015, 3, 20305–20312 RSC.
- T. Tang, J. Qu, K. Müllen and S. E. Webber, Langmuir, 2006, 22, 26–28 CrossRef CAS PubMed.
- P. Singh, P. Sharma, N. Sharma and S. Kaur, J. Mater. Chem. B, 2022, 10, 107–119 RSC.
- P. Chal, A. Shit and A. K. Nandi, ChemistrySelect, 2018, 3, 3993–4003 CrossRef CAS.
- P. Moharana and G. Santosh, Spectrochim. Acta, Part A, 2023, 297, 122696 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4py00078a |
| This journal is © The Royal Society of Chemistry 2024 |