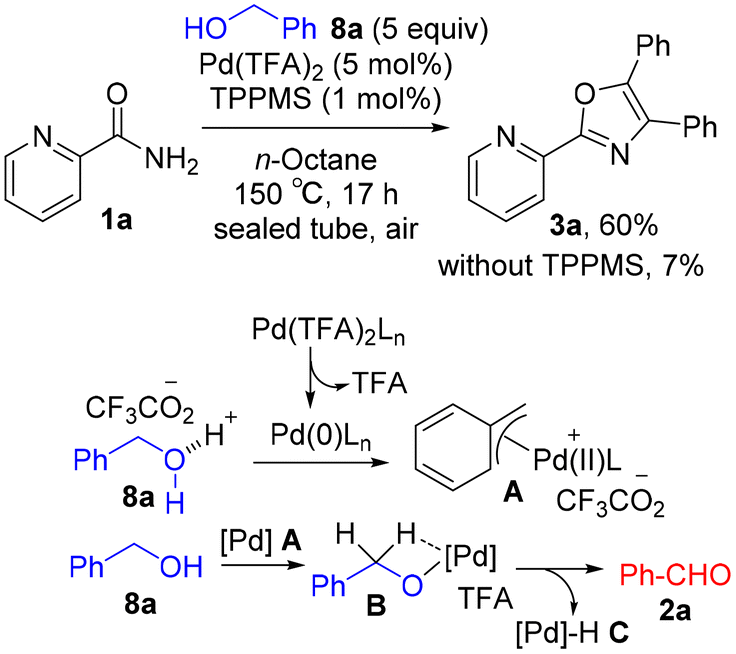
A sequential reaction of picolinamide with benzaldehydes promoted by Pd(TFA)2: rapid access to 4,5-disubstituted 2-(pyridin-2-yl)oxazoles in n-octane†
Received
6th November 2023
, Accepted 16th December 2023
First published on 19th December 2023
Abstract
We developed a synthetic method for obtaining 4,5-disubstituted 2-(pyridin-2-yl)oxazoles from picolinamide and aldehydes by employing Pd(TFA)2 as the catalyst in n-octane. This cascade reaction involves the condensation of picolinamide and two aldehyde molecules promoted by trifluoroacetic acid (TFA) generated in situ from Pd(TFA)2. This one-pot protocol provides rapid access to synthetically valuable triaryloxazoles from readily available starting materials under mild conditions. An 18O labeling study revealed that this tandem reaction proceeded via a different reaction mechanism compared to the Robinson–Gabriel oxazole synthesis.
Introduction
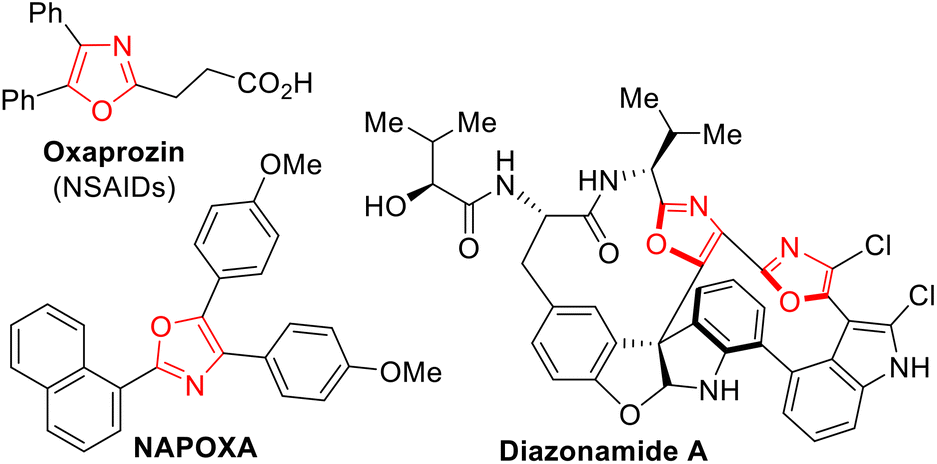
Oxazole moieties, five-membered aromatic heterocycles containing one oxygen and one nitrogen atom, are found in a wide range of pharmaceuticals and fine chemicals including non-steroidal anti-inflammatory drugs (NSAIDs),1 blue organic LEDs,2 and antibacterial peptides3 (Fig. 1).
 |
| | Fig. 1 Examples of drugs, a blue organic LED, and a marine natural product containing the oxazole moiety. | |
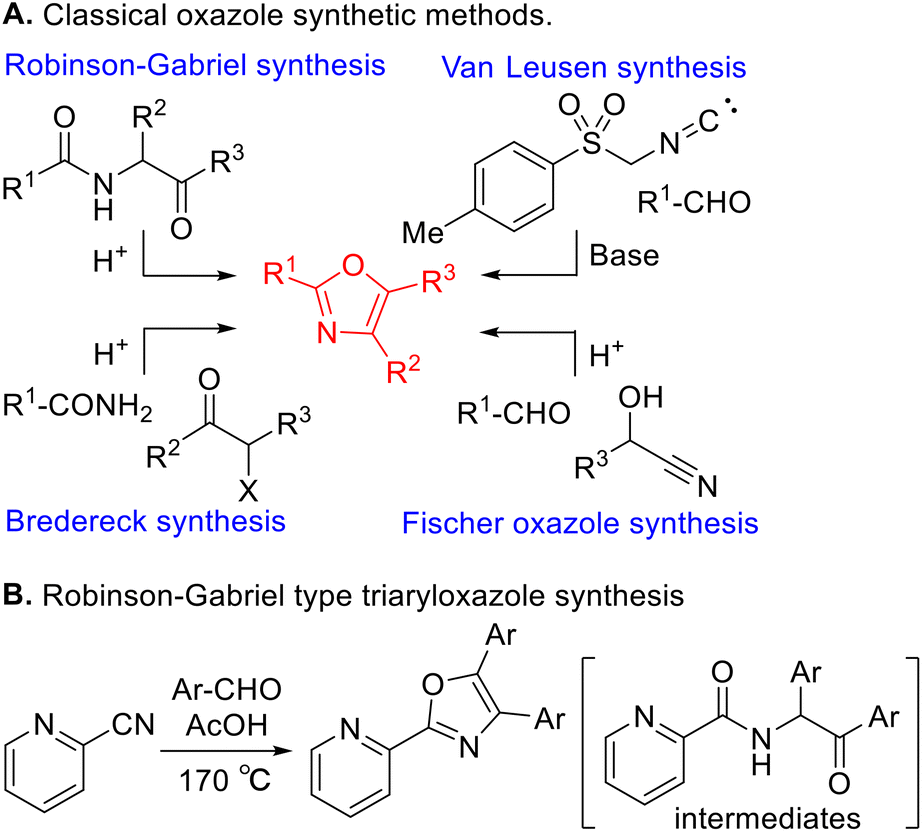
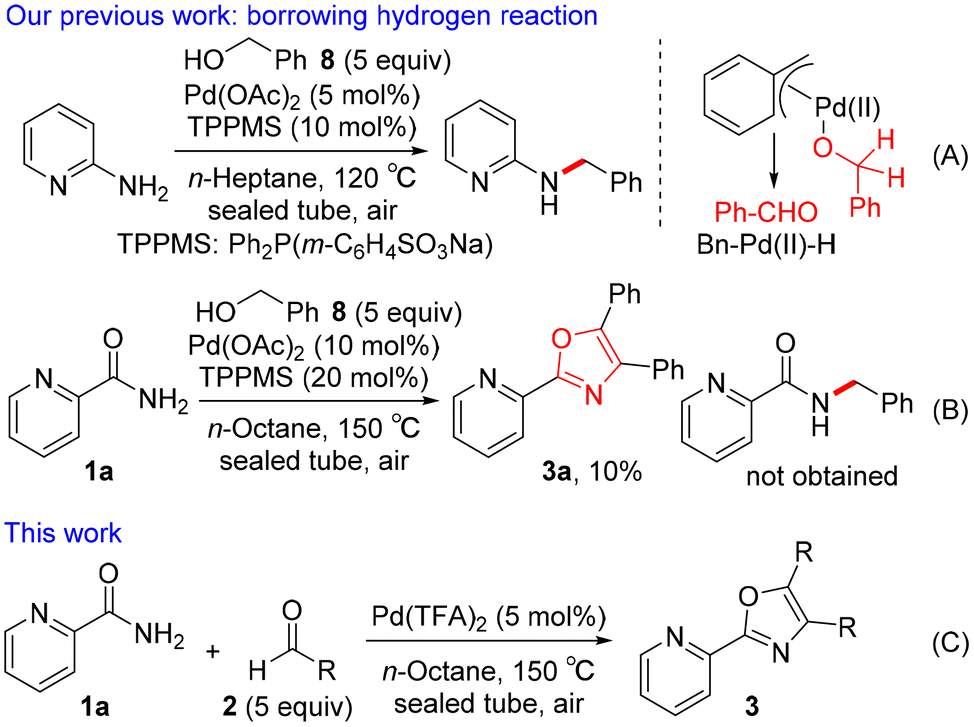
Various classical methods for oxazole ring construction have been reported so far (Scheme 1A).4–15 Although many efficient protocols have been developed, to the best of our knowledge, few methods exist to synthesize a range of highly functionalized oxazoles through one-pot tandem reactions using commercially available substrates. In 2015, Meng et al. achieved a Robinson–Gabriel type triaryloxazole synthesis from 2-cyanopyridine and benzaldehydes via α-acylaminoketone intermediates (Scheme 1B).16 However, this method requires the use of acetic acid as a solvent under harsh reaction conditions. Our group recently developed a water-promoted borrowing hydrogen reaction between 2-aminopyridines and benzylic alcohols utilizing a π-benzylpalladium(II) species in n-heptane, leading to a series of N-benzylpyridin-2-amines17 (Scheme 2A). Inspired by this discovery, we attempted to extend the method to more challenging electron-deficient amide nucleophiles for the direct substitution of alcohols (Scheme 2B). To our surprise, however, the attempted Pd-catalyzed N-benzylation of substrate 1a yielded triaryloxazole 3a instead of the N-benzylated product. Based on this result and our previous work, we hypothesized that the amide nucleophile 1a reacted with the in situ generated aldehydes 2, forming oxazoles 3. To the best of our knowledge, the straightforward synthesis of multi-substituted oxazoles from readily available picolinamides and aldehydes without the use of stoichiometric amounts of acid has not been reported previously.
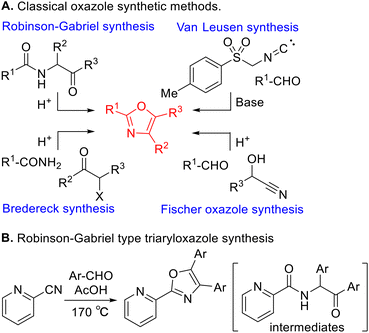 |
| | Scheme 1 Classical oxazole synthetic methods. | |
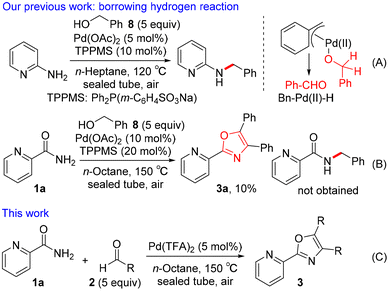 |
| | Scheme 2 Straightforward Pd-catalyzed synthesis of triaryloxazoles 3. | |
We herein present an example of the synthesis of 4,5-disubstituted 2-(pyridin-2-yl)oxazoles from picolinamide and aldehydes using Pd(TFA)2 in n-octane (Scheme 2C). This cascade reaction was performed using TFA as the catalyst generated in situ from Pd(TFA)2 under neutral reaction conditions, allowing rapid access to the valuable triaryloxazoles 3. A plausible mechanism different from the Robinson–Gabriel reaction pathway was proposed based on an 18O labeling study and several control experiments.
Results and discussion
Reaction optimization
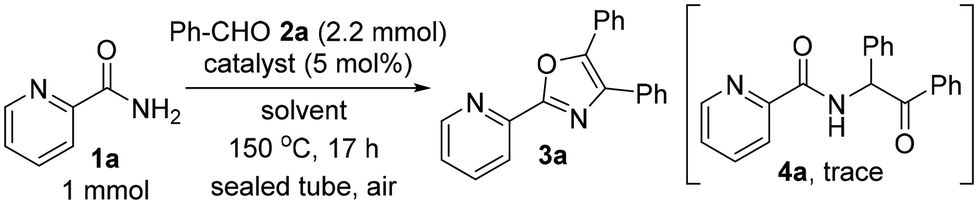
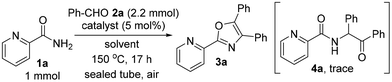
Initially, a mixture of picolinamide (1a), benzaldehyde (2a, 2.2 equiv.) and Pd(TFA)2 (5 mol%) was heated at 150 °C in n-octane in a sealed tube in air, furnishing the desired triaryloxazole 3a in 62% yield (Table 1, entry 1). Replacing n-octane with o-xylene slightly diminished the yield (50%, entry 2). Polar solvents such as DMF or n-pentanol were not suitable for the oxazole synthesis (entries 3 and 4). No reaction occurred when using (CHCl2)2 as a solvent (entry 5). To investigate the effect of Pd(TFA)2 on oxazole synthesis, the reaction using TFA as a catalyst was carried out. Surprisingly, TFA showed almost the same effect as Pd(TFA)2 (54%, entry 6), suggesting that the in situ generated Brønsted acid catalyst from Pd(TFA)2 promoted this sequential reaction. Screening of Brønsted acid catalysts showed no linear correlation between the pKa values and the yield of 3a, and the use of TFA gave the best result (entries 6–11).
Table 1 Optimization of the reaction conditionsa
|
|
| Entry |
Catalyst |
pKab |
Solvent |
Yieldc (%) |
|
Reaction conditions: amide 1a (1.0 mmol), aldehyde 2a (2.2 mmol), catalyst (5 mol%), solvent (4 mL), 150 °C, 17 h, sealed tube, in air.
pKa values in H2O.
The conversion was determined by 1H NMR analysis of the crude product using 1,3,5-trimethoxybenzene (1 mmol) as an internal standard.
5 equiv. of aldehyde 2a was used.
Isolated yield in parenthesis.
|
| 1 |
Pd(TFA)2 |
— |
n-Octane |
62 |
| 2 |
Pd(TFA)2 |
— |
o-Xylene |
50 |
| 3 |
Pd(TFA)2 |
— |
DMF |
0 |
| 4 |
Pd(TFA)2 |
— |
n-Pentanol |
0 |
| 5 |
Pd(TFA)2 |
— |
(CHCl2)2 |
0 |
| 6 |
TFA |
−0.25 |
n-Octane |
54 (70)d |
| 7 |
TfOH |
−14 |
n-Octane |
22 |
| 8 |
TsOH·H2O |
−6.5 |
n-Octane |
33 |
| 9 |
H2SO4 |
−3.0 |
n-Octane |
32 |
| 10 |
MsOH |
−2.6 |
n-Octane |
40 |
| 11 |
AcOH |
4.76 |
n-Octane |
0 |
| 12 |
NaTFA |
— |
n-Octane |
0 |
| 13 |
Zn(TFA)2 |
— |
n-Octane |
26 |
| 14 |
AgTFA |
— |
n-Octane |
56 |
| 15d |
Pd(TFA)2 |
— |
n-Octane |
86 (77)e |
Although the use of other salts such as NaTFA and Zn(TFA)2 was not effective (entries 12 and 13), AgTFA showed almost the same result as Pd(TFA)2 (56%, entry 14). To our delight, increasing the amount of aldehyde 2a to 5 equiv. was shown to increase the yield of triaryloxazole 3a (86%, entry 15), although a trace amount of by-product 4a was formed.
Reaction scope
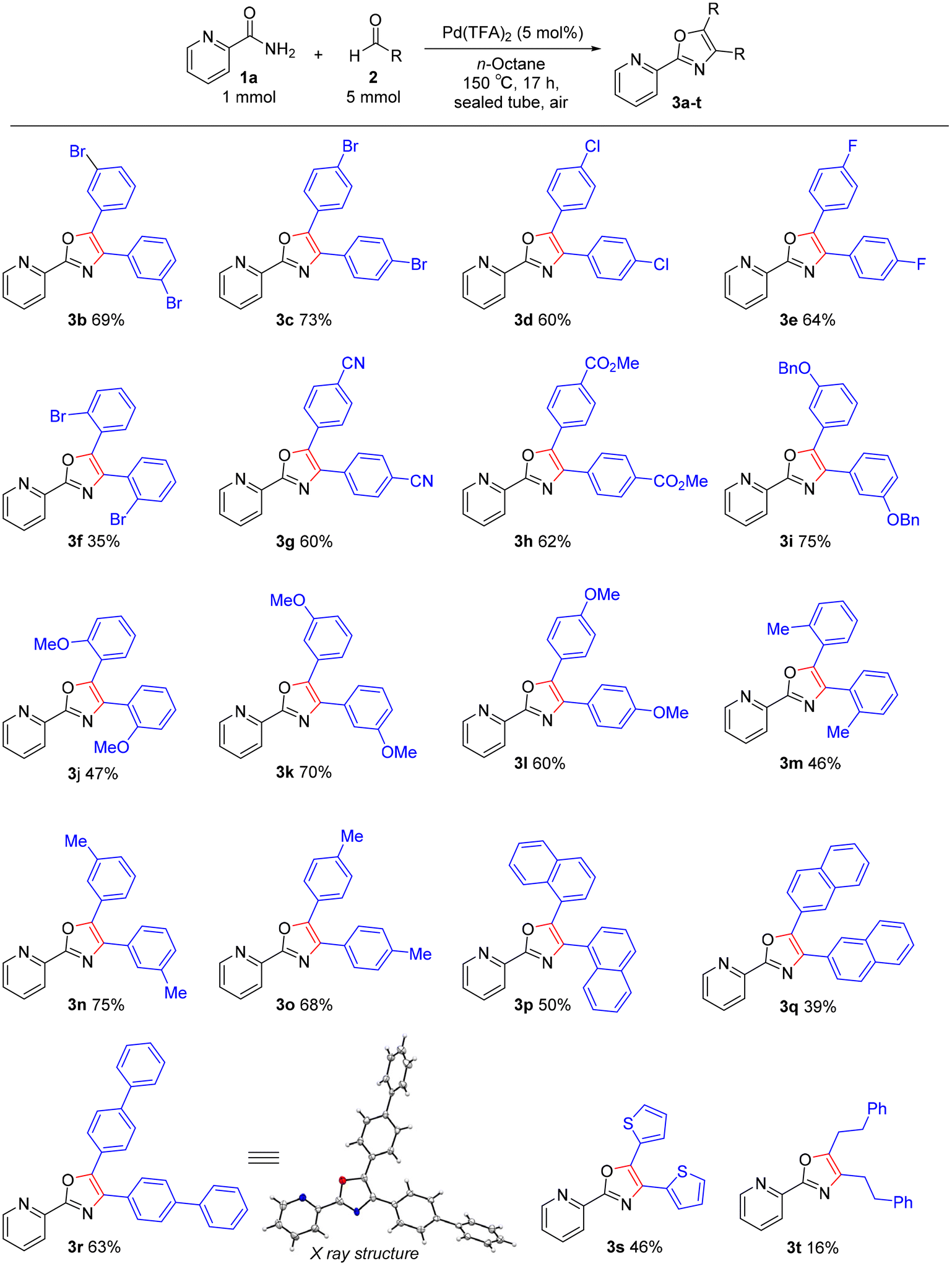
The reaction scope of aldehydes 2 was explored under the optimal conditions for triaryloxazole synthesis (Scheme 3). Several benzaldehydes with halogen groups could be converted to oxazole products with the carbon–halogen moieties left intact, which would be useful for further synthetic conversions (3b–e). The use of 2-bromobenzaldehyde led to a lower yield of 3f, probably due to steric hindrance. A wide variety of functional groups including electron-withdrawing groups (cyano and ester) and electron-donating groups (benzyloxy, methoxy, and methyl) were tolerated under our catalytic conditions, furnishing a series of oxazoles in moderate yields (3g–o). Advantageously, aldehydes containing acid-sensitive cyano or benzyloxy group led to the desired products. Even the hydrophobic and sterically hindered 4-phenylbenzaldehyde and naphthaldehydes led to the corresponding desired products (3p–r). The structure of 3r was unambiguously confirmed by single-crystal X-ray diffraction analysis. Heterocyclic and aliphatic aldehydes were also converted to the corresponding oxazoles 3s and t, albeit in poor yields. Unfortunately, 2-pyrazinecarboxamide and N,N-dimethylaminobenzaldehyde were not applicable to the TFA-catalyzed oxazole synthesis. These substrates are considered unsuitable for acid-catalyzed reactions due to their basicity.
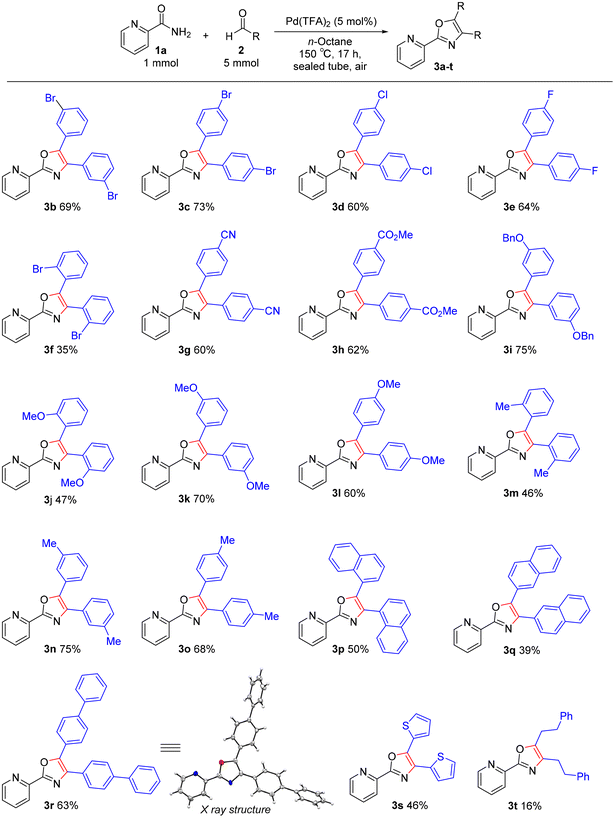 |
| | Scheme 3 Substrate scope of oxazole synthesis. Yields are those of isolated products 3. Reaction conditions: amide 1a (1.0 mmol), aldehyde 2 (5 mmol), Pd(TFA)2 (5 mol%), n-octane (4 mL), 150 °C, 17 h, sealed tube, in air. | |
Mechanistic investigations
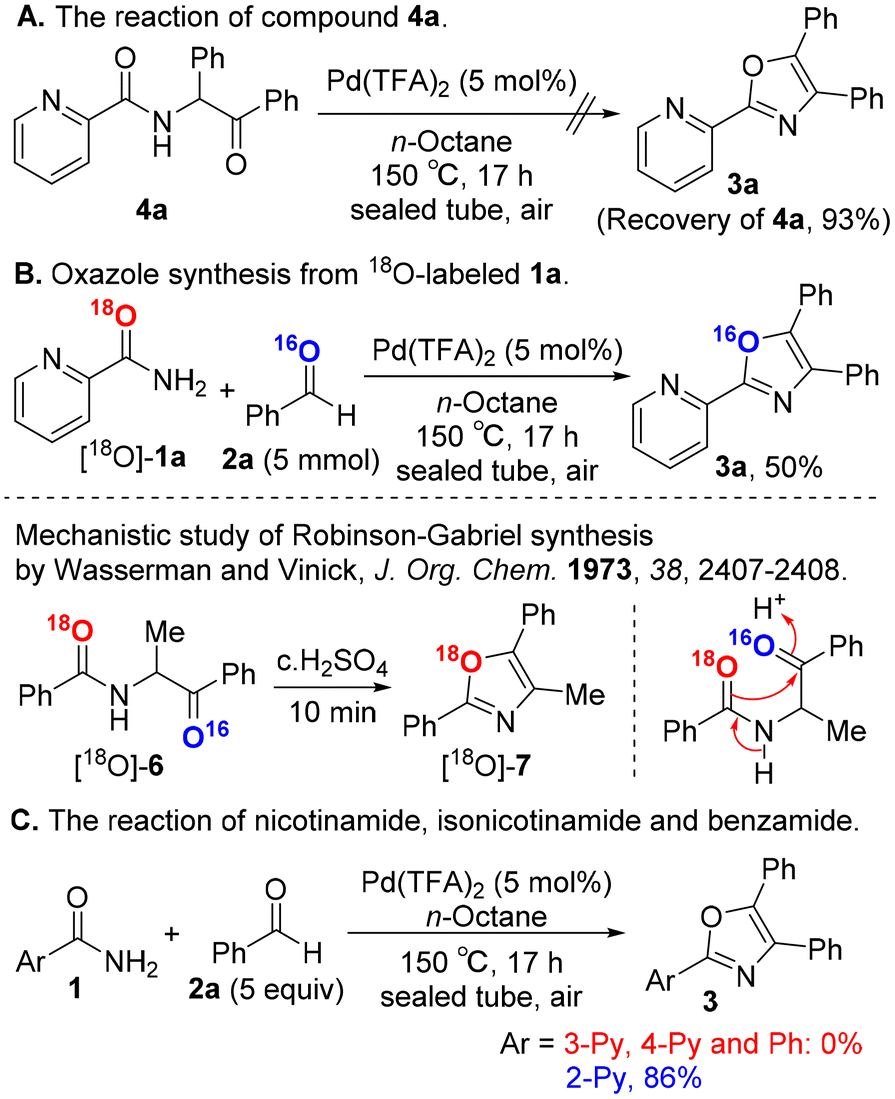
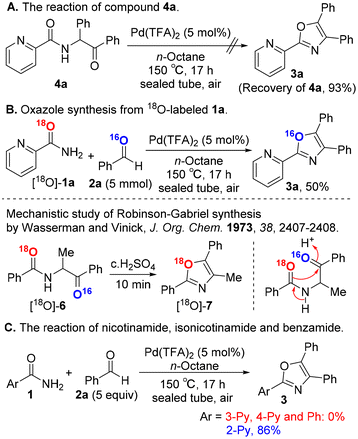
To gain mechanistic insights into our oxazole synthesis, we performed several control experiments. The Robinson–Gabriel reaction generally proceeds via α-acylaminoketone intermediates 4 to afford oxazoles 3. Surprisingly, compound 4a was not converted to oxazole 3a in our catalytic system (Scheme 4A). Next, we conducted an oxygen-18 tracer examination using 18O-labeled picolinamide, and the resulting oxazole product was measured by high resolution mass spectrometry. The 18O-labeled substrate [18O]-1a was prepared from picolinonitrile and H218O, based on Sharley's method18 (see the ESI†). The substrate [18O]-1a was successfully transformed into the corresponding oxazole compound, leading to the non-18O-labeled 3a with a corresponding m/z value of 298.1106 (calcd mass for [M]+: 298.1106) (Scheme 4B). In contrast, Wasserman and Vinick reported that the cyclization of substrate [18O]-6 gave oxazole [18O]-7, clearly showing that the amide oxygen is incorporated in the oxazole ring.19 These results exclude the Robinson–Gabriel reaction pathway via intermediate 4a in our oxazole synthesis.
 |
| | Scheme 4 Control experiments. | |
When replacing picolinamide (1a) with other amide substrates 1 such as nicotinamide, isonicotinamide and benzamide, the corresponding oxazole products 3 were not obtained under the standard conditions,20 suggesting that the nitrogen atom in the pyridine ring of 1a plays an important role in oxazole synthesis (Scheme 4C).
Reaction progress
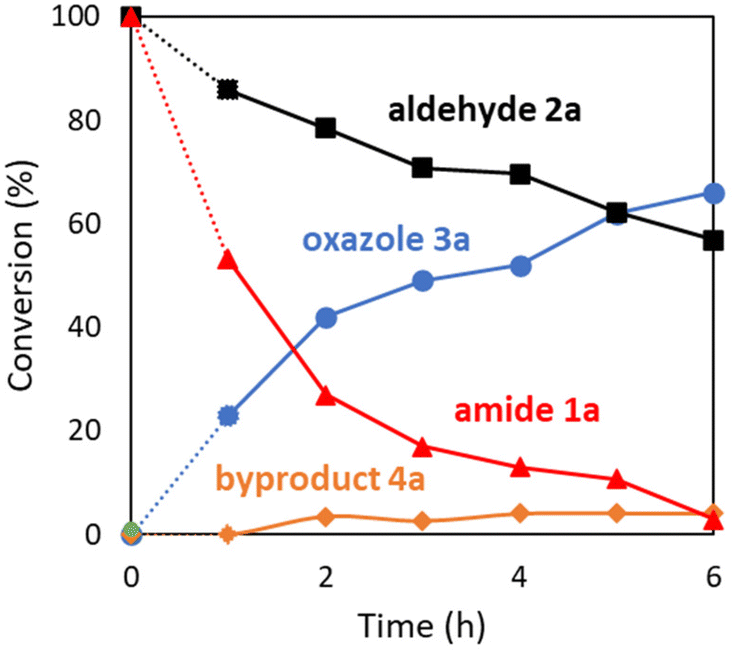
The oxazole synthesis of 3a was monitored over time by 1H NMR spectroscopy to understand reaction progress (Fig. 2). The coupling reaction of amide 1a with aldehyde 2a proceeded smoothly to generate the triaryloxazole 3a. Notably, the Robinson–Gabriel intermediate 4a was not formed (<5%), ruling out the possibility of the Robinson–Gabriel reaction pathway (see Scheme 4B).
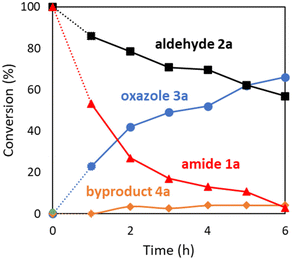 |
| | Fig. 2 Time course of the reaction for oxazole synthesis: amide 1a (1 mmol), aldehyde 2a (5 mmol), Pd(TFA)2 (5 mol%), n-octane (4 mL), 150 °C, sealed tube, in air. | |
Proposed reaction mechanism
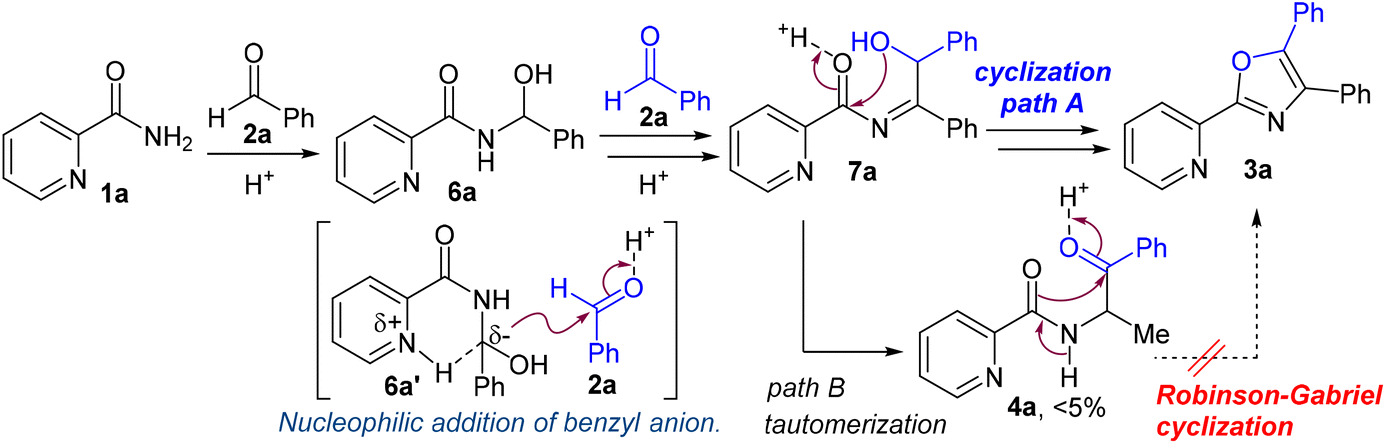
On the basis of several control experiments and previous reports, a plausible mechanism for the 4,5-disubstituted 2-(pyridin-2-yl)oxazole synthesis between picolinamide (1a) and benzaldehyde (2a) was proposed, as illustrated in Scheme 5. First, the nucleophilic amide nitrogen of 1a attacks the electrophilic carbon of aldehyde 2a to generate the aminal 6a.
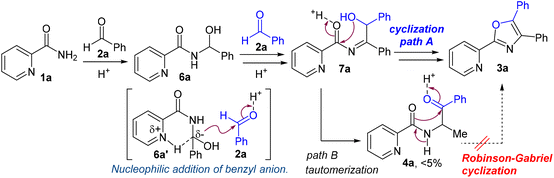 |
| | Scheme 5 Proposed mechanism for triaryloxazole synthesis. | |
The benzylic proton of 6a is removed by the neighboring pyridine base. The resulting benzyl anion 6a′ is stabilized by the adjacent substituents (carboxamide, hydroxy and phenyl groups) and the hydrogen bond in the pyridine ring. Subsequently, the nucleophilic addition of nucleophile 6a′ to a second aldehyde followed by dehydration proceeds to form intermediate 7a. Finally, the cyclocondensation of 7a affords the desired oxazole 3a (path A). Following path B, the tautomerization of intermediate 7a generates the Robinson–Gabriel intermediate 4a as a minor product (<5% yield), which cannot be converted to the desired oxazole 3a in our catalytic system.
Direct use of benzyl alcohol 8a for the construction of oxazole 3a
Pd-catalyzed oxidative coupling of picolinamide 1a with the in situ generated benzaldehyde 2a from benzyl alcohol 8a enables the atom-economical synthesis of triaryloxazole 3a along with H2 and H2O as the co-products. Encouraged by the finding of this oxazole synthesis (Scheme 2B), we examined the optimization of the Pd-catalyzed oxidative coupling of amide 1a with alcohol 8a. To our delight, decreasing the amount of TPPMS to 1 mol% improved the yield of 3a to 60% (Scheme 6). In contrast, a lower yield was obtained in the absence of TPPMS (7%). The mechanism for Pd-catalyzed dehydrogenation of alcohol 8a to aldehyde 2a is proposed as follows: (1) reduction of Pd(TFA)2Ln (L = TPPMS) with alcohol 8a leads to an active Pd(0)Ln species along with TFA; (2) alcohol 8a undergoes oxidative addition to Pd(0)Ln (the C–O bond of 8a is activated by the in situ generated TFA catalyst), forming π-benzylPd(II) A; and (3) β-hydride elimination of Pd(II)–alkoxide B generates aldehyde 2a.
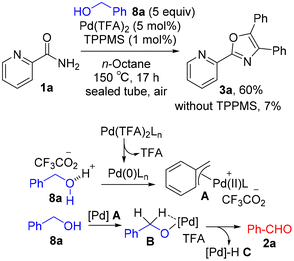 |
| | Scheme 6 Pd-catalyzed dehydrogenative coupling of 1a with 8a. | |
Conclusions
In summary, we developed a sequential reaction between picolinamide and aldehydes, which affords a series of 4,5-disubstituted 2-(pyridin-2-yl)oxazoles in n-octane. This one-pot protocol features, namely, practical simplicity, broad substrate scope, and easily available starting materials. Based on an 18O labeling study, it was shown that the reaction mechanism differs from the Robinson–Gabriel synthetic pathway that relies on a stoichiometric amount of Brønsted acid under harsh reaction conditions. Therefore, this mild protocol is compatible with the substrate scope of aldehydes containing acid-sensitive functional groups. Further investigations on the detailed mechanism and extension towards other motifs are underway in our laboratory.
Experimental section
General comments
All starting materials and solvents were purchased from Aldrich, Wako, nacalai, and TCI Co., Ltd, Tokyo, Japan. All commercially available reagents and solvents were used without further purification. FT-IR spectra were recorded on a JASCO FT/IR-4100 spectrometer using KBr tablets. 1H-NMR spectra were recorded on a JEOL JNM-ECS400 (400 MHz) spectrometer. Chemical shifts (δ) are given from TMS (0 ppm) in CDCl3 and coupling constants are expressed in hertz (Hz). The following abbreviations are used: s = singlet, d = doublet, t = triplet, q = quartet, dd = double doublet, ddd = double double doublet, dt = double triplet, td = triple doublet and m = multiplet. 13C-NMR spectra were recorded on a JEOL ECS400 (100 MHz) spectrometer. Chemical shifts (δ) are given from 13CDCl3 (77.0 ppm). Mass spectra and high-resolution mass spectra were measured on a JEOL JMS700 MStation.
Synthesis and spectroscopic and analytical data of 3a–3r
General procedure.
A mixture of 2-picolinamide (1) (1 mmol), palladium(II) trifluoroacetate (16 mg, 0.05 mmol) and benzaldehyde 2 (5 mmol) in n-octane (4 mL) was heated for 17 h in a sealed tube in air. After cooling, CHCl3 was added to the reaction mixture and concentrated in vacuo. The residue was purified by flash column chromatography (silica gel, hexane/EtOAc) to give the desired product 3.
4,5-Diphenyl-2-(pyridin-2-yl)oxazole (3a)16.
Yield: 230 mg (0.77 mmol), 77%; light yellow solid; mp: 112.0–113.5 °C; 1H NMR (400 MHz, CDCl3): δ 7.34–7.42 (m, 7H), 7.71–7.77 (m, 4H), 7.84 (td, J = 7.5, 1.8 Hz, 1H), 8.24 (ddd, J = 8.0, 1.1, 0.9 Hz, 1H), 8.78 (ddd, J = 4.8, 1.8, 0.9 Hz, 1H); 13C NMR (100 MHz, CDCl3): δ 122.3, 124.7, 127.2, 128.2, 128.4, 128.7, 128.7, 128.8, 129.0, 132.3, 137.0, 137.2, 146.2, 147.0, 150.2, 159.1; FT–IR (KBr, cm−1): 3051, 1586, 1551; MS (FAB): m/z 299 [M + H]+.
4,5-Bis(3-bromophenyl)-2-(pyridin-2-yl)oxazole (3b)16.
Yield: 314 mg (0.69 mmol), 69%; white solid; mp: 156.2–157.2 °C; 1H NMR (400 MHz, CDCl3): δ 7.26 (t, J = 8.0 Hz, 2H), 7.41 (ddd, J = 7.5, 4.8, 1.1 Hz, 1H), 7.50–7.53 (m, 2H), 7.59–7.64 (m, 2H), 7.87 (td, J = 7.7, 1.8 Hz, 1H), 7.91 (t, J = 1.83 Hz, 1H), 7.97 (t, 1.8 Hz, 1H), 8.24 (ddd J = 7.7, 1.1 0.9 Hz, 1H), 8.80 (ddd, J = 4.8, 1.8, 0.9 Hz, 1H); 13C NMR (100 MHz, CDCl3): δ 122.5, 123.0, 123.0, 125.1, 125.7, 126.6, 130.0, 130.2, 130.2, 130.4, 131.2, 131.8, 132.3, 133.8, 136.5, 137.1, 145.8, 145.8, 150.3, 159.6; FT–IR (KBr, cm−1): 3058, 1554; MS (FAB): m/z 457 [M + H]+.
4,5-Bis(4-bromophenyl)-2-(pyridin-2-yl)oxazole (3c)16.
Yield: 333 mg (0.73 mmol), 73%; white solid; mp: 154.0–155.1 °C. 1H NMR (400 MHz, CDCl3): δ 7.43 (ddd, J = 7.5, 4.8, 1.1 Hz, 1H), 7.65–7.55 (m, 8H), 7.88 (td, J = 7.8, 1.8 Hz, 1H), 8.24 (dt, J = 8.0, 1.1 Hz, 1H), 8.81 (ddd, J = 4.8, 1.6, 0.9 Hz, 1H); 13C NMR (100 MHz, CDCl3): δ 122.3, 122.7, 123.3, 124.8, 127.2, 128.5, 129.6, 130.8, 131.9, 132.1, 136.5, 137.0, 145.8, 146.0, 150.2, 159.3; FT–IR (KBr, cm−1): 3060, 1587 MS (FAB): m/z 457 [M + H]+.
4,5-Bis(4-chlorophenyl)-2-(pyridin-2-yl)oxazole (3d)16.
Yield: 221 mg (0.6 mmol), 60%; white solid; mp: 151.3–152.4 °C; 1H NMR (400 MHz, CDCl3): δ 7.36–7.42 (m, 5H), 7.62–7.69 (m, 4H), 7.85 (td, 7.7, 1.8 Hz, 1H), 8.22 (ddd, J = 8.0, 1.1, 0.9 Hz, 1H), 8.79 (ddd, J = 4.8, 1.6, 0.9 Hz, 1H); 13C NMR (100 MHz, CDCl3): δ 122.4, 125.0, 126.9, 128.4, 129.0, 129.2, 129.5, 130.5, 134.5, 135.2, 136.5, 137.1, 145.9, 146.1, 150.3, 159.3; FT–IR (KBr, cm−1): 3067, 3049, 1588; MS (FAB): m/z 367 [M + H]+.
4,5-Bis(4-fluorophenyl)-2-(pyridin-2-yl)oxazole (3e)16.
Yield: 214 mg (0.64 mmol), 64%; white solid; mp: 172.0–172.8 °C; 1H NMR (400 MHz, CDCl3): δ 7.13–7.07 (m, 4H), 7.39 (ddd, J = 7.5, 4.8, 1.1 Hz, 1H), 7.65–7.73 (m, 4H), 7.85 (td, J = 7.5, 1.8 Hz, 1H), 8.22 (dt, J = 8.0, 1.1 Hz, 1H), 8.78 (ddd, J = 4.8, 1.6, 0.9 Hz, 1H); 13C NMR (100 MHz, CDCl3): δ 115.7 (d, JF = 22.0 Hz), 116.0 (d, JF = 22.0 Hz), 122.2, 124.6, 124.7, 128.0, 129.1 (d, JF = 8.6 Hz), 129.9 (d, JF = 8.6 Hz), 136.0, 136.9, 145.8, 145.9, 150.2, 159.0, 162.7 (d, JF = 248.2 Hz), 163.0 (d, JF = 250.2 Hz); FT–IR (KBr, cm−1): 3055, 1587, 1552; MS (FAB): m/z 335 [M + H]+.
4,5-Bis(2-bromophenyl)-2-(pyridin-2-yl)oxazole (3f)16.
Yield: 159 mg (0.35 mmol), 35%; white solid; mp: 139.5–140.2 °C; 1H NMR (400 MHz, CDCl3): δ 7.19–7.34 (m, 4H), 7.38–7.41 (m, 2H), 7.49 (dd, J = 7.6, 1.6 Hz, 1H), 7.60 (dd, J = 8.0, 1.1 Hz, 1H), 7.64 (dd, J = 7.7, 0.9 Hz, 1H), 7.85 (dt, J = 7.6, 1.8 Hz, 1H), 8.26 (ddd, J = 7.7, 1.1, 0.9 Hz, 1H), 8.78 (ddd, J = 4.8, 1.6, 0.9 Hz, 1H); 13C NMR (100 MHz, CDCl3): δ 122.4, 123.4, 123.5, 124.9, 127.3, 127.4, 129.9, 130.2, 131.0, 132.2, 132.2, 133.1, 133.3, 133.6, 137.1, 138.5, 146.1, 147.3, 150.3, 159.6; FT–IR (KBr, cm−1): 3062, 1586, 1560; MS (FAB): m/z 457 [M + H]+.
4,4′-(2-(Pyridin-2-yl)oxazole-4,5-diyl)dibenzonitrile (3g).
Yield: 210 mg (0.6 mmol), 60%; white solid; mp: 256–257 °C; 1H NMR (400 MHz, CDCl3): δ 7.46 (ddd, J = 7.5, 4.8, 1.1 Hz, 1H), 7.75–7.70 (m, 4H), 7.81–7.92 (m, 5H), 8.25 (ddd, J = 8.0, 1.1, 0.9 Hz, 1H), 8.81 (ddd, J = 4.8, 1.8, 0.9 Hz, 1H); 13C NMR (100 MHz, CDCl3): δ 122.7, 113.0, 118.3, 118.5, 122.8, 125.5, 127.5, 128.8, 132.2, 132.7, 132.8, 136.1, 137.3, 137.6, 145.3, 146.1, 150.5, 160.4; FT–IR (KBr, cm−1): 3062, 2225, 1735, 1608; MS (FAB): m/z 349 [M + H]+; anal. calcd for C22H12N4O·0.2H2O·0.3CHCl3: C, 69.07; H, 3.30; N, 14.45. Found: C, 69.47; H, 3.67; N, 14.27.
Dimethyl 4,4′-(2-(pyridin-2-yl)oxazole-4,5-diyl)dibenzoate (3h).
Yield: 258 mg (0.62 mmol), 62%; white solid; mp: 198.5–199.0 °C; 1H NMR (400 MHz, CDCl3): δ 3.95 (s, 3H), 3.95 (s, 3H), 7.43 (ddd, J = 7.5, 4.8, 1.1 Hz, 1H), 7.78–7.90 (m, 5H), 8.08 (td, J = 8.2, 1.8 Hz, 4H), 8.26 (dt, J = 8.3, 1.1 Hz, 1H), 8.81 (ddd, J = 4.8, 1.6, 0.9 Hz, 1H); 13C NMR (100 MHz, CDCl3): δ 52.2, 52.3, 122.5, 125.0, 126.8, 128.1, 129.9, 130.0, 130.1, 132.3, 136.2, 137.0, 137.6, 145.6, 146.7, 150.3, 159.8, 166.4, 166.7; FT–IR (KBr, cm−1): 2954, 1716, 1609; HRMS (FAB): m/z [M + H]+ calcd for C24H19 415.1294; found: 415.1294.
4,5-Bis(3-(benzyloxy)phenyl)-2-(pyridin-2-yl)oxazole (3i).
Yield: 383 mg (0.75 mmol), 75%; brown solid; mp: 135.5–135.8 °C; 1H NMR (400 MHz, CDCl3): δ 5.01 (s, 2H), 5.06 (s, 2H), 6.96–7.00 (m, 2H), 7.28–7.44 (m, 17H), 7.85 (td, J = 7.7, 1.8 Hz, 1H), 8.23 (ddd, J = 7.7, 1.1, 0.9 Hz, 1H), 8.78 (ddd, J = 4.8, 1.1, 0.9 Hz, 1H); 13C NMR (100 MHz, CDCl3): δ 70.2, 70.2, 113.3, 114.4, 115.5, 116.0, 120.0, 121.1, 122.4, 124.7, 127.6, 128.1, 128.1, 128.7, 128.7, 129.7, 129.8, 129.9, 133.6, 136.8, 137.0, 137.2, 146.2, 146.9, 150.3, 159.0, 159.0, 159.1; FT–IR (KBr, cm−1): 3032, 2916, 1588, 1496; MS (FAB): m/z 511 [M + H]+; anal. calcd for C34H26N2O3·0.9H2O: C, 77.52; H, 5.32; N, 5.32, found: C, 77.45; H, 5.07; N, 5.21.
4,5-Bis(2-methoxyphenyl)-2-(pyridin-2-yl)oxazole (3j).
Yield: 169 mg (0.47 mmol), 47%; white solid; mp: 142.1–143.5 °C; 1H NMR (400 MHz, CDCl3): δ 3.38 (s, 3H), 3.43 (s, 3H), 6.81–6.86 (m, 2H), 6.96–7.06 (m, 2H), 7.27–7.36 (m, 3H), 7.59 (ddd J = 7.6, 1.8, 1.6 Hz, 1H), 7.71 (dd J = 7.5, 1.8 Hz, 1H), 7.81 (td J = 7.7, 1.8 Hz, 1H), 8.23 (ddd J = 8.0, 1.1, 0.9 Hz, 1H), 8.76 (ddd J = 4.8, 1.8, 0.9 Hz, 1H); 13C NMR (100 MHz, CDCl3): δ 54.8, 55.0, 110.3, 110.5, 119.7, 120.2, 120.3, 122.2, 123.0, 124.3, 129.2, 129.8, 130.1, 130.5, 135.5, 136.8, 146.1, 146.6, 150.1, 156.8, 156.8, 159.2; FT–IR (KBr, cm−1): 3068, 1584, 1502; HRMS (FAB): m/z [M + H]+ calcd for C22H19N2O3 359.1; found: 359.1.
4,5-Bis(3-methoxyphenyl)-2-(pyridin-2-yl)oxazole (3k).
Yield: 250 mg (0.7 mmol), 70%; yellow oil; 1H NMR (400 MHz, CDCl3): δ 3.77 (s, 3H), 3.80 (s, 3H), 6.88–6.91 (m, 1H), 6.92 (ddd J = 2.7, 1.6 Hz, 1H), 7.26–7.34 (m 6H), 7.38 (ddd J = 7.5, 4.8, 1.1 Hz, 1H); 13C NMR (100 MHz, CDCl3): δ 55.4, 112.3, 113.3, 114.8, 115.2, 119.8, 120.8, 122.4, 124.7, 129.6, 129.8, 129.8, 133.5, 137.0, 137.2, 146.2, 146.9, 150.2, 158.9, 159.7, 159.8; FT–IR (KBr, cm−1): 3057, 3001, 2938, 2835, 1589; MS (FAB): m/z 359 [M + H]+: anal. calcd for C22H18N2O3·0.2CHCl3: C, 69.75; H, 4.80; N, 7.33, found: C, 69.77; H, 4.97; N, 7.31.
4,5-Bis(4-methoxyphenyl)-2-(pyridin-2-yl)oxazole (3l)16.
Yield: 215 mg (0.6 mmol), 60%; light yellow solid; mp: 129.5–130.2 °C; 1H NMR (400 MHz, CDCl3): δ 3.85 (s, 6H), 6.90–6.94 (m, 4H), 7.36 (ddd, J = 7.5, 4.8, 1.1 Hz, 1H), 7.63–7.68 (m, 4H), 7.82 (td, J = 7.7, 1.8 Hz, 1H), 8.72 (dt, J = 7.9, 0.9 Hz, 1H), 8.77 (ddd, J = 4.8, 1.8, 0.9 Hz, 1H); 13C NMR (100 MHz, CDCl3): δ 55.2, 55.3, 114.0, 114.1, 121.3, 122.0, 124.3, 124.8, 128.5, 129.3, 135.8, 129.3, 135.8, 136.8, 146.2, 146.3, 150.1, 158.4, 159.5, 160.0; FT–IR (KBr, cm−1): 2960, 1597, 1578; MS (FAB): m/z 359 [M + H]+.
2-(Pyridin-2-yl)-4,5-di-o-tolyloxazole (3m).
Yield: 150 mg (0.46 mmol), 46%; yellow oil; 1H NMR (400 MHz, CDCl3): δ 2.20 (s, 3H), 2.21 (s, 3H), 7.17–7.12 (m, 2H), 7.20–7.33 (m, 6H), 7.37 (ddd, J = 7.5, 4.8, 1.1 Hz, 1H), 7.83 (td, J = 7.7, 1.8 Hz, 1H), 8.22 (ddd, J = 8.0, 1.1, 0.9 Hz, 1H), 8.76 (ddd, J = 4.8, 1.8, 0.9 Hz, 1H); 13C NMR (100 MHz, CDCl3): δ 20.2, 20.5, 122.2, 124.6, 125.9, 128.1, 128.5, 129.4, 130.2, 130.3, 130.8, 131.6, 137.0, 137.2, 138.4, 146.4, 148.2, 150.2, 159.3; FT–IR (KBr, cm−1): 3058, 1589, 1457; MS (FAB): m/z 327 [M + H]+; anal. calcd for C22H18N2O·0.3CHCl3: C, 73.95; H, 5.09; N, 7.73, found: C, 74.17; H, 5.14; N, 7.62.
2-(Pyridin-2-yl)-4,5-di-m-tolyloxazole (3n).
Yield: 246 mg (0.75 mmol), 75%; white solid; mp: 125.8–127.1 °C; 1H NMR (400 MHz, CDCl3): δ 2.36 (s, 1H), 2.38 (s, 1H), 7.16–7.18 (m, 2H), 7.25 (t, J = 7.5 Hz 1H), 7.26 (t, J = 7.5 Hz, 1H), 7.38 (ddd, J = 7.5, 4.8, 1.1 Hz, 1H), 7.48–7.52 (m, 2H), 7.59–7.60 (m, 1H), 7.64–7.65 (m, 1H), 7.84 (td, J = 7.7, 1.8 Hz, 1H), 8.24 (ddd, J = 8.0, 1.1 Hz, 1H), 8.79 (ddd, J = 4.8, 1.6, 0.9 Hz, 1H); 13C NMR (100 MHz, CDCl3): δ 21.5, 21.5, 122.3, 124.3, 124.6, 125.2, 127.7, 128.4, 128.5, 128.6, 128.9, 129.1, 129.8, 132.2, 137.0, 137.2, 138.3, 138.5, 146.3, 147.1, 150.2, 158.9; FT–IR (KBr, cm−1): 2919, 2345, 1589; MS (FAB): m/z 327 [M + H]+; anal. calcd for C32H22N2O: C, 80.96; H, 5.56; N, 8.58, found: C, 80.70; H, 5.56; N, 8.47.
2-(Pyridin-2-yl)-4,5-di-p-tolyloxazole (3o).
Yield: 221 mg (0.68 mmol), 68%; white solid; mp: 118.0–119.2 °C; 1H NMR (400 MHz, CDCl3): δ 2.38 (s, 6H), 7.17–7.20 (m, 4H), 7.35 (ddd, J = 7.6, 4.8, 1.1 Hz, 1H), 7.61 (d, J = 8.2 Hz, 2H), 7.63 (d, J = 8.2 Hz, 2H), 7.82 (td, J = 7.8, 1.1 Hz, 1H), 8.77 (ddd, J = 4.8, 1.6, 0.9 Hz, 1H); 13C NMR (100 MHz, CDCl3): δ 21.3, 21.4, 122.1, 124.4, 125.9, 127.0, 128.0, 129.2, 129.3, 129.4, 136.6, 136.8, 138.0, 138.9, 146.2, 146.8, 150.1, 158.6; FT–IR (KBr, cm−1): 2918, 2860, 1699; HRMS (FAB): m/z [M + H]+ calcd for C22H19N2O: 327.1497; found: 327.1496.
4,5-Di(naphthalen-1-yl)-2-(pyridin-2-yl)oxazole (3p).
Yield: 199 mg (0.5 mmol), 50%; white solid; mp: 174.5–175.0 °C; 1H NMR (400 MHz, CDCl3): δ 7.28–7.50 (m, 9H), 7.79–7.90 (m, 5H), 8.05 (d, J = 8.5 Hz, 1H), 8.26 (d, J = 8.9 Hz, 1H), 8.34 (dt, J = 8.0, 0.9 Hz, 1H), 8.81 (ddd, J = 4.8, 1.8, 0.9 Hz, 1H); 13C NMR (100 MHz, CDCl3): δ 122.3, 124.7, 125.1, 125.2, 125.5, 125.9, 126.1, 126.4, 126.9, 128.2, 128.3, 128.4, 129.0, 130.0, 133.6, 133.9, 137.0, 138.6, 146.3, 148.5, 150.2, 159.8; FT–IR (KBr, cm−1): 3053, 2362, 1698; HRMS (FAB): m/z [M + H]+ calcd for C28H19N2O: 399.1497; found: 399.1498.
4,5-Di(naphthalen-2-yl)-2-(pyridin-2-yl)oxazole (3q).
Yield: 155 mg (0.39 mmol), 39%; white solid; mp: 169.8–170.2 °C; 1H NMR (400 MHz, CDCl3): δ 7.42 (ddd, J = 7.5, 4.8, 1.1 Hz, 1H), 7.47–7.55 (m, 4H), 7.76–7.91 (m, 9H), 8.31–8.37 (m, 3H), 8.83 (ddd, J = 4.8, 1.8, 0.9 Hz, 1H); 13C NMR (100 MHz, CDCl3) (ppm): δ 122.5, 124.5, 126.0, 126.5, 126.8, 127.6, 127.8, 127.9, 128.2, 128.4, 128.5, 133.3, 133.4, 133.6, 137.0, 137.6, 146.2, 147.4, 150.3, 159.4; FT–IR (KBr, cm−1): 3052, 1698, 1587; HRMS (FAB): m/z [M + H]+ calcd for C28H19N2O: 399.1497; found: 399.1497.
4,5-Di([1,1′-biphenyl]-4-yl)-2-(pyridin-2-yl)oxazole (3r).
Yield: 285 mg (0.63 mmol), 63%; white solid; mp: 219.5–220.4 °C; 1H NMR (400 MHz, CDCl3): δ 7.34–7.41 (m, 3H), 7.44–7.48 (m, 4H), 7.63–7.68 (m, 8H), 7.84–7.90 (m, 5H), 8.27 (ddd, J = 7.79, 1.14, 0.92 Hz, 1H), 8.80 (ddd, J = 4.8, 1.8, 0.9 Hz, 1H); 13C NMR (100 MHz, CDCl3): δ 122.4, 124.7, 127.1, 127.1, 127.4, 127.4, 127.5, 127.6, 127.8, 128.7, 128.9, 129.0, 131.3, 137.0, 137.1, 140.3, 140.7, 141.2, 141.7, 146.2, 146.9, 150.3, 159.2; FT–IR (KBr, cm−1): 3647, 3035, 2355, 1588, 1483; MS (FAB): m/z 451 [M + H]+; anal. calcd for C32H22N2O: C, 85.31; H, 4.92; N, 6.22, found: C, 85.47; H, 5.00; N, 6.27.
2-(Pyridin-2-yl)-4,5-di(thiophen-2-yl)oxazole (3s).
Yield: 143 mg (0.46 mmol), 46%; brown solid; mp: 128.6–129.5 °C; 1H NMR (400 MHz, CDCl3): δ 7.10 (dd, J = 5.2, 3.6 Hz, 1H), 7.13 (dd, J = 5.0, 3.6 Hz, 1H), 7.37–7.41 (m, 2H), 7.45 (dd, J = 5.2, 1.1 Hz, 1H), 7.58 (dd, J = 3.6, 1.1 Hz, 1H), 7.61 (dd, J = 3.6, 1.1 Hz, 1H), 7.84 (td, J = 7.7, 1.6 Hz, 1H), 8.22 (ddd, J = 8.0, 1.1, 0.9 Hz, 1H), 8.78 (ddd, J = 4.8, 1.6, 0.9 Hz, 1H); 13C NMR (100 MHz, CDCl3): δ 122.6, 124.9, 126.5, 126.6, 127.5, 127.5, 127.7, 127.9, 129.1, 132.1, 133.6, 137.0, 141.6, 145.7, 150.3, 158.7; FT–IR (KBr, cm−1): 3078, 1588, 1455; MS (FAB): m/z 311 [M + H]+; anal. calcd for C16H10N2O1S2·0.15H2O: C, 61.38; H, 3.32; N, 8.95, found: C, 61.45; H, 3.37; N, 8.73.
4,5-Diphenethyl-2-(pyridin-2-yl)oxazole (3t).
Yield: 57 mg (0.16 mmol), 16%; brown solid; mp: 98.8–99.5 °C; 1H NMR (400 MHz, CDCl3): δ 2.61–2.65 (m, 2H), 2.77 (s, 4H), 2.80–2.84 (m, 2H), 7.07–7.12 (m, 4H), 7.15–7.21 (m, 2H), 7.23–7.28 (m, 4H), 7.34 (ddd, J = 7.5, 4.8, 1.1 Hz, 1H), 7.82 (td, J = 7.9, 1.8 Hz, 1H), 8.09 (ddd, J = 8.0, 1.1, 0.9 Hz, 1H), 8.74 (ddd, J = 4.8, 1.6, 0.9 Hz, 1H); 13C NMR (100 MHz, CDCl3): δ 27.0, 28.1, 34.6, 35.2, 121.7, 124.2, 126.1 126.3, 128.4, 128.5, 128.6, 128.7, 136.6, 137.0, 140.8, 141.7, 146.5, 148.4, 150.1, 158.6; FT–IR (KBr, cm−1): 3029, 2920, 1634, 1591, 1456; MS (FAB): m/z 355 [M + H]+; anal. calcd for C24H22N2O: C, 81.33; H, 6.26; N, 7.90, found: C, 81.10; H, 6.30; N, 7.70.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We thank Dr Katsuyoshi Mitsunaga (Faculty of Pharmaceutical Sciences, Toho University) for recording the high-resolution mass spectra. This work was supported by JSPS KAKENHI, Grant Number 19K07003. The authors thank FORTE Science Communications (https://www.forte-science.co.jp/) for English language editing.
References
- B. Cryer and M. Feldman, Am. J. Med., 1998, 104, 413–421 CrossRef CAS.
- A. Dodabalapur, Solid State Commun., 1997, 102, 259–267 CrossRef CAS.
- C. K. Nicolaou, M. Bella, K. D. Y. Chen, X. Huang, T. Ling and A. S. Snyder, Angew. Chem., 2002, 114, 3645–3649 CrossRef.
- R. Robinson, J. Chem. Soc., 1909, 95, 2167–2174 RSC.
- S. Gabriel, Berichte, 1910, 43, 134–138 CrossRef CAS.
- S. Gabriel, Berichte, 1910, 43, 1283–1287 CrossRef CAS.
- M. Pulici, F. Quartieri and E. R. Felder, J. Comb. Chem., 2005, 7, 463–473 CrossRef CAS.
-
(a) H. Bredereck and R. Bangert, Angew. Chem., Int. Ed. Engl., 1962, 1, 662–663 CrossRef. For details on the modified Bredereck method, see:
(b) H. Käsnänen, M. J. Myllymäki, A. Minkkilä, A. O. Kataja, S. M. Saario, T. Nevalainen, A. M. P. Koskinen and A. Poso, ChemMedChem, 2010, 5, 213–231 CrossRef.
- A. Aguado, J. Boulos, A. Carreras, A. Montoya and J. Rodriquez, J. Heterocycl. Chem., 2007, 44, 1517–1520 CrossRef CAS.
- E. Fischer, Berichte, 1896, 29, 205 CrossRef CAS.
- H. Kunz, Angew. Chem., Int. Ed., 2002, 41, 4439–4451 CrossRef CAS.
- A. Radspieler and J. Liebscher, Synthesis, 2001, 745–750 CrossRef CAS.
- A. M. Van Leusen, B. E. Hoogenboom and H. Siderius, Tetrahedron Lett., 1972, 13, 2369–2372 CrossRef.
- A. M. Van Leusen, H. Siderius, B. E. Hoogenboom and D. van Leusen, Tetrahedron Lett., 1972, 13, 5337–5340 CrossRef.
- A. Alizadeh, R. Ghanbaripour and L. G. Zhu, Synlett, 2013,(16), 2124–2126 CrossRef CAS.
- H. Meng, Y. Zi, X.-P. Xu and S.-J. Ji, Tetrahedron, 2015, 71, 3819–3826 CrossRef CAS.
-
(a) T. Nakayama, H. Hikawa, S. Kikkawa and I. Azumaya, RSC Adv., 2021, 11, 23144–23150 RSC;
(b) W. Yao, Y. Zhang, H. Zhu, C. Ge and D. Wang, Chin. Chem. Lett., 2020, 31, 701–705 CrossRef CAS.
- D. D. S. Sharley and W. M. J. Jonathan, Tetrahedron Lett., 2017, 58(43), 4090–4093 CrossRef.
- H. H. Wasserman and F. J. Vinick, J. Org. Chem., 1973, 38, 2407–2408 CrossRef CAS.
- B. Banerji, S. Adhikary, L. Majumder and S. Ghosh, Asian J. Org. Chem., 2019, 8, 514–525 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ,
Hidemasa
Hikawa
,
Hidemasa
Hikawa